

Abstract
In addition to mediating sexual maturation and reproduction through stimulation of classical intracellular receptors that bind DNA and regulate gene expression, estradiol is also thought to influence various brain functions by acting on receptors localized to the neuronal membrane surface. Many intracellular signaling pathways and modulatory proteins are affected by estradiol via this unconventional route, including regulation of the transcription factor cAMP response element-binding protein (CREB). However, the mechanisms by which estradiol acts at the membrane surface are poorly understood. Because both estradiol and CREB have been implicated in regulating learning and memory, we characterized the effects of estradiol on this transcription factor in cultured rat hippocampal neurons. Within minutes of administration, estradiol triggered mitogen-activated protein kinase (MAPK)-dependent CREB phosphorylation in unstimulated neurons. Furthermore, after brief depolarization, estradiol attenuated L-type calcium channel-mediated CREB phosphorylation. Thus, estradiol exhibited both positive and negative influences on CREB activity. These effects of estradiol were sex specific and traced to membrane-localized estrogen receptors that stimulated group I and II metabotropic glutamate receptor (mGluR) signaling. Activation of estrogen receptor α (ERα) led to mGluR1a signaling, triggering CREB phosphorylation through phospholipase C regulation of MAPK. In addition, estradiol stimulation of ERα or ERβ triggered mGluR2/3 signaling, decreasing L-type calcium channel-mediated CREB phosphorylation. These results not only characterize estradiol regulation of CREB but also provide two putative signaling mechanisms that may account for many of the unexplained observations regarding the influence of estradiol on nervous system function.
Keywords: estrogen, hippocampus, CREB, mGluR, MAPK, IP3, PLC
Introduction
Estradiol was characterized initially as binding to a single intracellular receptor [estrogen receptor α (ERα)], regulating gene expression through interactions with estrogen response elements (EREs) located within gene promoter regions (Couse and Korach, 1999; Klinge, 2001). With ERα highly expressed in brain regions that control female sexual behavior (Pfaff and Keiner, 1973), it was believed this was the sole mechanism of estrogen action. However, we now know the actions of estradiol in the brain are multifaceted (Pfaff et al., 1994). For instance, a second estrogen receptor (ERβ) has been identified (Kuiper et al., 1996; Tremblay et al., 1997), and ERα and ERβ also influence the expression of genes independent of EREs, through opposing influences on the activator protein-1 (AP-1) class of transcription factor (Gaub et al., 1990; Philips et al., 1993; Umayahara et al., 1994; Paech et al., 1997; Webb et al., 1999).
Further revision of the classical model of estrogen action (i.e., direct influences on gene expression) resulted from observations that estradiol mediates a variety of responses that are initiated at the neuronal membrane surface and occur within seconds to minutes of steroid exposure (McEwen and Alves, 1999). Through unidentified mechanisms, estradiol regulates multiple signaling pathways, including those involving cAMP/protein kinase A (PKA), mitogen-activated protein kinase (MAPK), and phospholipase C (PLC) (Nabekura et al., 1986; Minami et al., 1990; Mobbs et al., 1991; Aronica et al., 1994; Gu et al., 1996; Zhou et al., 1996; Watters et al., 1997; Singh et al., 1999; Toran-Allerand et al., 1999; Qiu et al., 2003; Chaban et al., 2004). These intracellular signaling cascades modulate receptors, channels, other signaling enzymes, and various transcription factors (Weng et al., 1999; Deisseroth et al., 2003; Waltereit and Weller, 2003; Thomas and Huganir, 2004). Thus, although often termed “nongenomic,” these rapid actions of estradiol can also exert long-term effects on neuronal function via changes in gene expression. Not surprisingly, novel actions of estradiol affect synaptic strength and neuronal structure (Gould et al., 1990; Weiland, 1992; Wong and Moss, 1992; Woolley and McEwen, 1992, 1993; Brinton, 1993; Woolley et al., 1997; Foy et al., 1999) and modulate processes including learning and memory, motor control, pain sensitivity, and cognition (McEwen and Alves, 1999).
Interestingly, each of these behaviors is also regulated by the transcription factor cAMP response element-binding protein (CREB) (Lonze and Ginty, 2002). Thus, modulation of CRE-dependent gene expression by estradiol is a candidate for explaining the influence of estrogens on a variety of behaviors. Recent work demonstrating that estradiol triggers CREB phosphorylation (Gu et al., 1996; Zhou et al., 1996; Wade and Dorsa, 2003; Lee et al., 2004) and that dominant-negative CREB inhibits estradiol-induced hippocampal spine formation (Murphy and Segal, 1997) supports this hypothesis. Although a link between estradiol and CREB has been established, the mechanism by which estradiol influences CREB nevertheless remains unclear. Thus, the aims of this study were to characterize the regulation of CREB by estradiol in hippocampal neurons and to identify the underlying cellular mechanisms by which this process occurs. We found estradiol can both increase and decrease CREB activation, with directional changes dependent on the absence or presence of additional neuronal stimuli. Furthermore, estradiol influenced CREB through activation of two distinct signaling pathways, triggered by stimulation of group I and II metabotropic glutamate receptors (mGluRs). Given that mGluRs regulate a variety of intracellular signaling pathways, our data not only describe the means by which estradiol influences CREB function but may also explain the underlying mechanisms by which estradiol rapidly influences a variety of other cellular behaviors.
Materials and Methods
Cell culture. CA3-CA1 Hippocampal pyramidal neurons were cultured from male and female 1- to 2-d-old rat pups as described previously (Mermelstein et al., 2000), using a protocol approved by the Animal Care and Use Committee at the University of Minnesota. Chemicals were obtained from Sigma (St. Louis, MO) unless stated otherwise. After decapitation, the CA3-CA1 region of the hippocampus was isolated (after removal of the dentate gyrus) in ice-cold modified HBSS containing 20% fetal bovine serum (FBS; HyClone, Logan, UT), 4.2 mm NaHCO3, and 1 mm HEPES, pH 7.35, at 300 mOsm. The tissue was washed, followed by a 5 min digestion in a trypsin solution (type XI; 10 mg/ml) containing 137 mm NaCl, 5 mm KCl, 7 mm Na2HPO4, 25 mm HEPES, and 1500 U of DNase, pH 7.2, at 300 mOsm. After an additional wash, the tissue was dissociated, and the cell suspension was pelleted twice to remove contaminants, plated (∼6.4 × 104 viable cells/well) on 10 mm coverslips (treated previously with Matrigel to promote adherence; BD Biosciences, San Jose, CA), and incubated for 15 min at room temperature. Two milliliters of Minimum Essential Media (MEM; Invitrogen, Grand Island, NY) containing 28 mm glucose, 2.4 mm NaHCO3, 0.0013 mm transferrin (Calbiochem, La Jolla, CA), 2 mm glutamine, 0.0042 mm insulin, 1% B-27 supplement (Invitrogen), and 10% FBS, pH 7.35, at 300 mOsm, were added to each coverslip. Twenty-four hours later, 1 ml of media was replaced with a similar solution containing 4 μm cytosine 1-β-d-arabinofuranoside and 5% FBS. Seventy-two hours later, 1 ml of media was replaced with modified MEM solution containing 5% FBS. Media solutions contained 2 μg/ml gentamicin (Invitrogen) to eliminate bacterial growth. For low-density cultures, ∼4 × 103 viable cells/well were plated on 10 mm coverslips. Cultures were treated similarly as outlined above, except that on day 6 they were also fed a supplement of glial-conditioned media (Banker and Goslin, 1998).
Drugs. The drugs used in these studies were as follows: tetrodotoxin (TTX; 1 μm), d(-)-2-amino-5-phosphonopentanoic acid (AP-5; 25 μm; Tocris, Ellisville, MO), nifedipine (5 μm), 17β-estradiol (17βE; 1 nm unless stated otherwise), 4-hydroxy-tamoxifen (1 nm), 7a, 17b-[9[(4,4,5,5,5-pentafluoropentyl)sulfinyl]nonyl]estra-1,3,5(10)-triene-3,17-diol (ICI 182,780) (100 nm; Tocris), 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene (U0126) (10 μm; Calbiochem), 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98059) (25 μm; Promega, Madison, WI), 1-[6-[[(17b)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione (U73122) (1 μm), bisindolylmaleimide I (500 nm; Calbiochem), xestospongin C (XC; 1 μm; Sigma), (S)-(+)-a-amino-4-carboxy-2-methylbenzeneacetic acid (LY367385; 100 μm; Tocris), 7-(hydroxyimino) cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPPCCOEt; 50 μm; Tocris), (RS)-3,5-dihydroxyphenylglycine (DHPG; 50 μm; Tocris), (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid (LY341495; 100 nm; Tocris), (RS)-a-cyclopropyl-4-phosphonophenylglycine (CPPG; 100 nm; Tocris), (2R,4R)-4-aminopyrrolidine-2,4-dicarboxylate (APDC; 10 μm; Tocris), 4,4′,4″-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol (PPT; 1 nm; Tocris), 2,3-bis(4-hydroxyphenyl)-propionitrile (DPN; 10 nm; Tocris), 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP; 5 μm; Tocris), pilocarpine (1 μm; Tocris), pertussis toxin (PTX; 500 ng/ml), 8-bromoadenosine-3′,5′-monophosphorothioate, Rp isomer (Rp-8-Br-cAMPs; Rp-cAMPs; 50 μm; Calbiochem), cyclosporin A (CsA; 1 μg/ml), FK506 (200 ng/ml; Calbiochem), and 17β-estradiol, 17-hemisuccinate-BSA (EBSA; 1 nm, Sigma or Steraloids, Newport, RI). As described previously, the EBSA was passed through a 3 kDa cutoff filter (Millipore, Billerica, MA) to remove free estrogens, and the retentate was resuspended immediately before use (Chaban et al., 2004).
Immunocytochemistry. Immunocytochemistry procedures were similar to those described previously (Mermelstein et al., 2001). Cultured neurons [9 d in vitro (d.i.v.)] were preincubated for 3 h in a Tyrode's solution containing 1 μm TTX and 25 μm AP-5, with cells being washed halfway through the preincubation period. Afterward, cells were stimulated for 5 min with estradiol or mock treated. For experiments using 20 mm K+, the 3 min depolarization occurred 5 min into the estradiol treatment period, with steroid also present in the high-K+ solution. Experiments using the mGluR agonists followed a similar time course. When applicable, inhibitors/antagonists used in this study were applied 30 min before stimulation. Immediately after treatments, cells were fixed for 20 min with ice-cold 4% paraformaldehyde (Electron Microscopy Sciences, Ft. Washington, PA) in PBS containing 4 mm EGTA. After being washed three times in PBS, cells were permeabilized via a 5 min incubation in a 0.1% Triton X-100 (VWR Scientific, West Chester, PA) solution. After an additional three washes, cells were incubated at 37°C for 30 min in a PBS-based block solution containing 1% BSA and 2% goat serum (Jackson ImmunoResearch, West Grove, PA). Coverslips were then incubated at 4°C overnight in a block solution containing a monoclonal antibody directed against serine 133 phosphorylated CREB (pCREB; 1:1000; Upstate Biotechnology, Lake Placid, NY), and, to identify individual cell morphology, a polyclonal antibody targeting microtubule-associated protein 2 (MAP2, 1:1000; Calbiochem). The next morning, cells were washed three times with PBS and incubated for 1 h at 37°C in a block solution containing FITC- and rhodamine RedX-conjugated secondary antibodies for visualization of MAP2 and pCREB, respectively (Jackson ImmunoResearch). After another wash, coverslips were mounted using the antiquenching agent Citifluor (Ted Pella, Redding, CA). Staining for ERα (1:500; Upstate Biotechnology) and ERβ (1:500; Affinity Bioreagents, South San Francisco, CA) was performed similarly, except using α-tubulin (1:2000; Molecular Probes, Eugene, OR) as the cytoskeletal stain, and cells were not stimulated. Acquisition of the nuclear fluorescent intensities for pCREB (n =∼20 cells/group) was performed using a Yokogawa (Tokyo, Japan) spinning-disc confocal system mounted to an Olympus (Tokyo, Japan) IX-70 inverted microscope and attached to a 12-bit digital camera (Hamamatsu Photonics, Hamamatsu City, Japan). Data were quantified using MetaMorph software (version 6.0; Universal Imaging, Downington, PA). Briefly, for each experimental run, there were two coverslips for each treatment group to account for potential inter-coverslip variability. After the confocal settings were first determined using neurons stimulated with 20 mm K+ as a guide, they remained constant throughout the experiment. Of note, although not always presented, each experiment contained 20 mm K+-treated cells to serve in this capacity. The only exception was for the immunocytochemistry experiments using PTX, in which the PTX plus 20 mm K+ group was used as a guide because PTX induced CREB phosphorylation. The coverslip order in which data were acquired was random. For data acquisition, cells were selected randomly (∼10 per coverslide) via MAP2 fluorescence, and a section through the approximate midline of each neuron was captured; the experimenter was blind to pCREB fluorescence. After all of the data were acquired, the experimenter drew a region of interest (ROI) outlining the nucleus of each pyramidal neuron using MAP2 staining. The software program then transferred that ROI to the pCREB image and provided the average fluorescence within each neuron. The data were background subtracted. Each experiment was repeated twice to verify results. Similar procedures were run for estrogen receptor staining, except a nuclear and cytoplasmic ROI was generated using α-tubulin staining as a guide. Because the confocal settings were set at the beginning of each experiment, comparing fluorescent intensities between trials is not appropriate.
Western blot. Cultured neurons (9 d.i.v.) were incubated for 3 h in a Tyrode's solution containing TTX (1 μm) and AP-5 (25 μm), with cells being washed halfway through the preincubation period. Afterward, cells were stimulated for 5 min with estradiol or mock treated. After stimulation, cells were lysed in a radioimmunoprecipitation assay buffer containing 1 mm NaF, 1 mm Na-orthovanadate, and a protease inhibitor mixture (Complete Mini EDTA-free; Roche, Indianapolis, IN). LDS loading buffer and β-mercaptoethanol were added to the lysate (25 and 10% of the total lysate volume, respectively) before the mixture was heated for 5 min to 95°C. Cellular protein was separated on a 4-12% Bis-Tris gel (Invitrogen) and transferred to a nitrocellulose membrane. The membrane was blocked at room temperature for 1 h in a Trisbuffered saline (TBS) solution containing 10% milk and 1% BSA. The membrane was incubated overnight at 4°C with an antibody for either pCREB (1:1000) or the loading control extracellular signal-regulated kinase (ERK; 1:10,000; Upstate Biotechnology), diluted in TBS containing 1% milk, 1% BSA, and 0.1% Tween 20. The next day, the membrane was washed with 0.1% Tween 20-TBS and incubated for 1 h at room temperature with a secondary antibody conjugated to horseradish peroxidase (1:25,000; Pierce, Rockford, IL). After incubation in a chemiluminescent substrate (Super Signal; Pierce), the membrane was exposed to X-Omat XB-1 film (Kodak, Rochester, NY). The experiment was repeated to verify the result.
Luciferase-based gene reporter assays. Cultured neurons were transfected 8 d.i.v. with a luciferase-based reporter (1 μg DNA/coverslip) of CRE-dependent transcription (three repeats of the CRE-binding site were incorporated into a minimal promoter expression vector; a gift from K. Heist, Stanford University, Stanford, CA) using a calcium-phosphate method that results in >95% of the transfected cells being neurons (Deisseroth et al., 1998; Weick et al., 2003). To verify that the effects of estradiol could not be attributed to a nonspecific alteration in gene expression or protein synthesis, sister cultures were transfected with a constitutively active luciferase vector (Groth and Mermelstein, 2003). After transfection, cells were placed in serum-free, DMEM (Invitrogen) containing an insulin-transferrin-selenium-A (ITS) supplement (Invitrogen) and 1% B-27 supplement, pH 7.35, at 300 mOsm. The DMEM/ITS/B-27 also contained 2 μg/ml gentamicin to prevent bacterial growth and 1 μm TTX to minimize CRE-dependent transcription triggered by synaptic activity. Twenty-four hours later, one-half of the neurons (n = 10 coverslips) were readministered TTX; the other half (n = 10) received TTX with the addition of 1 nm estradiol. Approximately 16 h after stimulation, cells were lysed and assayed for luciferase expression using a standard luminometer (Monolight 3010; PharMingen, San Diego, CA). Each experiment was replicated to verify the consistency of the results.
Electrophysiology. Miniature EPSCs (mEPSCs) were recorded from cultured hippocampal neurons (9 d.i.v.) at a holding potential of -70 mV using standard voltage-clamp techniques. Neurons were superfused with a room temperature solution containing the following (in mm): 119 NaCl, 2.5 KCl, 1.3 MgSO4-7 H2O, 11 glucose, 1 NaH2PO4-H2O, 2.5 CaCl2-2 H2O, 26.2 NaHCO3, 0.001 TTX, and 0.1 picrotoxin, pH 7.35, at 300 mOsm. Warner GC120T-10 (Warner Instruments, Hamden, CT) borosilicate glass electrodes (∼3 MΩ) contained the following (in mm): 117 cesium gluconate, 2.8 NaCl, 20 HEPES, 5 BAPTA, 4 Na2-ATP, 0.8 Na3-GTP, 12 phosphocreatine, and 5 tetraethyl-ammonium chloride, pH 7.2, at 285 mOsm. ATP and GTP were obtained from Boehringer Mannheim (Indianapolis, IN); BAPTA was purchased from Calbiochem. Recordings were made using an Axopatch 200B amplifier (Molecular Devices, Foster City CA), controlled by a personal computer running pClamp software (version 8.2). All recordings were filtered at 2 kHz, and the series resistance was not compensated. Experiments with a series resistance >20 MΩ or with a change in input resistance of >20% were discarded. A single recording sweep lasting 500 ms was sampled every 1 s. After acquisition of a stable baseline, cells were stimulated with 1 nm estradiol. Detection criteria included peak amplitudes >5 pA, with fast rise times and slow decay times, and all mEPSCs were confirmed visually. Thirty continuous sweeps were analyzed comparing mEPSC frequency and amplitude before and during estradiol application.
Whole-cell calcium channel currents were measured in cultured hippocampal neurons (9 d.i.v.) as described previously, using 5 mm Ba2+ as the charge carrier (Mermelstein et al., 1996, 2000; Weick et al., 2003). To verify the isolation of calcium channel currents, currents were also monitored after the addition of Cd2+ (200 μm) to the perfusate. Electrodes (Warner Instruments) were fire-polished with an MF-830 microforge (Narishige, Hempstead, NY). The intracellular recording solution contained the following (in mm): 190 N-methyl-d-glucamine, 40 HEPES, 5 BAPTA, 4 MgCl2, 12 phosphocreatine, 3 Na2ATP, and 0.2 Na3GTP, pH 7.2, at 275 mOsm. The bath solution contained the following (in mm): 140 NaCl, 2 KCl, 23 glucose, 15 HEPES, 1 CaCl2, 2 MgCl2, and 0.01 glycine. Drugs (nifedipine, APDC, and DPN) were added to the control external recording solution that contained the following (in mm): 135 NaCl, 20 CsCl, 1 MgCl2, 10 HEPES, 0.0001 TTX, and 5 BaCl2. The perfusion system consisted of a convergent single-barrel design that allowed for extracellular solution changes in <1 s. The junction potential (<2 mV) was not compensated. The series resistance was compensated ≥70%. Voltage sweeps were performed once every 5-7 s.
Statistics. Immunocytochemistry experiments were analyzed using ANOVAs (F values) and Fisher's protected least significant difference post hoc tests. Because of the required replication of controls for each immunocytochemistry experiment run and the large number of groups in several experiments, post hoc values are mainly provided for novel comparisons (i.e., not presented in a previous figure) and in which differences can be attributed to a single variable. Luciferase and mEPSC data were analyzed via a Student's t test. p values <0.05 were considered a priori as significant.
Results
Estradiol induces rapid nuclear CREB phosphorylation via MAPK signaling
Estrogens have been reported to rapidly induce CREB phosphorylation in both neurons and cell lines in as little as 15 min, with recent data indicating that steroid effects are mediated via activation of MAPK (Gu et al., 1996; Zhou et al., 1996; Wade and Dorsa, 2003; Lee et al., 2004). Our initial experiments were designed to test whether this signaling pathway was intact within cultured CA3-CA1 hippocampal neurons obtained from 1- to 2-d-old female rat pups. Indeed, a 5 min application of estradiol (1 nm) significantly increased nuclear CREB phosphorylation (hereafter termed CREB phosphorylation). This effect of estradiol was blocked by either of the MAPK/ERK kinase (MEK) inhibitors, PD98059 (25 μm) (Fig. 1A,B) or U0126 (10 μm) (data not shown). Heightened CREB phosphorylation can be caused by an enhancement of protein kinase activity or a reduction in protein phosphate 1 (PP1) activity. Estradiol has been reported to depress calcineurin (CaN) activity (Sharrow et al., 2002; Foster et al., 2003), which may lead to a reduction in PP1 signaling through disinhibition of inhibitor-1 (Bito et al., 1997; Groth et al., 2003). However, estradiol-induced CREB phosphorylation was unaffected after inhibition of CaN with FK506 (200 ng/ml) and CsA (1 μg/ml), indicating this effect of estradiol is primarily routed through activation of MAPK (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 1.
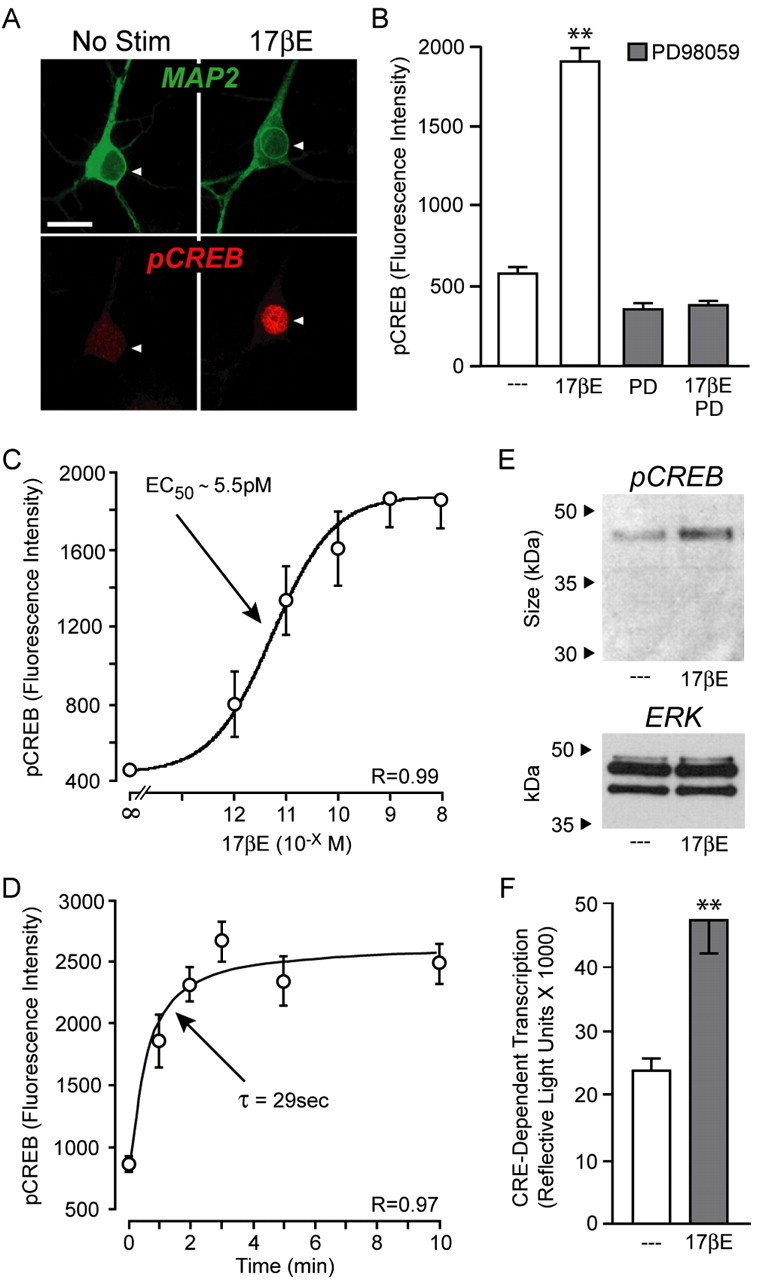
Estradiol rapidly stimulates MAPK-dependent CREB phosphorylation. A, Example confocal images of cultured CA3-CA1 female hippocampal neurons immunolabeled with MAP2 (green) and pCREB (red). Pyramidal neurons exhibited heightened nuclear staining for pCREB after a 5 min application of estradiol (17βE; 1 nm). No Stim, No stimulation. Scale bar, 20 μm. B, Quantification of the immunolabeling demonstrated that estradiol significantly increased CREB phosphorylation (F = 183.79; **p < 0.01). Furthermore, the effect of estradiol was eliminated after inhibition of MEK with PD98059 (PD; 25 μm). C, Estradiol increased CREB phosphorylation in a concentration-dependent manner (F = 14.73; p < 0.05 at 1 pm; p < 0.01 at all other concentrations when compared with control). D, Time course of estradiol-induced CREB phosphorylation. E, Western blot verification of estradiol-induced CREB phosphorylation (∼46 kDa band). ERK was used as the loading control. F, Application of estradiol also increased CRE-dependent transcription (T = 4.42; **p < 0.01). Error bars indicate SEM.
Estradiol elicited CREB phosphorylation in a dose-dependent manner (Fig. 1C) with an EC50 value of ∼5.5 pm (r = 0.99), indicating a physiologically relevant phenomenon. Of note, because 1 nm was maximally effective in eliciting CREB phosphorylation, this concentration was used for the remainder of the studies. Estradiol-induced CREB phosphorylation occurred rapidly; the time course data were well fitted (r = 0.97) with a single exponential (τ = 29 s) (Fig. 1D). Western blot experiments were performed to verify the immunocytochemistry studies indicating estradiol-induced CREB phosphorylation. As shown in Figure 1E, a 5 min exposure to estradiol elicited an increase in CREB phosphorylation. Because phosphorylation of CREB on serine 133 is necessary but not sufficient for activation of CRE-dependent transcription, we applied estradiol to neuronal cultures transfected with a luciferase-based, CRE reporter construct to monitor changes in CRE-dependent transcription. As shown in Figure 1F, estradiol significantly increased CRE-dependent transcription. Estradiol had no effect on a constitutively active luciferase reporter [control, 967,418 ± 72,834 (mean ± SEM) vs estradiol, 858,751 ± 43,238; p > 0.05]. These results suggest that along with its more defined actions on ERE- and AP-1-based transcription, estrogens may also regulate the expression of genes under control of the CREB family of transcription factors.
Estradiol attenuates L-type calcium channel-mediated CREB phosphorylation
Because of its importance in activity-dependent neuronal plasticity, the regulation of CREB has been well characterized. Increased periods of synaptic activity are a principal mechanism underlying CREB activation. Synaptic stimulation results in pronounced CREB phosphorylation via depolarization and subsequent activation of L-type voltage-gated calcium channels (Deisseroth et al., 1996; Mermelstein et al., 2000). Notably, high-K+ stimulations of brief duration will also stimulate L-type calcium channel-mediated CREB phosphorylation (Sheng et al., 1991; Dolmetsch et al., 2001; Mermelstein et al., 2001; Weick et al., 2003) through activation of calmodulin-dependent protein kinase IV (Bito et al., 1996; Ho et al., 2000; Ribar et al., 2000). Thus, we next determined the effect of estradiol on L-type calcium channel-mediated CREB phosphorylation, because estrogens have been demonstrated to rapidly decrease L-type calcium channel currents in other neuronal systems (Mermelstein et al., 1996; Chaban et al., 2003). As established in Figure 1, estradiol alone rapidly increased CREB phosphorylation (Fig. 2A, left two bars). However, in neurons that were stimulated for 3 min with 20 mm K+, estradiol (applied 5 min before and present during 20 mm K+) significantly decreased depolarization-induced CREB phosphorylation (Fig. 2A, middle two bars). Preincubation of cultures with the L-type calcium channel blocker Nif (5 μm) both eliminated depolarization-induced CREB phosphorylation and unmasked estradiol-mediated CREB phosphorylation (Fig. 2A, right two bars). These data indicate that estradiol decreases L-type calcium channel-mediated CREB phosphorylation and that estradiol-triggered CREB phosphorylation occurs independently of L-type calcium channels. Furthermore, estradiol can both positively and negatively regulate CREB signaling (see Discussion).
Figure 2.

Estradiol attenuates L-type calcium channel-mediated CREB phosphorylation. A, Estradiol alone resulted in an increase (F = 16.97; **p < 0.01) in CREB phosphorylation (left 2 bars). However, estradiol treatment in the presence of a 20 mm K+ stimulus led to a significant (**p < 0.01) attenuation of depolarization-induced CREB phosphorylation (middle 2 bars). Pretreatment with the L-type calcium channel blocker nifedipine (NIF; 5 μm) eliminated depolarization-dependent CREB phosphorylation and unmasked estradiol-induced CREB phosphorylation (right 2 bars; **p < 0.01). B, Estradiol produced a rightward shift in the cumulative percentile of CREB fluorescence under basal conditions. C, Conversely, estradiol generated a leftward population shift in CREB phosphorylation after a 20 mm K+ depolarization. Error bars indicate SEM.
Estradiol produced an observable shift in the neuronal population when measuring CREB phosphorylation. To illustrate this point, nuclear phospho-CREB fluorescence within individual neurons was plotted as a cumulative percentile, providing the population distribution for each treatment group (Fig. 2B,C). As shown in Figure 2B, estradiol induced a rightward shift in CREB phosphorylation within the population of hippocampal pyramidal cells under basal conditions. Conversely, a leftward shift in CREB phosphorylation was attributable to estradiol administration when cells were depolarized for 3 min with 20 mm K+ (Fig. 2C).
The effects of estradiol on CREB phosphorylation are sex specific and occur via a membrane receptor
The previous experiments were performed on hippocampal cultures generated solely from female animals, because many of the characterized actions of estradiol in the brain are sex specific (Arnold and Breedlove, 1985; Williams, 1986). Thus, we next investigated whether the bidirectional effects of estradiol observed in female-derived cultures would also occur in those prepared from male rats. As shown in Figure 3, age-matched cultured hippocampal neurons from male animals did not exhibit either the estradiol-induced increase in MAPK-dependent CREB phosphorylation, or the estradiol-mediated decrease in CREB phosphorylation after activation of L-type calcium channels. As another control, cultures from male rats did not exhibit a change in CRE-dependent transcription after estradiol treatment (control, 17,913 ± 1,934 vs estradiol, 15,511 ± 2,835; p > 0.05). Estradiol was used at 1 nm for comparisons between male- and female-derived cultures. We did not examine whether higher concentrations of steroid would be effective on male-derived tissue.
Figure 3.

The bidirectional effects of estradiol on CREB phosphorylation are sex specific. A, B, Estradiol, in the absence of depolarization, increased (F = 26.82; **p < 0.01) CREB phosphorylation and decreased (**p < 0.01) L-type calcium channel-dependent CREB phosphorylation in neurons derived from female but not male animals. Error bars indicate SEM.
We were also interested in determining the mechanism(s) by which estradiol influenced CREB activation. Because the observed responses were rapid (i.e., seconds to 8 min after steroid application), the classical mechanism of estrogen action appeared unlikely. In support of this hypothesis, both effects of estradiol were mimicked by the membrane-impermeable estrogen analog EBSA (1 nm) (Fig. 4A,B), suggesting that the regulation of CREB phosphorylation occurred via activation of an estrogen receptor localized to the membrane surface. In support of this hypothesis, the mixed estrogen receptor agonist/antagonist 4-hydroxy-tamoxifen (1 nm) acted as an agonist in this paradigm, both increasing and decreasing CREB phosphorylation (Fig. 4C,D). Furthermore, both responses were blocked by the pure estrogen receptor antagonist ICI 182,780 (100 nm) (Fig. 4E).
Figure 4.
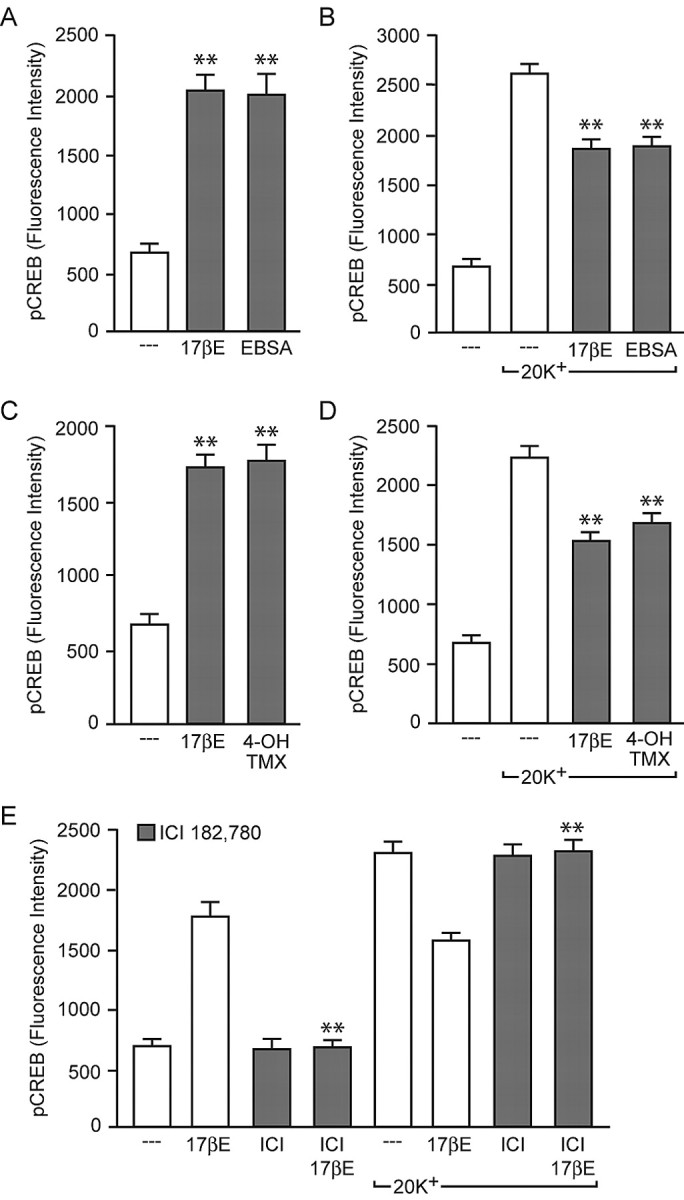
The effects of estradiol occur via action at the plasma membrane, are mimicked by the mixed estrogen receptor agonist/antagonist 4-hydroxy-tamoxifen, and are blocked by the estrogen receptor antagonist ICI 182,780. A, B, The membrane-impermeable estrogen analog EBSA mimicked the bidirectional effects of estradiol on CREB phosphorylation [F = 42.02 and 79.60; **p < 0.01 vs basal (A) and 20 mm K+ (B)]. C, D, The effect of 4-hydroxy-tamoxifen (4-OH-TMX) also paralleled the effect of estradiol [F = 31.15 and 41.27; **p < 0.01 vs basal (C) and 20 mm K+ (D)]. E, ICI 182,780 (ICI; 100 nm) eliminated both the estradiol-induced increase in CREB phosphorylation (F = 81.28; **p < 0.01 vs 17βE) and the estradiol-induced attenuation of L-type calcium channel-mediated CREB phosphorylation (**p < 0.01 vs 20 mm K+ plus 17βE). Error bars indicate SEM.
Estradiol regulates CREB phosphorylation via activation of mGluRs
Unable to distinguish the opposing effects of estradiol using estrogen receptor ligands, we decided on a different strategy. Switching to a “bottom-up” approach, we instead tried to isolate the two signaling pathways by targeting proteins known to influence MAPK. Inhibition of PKC with bisindolylmaleimide (500 nm) or inositol trisphosphate receptors (IP3Rs) with XC (1 μm) resulted in the attenuation of estradiol-induced CREB phosphorylation (Fig. 5A,B). Coapplication of both drugs eliminated the effect (Fig. 5C). PLC signaling is linked to MAPK activation via both PKC and IP3R. This particular pathway appeared important for estradiol-induced CREB phosphorylation, because inhibition of PLC with U73122 (1 μm) resulted in the blockade of estradiol-induced CREB phosphorylation (Fig. 5D). Inhibition of the PLC signaling pathway did not affect the influence of estradiol on L-type calcium channel CREB phosphorylation (data not shown).
Figure 5.

Estradiol-induced CREB phosphorylation is dependent on activation of PLC, PKC, and IP3R signaling. A, The effect of estradiol was reduced (F = 16.76; **p < 0.01) by inhibition of PKC with bisindolylmaleimide (500 nm). B, Similar results (F = 11.37; **p < 0.01) were observed after inhibition of IP3R with XC (1 μm). C, Pretreatment with both bisindolylmaleimide (Bis) and XC eliminated estradiol-induced CREB phosphorylation (F = 25.38; **p < 0.01). D, Consistent with estradiol activation of PLC, pretreatment with the inhibitor U73122 (1 μm) significantly (F = 19.89; **p < 0.01) attenuated estradiol-induced CREB phosphorylation. Error bars indicate SEM.
PLC, PKC, and IP3R can be activated by several mechanisms, including Gq G-protein-coupled receptors (GPCRs) (Gutkind, 2000). Kelly and colleagues (Qiu et al., 2003) recently determined that estradiol can stimulate Gq signaling in neurons; this signaling pathway also appears to be present in astrocytes (Chaban et al., 2004). Based on these data, we hypothesized that estradiol may be acting through a previously defined Gq signaling pathway to induce CREB phosphorylation. Group I mGluRs are linked to Gq G-proteins. As shown in Figure 6A, the mGluR1a antagonist LY367385 (100 μm; see Discussion) blocked estradiol-induced CREB phosphorylation without affecting estradiol attenuation of L-type calcium channel-mediated CREB phosphorylation. mGluR1a specificity was verified with a separate antagonist, CPPCCOEt (50 μm) (data not shown). Along with mGluR1a, mGluR5 comprises the Gq-coupled group I mGluRs. Application of the mGluR5 antagonist MPEP (5 μm) (data not shown) had no effect on estradiol signaling to CREB, demonstrating the actions of estradiol within hippocampal neurons are specific to mGluR1a. Additionally, 5 min administration of the group I mGluR agonist DHPG (50 μm) in the presence of MPEP resulted in CREB phosphorylation, similar to estradiol and occluding the effect of the steroid (Fig. 6B). We also applied DHPG (and MPEP) to hippocampal cultures generated from male animals (Fig. 6C). Whereas estradiol had no effect on CREB in male neurons, DHPG significantly increased CREB phosphorylation. These data suggest that the “breakpoint” in the signaling link between estradiol and CREB in cultured neurons from male rats is localized to actions between estradiol and activation of mGluRs (i.e., upstream of mGluRs). Finally, LY367385 had no effect on CREB phosphorylation mediated by the muscarinic agonist pilocarpine (1 μm), indicating application of LY367385 was not globally disrupting Gq signaling (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 6.

Estradiol-mediated CREB phosphorylation occurs via activation of mGluR1a signaling, independent of glutamate release. A, The mGluR1a antagonist/inverse agonist LY367385 (100 μm) eliminated estradiol-mediated CREB phosphorylation, without affecting estradiol inhibition of depolarization-induced CREB phosphorylation (F = 45.18; **p < 0.01 vs 17βE). B, The mGluR1/5 agonist DHPG (50 μm) in the presence of the mGluR5 antagonist MPEP (5 μm) increased CREB phosphorylation in cultures derived from female animals similar to estradiol and occluded the steroid effect (F = 49.98; **p < 0.01). C, DHPG (in the presence of MPEP) also resulted in an increase in CREB phosphorylation in cultures derived from male animals (F = 67.87; **p < 0.01). D, E, Whole-cell recordings of female hippocampal neurons (n = 9) revealed that estradiol does not evoke glutamate release or alter the frequency of mEPSCs. Calibration: 50 pA, 25 ms. F, Estradiol increased CREB phosphorylation after the removal of extracellular calcium, further supporting the notion that estradiol activation of mGluR1a signaling is independent of presynaptic glutamate release. Error bars indicate SEM.
We theorized that estradiol activation of mGluR signaling could either occur by a postsynaptic effect, leading to activation of mGluR1, or by a presynaptic effect, in which estradiol mediated the release of glutamate. To distinguish between these two hypotheses, we first monitored spontaneous glutamatergic activity via whole-cell recordings of hippocampal neurons (n = 9). We hypothesized that if estradiol was inducing neurotransmitter release, the effect would be detected by postsynaptic AMPA receptors. Nevertheless, a 5 min application of estradiol did not evoke measurable glutamate release nor influence mEPSC frequency (Fig. 6D,E) or amplitude (control, 41.5 ± 4.9 pA vs estradiol, 40.4 ± 4.7 pA). In the next experiment, we monitored the effect of estradiol in the absence of extracellular calcium, which eliminates evoked neurotransmitter release. Although there was a reduction in basal pCREB levels, estradiol significantly increased CREB phosphorylation after removal of extracellular calcium (Fig. 6F). In a third experiment, the effects of estradiol were monitored in low-density cultures, reducing the number of cell-cell contacts and extracellular glutamate. Under these conditions, estradiol still elicited CREB phosphorylation (fluorescence intensity: control, 977.0 ± 69.2 vs estradiol, 2179.7 ± 164.4; p < 0.01). In combination, these data strongly suggest that the effects of estradiol are initiated postsynaptically.
Whereas group I mGluRs are coupled to Gq, group II and III mGluRs are coupled to Gi/o G-proteins. Gi/o signaling typically results in inhibition of adenylyl cyclase (AC) (Neves et al., 2002). Previous work has demonstrated that activation of Gs GPCRs results in an enhancement of L-type calcium channel currents, through activation of AC and PKA (Surmeier et al., 1995). PKA-mediated phosphorylation of the L-type calcium channel α1C subunit at Ser1928 is responsible for increased channel activity (De Jongh et al., 1996; Gao et al., 1997). Thus, activation of Gi/o may result in a decrease in L-type calcium channel phosphorylation and currents, thereby reducing L-type calcium channel-mediated CREB phosphorylation. We hypothesized that similar to its actions on mGluR1a, estradiol can stimulate group II and/or group III mGluRs, ultimately resulting in decreased L-type calcium channel CREB phosphorylation. Consistent with this theory, application of the group II mGluR antagonist LY341495 (100 nm) eliminated the effect of estradiol on L-type calcium channel-mediated CREB phosphorylation (Fig. 7A). Similar results were observed using the group II/III mGluR antagonist CPPG (100 nm) (data not shown). Neither drug blocked estradiol-mediated CREB phosphorylation. Interestingly, inhibition of group II mGluRs with LY341495 exposed estradiol-mediated CREB phosphorylation normally obscured after depolarization.
Figure 7.

Estradiol attenuates L-type calcium channel-mediated CREB phosphorylation via activation of group II mGluR signaling. A, The group II mGluR antagonist LY341495 (100 nm) eliminated the effect of estradiol on L-type calcium channel-mediated CREB phosphorylation (F = 38.19; **p < 0.01 vs 20K+ plus LY341495 and 20K+ plus 17βE) but had no effect on estradiol-induced CREB phosphorylation. B, The group II mGluR agonist APDC (10 μm) significantly attenuated L-type calcium channel-mediated CREB phosphorylation (F = 126.56) similar to estradiol and also occluded the steroid effect (**p < 0.01). C, The effect of APDC was also observed in cultures derived from male animals (F = 57.74; **p < 0.01). Error bars indicate SEM.
Just as the group I mGluR agonist mimicked the effect of estradiol on stimulating CREB phosphorylation, application of the mGluR2/3 agonist APDC (10 μm) resulted in a significant attenuation of L-type calcium channel-mediated CREB phosphorylation and occluded the effect of estradiol (Fig. 7B). APDC had no effect on basal levels of CREB activation. Application of APDC to hippocampal cultures derived from male animals also significantly decreased depolarization-induced CREB phosphorylation, demonstrating that the lack of an effect of estradiol in these cells is not attributable to deficient mGluR2/3 signaling (Fig. 7C).
Estradiol activation of mGluRs is mediated via surface expression of classical estrogen receptors
It was recently reported that estradiol-induced CREB phosphorylation is eliminated in ERα and ERβ knock-out mice (Abraham et al., 2004). Based on the fact that ERα and ERβ have been identified within the plasma membrane (Razandi et al., 1999, 2004; Watson et al., 1999; Wade et al., 2001) and that CREB signaling is influenced by the estrogen receptor ligands 4-hydroxy-tamoxifen and ICI 182,780, we hypothesized that it is, in fact, these receptors that are directly linked to mGluR activation. To test this hypothesis, we used agonists specific to either ERα (PPT; 1 nm) or ERβ (DPN; 10 nm) (Stauffer et al., 2000). As shown in Figure 8A, PPT application led to a significant increase in CREB phosphorylation, whereas DPN was without effect, indicating that in hippocampal neurons, ERα mediates estradiol-induced CREB phosphorylation. Conversely, both PPT and DPN reduced L-type calcium channel-dependent CREB phosphorylation (Fig. 8B), suggesting that both estrogen receptors are capable of signaling to mGluR2/3.
Figure 8.

Estradiol activation of mGluR signaling is mediated by classical estrogen receptors located at the membrane surface. A, The ERα-specific agonist PPT (1 nm) led to an increase (F = 22.51; **p < 0.01) in CREB phosphorylation, similar to the actions of estradiol. The ERβ-specific agonist DPN (10 nm) was without effect. B, Conversely, both PPT and DPN were equally effective to estradiol in the attenuation of L-type calcium channel-dependent CREB phosphorylation (F = 74.17; **p < 0.01). Error bars indicate SEM.
Estradiol and mGluR2/3-mediated diminution of L-type calcium channel currents is routed through Gi/o signaling
As described above, our working hypothesis is that estradiol and mGluR2/3 attenuate L-type calcium channel-mediated CREB phosphorylation in hippocampal neurons via modulation of the calcium channel. Previous work has shown a reduction in L-type calcium channel currents after estradiol administration in both striatal and DRG cells (Mermelstein et al., 1996; Chaban et al., 2003). Furthermore, mixed mGluR agonists reduce L-type calcium channel currents in hippocampal, neocortical, and cerebellar neurons (Sayer et al., 1992; Sahara and Westbrook, 1993; Chavis et al., 1994). Thus, as an initial foray into the signaling pathway by which estradiol and mGluR2/3 reduce L-type calcium channel-mediated CREB phosphorylation, we performed whole-cell recordings of hippocampal neurons. Application of APDC resulted in a decrease in the whole-cell current (Fig. 9A). The actions of APDC were attributable to a reduction in L-type calcium channel activity, because the effect was eliminated in the presence of Nif (Fig. 9B). In this experimental paradigm, APDC was found to reduce the L-type calcium current by 52.6 ± 14.6% (n = 9). Because application of the ERβ agonist DPN only reduced L-type calcium channel-mediated CREB phosphorylation and did not elicit MAPK-dependent CREB phosphorylation (Fig. 8), we next decided to examine the effect of this drug (Fig. 9C). Consistent with our hypothesis, DPN was also found to reduce L-type calcium channel currents by 23.7 ± 9.1% (n = 7).
Figure 9.

Activation of mGluR2/3 and ERβ reduce L-type calcium channel currents. Furthermore, estradiol attenuation of L-type calcium channel-dependent CREB phosphorylation is mediated through Gi/o and PKA. A, Example traces of the whole-cell calcium channel current under control conditions and in the presence of the mGluR2/3 agonist APDC (10 μm). Calibration: 400 pA, 20 ms. B, Plot of the peak current as a function of time. The application of APDC produced a reduction in the whole-cell calcium current. The effect was eliminated in the presence of the L-type calcium channel blocker nifedipine (5 μm), indicating that activation of mGluR2/3 results in a decrease in the L-type calcium channel current. C, Both APDC (n = 9) and the ERβ agonist DPN (10 nm, n = 7) resulted in a decrease in the L-type calcium channel current. D, Estradiol reduction of L-type calcium channel-mediated CREB phosphorylation was eliminated after a 24 h pretreatment with the Gi/o inhibitor PTX (500 ng/ml). E, Inhibition of PKA with Rp-cAMPs (50 μm) also occluded the effect of estradiol on L-type calcium channel-mediated CREB phosphorylation. Neither PTX nor Rp-cAMPs affected estradiol-induced CREB phosphorylation (F = 47.79 and 216.05; **p < 0.01 vs baseline). Error bars indicate SEM.
Previous reports have established that micromolar concentrations of estradiol can directly inhibit L-type calcium channels (Kurata et al., 2001). In the final set of experiments, the effect of estradiol on CREB phosphorylation was examined in the presence of the Gi/o inhibitor PTX and the PKA inhibitor Rp-cAMPs to verify the actions of estradiol in our preparation were mediated through an intracellular signaling pathway. Although overnight treatment with PTX (500 ng/ml) was found to increase basal levels of CREB phosphorylation (data not shown), significant increases in CREB phosphorylation were still observed after application of either estradiol or depolarization with 20 mm K+. However, the reduction in L-type calcium channel-mediated CREB phosphorylation by estradiol was eliminated (Fig. 9D). Furthermore, a 3 h preincubation of the PKA inhibitor Rp-cAMPs (50 μm) occluded estradiol attenuation of L-type calcium channel-mediated CREB phosphorylation (Fig. 9E). After application of both PTX and Rp-cAMPs, estradiol tended to enhance 20 mm K+ CREB phosphorylation, although the effects never reached significance. In the last series of experiments, PTX eliminated the APDC-induced reduction in CREB phosphorylation (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). Moreover, Rp-cAMPs occluded the effect of APDC (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material). These experiments confirm that both estradiol and mGluR2/3 stimulation reduce L-type calcium channel-dependent CREB phosphorylation through Gi/o and AC/PKA signaling.
Discussion
Previous research has determined that estradiol will induce CREB phosphorylation via activation of MAPK. We have expanded on these studies, uncovering that in hippocampal neurons, estradiol has bidirectional influences on CREB phosphorylation, initiated through activation of membrane-localized estrogen receptors. We hypothesize that estradiol binds to membrane-associated estrogen receptors, leading to activation of both group I and II mGluR signaling in a glutamate-independent manner. mGluR1a stimulation through activation of ERα leads to PLC, MAPK, and IP3R signaling, resulting in CREB phosphorylation. Concurrently, activation of mGluR2 and/or mGluR3 via ERα or ERβ leads to diminished cAMP concentrations and a reduction in PKA activity, ultimately resulting in dephosphorylation of L-type calcium channels and a reduction in L-type calcium channel-mediated CREB phosphorylation (Fig. 10). Of note, without specific mGluR2 or mGluR3 agonists/antagonists, we were unable to definitively determine whether this effect was mediated by both or just one of these receptors, although within hippocampal pyramidal neurons, mGluR2 expression appears predominant (Ohishi et al., 1993a,b). The actions of estradiol on mGluR signaling were found to be sex specific and occurred with physiological concentrations of hormone. Collectively, these results not only clarify the actions of estradiol on CREB signaling but may also provide insight into the major signaling mechanisms by which estrogens rapidly affect brain function through processes initiated at the membrane surface.
Figure 10.

Proposed mechanism by which estradiol produces bidirectional effects on CREB phosphorylation. Via interactions with classical estrogen receptors localized at the neuronal membrane, estradiol activates both group I and group II mGluR signaling. In one case, estradiol stimulation of mGluR1a leads to PLC/MAPK-induced CREB phosphorylation. Concurrently, activation of group II mGluR signaling decreases L-type calcium channel-dependent CREB phosphorylation through Gi/o inhibition of AC and PKA. The shaded arrows indicate decreased activity. CaM, Calmodulin; RSK, p90 ribosomal protein S6 kinase; CaMKIV, calmodulin-dependent protein kinase IV.
Regulation of CREB is believed to play a principal role in hippocampal learning and memory (Silva et al., 1998; Lonze and Ginty, 2002; Balschun et al., 2003). Previous work describing estradiol-mediated CREB phosphorylation outlined a potential mechanism by which estrogens could augment gene expression and, presumably, performance on learning and memory tasks. In fact, the effects of estradiol on these behaviors have been examined in great detail. Yet, these studies have provided inconsistent results, with some groups suggesting that estrogens enhance performance on hippocampal-dependent learning and memory tasks, whereas others report no effect or even cognitive deficits (Dohanich, 2002). The demonstration that estradiol exerts bidirectional effects on CREB phosphorylation may be one cause for these conflicting reports, because estradiol will either promote or attenuate CREB signaling reliant on the magnitude of concurrent excitatory inputs.
CREB is a stimulus-induced transcription factor that, although frequently studied in the context of hippocampal learning and memory, acts throughout the nervous system to regulate various behaviors (Lonze and Ginty, 2002). For instance, in areas such as the nucleus accumbens and DRG of the spinal cord, CREB regulates properties related to addiction and chronic pain (Blendy and Maldonado, 1998; Berke and Hyman, 2000; Nestler, 2001; Ji et al., 2003). Other examples of CREB-dependent cellular processes include neuronal survival, proliferation, and differentiation (Rudolph et al., 1998; Lonze et al., 2002; Redmond et al., 2002). Intriguingly, considerable overlap exists between behaviors and processes reliant on CREB and those that are influenced by estradiol (Lee and McEwen, 2001; Aloisi, 2003; Carroll et al., 2004; Hu et al., 2004). Thus, the present study may provide insight into a functional means by which estradiol, by acting through CREB, extensively modulates nervous system function.
Although rapid actions of estradiol have been studied for >20 years (Szego and Pietras, 1981), the underlying mechanisms by which they occur have remained elusive (McEwen, 2002). Although a membrane-specific estrogen receptor has been identified recently (ER-X), this protein seems only to be expressed during development or after neuronal injury (Toran-Allerand et al., 2002; Toran-Allerand, 2004). Because ER-X is unable to account for many of the actions of estradiol within adult tissue, two major theories suggesting the mechanism by which estrogens rapidly influence cell function have continued to endure. The first hypothesis is that classical estrogen receptors can have rapid effects on neuronal excitability. Consistent with this hypothesis, ERα and ERβ are found within neuronal membranes where they appear capable of influencing cell signaling (Razandi et al., 1999; Watson et al., 1999; Belcher and Zsarnovszky, 2001; Wade et al., 2001; Levin, 2002). Alternatively, because many of the novel effects of estradiol are sensitive to modulation of G-proteins, the opposing hypothesis suggests estradiol activates GPCRs (Kelly and Wagner, 1999; Kelly et al., 1999). Our results suggest a parsimonious explanation, incorporating major aspects from each of the previous theories. That is, estradiol acts with classical estrogen receptors localized to the neuronal membrane, which in turn stimulate several different classes of mGluRs, modulating cell function through activation of G-proteins.
An appealing feature to this revised model is that mGluR signaling can account for the ability of estradiol to regulate a diverse array of intracellular signaling cascades. These estrogen-sensitive pathways include PKA, MAPK, phosphoinositide 3-kinase, protein kinase B, PLC, and IP3 (Nabekura et al., 1986; Minami et al., 1990; Mobbs et al., 1991; Aronica et al., 1994; Gu et al., 1996; Zhou et al., 1996; Watters et al., 1997; Wiebe, 1997; Marino et al., 1998; Singh et al., 1999; Cardona-Gomez et al., 2002; Qiu et al., 2003). With promiscuous but also differential expression of mGluRs throughout the brain (Shigemoto et al., 1992; Ohishi et al., 1993a), activation of surface-expressed estrogen receptors can potentially have varied responses in nervous system tissue. Thus, our results may also impact other studies regarding nonclassical mechanisms of estrogen action. Of note, we focused on the major signaling pathways activated by group I and II mGluRs. Yet, there are mechanisms by which cross talk may occur between mGluR signaling. For example, activation of mGluR1a can affect L-type calcium channels (Sayer, 1998), possibly through IP3R (Lester and Jahr, 1990). Thus, estradiol regulation of intracellular signaling is most likely an assorted integration of numerous signaling pathways.
Because estradiol triggered CREB phosphorylation in the absence of depolarization, it was initially surprising that a significant attenuation of CREB signaling was observed after 20 mm K+ depolarization. Stated another way, why did the competing process of MAPK activation not obscure estradiol inhibition of L-type calcium channel-dependent CREB phosphorylation? The answer most likely is a result of depolarization-induced activation of CaN and PP1, which act in concert to counteract CREB phosphorylation (Bito et al., 1996, 1997). By attenuating calmodulin-dependent protein kinase IV-dependent CREB phosphorylation, estradiol may have unmasked depolarization-induced activation of CaN and PP1, resulting in decreased CREB activation.
A recent study using estrogen receptor knock-out mice demonstrated that expression of ERα and ERβ is necessary for estradiol-induced CREB phosphorylation (Abraham et al., 2004). Although these results strongly suggested a direct effect of estrogen receptors on CREB activation, they did not rule out the possibility that classical estrogen receptors are responsible for the expression of another estrogen-sensitive protein that directly initiates CREB phosphorylation. Our results help support the hypothesis that classical estrogen receptors, without activation of gene expression, can regulate CREB functioning.
One avenue of future research will be to pursue the additional discovery and quantification of classical estrogen receptors on the membrane surface. Consistent with previous reports (Su et al., 2001; Prange-Kiel et al., 2003), we readily observed expression of ERα and ERβ within the cytosol and nucleus of cultured hippocampal neurons (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Interestingly, ERα and ERβ expression was comparable between cultures derived from male versus female animals, consistent with previous reports (Weiland et al., 1997; Kalita et al., 2005) and highlighting the mystery surrounding the sexually dimorphic actions of estradiol. Detection of membrane estrogen receptors has proven to be substantially more difficult using conventional methods, although several recent reports have begun to identify localization of ERα and ERβ within the plasma membrane (Razandi et al., 1999; Watson et al., 1999; Wade et al., 2001). However, these results are still somewhat controversial, and membrane expression is not readily quantifiable (McEwen and Alves, 1999; Toran-Allerand, 2004). Hopefully, future studies will allow correlations between the expression of membrane estrogen receptors and physiological responses to the steroid hormone. Related, with the development of estrogen receptor-specific ligands, functional effects of these presumptive membrane receptors are just now being characterized, possibly leading to increased acceptance that detection of ERα and ERβ on the membrane surface is of physiological relevance.
Future research will also need to determine how membrane-localized ERα and ERβ stimulate mGluR signaling. The protein structure of classical estrogen receptors (ligand-binding domain, DNA-binding domain, etc.) does not lend itself to many clues regarding this process. However, in non-neuronal cells, estrogen receptors have been found to interact and activate the epidermal growth factor receptor and insulin-like growth factor-1 receptor (Kahlert et al., 2000; Razandi et al., 2003). These interactions between estrogen receptors and surface receptors appear to occur within caveolae (Wyckoff et al., 2001; Razandi et al., 2002). Interestingly, mGluRs are also concentrated within caveolae (Burgueno et al., 2003, 2004). Thus, through a similar process, estrogen receptors may activate mGluR signaling via direct protein-protein interactions. Experiments are under-way to test this hypothesis. These studies may also clarify whether one or both group II mGluRs are activated after estradiol treatment.
Three lines of evidence (mEPSC analysis and estradiol-induced CREB phosphorylation in both zero external calcium and in low-density cultures) suggest estradiol activates mGluRs via a postsynaptic mechanism. Thus, our working hypothesis is that estradiol binds to membrane-localized estrogen receptors, leading to activation of mGluRs. However, this raises an important issue regarding the ability of mGluR antagonists to eliminate estradiol-induced responses. Because antagonists are classically defined as preventing the binding of agonist, and not altering rates of transition between inactive and active states, why do these drugs block estradiol responses? Most likely, the answer is attributable to the fact that many GPCR antagonists also have the ability to dampen the activity of their targeted receptor (Lefkowitz et al., 1993; Milligan et al., 1995) and thus act as inverse agonists (Chen et al., 2000). Only modest work has been done to examine inverse agonist properties of antagonists, although there is some recent evidence implicating LY367385 (Pula et al., 2004). Notably, studies examining inverse agonists typically measure changes in basal rates of activity, and thus stimulus-induced activation may not be equally affected. Nevertheless, it is suggestive that estradiol-induced changes in CREB phosphorylation are blocked by the mGluR “antagonists” because of the capability of these drugs to generate hindrance in the transition of the receptor to an active confirmation.
Another area that will require additional research is determining the cause for the observed sex differences in response to estradiol. Given that postnatal animals were used in the present study, the testosterone surge in male animals occurring at birth may have affected estrogen sensitivity. Future use of prenatal animals, castration of newborn male pups, and injection of female pups with testosterone may determine whether the masculinization and defeminization of male animals at birth is responsible for this outcome, or whether an earlier developmental component is responsible. Other avenues of future research will include the use of knock-out mice (mGluR and estrogen receptor) in our paradigm to determine the overall significance of estradiol regulation of CREB activity, and to more fully characterize estradiol and mGluR2/3 inhibition of L-type calcium channel-mediated CREB phosphorylation.
In conclusion, we provide evidence that estradiol can trigger mGluR signaling through activation of classical estrogen receptors located at the plasma membrane. This previously undefined mechanism of estrogen action results in the stimulation of numerous intracellular signaling pathways, which may lead to both rapid and enduring changes in cell function. These novel estrogen actions most likely work synergistically with previously defined estrogen receptor responses to influence a diverse array of brain functions.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS41302 and by a grant from the Whitehall Foundation to P.G.M. J.P.W. was supported by a predoctoral National Research Service Award. R.D.G. was supported by NIH Training Grant DA07234. We thank Drs. Roger Albin, Bill Engeland, Martin Wessendorf, and Lance Zirpel for their scientific input and Drs. Robert Meisel and Geoff Pitt for suggestions on this manuscript. We also thank the reviewer who noticed the discrepancy in the concentrations listed for PPT and DPN in a previous version of this manuscript.
Correspondence should be addressed to Dr. Paul G. Mermelstein, Department of Neuroscience, University of Minnesota, 6-145 Jackson Hall, 321 Church Street Southeast, Minneapolis, MN 55455. E-mail: pmerm@umn.edu.
B. R. Becklund's present address: Department of Biochemistry, University of Wisconsin, Madison, WI 53706.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255066-13$15.00/0
References
- Abraham IM, Todman MG, Korach KS, Herbison AE (2004) Critical in vivo roles for classical estrogen receptors in rapid estrogen actions on intracellular signaling in mouse brain. Endocrinology 145: 3055-3061. [DOI] [PubMed] [Google Scholar]
- Aloisi AM (2003) Gonadal hormones and sex differences in pain reactivity. Clin J Pain 19: 168-174. [DOI] [PubMed] [Google Scholar]
- Arnold AP, Breedlove SM (1985) Organizational and activational effects of sex steroids on brain and behavior: a reanalysis. Horm Behav 19: 469-498. [DOI] [PubMed] [Google Scholar]
- Aronica SM, Kraus WL, Katzenellenbogen BS (1994) Estrogen action via the cAMP signaling pathway: stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc Natl Acad Sci USA 91: 8517-8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balschun D, Wolfer DP, Gass P, Mantamadiotis T, Welzl H, Schutz G, Frey JU, Lipp HP (2003) Does cAMP response element-binding protein have a pivotal role in hippocampal synaptic plasticity and hippocampus-dependent memory? J Neurosci 23: 6304-6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker G, Goslin K (1998) Culturing nerve cells, Ed 2. Cambridge, MA: MIT.
- Belcher SM, Zsarnovszky A (2001) Estrogenic actions in the brain: estrogen, phytoestrogens, and rapid intracellular signaling mechanisms. J Pharmacol Exp Ther 299: 408-414. [PubMed] [Google Scholar]
- Berke JD, Hyman SE (2000) Addiction, dopamine, and the molecular mechanisms of memory. Neuron 25: 515-532. [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW (1996) CREB phosphorylation and dephosphorylation: a Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell 87: 1203-1214. [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW (1997) Ca2+-dependent regulation in neuronal gene expression. Curr Opin Neurobiol 7: 419-429. [DOI] [PubMed] [Google Scholar]
- Blendy JA, Maldonado R (1998) Genetic analysis of drug addiction: the role of cAMP response element binding protein. J Mol Med 76: 104-110. [DOI] [PubMed] [Google Scholar]
- Brinton RD (1993) 17β-Estradiol induction of filopodial growth in cultured hippocampal neurons within minutes of exposure. Mol Cell Neurosci 4: 36-46. [DOI] [PubMed] [Google Scholar]
- Burgueno J, Enrich C, Canela EI, Mallol J, Lluis C, Franco R, Ciruela F (2003) Metabotropic glutamate type 1alpha receptor localizes in low-density caveolin-rich plasma membrane fractions. J Neurochem 86: 785-791. [DOI] [PubMed] [Google Scholar]
- Burgueno J, Canela EI, Mallol J, Lluis C, Franco R, Ciruela F (2004) Mutual regulation between metabotropic glutamate type 1alpha receptor and caveolin proteins: from traffick to constitutive activity. Exp Cell Res 300: 23-34. [DOI] [PubMed] [Google Scholar]
- Cardona-Gomez GP, Mendez P, Garcia-Segura LM (2002) Synergistic interaction of estradiol and insulin-like growth factor-I in the activation of PI3K/Akt signaling in the adult rat hypothalamus. Brain Res Mol Brain Res 107: 80-88. [DOI] [PubMed] [Google Scholar]
- Carroll ME, Lynch WJ, Roth ME, Morgan AD, Cosgrove KP (2004) Sex and estrogen influence drug abuse. Trends Pharmacol Sci 25: 273-279. [DOI] [PubMed] [Google Scholar]
- Chaban VV, Mayer EA, Ennes HS, Micevych PE (2003) Estradiol inhibits atp-induced intracellular calcium concentration increase in dorsal root ganglia neurons. Neuroscience 118: 941-948. [DOI] [PubMed] [Google Scholar]
- Chaban VV, Lakhter AJ, Micevych P (2004) A membrane estrogen receptor mediates intracellular calcium release in astrocytes. Endocrinology 145: 3788-3795. [DOI] [PubMed] [Google Scholar]
- Chavis P, Shinozaki H, Bockaert J, Fagni L (1994) The metabotropic glutamate receptor types 2/3 inhibit L-type calcium channels via a pertussis toxin-sensitive G-protein in cultured cerebellar granule cells. J Neurosci 14: 7067-7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Way J, Armour S, Watson C, Queen K, Jayawickreme CK, Chen WJ, Kenakin T (2000) Use of constitutive G protein-coupled receptor activity for drug discovery. Mol Pharmacol 57: 125-134. [PubMed] [Google Scholar]
- Couse JF, Korach KS (1999) Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev 20: 358-417. [DOI] [PubMed] [Google Scholar]
- De Jongh KS, Murphy BJ, Colvin AA, Hell JW, Takahashi M, Catterall WA (1996) Specific phosphorylation of a site in the full-length form of the alpha 1 subunit of the cardiac L-type calcium channel by adenosine 3′,5′-cyclic monophosphate-dependent protein kinase. Biochemistry 35: 10392-10402. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Bito H, Tsien RW (1996) Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron 16: 89-101. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Heist EK, Tsien RW (1998) Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature 392: 198-202. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Mermelstein PG, Xia H, Tsien RW (2003) Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol 13: 354-365. [DOI] [PubMed] [Google Scholar]
- Dohanich GP (2002) Gonadal steroids, learning and memory. San Diego: Academic.
- Dolmetsch RE, Pajvani V, Fife K, Spotts J, Greenberg ME (2001) Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294: 333-339. [DOI] [PubMed] [Google Scholar]
- Foster TC, Sharrow KM, Kumar A, Masse J (2003) Interaction of age and chronic estradiol replacement on memory and markers of brain aging. Neurobiol Aging 24: 839-852. [DOI] [PubMed] [Google Scholar]
- Foy MR, Xu J, Xie X, Brinton RD, Thompson RF, Berger TW (1999) 17β-Estradiol enhances NMDA receptor-mediated EPSPs and long-term potentiation. J Neurophysiol 81: 925-929. [DOI] [PubMed] [Google Scholar]
- Gao T, Yatani A, Dell'Acqua ML, Sako H, Green SA, Dascal N, Scott JD, Hosey MM (1997) cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron 19: 185-196. [DOI] [PubMed] [Google Scholar]
- Gaub MP, Bellard M, Scheuer I, Chambon P, Sassone-Corsi P (1990) Activation of the ovalbumin gene by the estrogen receptor involves the fos-jun complex. Cell 63: 1267-1276. [DOI] [PubMed] [Google Scholar]
- Gould E, Woolley CS, Frankfurt M, McEwen BS (1990) Gonadal steroids regulate dendritic spine density in hippocampal pyramidal cells in adulthood. J Neurosci 10: 1286-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth RD, Mermelstein PG (2003) Brain-derived neurotrophic factor activation of NFAT (nuclear factor of activated T-cells)-dependent transcription: a role for the transcription factor NFATc4 in neurotrophin-mediated gene expression. J Neurosci 23: 8125-8134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth RD, Dunbar RL, Mermelstein PG (2003) Calcineurin regulation of neuronal plasticity. Biochem Biophys Res Commun 311: 1159-1171. [DOI] [PubMed] [Google Scholar]
- Gu G, Rojo AA, Zee MC, Yu J, Simerly RB (1996) Hormonal regulation of CREB phosphorylation in the anteroventral periventricular nucleus. J Neurosci 16: 3035-3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutkind JS (2000) Regulation of mitogen-activated protein kinase signaling networks by G protein-coupled receptors. Sci STKE 2000: RE1. [DOI] [PubMed] [Google Scholar]
- Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM, Wozniak DF, Nardi A, Arvin KL, Holtzman DM, Linden DJ, Zhuo M, Muglia LJ, Chatila TA (2000) Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci 20: 6459-6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Crombag HS, Robinson TE, Becker JB (2004) Biological basis of sex differences in the propensity to self-administer cocaine. Neuropsychopharmacology 29: 81-85. [DOI] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ (2003) Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci 26: 696-705. [DOI] [PubMed] [Google Scholar]
- Kahlert S, Nuedling S, van Eickels M, Vetter H, Meyer R, Grohe C (2000) Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway. J Biol Chem 275: 18447-18453. [DOI] [PubMed] [Google Scholar]
- Kalita K, Szymczak S, Kaczmarek L (2005) Non-nuclear estrogen receptor beta and alpha in the hippocampus of male and female rats. Hippocampus 15: 404-412. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Wagner EJ (1999) Estrogen modulation of G-protein-coupled receptors. Trends Endocrinol Metab 10: 369-374. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Lagrange AH, Wagner EJ, Ronnekleiv OK (1999) Rapid effects of estrogen to modulate G protein-coupled receptors via activation of protein kinase A and protein kinase C pathways. Steroids 64: 64-75. [DOI] [PubMed] [Google Scholar]
- Klinge CM (2001) Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res 29: 2905-2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA (1996) Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA 93: 5925-5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata K, Takebayashi M, Kagaya A, Morinobu S, Yamawaki S (2001) Effect of beta-estradiol on voltage-gated Ca2+ channels in rat hippocampal neurons: a comparison with dehydroepiandrosterone. Eur J Pharmacol 416: 203-212. [DOI] [PubMed] [Google Scholar]
- Lee SJ, McEwen BS (2001) Neurotrophic and neuroprotective actions of estrogens and their therapeutic implications. Annu Rev Pharmacol Toxicol 41: 569-591. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Campomanes CR, Sikat PT, Greenfield AT, Allen PB, McEwen BS (2004) Estrogen induces phosphorylation of cyclic AMP response element binding (pCREB) in primary hippocampal cells in a time-dependent manner. Neuroscience 124: 549-560. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Cotecchia S, Samama P, Costa T (1993) Constitutive activity of receptors coupled to guanine nucleotide regulatory proteins. Trends Pharmacol Sci 14: 303-307. [DOI] [PubMed] [Google Scholar]
- Lester RA, Jahr CE (1990) Quisqualate receptor-mediated depression of calcium currents in hippocampal neurons. Neuron 4: 741-749. [DOI] [PubMed] [Google Scholar]
- Levin ER (2002) Cellular functions of plasma membrane estrogen receptors. Steroids 67: 471-475. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35: 605-623. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Riccio A, Cohen S, Ginty DD (2002) Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron 34: 371-385. [DOI] [PubMed] [Google Scholar]
- Marino M, Pallottini V, Trentalance A (1998) Estrogens cause rapid activation of IP3-PKC-alpha signal transduction pathway in HEPG2 cells. Biochem Biophys Res Commun 245: 254-258. [DOI] [PubMed] [Google Scholar]
- McEwen B (2002) Estrogen actions throughout the brain. Recent Prog Horm Res 57: 357-384. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Alves SE (1999) Estrogen actions in the central nervous system. Endocr Rev 20: 279-307. [DOI] [PubMed] [Google Scholar]
- Mermelstein PG, Becker JB, Surmeier DJ (1996) Estradiol reduces calcium currents in rat neostriatal neurons via a membrane receptor. J Neurosci 16: 595-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermelstein PG, Bito H, Deisseroth K, Tsien RW (2000) Critical dependence of cAMP response element-binding protein phosphorylation on L-type calcium channels supports a selective response to EPSPs in preference to action potentials. J Neurosci 20: 266-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermelstein PG, Deisseroth K, Dasgupta N, Isaksen AL, Tsien RW (2001) Calmodulin priming: nuclear translocation of a calmodulin complex and the memory of prior neuronal activity. Proc Natl Acad Sci USA 98: 15342-15347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G, Bond RA, Lee M (1995) Inverse agonism: pharmacological curiosity or potential therapeutic strategy? Trends Pharmacol Sci 16: 10-13. [DOI] [PubMed] [Google Scholar]
- Minami T, Oomura Y, Nabekura J, Fukuda A (1990) 17β-Estradiol depolarization of hypothalamic neurons is mediated by cyclic AMP. Brain Res 519: 301-307. [DOI] [PubMed] [Google Scholar]
- Mobbs CV, Kaplitt M, Kow LM, Pfaff DW (1991) PLC-alpha: a common mediator of the action of estrogen and other hormones? Mol Cell Endocrinol 80: C187-C191. [DOI] [PubMed] [Google Scholar]
- Murphy DD, Segal M (1997) Morphological plasticity of dendritic spines in central neurons is mediated by activation of cAMP response element binding protein. Proc Natl Acad Sci USA 94: 1482-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabekura J, Oomura Y, Minami T, Mizuno Y, Fukuda A (1986) Mechanism of the rapid effect of 17 beta-estradiol on medial amygdala neurons. Science 233: 226-228. [DOI] [PubMed] [Google Scholar]
- Nestler EJ (2001) Molecular neurobiology of addiction. Am J Addict 10: 201-217. [DOI] [PubMed] [Google Scholar]
- Neves SR, Ram PT, Iyengar R (2002) G protein pathways. Science 296: 1636-1639. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Shigemoto R, Nakanishi S, Mizuno N (1993a) Distribution of the messenger RNA for a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat. Neuroscience 53: 1009-1018. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Shigemoto R, Nakanishi S, Mizuno N (1993b) Distribution of the mRNA for a metabotropic glutamate receptor (mGluR3) in the rat brain: an in situ hybridization study. J Comp Neurol 335: 252-266. [DOI] [PubMed] [Google Scholar]
- Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS (1997) Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science 277: 1508-1510. [DOI] [PubMed] [Google Scholar]
- Pfaff D, Keiner M (1973) Atlas of estradiol-concentrating cells in the central nervous system of the female rat. J Comp Neurol 151: 121-158. [DOI] [PubMed] [Google Scholar]
- Pfaff DW, Schwartz-Giblin S, McCarthy MM, Kow L-M (1994) Cellular and molecular mechanisms of female reproductive behaviors, Ed 2. New York: Raven.
- Philips A, Chalbos D, Rochefort H (1993) Estradiol increases and antiestrogens antagonize the growth factor-induced activator protein-1 activity in MCF7 breast cancer cells without affecting c-fos and c-jun synthesis. J Biol Chem 268: 14103-14108. [PubMed] [Google Scholar]
- Prange-Kiel J, Wehrenberg U, Jarry H, Rune GM (2003) Para/autocrine regulation of estrogen receptors in hippocampal neurons. Hippocampus 13: 226-234. [DOI] [PubMed] [Google Scholar]
- Pula G, Mundell SJ, Roberts PJ, Kelly E (2004) Agonist-independent internalization of metabotropic glutamate receptor 1a is arrestin- and clathrin-dependent and is suppressed by receptor inverse agonists. J Neurochem 89: 1009-1020. [DOI] [PubMed] [Google Scholar]
- Qiu J, Bosch MA, Tobias SC, Grandy DK, Scanlan TS, Ronnekleiv OK, Kelly MJ (2003) Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase C. J Neurosci 23: 9529-9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razandi M, Pedram A, Greene GL, Levin ER (1999) Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERalpha and ERbeta expressed in Chinese hamster ovary cells. Mol Endocrinol 13: 307-319. [DOI] [PubMed] [Google Scholar]
- Razandi M, Oh P, Pedram A, Schnitzer J, Levin ER (2002) ERs associate with and regulate the production of caveolin: implications for signaling and cellular actions. Mol Endocrinol 16: 100-115. [DOI] [PubMed] [Google Scholar]
- Razandi M, Pedram A, Park ST, Levin ER (2003) Proximal events in signaling by plasma membrane estrogen receptors. J Biol Chem 278: 2701-2712. [DOI] [PubMed] [Google Scholar]
- Razandi M, Pedram A, Merchenthaler I, Greene GL, Levin ER (2004) Plasma membrane estrogen receptors exist and functions as dimers. Mol Endocrinol 18: 2854-2865. [DOI] [PubMed] [Google Scholar]
- Redmond L, Kashani AH, Ghosh A (2002) Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 34: 999-1010. [DOI] [PubMed] [Google Scholar]
- Ribar TJ, Rodriguiz RM, Khiroug L, Wetsel WC, Augustine GJ, Means AR (2000) Cerebellar defects in Ca2+/calmodulin kinase IV-deficient mice. J Neurosci 20: RC107(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph D, Tafuri A, Gass P, Hammerling GJ, Arnold B, Schutz G (1998) Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc Natl Acad Sci USA 95: 4481-4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara Y, Westbrook GL (1993) Modulation of calcium currents by a metabotropic glutamate receptor involves fast and slow kinetic components in cultured hippocampal neurons. J Neurosci 13: 3041-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayer RJ (1998) Group I metabotropic glutamate receptors mediate slow inhibition of calcium current in neocortical neurons. J Neurophysiol 80: 1981-1988. [DOI] [PubMed] [Google Scholar]
- Sayer RJ, Schwindt PC, Crill WE (1992) Metabotropic glutamate receptor-mediated suppression of L-type calcium current in acutely isolated neocortical neurons. J Neurophysiol 68: 833-842. [DOI] [PubMed] [Google Scholar]
- Sharrow KM, Kumar A, Foster TC (2002) Calcineurin as a potential contributor in estradiol regulation of hippocampal synaptic function. Neuroscience 113: 89-97. [DOI] [PubMed] [Google Scholar]
- Sheng M, Thompson MA, Greenberg ME (1991) CREB: a Ca2+-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science 252: 1427-1430. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Nakanishi S, Mizuno N (1992) Distribution of the mRNA for a metabotropic glutamate receptor (mGluR1) in the central nervous system: an in situ hybridization study in adult and developing rat. J Comp Neurol 322: 121-135. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S (1998) CREB and memory. Annu Rev Neurosci 21: 127-148. [DOI] [PubMed] [Google Scholar]
- Singh M, Setalo Jr G, Guan X, Warren M, Toran-Allerand CD (1999) Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: convergence of estrogen and neurotrophin signaling pathways. J Neurosci 19: 1179-1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS, Katzenellenbogen JA (2000) Pyrazole ligands: structure-affinity/activity relationships and estrogen receptor-alpha-selective agonists. J Med Chem 43: 4934-4947. [DOI] [PubMed] [Google Scholar]
- Su JD, Qiu J, Zhong YP, Li XY, Wang JW, Chen YZ (2001) Expression of estrogen receptor (ER)-alpha and -beta immunoreactivity in hippocampal cell cultures with special attention to GABAergic neurons. J Neurosci Res 65: 396-402. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Bargas J, Hemmings Jr HC, Nairn AC, Greengard P (1995) Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron 14: 385-397. [DOI] [PubMed] [Google Scholar]
- Szego CM, Pietras RJ (1981) Membrane recognition and effector sites in steroid hormone action. In: Biochemical actions of hormones, Vol 8 (Litwack G, ed), pp 307-464. New York: Academic. [Google Scholar]
- Thomas GM, Huganir RL (2004) MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 5: 173-183. [DOI] [PubMed] [Google Scholar]
- Toran-Allerand CD (2004) Minireview: A plethora of estrogen receptors in the brain: where will it end? Endocrinology 145: 1069-1074. [DOI] [PubMed] [Google Scholar]
- Toran-Allerand CD, Singh M, Setalo Jr G (1999) Novel mechanisms of estrogen action in the brain: new players in an old story. Front Neuroendocrinol 20: 97-121. [DOI] [PubMed] [Google Scholar]
- Toran-Allerand CD, Guan X, MacLusky NJ, Horvath TL, Diano S, Singh M, Connolly Jr ES, Nethrapalli IS, Tinnikov AA (2002) ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J Neurosci 22: 8391-8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay GB, Tremblay A, Copeland NG, Gilbert DJ, Jenkins NA, Labrie F, Giguere V (1997) Cloning, chromosomal localization, and functional analysis of the murine estrogen receptor beta. Mol Endocrinol 11: 353-365. [DOI] [PubMed] [Google Scholar]
- Umayahara Y, Kawamori R, Watada H, Imano E, Iwama N, Morishima T, Yamasaki Y, Kajimoto Y, Kamada T (1994) Estrogen regulation of the insulin-like growth factor I gene transcription involves an AP-1 enhancer. J Biol Chem 269: 16433-16442. [PubMed] [Google Scholar]
- Wade CB, Dorsa DM (2003) Estrogen activation of cyclic adenosine 5′-monophosphate response element-mediated transcription requires the extracellularly regulated kinase/mitogen-activated protein kinase pathway. Endocrinology 144: 832-838. [DOI] [PubMed] [Google Scholar]
- Wade CB, Robinson S, Shapiro RA, Dorsa DM (2001) Estrogen receptor (ER)alpha and ERbeta exhibit unique pharmacologic properties when coupled to activation of the mitogen-activated protein kinase pathway. Endocrinology 142: 2336-2342. [DOI] [PubMed] [Google Scholar]
- Waltereit R, Weller M (2003) Signaling from cAMP/PKA to MAPK and synaptic plasticity. Mol Neurobiol 27: 99-106. [DOI] [PubMed] [Google Scholar]
- Watson CS, Campbell CH, Gametchu B (1999) Membrane oestrogen receptors on rat pituitary tumour cells: immuno-identification and responses to oestradiol and xenoestrogens. Exp Physiol 84: 1013-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters JJ, Campbell JS, Cunningham MJ, Krebs EG, Dorsa DM (1997) Rapid membrane effects of steroids in neuroblastoma cells: effects of estrogen on mitogen activated protein kinase signalling cascade and c-fos immediate early gene transcription. Endocrinology 138: 4030-4033. [DOI] [PubMed] [Google Scholar]
- Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, Katzenellenbogen BS, Enmark E, Gustafsson JA, Nilsson S, Kushner PJ (1999) The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol Endocrinol 13: 1672-1685. [DOI] [PubMed] [Google Scholar]
- Weick JP, Groth RD, Isaksen AL, Mermelstein PG (2003) Interactions with PDZ proteins are required for L-type calcium channels to activate cAMP response element-binding protein-dependent gene expression. J Neurosci 23: 3446-3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiland NG (1992) Estradiol selectively regulates agonist binding sites on the N-methyl-d-aspartate receptor complex in the CA1 region of the hippocampus. Endocrinology 131: 662-668. [DOI] [PubMed] [Google Scholar]
- Weiland NG, Orikasa C, Hayashi S, McEwen BS (1997) Distribution and hormone regulation of estrogen receptor immunoreactive cells in the hippocampus of male and female rats. J Comp Neurol 388: 603-612. [DOI] [PubMed] [Google Scholar]
- Weng G, Bhalla US, Iyengar R (1999) Complexity in biological signaling systems. Science 284: 92-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiebe JP (1997) Nongenomic actions of steroids on gonadotropin release. Recent Prog Horm Res 52: 71-99; discussion 99-101. [PubMed] [Google Scholar]
- Williams CL (1986) A reevaluation of the concept of separable periods of organizational and activational actions of estrogens in development of brain and behavior. Ann NY Acad Sci 474: 282-292. [DOI] [PubMed] [Google Scholar]
- Wong M, Moss RL (1992) Long-term and short-term electrophysiological effects of estrogen on the synaptic properties of hippocampal CA1 neurons. J Neurosci 12: 3217-3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley CS, McEwen BS (1992) Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J Neurosci 12: 2549-2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley CS, McEwen BS (1993) Roles of estradiol and progesterone in regulation of hippocampal dendritic spine density during the estrous cycle in the rat. J Comp Neurol 336: 293-306. [DOI] [PubMed] [Google Scholar]
- Woolley CS, Weiland NG, McEwen BS, Schwartzkroin PA (1997) Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor-mediated synaptic input: correlation with dendritic spine density. J Neurosci 17: 1848-1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff MH, Chambliss KL, Mineo C, Yuhanna IS, Mendelsohn ME, Mumby SM, Shaul PW (2001) Plasma membrane estrogen receptors are coupled to endothelial nitric-oxide synthase through Galpha(i). J Biol Chem 276: 27071-27076. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Watters JJ, Dorsa DM (1996) Estrogen rapidly induces the phosphorylation of the cAMP response element binding protein in rat brain. Endocrinology 137: 2163-2166. [DOI] [PubMed] [Google Scholar]