

Abstract
Methylation-induced transcriptional silencing of the fragile X mental retardation-1 (Fmr1) gene leads to absence of the gene product, fragile X mental retardation protein (FMRP), and consequently fragile X syndrome (FrX), an X-linked inherited form of mental retardation. Absence of FMRP in Fmr1 null mice imparts some characteristics of the FrX phenotype, but the precise role of FMRP in neuronal function remains unknown. FMRP is an RNA-binding protein that has been shown to suppress translation of certain mRNAs in vitro. We applied the quantitative autoradiographic l-[1-14C]leucine method to the in vivo determination of regional rates of cerebral protein synthesis (rCPS) in adult wild-type (WT) and Fmr1 null mice at 4 and 6 months of age. Our results show a substantial decrease in rCPS in all brain regions examined between the ages of 4 and 6 months in both WT and Fmr1 null mice. Superimposed on the age-dependent decline in rCPS, we demonstrate a regionally selective elevation in rCPS in Fmr1 null mice. Our results suggest that the process of synaptic pruning during young adulthood may be reflected in decreased rCPS. Our findings support the hypothesis that FMRP is a suppressor of translation in brain in vivo.
Keywords: fragile X, age, synaptic pruning, synaptic plasticity, FMRP, protein synthesis
Introduction
The requirement for protein synthesis in long-term adaptive change in the nervous system is well documented (West et al., 2001). Inhibition of protein synthesis blocks long-term memory storage in both vertebrates and invertebrates (Agranoff, 1981). Activity-driven changes in gene expression are essential for maintenance of the late phase of long-term potentiation (Nguyen et al., 1994) and long-term depression (Linden, 1996) and promote neuronal survival during development (Mao et al., 1999). Molecular mechanisms underlying these processes may involve translation of newly transcribed targeted genes in neuronal soma and local translation of selected mRNAs in dendrites (Steward and Schuman, 2001).
One of the mRNAs translated in dendrites in response to activation of metabotropic glutamate receptors (mGluRs) is fragile X mental retardation-1 (Fmr1) (Weiler et al., 1997). Silencing of the gene for Fmr1 by a repeat expansion of (CGG)n trinucleotide in the 5′-untranslated region results in fragile X syndrome (FrX), an X-linked disorder characterized by mental retardation. In patients with FrX, fragile X mental retardation protein (FMRP), the gene product of Fmr1, is absent. FrX phenotype includes cognitive impairments ranging from learning disabilities to severe mental retardation (Rousseau et al., 1994), behavioral dysfunction such as hyperactivity, social anxiety, attention problems, and autistic-like behavior (Miller et al., 1999), and subtle physical abnormalities, including macroorchidism in males (Hagerman, 2002). A striking neuropathological feature of FrX syndrome is the long, thin, and tortuous appearance of dendritic spines (Rudelli et al., 1985; Hinton et al., 1991), a morphology similar to that seen early in development (Marin-Padilla, 1967).
Normally, FMRP is widely expressed in fetal and adult tissues and is most abundant in brain and testes (Devys et al., 1993). It is highly expressed in neurons but not in glia (Feng et al., 1997) and is found in cytoplasm, dendrites, and dendritic spines. FMRP is an RNA-binding protein that associates with translating polyribosomes (Feng et al., 1997), and accumulating evidence suggests that FMRP may act as a negative regulator of translation. Mammalian FMRP inhibits mRNA translation in rabbit reticulocyte lysates (Li et al., 2001) and in microinjected Xenopus oocytes (Laggerbauer et al., 2001). In Drosophila, the homolog of FMRP (dFMRP) binds futsch [the Drosophila homolog of MAP-1B (microtubule-associated protein 1B)] mRNA and negatively regulates futsch expression (Zhang et al., 2001). Recent evidence suggests that dFMRP is involved in the micro RNA (miRNA) and RNA interference (RNAi) pathways (Caudy et al., 2002; Ishizuka et al., 2002), implicating dFMRP in the RNAi/miRNA-directed processes of mRNA degradation, translation inhibition, and/or chromatin silencing. To what extent these effects of FMRP on gene expression are operating in brain in vivo has not been shown.
We used the Fmr1 null mouse (Dutch-Belgian Fragile X Consortium, 1994) to study cerebral protein synthesis in an animal model of FrX. In this study, we report that in adult Fmr1 null mice regional rates of cerebral protein synthesis (rCPS) measured in vivo are increased in selective brain regions compared with wildtype (WT) littermates, supporting the idea that FMRP acts as a suppressor of translation in brain in vivo.
Parts of this work have been published previously in abstract form (Qin et al., 2003).
Materials and Methods
Chemicals and materials were obtained from the following sources: l-[1-14C]leucine [specific activity (SA), 60 mCi/mmol] and l-[3,4,5-3H]leucine (SA, 50 Ci/mmol) from PerkinElmer Life Sciences (Boston, MA); Escherichia coli tRNA from Sigma (St. Louis, MO); vanadyl ribonucleoside complex and redistilled nucleic acid-grade phenol from Bethesda Research Laboratories (Gaithersburg, MD); and l-norleucine and 5-sulfosalicylic acid from Fluka (Buchs, Switzerland).
Animals. All procedures were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Animals and an animal study protocol approved by the National Institute of Mental Health Animal Care and Use Committee. FVB/NJ-Fmr1tm1Cgr breeding pairs (heterozygous females and hemizygous males) were obtained from The Jackson Laboratory (Bar Harbor, ME). Heterozygous female and wild-type male offspring were mated to provide offspring in two experimental groups: hemizygous males and wild-type males. All mice were housed in a central facility and maintained under controlled conditions of normal humidity and temperature with standard alternating 12 h periods of light and darkness. Food (NIH-31 rodent chow) and water were provided ad libitum. Mature mice between 3 and 6 months of age were studied.
Genotyping. Genomic DNA was extracted from a section of tail taken from each animal (Puregene; Gentra System, Minneapolis, MN). The M2 (5′-ATCTAGTCATGCTATGGATATCAGC-3′) and N2 (5′-GTGGG CTCTATGGCTTCTGAGG-3′) primers (12 pmol) were used to screen for the presence or absence of the mutant allele in a PCR buffer containing 10 mm Tris-HCl, pH 8.3, 50 mm KCl, 2.5 mm MgCl2, 0.2 mm dNTPs, and 2 U TaqDNA polymerase (AmpliTaq Gold; Applied Biosystems, Branchburg, NJ) as described previously (Qin et al., 2002). The PCR product at ∼800 bp indicated the presence of the knock-out allele.
Surgical preparation of animals. Mice were prepared for studies by insertion under light halothane anesthesia of polyethylene catheters (PE-10) into one femoral artery and one or two femoral veins as described previously (Smith and Kang, 2000). Mice were allowed to recover from the surgery overnight in clear plastic cylinders 13 cm in diameter. This apparatus permitted mice to move freely throughout the recovery and experimental periods but not gain access to the tubing. Food and water were available ad libitum.
Measurement of physiological variables. Mean arterial blood pressure, hematocrit, and arterial plasma glucose concentrations were measured to evaluate each animal's physiological state (Qin et al., 2002). Arterial plasma corticosterone concentration was determined by radioimmunoassay (MP Biomedicals, Costamesa, CA).
Determination of rCPS. rCPS was determined with l-[1-14C]leucine as the radiolabeled tracer used in conjunction with quantitative autoradiography to achieve spatial localization of the labeled protein in the brain (Smith et al., 1988). The operational equation of the method is as follows:
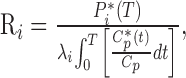 |
in which Ri is the rate of leucine incorporation into protein in tissue i; Pi* (T) is the concentration of 14C fixed in tissue i at any time, T, after introduction of the tracer into the circulation; λi is equal to the fraction of leucine in the precursor pool for protein synthesis in tissue i that is derived from plasma; Cp*(t) and Cp are the concentrations of labeled and unlabeled leucine in the arterial plasma, respectively; and t is the variable time. The evaluation of λi is performed in a separate series of experiments by measuring the steady-state ratio of leucine SA in the tissue tRNA-bound pool to that of the arterial plasma.
Mice were surgically prepared and catheterized, and their physiological states were monitored as described above. The experimental period was initiated by an intravenous pulse of 100 μCi/kg l-[1-14C]leucine contained in ∼40 μl of physiological saline. Timed arterial samples were collected during the following 60 min for determination of the time courses of plasma concentrations of leucine and [14C]leucine as described previously (Smith and Kang, 2000). At the end of the experimental interval, mice were killed by an intravenous injection of sodium pentobarbital, and brains were rapidly removed and frozen in isopentane cooled to -40°C with dry ice. Serial sections, 20 μm thick, were cut in a Leica (Deerfield, IL) 1850 cryostat at -18°C, thaw mounted on gelatin-coated slides, dried in a stream of air, fixed and washed in 10% formalin, and exposed to EMC-1 film (Eastman Kodak, Rochester, NY) along with calibrated [14C]methylmethacrylate standards as described previously (Smith and Kang, 2000). Autoradiograms were digitized with a pixel size of 11 μm by means of a 10-bit DVC-1310 digital camera (DVC, Austin, TX). Images were analyzed with an MCID Elite image processing system (Imaging Research, St. Catharine's, Ontario, Canada). The concentration of 14C in each region of interest was determined from the optical density versus 14C concentration curve determined from the calibrated plastic standards, and rCPS was calculated by means of the above equation. Brain regions were identified by reference to a mouse brain atlas (Paxinos and Franklin, 2001). Global CPS and brain volume were determined by analysis of autoradiograms of all sections of the entire brain digitized (42 μm pixel size) by means of a Multirad 850 Howtek Film Digitzer (Howtek, Hudson, NH). Weighted average CPS was determined as described above, and brain volume was determined from the total number of pixels and the calibrated pixel size.
Evaluation of λi. λi for whole brain (λWB) was evaluated in a series of steady-state experiments as described previously (Smith et al., 1988). A constant arterial plasma SA for [3H]leucine was maintained for 120 min by means of a programmed intravenous infusion of [3H]leucine designed to achieve and maintain a constant arterial plasma [3H]leucine concentration. The total infusion contained 3-8 mCi of [3H]leucine in a volume of ∼0.4 ml. The SAs of [3H]leucine in the arterial plasma and tRNA-bound pools in brain were determined as described below. Timed arterial blood samples (∼20 μl) were collected every 15 or 30 min during the infusion and centrifuged immediately to separate plasma, which was then deproteinized by addition of one-third of a volume of 16% (W/V) sulfosalicylic acid containing l-norleucine (0.04 mm) as an internal standard for amino acid analyses. The deproteinized samples were stored at -70°C until assayed for leucine and [3H]leucine concentrations. At the end of the infusions, the mice were decapitated, and their brains were quickly removed and chilled to 0°C in ice-cold 0.25 m sucrose.
Each brain was weighed and homogenized in 5 ml of 0.25 m sucrose (0°C) containing 10 mm vanadyl ribonucleoside complex to inhibit ribonuclease, 6 mg of uncharged tRNA as carrier, and l-norleucine (0.02 mm) added as an internal standard and centrifuged at 100,000 × g for 1 h. The tRNA-bound amino acids were purified as described previously (Smith et al., 1988). The pure tRNA fraction was dissolved in 50 mm sodium carbonate, pH 10, and incubated at 37°C for 90 min to deacylate the aminoacyl-tRNA. Deacylated tRNA was precipitated overnight at -20°C in ethanol and removed by centrifugation (12,000 × g, 20 min) (Smith et al., 1988). The supernatant solution, which contained the previously tRNA-bound but now free amino acids, was dried in a stream of N2 and redissolved in 40 μl of 0.2 m sodium citrate, pH 2.2.
Deproteinized plasma samples and tissue fractions of deacylated aminoacyl-tRNA pools were assayed for leucine concentration by postcolumn derivatization with o-phthaldehyde and fluorometric assay with a Beckman model 7300 Amino Acid Analyzer (Beckman Instruments, Fullerton, CA), and column eluates were collected every 0.5 min and assayed for 3H with a model 2250CA Packard Liquid Scintillation Counter (PerkinElmer Life Sciences). SA of each sample was calculated from total 3H in all fractions in the leucine peak and the total measured leucine content in the peak.
Values of λWB were calculated as the ratios of the measured steady-state SA of leucine in the tissue tRNA-bound pool to that of the free leucine in arterial plasma. The time course of the SA in arterial plasma and the SA of tRNA-bound leucine in whole brain at the end of the experimental interval were determined as described above. Only animals with relatively constant plasma leucine SAs were included in the series.
Immunohistochemistry. For FMRP-immunohistochemical staining, mice were deeply anesthetized with halothane and perfused via the ascending aorta with 0.9% normal saline, followed by 4% paraformaldehyde. Brains were postfixed in 4% paraformaldehyde for 6 h at 4°C, immersed in 0.1 m PBS, pH 7.4, containing 20% sucrose for 48 h and frozen and stored at -75°C. Coronal sections 20 μm in thickness were cut with a cryostat and thaw mounted on gelatin-coated glass slides. Alternate sections were taken for thionin staining and immunohistochemistry. We used the immunoperoxidase procedure (M.O.M. kit; Vector Laboratories, Burlingame, CA) (Hsu et al., 1981) for FMRP immunohistochemistry with a monoclonal mouse anti-FMRP IgG (Chemicon, Temecula, CA) at 1:50.
The specificity of the antibody (monoclonal antibody 1a) was originally demonstrated by Devys et al. (1993). In the present study, the specificity of the primary antibody was confirmed with the following controls: (1) omission of the specific antibody and (2) use of normal mouse IgG instead of the specific antibody. We also used Western blot analysis of tissue extracts prepared from WT and Fmr1 null mouse brains to confirm the specificity of the primary antibody (Fig. 1). Equal amounts of protein (50 μg) were separated by PAGE and blotted onto polyvinylidene difluoride membranes (Invitrogen, Carlsbad, CA). Membranes were incubated overnight at 4°C with FMRP antibody (1:1000), followed by biotinylated secondary antibody (1:500) (Amersham Biosciences, Piscataway, NJ). The blot was then reacted with streptavidin-HRP-conjugated antibody (1: 5000), and immunoreactive bands were visualized with ECL Western blotting detection reagents (Amersham Biosciences).
Figure 1.

Western blots of protein extracts from cortex and hippocampus of WT and Fmr1 null mice demonstrating the specificity of the FMRP antibody.
Statistical analyses. Measurements were analyzed by means of two-way ANOVA with post hoc t tests.
Results
Determination of λWB
To correct for possible differences in the effects of recycling of leucine between the two genotypes, we determined the fraction of leucine in the precursor pool derived from the arterial plasma in three WT and three Fmr1 null mice between 76 and 104 d of age. For the determination of λWB, only studies with fairly constant arterial plasma leucine SA (i.e., all samples taken during the last 30 min of the study within ±10% of the mean of those samples) were included in the series (Fig. 2). One animal was eliminated because arterial plasma leucine SA varied by up to 20% during the 90-120 min interval. Mean ± SEM steady-state SA ratios for the tRNA-bound pool were 0.616 ± 0.006 and 0.595 ± 0.012 for two WT and three Fmr1 null mice, respectively, indicating that ∼60% of leucine in the precursor pool in brain comes from plasma and 40% comes from protein breakdown in the tissue. Because the values in the two groups were not statistically significantly different from each other, we used the pooled value of 0.603 ± 0.009 (mean ± SEM) for the calculation of the integrated tissue precursor pool SA for both WT and Fmr1 null mice.
Figure 2.

Typical steady-state experiments for the determination of λ in WT (A) and Fmr1 null (B) mice illustrating the achievement and maintenance of a constant SA of [3H]leucine (□) over 120 min and the relatively constant arterial plasma leucine concentration (▵) over this interval. The figure also shows the SA of the tRNA-bound leucine (•) in brain at the end of the experiment. In both genotypes, the tRNA-bound leucine SA is considerably below that of the arterial plasma, indicating that there is significant recycling of unlabeled leucine derived from protein breakdown back into the precursor amino acid pool.
Cerebral protein synthesis
Initially, our study design was a comparison of rCPS in mature male mice of the two genotypes, and we chose to study “young adult” mice between the ages of 4 and 6 months. In our preliminary analysis of nine WT mice between 118 and 197 d of age, we observed an effect of age on the global rate of CPS (Fig. 3). The effect was large, as much as a 40% decrease over this 79 d interval, and the regression of CPS versus age reached statistical significance.
Figure 3.

Correlation of CPS with age in young adult WT mice. Each point represents a measured value of global CPS in a single animal. The solid line is the best-fitting regression line. Dashed lines are the 95% confidence limits.
Because of this age effect, we redesigned our study to compare WT and Fmr1 null mice at two ages, 4 and 6 months. The groups were well matched with respect to physiology (Table 1), except for the weight of the testes. Testes of the Fmr1 null mice were 16 and 25% larger than those in the WT at 4 and 6 months of age, respectively. Arterial plasma leucine concentrations were lower in the 6-month-old mice for both WT and Fmr1 null mice (F(1,22) = 5.81; p = 0.02; two-way ANOVA, age by genotype); there was no main effect of genotype (F(1,22) = 0.15; p = 0.7).
Table 1.
Physiological variables in rCPS studies
|
|
4 months of age |
6 months of age |
||||
|---|---|---|---|---|---|---|
|
|
WT (6) |
Fmr1 null (8) |
WT (6) |
Fmr1 null (6) |
||
| Age (d) | 130 ± 2 | 131 ± 1 | 190 ± 2 | 194 ± 2 | ||
| Body weight (g) | 32 ± 2 | 33 ± 1 | 36 ± 2 | 33 ± 1 | ||
| Testes weight (g) | 0.19 ± 0.01 | 0.22 ± 0.01* | 0.18 ± 0.00 | 0.23 ± 0.01* | ||
| Body temperature (°C) | 37.4 ± 0.1 | 37.7 ± 0.2 | 37.5 ± 0.2 | 37.5 ± 0.2 | ||
| Hematocrit (%) | 41.8 ± 0.8 | 40.8 ± 0.5 | 42.2 ± 1.0 | 42.3 ± 1.4 | ||
| Arterial plasma glucose concentration (mm) | 8.10 ± 0.37 | 8.25 ± 0.44 | 8.28 ± 0.52 | 8.39 ± 0.73 | ||
| Arterial plasma leucine concentration (μm) | 203 ± 16 | 200 ± 10 | 165 ± 10 | 178 ± 13 | ||
| Arterial plasma corticosterone (ng/ml)a
|
ND |
ND |
267 ± 35 |
217 ± 28 |
||
Values are the means ± SEM for the number of mice indicated in parentheses. ND, Not determined. Data were analyzed by means of two-way ANOVA (age by genotype) with post hoc t tests. There were no statistically significant interactions. Testes weight showed a statistically significant main effect of genotype: F(1,22) = 19.27; p = 0.0002. Arterial plasma leucine concentration showed a statistically significant main effect of age: F(1,22) = 5.81; p = 0.0247.
Statistically significantly different from age-matched WT;p < 0.05; t test.
Measured in a separate series of five WT and five Fmr1 null mice 15 min after the initiation of sampling of arterial blood.
We found no interaction between genotype and age and no effect of genotype on either global CPS or brain volume (Table 2), but the effect of age on global CPS was statistically significant (p < 0.0001). Brain volume was similar in all four groups.
Table 2.
Effects of null mutation for Fmr1 on whole brain at two ages
|
|
4 months of age |
6 months of age |
|
|
||||
|---|---|---|---|---|---|---|---|---|
|
|
WT (6) |
Fmr1 null (8) |
WT (6) |
Fmr1 null (6) |
F(1,22) age |
F(1,22) genotype |
||
| CPS (nmol · g−1 · min−1) | 4.1 ± 0.2 | 4.4 ± 0.1 | 3.0 ± 0.2 | 3.4 ± 0.3 | 26.88* | 2.13 | ||
| Volume (ml) |
0.46 ± 0.01 |
0.46 ± 0.02 |
0.45 ± 0.01 |
0.45 ± 0.01 |
0.45 |
0.00 |
||
Values are the means ± SEM for the number of mice indicated in parentheses. Results were analyzed by means of two-way ANOVA (age by genotype). There were no statistically significant interactions.
p < 0.0001; statistically significant main effect.
We determined rCPS in 75 regions of brain (Tables 3, 4, 5) and found a statistically significant interaction between age and genotype in only one region, the locus ceruleus (F(1,22) = 5.01; p = 0.036). We found statistically significant main effects of age in all of the regions except the arcuate nucleus and median eminence. On average rCPS values were 24 and 20% lower in 6-month-old mice compared with the 4-month-old animals in WT and Fmr1 null, respectively. Statistically significant main effects of genotype were not as widespread, occurring in 26 of the regions examined, mostly in thalamus, hypothalamus, and hippocampus. In all cases, mean rCPS values were higher in Fmr1 null mice compared with age-matched WT. Of the areas of cortex analyzed, only frontal association and posterior parietal cortex were statistically significantly affected by genotype (Table 3). In the hippocampus, statistically significant effects of genotype occurred in all sectors (CA1-CA3) of the pyramidal cell layer (Fig. 4), stratum oriens, fimbria, and in the molecular layer of the dentate gyrus. In CA1 and stratum oriens, increases in rCPS were 28 and 23%, respectively, in the 6-month-old Frm1 null compared with age-matched WT. We also found a statistically significant effect of genotype in the presubiculum but not in the subiculum (Table 3). In five of the seven areas of thalamus analyzed, we found statistically significant effects of genotype. Some of the largest effects occurred in regions of hypothalamus (Table 4, Fig. 5). In the supraoptic, paraventricular, lateral hypothalamic, and periventricular nuclei, rCPS values were 24-37% higher in the 6-month-old Frm1 null compared with age-matched WT. Statistically significant effects of genotype also occurred in basolateral amygdala (Table 3), mammillary bodies, median raphe, and dorsal motor nucleus of the vagus (Table 5).
Table 3.
Effects of null mutation for Fmr1 on rCPS (nmol · g−1 · min−1) in regions of cortex, hippocampal formation, and subcortical areas
|
|
4 months of age |
6 months of age |
|
|
||||
|---|---|---|---|---|---|---|---|---|
| Region |
WT (6) |
Fmr1 null (8) |
WT (6) |
Fmr1 null (6) |
F(1,22) age |
F(1,22) genotype |
||
| Cortex | ||||||||
| Frontal association | 4.2 ± 0.3 | 4.8 ± 0.2 | 3.4 ± 0.2 | 4.0 ± 0.4 | 11.38** | 4.67* | ||
| Prelimbic | 5.2 ± 0.3 | 5.7 ± 0.2 | 4.3 ± 0.3 | 4.7 ± 0.4 | 11.14** | 1.79 | ||
| Piriform | 7.0 ± 0.4 | 7.7 ± 0.3 | 5.6 ± 0.3 | 6.1 ± 0.5 | 16.58*** | 2.52 | ||
| Primary motor | 4.8 ± 0.3 | 5.2 ± 0.2 | 3.8 ± 0.2 | 4.3 ± 0.4 | 11.53** | 3.07 | ||
| Primary somatosensory | 5.2 ± 0.3 | 5.6 ± 0.2 | 4.2 ± 0.2 | 4.7 ± 0.4 | 10.86** | 1.80 | ||
| Barrel | 5.4 ± 0.3 | 5.9 ± 0.2 | 4.3 ± 0.3 | 4.6 ± 0.4 | 18.38*** | 2.01 | ||
| Cingulate | 5.7 ± 0.4 | 6.5 ± 0.2 | 4.8 ± 0.2 | 5.0 ± 0.4 | 13.45** | 2.89 | ||
| Posterior parietal | 5.0 ± 0.3 | 5.9 ± 0.2† | 4.0 ± 0.2 | 4.7 ± 0.3 | 18.23*** | 8.40** | ||
| Auditory | 5.3 ± 0.4 | 5.7 ± 0.2 | 4.0 ± 0.3 | 4.3 ± 0.3 | 23.09*** | 1.81 | ||
| Visual | 5.4 ± 0.4 | 5.8 ± 0.2 | 4.0 ± 0.3 | 4.4 ± 0.4 | 22.14*** | 2.02 | ||
| Hippocampal formation | ||||||||
| Hippocampus | ||||||||
| Stratum oriens | 3.1 ± 0.2 | 3.7 ± 0.2 | 2.5 ± 0.2 | 3.2 ± 0.4 | 5.06* | 6.22* | ||
| Pyramidal cell layer, CA1 | 6.7 ± 0.4 | 7.6 ± 0.2 | 5.0 ± 0.3 | 6.2 ± 0.4† | 22.39*** | 9.95** | ||
| Pyramidal cell layer, CA2 | 10.2 ± 0.5 | 11.2 ± 0.3 | 7.4 ± 0.6 | 8.6 ± 0.6 | 33.01*** | 5.42* | ||
| Pyramidal cell layer, CA3 | 10.1 ± 0.6 | 11.1 ± 0.2 | 7.7 ± 0.3 | 8.9 ± 0.7 | 23.31*** | 5.57* | ||
| Stratum lucidem | 4.9 ± 0.2 | 5.2 ± 0.2 | 4.0 ± 0.2 | 4.3 ± 0.3 | 16.90*** | 2.40 | ||
| Stratum radiatum | 2.6 ± 0.2 | 2.9 ± 0.2 | 1.9 ± 0.2 | 2.3 ± 0.1 | 14.16** | 3.49 | ||
| Stratum lacunosum moleculare | 2.2 ± 0.2 | 2.5 ± 0.2 | 1.6 ± 0.1 | 1.9 ± 0.1 | 12.17** | 3.46 | ||
| Dentate gyrus | ||||||||
| Molecular layer | 2.6 ± 0.2 | 3.0 ± 0.1 | 1.9 ± 0.2 | 2.3 ± 0.1 | 18.66*** | 4.47* | ||
| Granular layer | 7.6 ± 0.6 | 8.1 ± 0.2 | 5.9 ± 0.4 | 6.6 ± 0.5 | 14.20** | 2.56 | ||
| Polymorph layer | 5.2 ± 0.5 | 5.7 ± 0.2 | 4.1 ± 0.2 | 4.6 ± 0.2 | 13.22** | 2.59 | ||
| Presubiculum | 5.7 ± 0.3 | 6.4 ± 0.3 | 3.5 ± 0.8 | 5.0 ± 0.4 | 14.89*** | 5.40* | ||
| Subiculum | 5.7 ± 0.3 | 6.2 ± 0.3 | 4.2 ± 0.3 | 4.9 ± 0.4 | 19.53*** | 3.31 | ||
| Subcortical areas | ||||||||
| Basolateral amygdala | 5.7 ± 0.4 | 6.4 ± 0.2 | 4.5 ± 0.2 | 5.2 ± 0.4 | 18.97*** | 5.62* | ||
| Bed nucleus of the stria terminalis | 3.3 ± 0.2 | 3.6 ± 0.1 | 2.6 ± 0.1 | 2.8 ± 0.2 | 16.77*** | 2.17 | ||
| Nucleus accumbens | 3.4 ± 0.2 | 3.7 ± 0.1 | 2.8 ± 0.1 | 3.0 ± 0.3 | 10.59** | 2.04 | ||
| Medial septal nucleus | 5.2 ± 0.4 | 5.7 ± 0.2 | 4.4 ± 0.2 | 4.9 ± 0.4 | 7.12* | 2.69 | ||
| Lateral septal nucleus |
4.1 ± 0.3 |
4.4 ± 0.1 |
3.5 ± 0.1 |
3.9 ± 0.4 |
5.52*
|
2.08 |
||
Values are the means ± SEM for the number of mice indicated in parentheses. Results were analyzed by means of two-way ANOVA (age by genotype). There were no statistically significant interactions.
p < 0.05; statistically significant main effect.
p < 0.01; statistically significant main effect.
p < 0.001; statistically significant main effect.
p < 0.05; statistically significantly different from age-matched WT; post hoc t test.
Table 4.
Effects of null mutation for Fmr1 on rCPS (nmol · g−1 · min−1) in thalamus and hypothalamus
|
|
4 months of age |
6 months of age |
|
|
||||
|---|---|---|---|---|---|---|---|---|
| Region |
WT (6) |
Fmr1 null (8) |
WT (6) |
Fmr1 null (6) |
F(1,22) age |
F(1,22) genotype |
||
| Thalamus | ||||||||
| Lateral dorsal nucleus | 4.4 ± 0.3 | 5.0 ± 0.2 | 3.4 ± 0.2 | 3.9 ± 0.3 | 16.89*** | 5.10* | ||
| Medial dorsal nucleus | 4.2 ± 0.3 | 5.0 ± 0.2 | 3.4 ± 0.2 | 4.1 ± 0.3 | 11.17** | 6.97* | ||
| Ventral medial posterior nucleus | 4.6 ± 0.3 | 5.4 ± 0.2 | 3.7 ± 0.2 | 4.3 ± 0.3 | 13.47** | 5.84* | ||
| Anterior dorsal nucleus | 8.5 ± 0.4 | 9.4 ± 0.3 | 6.9 ± 0.6 | 7.6 ± 0.6 | 13.88** | 2.77 | ||
| Anterior ventral nucleus | 5.8 ± 0.2 | 6.5 ± 0.2 | 4.6 ± 0.3 | 5.0 ± 0.4 | 23.86*** | 3.45 | ||
| Medial habenula | 8.2 ± 0.6 | 9.7 ± 0.2 | 6.7 ± 0.4 | 7.6 ± 0.4 | 22.16*** | 10.01** | ||
| Lateral habenula | 5.7 ± 0.4 | 6.7 ± 0.2† | 4.6 ± 0.2 | 5.2 ± 0.4 | 18.72*** | 8.00** | ||
| Hypothalamus | ||||||||
| Supraoptic nucleus | 8.6 ± 0.6 | 9.7 ± 0.4 | 6.5 ± 0.4 | 8.7 ± 0.8† | 7.85* | 8.78** | ||
| Paraventricular nucleus | 8.3 ± 0.5 | 9.8 ± 0.4† | 6.2 ± 0.4 | 7.7 ± 0.6 | 17.33** | 9.05** | ||
| Lateral magnocellular | 9.2 ± 0.4 | 10.2 ± 0.4 | 6.6 ± 0.6 | 7.9 ± 0.8 | 20.45*** | 4.40* | ||
| Ventral | 7.5 ± 0.4 | 8.4 ± 0.3 | 4.7 ± 0.4 | 5.6 ± 0.5 | 46.64*** | 5.03* | ||
| Medial parvicellular | 6.3 ± 0.3 | 6.9 ± 0.3 | 4.1 ± 0.4 | 4.6 ± 0.4 | 42.45*** | 2.65 | ||
| Suprachiasmatic nucleus | 4.9 ± 0.3 | 5.8 ± 0.2† | 3.8 ± 0.3 | 4.8 ± 0.4 | 11.95** | 9.00** | ||
| Medial preoptic area | 5.6 ± 0.3 | 6.4 ± 0.3 | 3.2 ± 0.2 | 3.1 ± 0.3 | 95.28*** | 1.48 | ||
| Anterior area | 3.9 ± 0.1 | 4.3 ± 0.2 | 3.0 ± 0.2 | 3.4 ± 0.4 | 16.05*** | 3.84 | ||
| Lateral area | 3.9 ± 0.2 | 4.5 ± 0.1† | 3.0 ± 0.3 | 4.1 ± 0.3†† | 6.92* | 12.34** | ||
| Lateral anterior nucleus | 3.9 ± 0.2 | 4.4 ± 0.2 | 3.2 ± 0.2 | 3.7 ± 0.4 | 7.78* | 3.60 | ||
| Periventricular nucleus | 5.3 ± 0.3 | 6.0 ± 0.3 | 3.8 ± 0.3 | 5.0 ± 0.5† | 12.73** | 8.80** | ||
| Medial preoptic nucleus | 4.4 ± 0.2 | 4.8 ± 0.1 | 3.8 ± 0.2 | 3.9 ± 0.4 | 8.71** | 1.24 | ||
| Lateral preoptic nucleus | 3.9 ± 0.2 | 4.2 ± 0.1 | 3.1 ± 0.2 | 3.5 ± 0.4 | 11.22** | 3.16 | ||
| Arcuate nucleus | 6.2 ± 0.2 | 6.8 ± 0.3 | 5.0 ± 0.5 | 6.6 ± 0.6† | 2.90 | 6.64* | ||
| Median eminence | 3.7 ± 1.2 | 5.2 ± 1.2 | 4.1 ± 0.4 | 5.8 ± 0.4 | 0.28 | 2.80 | ||
| Dorsomedial nucleus | 4.6 ± 0.2 | 4.7 ± 0.2 | 3.7 ± 0.2 | 4.1 ± 0.4 | 7.16* | 0.66 | ||
| Ventromedial nucleus | 5.0 ± 0.2 | 5.1 ± 0.3 | 3.9 ± 0.2 | 4.5 ± 0.4 | 8.40** | 1.21 | ||
| Supramammillary body, medial |
5.1 ± 0.3 |
5.3 ± 0.2 |
3.7 ± 0.2 |
4.9 ± 0.4†
|
10.41**
|
5.82*
|
||
Values are the means ± SEM for the number of mice indicated in parentheses. Results were analyzed by means of two-way ANOVA (age by genotype). There were no statistically significant interactions.
p < 0.05; statistically significant main effect.
p < 0.01; statistically significant main effect.
p < 0.001; statistically significant main effect.
p < 0.05; statistically significantly different from age-matched WT; post hoc t test.
p < 0.01; statistically significantly different from age-matched WT; post hoc t test.
Table 5.
Effects of null mutation for Fmr1 on rCPS (nmol · g−1 · min−1) in regions of white matter, cerebellum, pons, and medulla
|
|
4 months of age |
6 months of age |
|
|
||||
|---|---|---|---|---|---|---|---|---|
| Region |
WT (6) |
Fmr1 null (8) |
WT (6) |
Fmr1 null (6) |
F(1,22) age |
F(1,22) genotype |
||
| White matter | ||||||||
| Anterior commissure | 2.1 ± 0.1 | 2.4 ± 0.1 | 1.7 ± 0.1 | 1.8 ± 0.2 | 19.02*** | 3.72 | ||
| Corpus callosum | 2.1 ± 0.1 | 2.3 ± 0.1 | 1.6 ± 0.1 | 1.7 ± 0.2 | 22.70*** | 2.17 | ||
| Cingulum | 2.5 ± 0.1 | 2.8 ± 0.1 | 2.0 ± 0.1 | 2.1 ± 0.2 | 20.03*** | 2.74 | ||
| Hippocampal fimbria | 2.0 ± 0.2 | 2.4 ± 0.2 | 1.5 ± 0.1 | 1.7 ± 0.1 | 16.03*** | 5.47* | ||
| Optic chiasm | 1.5 ± 0.2 | 1.9 ± 0.2 | 1.2 ± 0.1 | 1.3 ± 0.1 | 10.14** | 3.16 | ||
| Optic tract | 1.6 ± 0.2 | 2.1 ± 0.2 | 1.4 ± 0.1 | 1.5 ± 0.1 | 7.76* | 4.12 | ||
| Fornix | 2.4 ± 0.1 | 2.6 ± 0.1 | 2.2 ± 0.2 | 2.3 ± 0.2 | 4.31* | 2.05 | ||
| Cerebellum | ||||||||
| Vermis 6 | 6.7 ± 0.4 | 7.0 ± 0.2 | 5.1 ± 0.3 | 6.2 ± 0.8 | 7.59* | 2.76 | ||
| Vermis 7 | 6.4 ± 0.4 | 6.7 ± 0.2 | 4.7 ± 0.3 | 5.6 ± 0.6 | 14.08** | 2.90 | ||
| Vermis 8 | 6.4 ± 0.4 | 6.5 ± 0.2 | 4.6 ± 0.4 | 5.6 ± 0.5 | 14.08** | 2.12 | ||
| Vermis 9 | 7.0 ± 0.4 | 7.5 ± 0.2 | 5.3 ± 0.3 | 6.1 ± 0.6 | 17.10*** | 3.24 | ||
| Whole cerebellum | 6.8 ± 0.4 | 7.2 ± 0.2 | 4.8 ± 0.4 | 5.6 ± 0.5 | 24.88*** | 2.62 | ||
| Pons and medulla | ||||||||
| Dorsal raphe | 8.3 ± 0.6 | 8.7 ± 0.3 | 5.4 ± 0.5 | 7.1 ± 0.8 | 16.33*** | 3.76 | ||
| Median raphe | 7.3 ± 0.5 | 8.2 ± 0.4 | 5.2 ± 0.5 | 6.4 ± 0.6 | 16.79*** | 5.21* | ||
| Periaqueductal grey | 4.6 ± 0.2 | 4.8 ± 0.2 | 3.2 ± 0.3 | 3.8 ± 0.4 | 18.16*** | 2.09 | ||
| Ventral tegmental nucleus | 4.2 ± 0.3 | 4.8 ± 0.2 | 3.2 ± 0.2 | 3.6 ± 0.4 | 16.07*** | 3.10 | ||
| Pontine nucleus | 7.4 ± 0.4 | 7.9 ± 0.3 | 5.3 ± 0.5 | 6.1 ± 0.6 | 20.40*** | 2.56 | ||
| Locus ceruleus | 9.3 ± 0.7 | 9.2 ± 0.3 | 6.2 ± 0.4 | 8.5 ± 0.8† | 12.13** | 4.01 | ||
| Nucleus of the solitary tract | 5.9 ± 0.4 | 6.1 ± 0.2 | 4.2 ± 0.3 | 4.4 ± 1.0 | 10.55** | 0.21 | ||
| Dorsal motor nucleus of vagus | 8.0 ± 0.4 | 8.8 ± 0.3 | 6.2 ± 0.5 | 7.9 ± 0.6† | 9.71** | 7.59* | ||
| Hypoglossal nucleus |
7.2 ± 0.5 |
7.7 ± 0.2 |
5.3 ± 0.4 |
6.2 ± 0.5 |
17.36***
|
3.12 |
||
Values are the means ± SEM for the number of mice indicated in parentheses. Results were analyzed by means of two-way ANOVA (age by genotype). There were no statistically significant interactions.
p < 0.05; statistically significant main effect.
p < 0.01; statistically significant main effect.
p < 0.001; statistically significant main effect.
p < 0.05; statistically significantly different from age-matched WT; post hoc t test.
p < 0.01; statistically significantly different from age-matched WT; post hoc t test.
Figure 4.
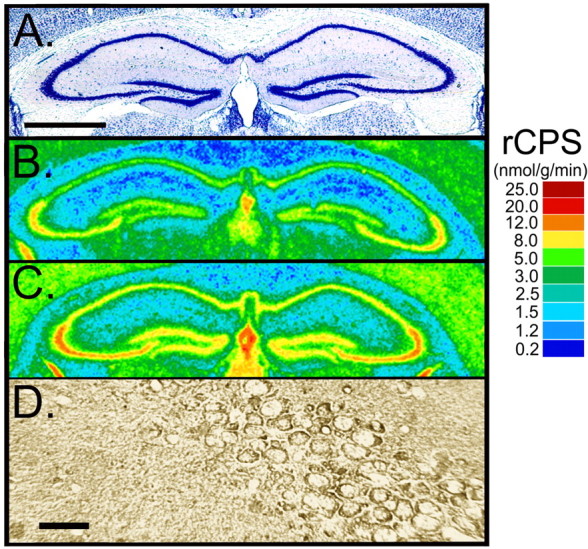
Digitized autoradiograms of coronal sections at the level of the dorsal hippocampus from representative 6-month-old WT (B) and Fmr1 null (C) mice. Images have been color coded for rCPS. The color bar (right) provides the calibration scale for the range of values of rCPS in nanomoles per gram per minute for each color. For comparison with the distribution of FMRP in a WT mouse brain, we used immunohistochemistry with a monoclonal antibody to FMRP. Immunostaining of FMRP in the CA1 sector of the pyramidal cell layer is shown in D. The CA1, CA2, and CA3 sectors of the pyramidal cell layer can be located in a thionin-stained section from a WT mouse (A). Scale bars: (in A) A-C, 1.0 mm; D, 35 μm. In A-D, dorsal is up and the left side of the brain is on the left.
Figure 5.

Digitized autoradiograms of coronal sections at the level of the anterior hypothalamus from representative 6-month-old WT (A) and Fmr1 null (B) mice. Images have been color coded for rCPS. The color bar (right) provides the calibration scale for the range of rCPS in nanomoles per gram per minute for each color. For comparison with the distribution of FMRP in a WT mouse brain, we used immunohistochemistry with a monoclonal antibody to FMRP (D). Supraoptic (SON), paraventricular (PVN), and suprachiasmatic (SCN) nuclei and optic chiasm (OC) can be located in a thionin-stained section from a WT mouse (C). Scale bars: (in C) A-C, 0.5 mm; D, 75 μm. In A-D, dorsal is up and the left side of the brain is on the left.
To compare the effects on rCPS with the regional distribution of FMRP, we used immunohistochemistry to localize FMRP in brain sections from 6-month-old WT animals (Figs. 4, 5). Immunohistochemical staining for FMRP in the anterior hypothalamus was most intense in the large cells of the supraoptic nucleus (Fig. 5D) in which the effect on rCPS was greatest. Immunolabeling for FMRP was also noteworthy in the pyramidal cells of the hippocampus (Fig. 4D).
Discussion
FMRP is thought to play a role in regulating experience-driven protein synthesis. Particularly provocative is the idea that it may function to repress translation primarily in postsynaptic terminals undergoing plasticity. Before the present study, the influence of FMRP on translation in vivo was not known. We addressed this question by measuring rCPS in conscious WT and Fmr1 null mice with the quantitative autoradiographic l-[1-14C]leucine method. Our results show that, in the absence of FMRP, rCPS is increased in selective brain regions. Effects of the mutation are noteworthy in hippocampus, thalamus, and hypothalamus.
We studied WT and Fmr1 null mice at 4 and 6 months of age and found large and widespread effects of age on rCPS. We chose this age range because it was well passed the developmental period and considerably before effects of aging are observed. Our results in the mouse are in keeping with our finding that mean CPS decreased by 12%, albeit not statistically significantly, between 35 and 60 d of age in rat (Sun et al., 1995). That rCPS continues to decrease after maturation may reflect the process of synaptic pruning that is a hallmark of the postadolescent period. In humans, decreases in gray matter volume between adolescence and young adulthood (Giedd et al., 1999) may also reflect synaptic pruning.
In Fmr1 null mice, effects of age on rCPS parallel those in WT but at a higher level, suggesting that pruning takes place even in the absence of FMRP. The effects of genotype show regional selectivity that may be a function of the regional distribution of FMRP. Immunohistochemistry for FMRP in WT showed note-worthy labeling in supraoptic nucleus and CA1 hippocampal pyramidal cells, areas in which the effect of genotype on rCPS at 6 months of age was >23%. There is also some correspondence between the regions most affected and the distribution of group 1 mGluRs (Shigemoto and Mizuno, 2000). This is of interest in light of the proposal that many of the symptoms of fragile X syndrome might be attributable to overactive signaling by group 1 mGluRs (Bear et al., 2004).
We measured rCPS with the l-[1-14C]leucine method, which is based on a compartmental model for the behavior of leucine in brain that includes recycling of unlabeled leucine from the break-down of tissue protein into the precursor pool (Smith et al., 1988). The constant λWB is a measure of recycling. Defined as the fraction of leucine in the precursor pool for protein synthesis derived from arterial plasma, λWB was determined in a separate series of experiments in both WT and Fmr1 null mice. Conditions were the same as those in which we measured rCPS, but the animals were somewhat younger. We have shown in rats that λWB is constant between 1 and 24 months (Smith et al., 1995; Sun et al., 1995), so this age difference should not confound our results. Because values of λWB measured in both genotypes were similar, we used the average value. We assumed that the fraction of recycled leucine in the precursor pool is constant from region to region and used λWB to calculate rCPS in all regions (Sun et al., 1992).
We surveyed 75 brain regions to determine the extent of effects on rCPS, and we considered the possibility that analysis of so many regions in the same series of animals may generate some false positives. If we apply the very conservative Bonferroni correction for multiple comparisons, a p ≤ 0.0007 would be required for statistical significance, and 35 regions would show a statistically significant effect of age and no region would show a statistically significant effect of genotype. Because this may result in false negatives, we reported the actual F and p values computed by the two-way ANOVA.
We considered a differential response of the two genotypes to stress as a possible explanation for our results. Effects of stress on rCPS in rats have been reported (Giaume et al., 1995; Baubet et al., 1996). Children with FrX may be more sensitive to stress than their siblings (Wisbeck et al., 2000; Hessl et al., 2002), and Fmr1 null mice may be more sensitive to stress than WT (Beckel-Mitchener et al., 2003). Our animals recovered from the surgical implantation of catheters and acclimated to the experiment environment for 24 h before rCPS measurement. They were freely moving, and, apart from the sampling of arterial blood, mice were not subjected to stressors during the study. Whereas measured plasma corticosterone concentrations were higher than basal levels (Muller et al., 2003), indicating that our mice were responding to stress during the study, both genotypes were similarly affected. It is, therefore, unlikely that a differential response to stress could account for differences in rCPS between the genotypes.
Some of the regions most affected in the Fmr1 null mice are consistent with the behavioral manifestations in the mouse and, to some extent, the clinical symptoms associated with FrX. In particular, changes in the hippocampus are consistent with learning and memory deficits in FrX and with deficits in performance on passive avoidance tests (Qin et al., 2002) and spatial learning tests (Kooy, 2003) in Fmr1 null mice. Effects in hypothalamus and brainstem may be related to increased sensory sensitivity and autistic-like behavior seen in FrX (Hagerman, 2002). The hypothalamus supplies a major input to the dorsal motor nucleus of the vagus, and, via the parasympathetic system, the dorsal motor nucleus of the vagus regulates visceral functions. Parasympathetic dysregulation, hyperarousal, and hyperactivity are all characteristic of FrX patients (Boccia and Roberts, 2000; Hagerman, 2002). Fmr1 null mice are hyperactive (Qin et al., 2002), are susceptible to audiogenic seizures, and have an increase in prepulse inhibition and auditory startle responses (Chen and Toth, 2001).
One interpretation of our results is that increased rCPS in Fmr1 null mice reflects an increase in the number of neurons and/or in the size of the dendritic arbor and number of spines in the mutant animals. Neuron density in neocortex in patients with FrX, however, does not differ from controls (Hinton et al., 1991). Furthermore, in sensory cortex of both FrX patients and adult Fmr1 null mice, the number of elongated immature spines is increased and the number of mature shaped spines is decreased (Hinton et al., 1991; Irwin et al., 2002), but changes in density of dendrites or spines are not evident. However, in our studies, rCPS was not significantly affected in areas of sensory cortex. It remains a possibility that changes do occur in spine and dendrite densities in brain regions particularly affected with respect to rCPS. A study of Timm staining in dentate gyrus mossy fiber terminals in Fmr1 null mice showed an increased staining in stratum oriens and the molecular layer (Ivanco and Greenough, 2002), both regions with statistically significant effects on rCPS.
The results of our study considered in the light of known RNA-binding properties of FMRP (Feng et al., 1997), the effects of FMRP on mRNA translation in vitro (Laggerbauer et al., 2001; Li et al., 2001), and the regulation by FMRP of translation of MAP-1B during synaptogenesis in cultured neurons (Lu et al., 2004) suggest that FMRP does indeed act to suppress protein synthesis in brain in vivo. Whereas certain messages may be preferentially affected by FMRP (Brown et al., 2001), our study points to an effect on protein synthesis that may be more general. It is highly unlikely that we could detect an effect on a small number of mRNAs with the autoradiographic method unless the small number of mRNAs could account for a major fraction of overall protein synthesis at the time of the study.
Whether the effects that we report are in soma or dendrites is difficult to discern at the level of resolution (∼50 μm) of the [14C]leucine autoradiographic method (Smith, 1983). There is increasing evidence of effects of FMRP in dendrites and synapses. Synaptic activation via mGluRs increases the synthesis of FMRP (Weiler et al., 1997) and affects the trafficking of FMRP and Fmr1 mRNA in dendrites (Antar et al., 2004). Studies of mRNA granules suggest that in vivo mGluR-activated translation from granules is ongoing and occurs at a higher rate in the absence of FMRP (Aschrafi et al., 2005). Enhanced mGluR-dependent long-term depression (Huber et al., 2002) and increased microtubule stability (Lu et al., 2004) in preparations from Fmr1 null mice suggest a role for FMRP in stabilization of synapses. Studies in synaptoneurosomes show increased assembly of polyribosomes in WT, but not Fmr1 null, mice in response to mGluR activation (Weiler et al., 2004), indicating that the regulatory effects of FMRP on translation in dendrites may be complex. Although we cannot draw any conclusions about FMRP and protein synthesis in dendrites from our studies, our results provide the first in vivo evidence of a role for FMRP in regulating translation and a map of where its effects are greatest.
Dysregulation of protein synthesis is very likely close to the underlying etiology of symptoms of FrX. Measurement of rCPS in patients with FrX syndrome may provide a means of measuring the progress of the disease and efficacy of treatments. With development of the l-[1-11C]leucine positron emission tomography method for measurement of rCPS in human subjects (Schmidt et al., 2005; Smith et al., 2005), we can now test this possibility in FrX patients.
Footnotes
We thank Robert Cohen for helpful discussions at the outset of this study.
Correspondence should be addressed to Carolyn Beebe Smith, Laboratory of Cerebral Metabolism, Building 36, Room 1A07, 36 Convent Drive, National Institutes of Health, Bethesda, MD 20892-4030. E-mail: beebec@intra.nimh.nih.gov.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255087-09$15.00/0
References
- Agranoff BW (1981) Learning and memory: biochemical approaches. In: Basic neurochemistry (Seigel GJ, Albers RW, Agranoff BW, Katzman R, eds), pp 801-820. Boston: Little, Brown and Company.
- Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ (2004) Metabotropic glutamate receptor activation regulates fragile X mental retardation protein and frma mRNA localization differentially in dendrites and at synapses. J Neurosci 24: 2648-2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschrafi A, Cunningham BA, Edelman GM, Vanderklish PW (2005) The fragile X mental retardation protein and group 1 metabotropic glutamate receptors regulate levels of mRNA granules in brain. Proc Natl Acad Sci USA 102: 2180-2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baubet V, Grange E, Sermet E, Giaume M, Gay N, Bobillier P (1996) Wide-spread increase in brain protein synthesis following acute immobilization stress in adult rat brain. Neurosci Lett 219: 187-190. [DOI] [PubMed] [Google Scholar]
- Bear M, Huber KM, Warren ST (2004) The mGluR theory of fragile X mental retardation. Trends Neurosci 27: 370-377. [DOI] [PubMed] [Google Scholar]
- Beckel-Mitchener AC, Churchill JD, Kim S, Estrada CM, Greenough WT (2003) Prolonged elevation of serum glucocorticoid (corticosterone) levels in fragile X knockout mice after acute stress. Soc Neurosci Abstr 29: 646.5. [Google Scholar]
- Boccia ML, Roberts JE (2000) Behavior and autonomic nervous system function assessed via heart period measures: the case of hyperarousal in boys with fragile X syndrome. Behav Res Methods Instrum Comput 32: 5-10. [DOI] [PubMed] [Google Scholar]
- Brown V, Vin P, Ceman S, Darnell JC, O'Donnell WR, Tennenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD (2001) Microarray identification of FMRP associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107: 477-487. [DOI] [PubMed] [Google Scholar]
- Caudy AA, Myers M, Hannon GJ, Hammond SM (2002) Fragile X related protein and VIG associate with the RNA interference machinery. Genes Dev 16: 2491-2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Toth M (2001) Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience 103: 1043-1050. [DOI] [PubMed] [Google Scholar]
- Devys D, Lutz Y, Rouyer N, Bellocq J-P, Mandel J-L (1993) The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X permutation. Nat Genet 4: 335-340. [DOI] [PubMed] [Google Scholar]
- Dutch-Belgian Fragile X Consortium (1994) Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 78: 23-33. [PubMed] [Google Scholar]
- Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST, Hersch SM (1997) Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci 17: 1539-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaume M, Grange E, Baubet V, Gay N, Sermet E, Sarda N, Bobillier P (1995) Cerebral protein synthesis alterations in response to acute and chronic immobilization stress in the rat. Brain Res 675: 121-126. [DOI] [PubMed] [Google Scholar]
- Giedd JN, Blumenthal J, Jeffries NO, Castellanos FX, Liu H, Zijdenbos A, Paus T, Evans AC, Rapoport JL (1999) Brain development during childhood and adolescence: a longitudinal MRI study. Nat Neurosci 2: 861-863. [DOI] [PubMed] [Google Scholar]
- Hagerman RJ (2002) The physical and behavioral phenotype. In: Fragile X syndrome: diagnosis, treatment, and research (Hagerman RJ, Hagerman PJ, eds) pp 3-109. Baltimore: Johns Hopkins UP.
- Hessl D, Glaser B, Dyer-Friedman J, Blasey C, Hastie T, Gunnar M, Reiss AL (2002) Cortisol and behavior in fragile X syndrome. Psychoneuroendocrinology 27: 855-872. [DOI] [PubMed] [Google Scholar]
- Hinton VJ, Brown WT, Wisniewski K, Rudelli RD (1991) Analysis of neocortex in three males with fragile X syndrome. Am J Med Genet 41: 289-294. [DOI] [PubMed] [Google Scholar]
- Hsu SM, Raine L, Fanger H (1981) Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem 29: 577-580. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF (2002) Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA 99: 7746-7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Idupulapati M, Gilbert ME, Harris JB, Chakravarti AB, Rogers EJ, Crisostomo R, Larsen BP, Mehta A, Alcantara CJ, Patel B, Swain R, Weiler IJ, Oostra BA, Greenough WT (2002) Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X mice. Am J Med Genet 111: 140-146. [DOI] [PubMed] [Google Scholar]
- Ishizuka A, Siomi MC, Siomi HA (2002) Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev 16: 2497-2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanco TL, Greenough WT (2002) Altered mossy fiber distributions in adult fmr1 (FVN) knockout mice. Hippocampus 12: 47-54. [DOI] [PubMed] [Google Scholar]
- Kooy RF (2003) Of mice and the fragile X syndrome. Trends Genet 19: 148-154. [DOI] [PubMed] [Google Scholar]
- Laggerbauer B, Ostareck D, Keidel E-M, Ostareck-Lederer A, Fischer U (2001) Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Gen 10: 329-338. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhang Y, Ku L, Wilkinson KD, Warren ST, Feng Y (2001) The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res 29: 2276-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DJ (1996) A protein synthesis-dependent late phase of cerebellar long-term depression. Neuron 17: 483-490. [DOI] [PubMed] [Google Scholar]
- Lu R, Wang H, Liang Z, Ku L, O'Donnell WT, Li W, Warren ST, Feng Y (2004) The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc Natl Acad Sci USA 101: 15201-15206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME (1999) Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 286: 785-790. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M (1967) Number and distribution of the apical dendritic spines of the layer V pyramidal cells in man. J Comp Neurol 131: 475-489. [DOI] [PubMed] [Google Scholar]
- Miller LJ, McIntosh DN, McGrath J, Shyu V, Lampe M, Taylor AK, Tassone F, Neitzel K, Stackhouse T, Hagerman RJ (1999) Electrodermal responses to sensory stimuli in individuals with fragile X syndrome: a preliminary report. Am J Med Genet 83: 268-279. [PubMed] [Google Scholar]
- Muller MB, Zimmermann S, Sillaber I, Hagemeyer TP, Deussing JM, Timpl P, Kormann SD, Droste SK, Kuhn R, Reul JMHM, Holsboer F, Wurst W (2003) Limbic corticotrophin-releasing hormone receptor 1 mediates anxiety-related behavior and hormonal adaptation to stress. Nat Neurosci 6: 1100-1107. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER (1994) Requirement of a critical period of transcription for induction of a late-phase of LTP. Science 265: 1104-1107. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2001) The mouse brain in stereotaxic coordinates. New York: Academic.
- Qin M, Kang J, Smith CB (2002) Increased rates of cerebral glucose metabolism in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA 99: 15758-15763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin M, Kang J, Smith CB (2003) Rates of cerebral protein synthesis increased in fragile X knockout mice in vivo Soc Neurosci Abstr 29: 646.8. [Google Scholar]
- Rousseau F, Heitz D, Tarleton J, MacPherson J, Malmgren H, Dahl N, Barnicoat A, Mathew C, Mornet E, Tejada I, Maddelena R, Spiegel R, Schnizel A, Marcos JAG, Schorderet DF, Schaap T, Maccioni L, Russo S, Jacobs PA, Schwartz C, et al. (1994) A multicenter study on genotype-phenotype correlations in fragile X syndrome, using direct diagnosis with probe StB12:3: the first 2253 cases. Am J Hum Genet 55: 225-237. [PMC free article] [PubMed] [Google Scholar]
- Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure-Kamionowska M, Connell F, Wisniewski HM (1985) Adult fragile X syndrome: cliniconeuropathologic findings. Acta Neuropath (Berl) 67: 289-295. [DOI] [PubMed] [Google Scholar]
- Schmidt KC, Cook MP, Qin M, Kang, Burlin TV, Smith CB (2005) Measurement of regional rates of cerebral protein synthesis with l-[1-11C]leucine and PET with correction for recycling of tissue amino acids. I. Kinetic modeling approach. J Cereb Blood Flow Metab 25: 617-628. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Mizuno N (2000) Metabotropic glutamate receptors-immunochemical and in situ hybridization analyses. In: Handbook of chemical neuroanatomy, Vol 18, Glutamate (Ottersen OP, Storm-Mathisen J, eds), pp 63-98. Amsterdam: Elsevier.
- Smith CB (1983) Localization of activity-associated changes in metabolism of the central nervous system with the deoxyglucose method: prospects for cellular resolution. In: Methods in cellular neurobiology, Vol 1 (Barker JL, McKelvey JF, eds), pp 269-317. New York: Wiley. [Google Scholar]
- Smith CB, Kang J (2000) Cerebral protein synthesis in a genetic mouse model of phenylketonuria. Proc Natl Acad Sci USA 97: 11014-11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CB, Deibler GE, Eng N, Schmidt K, Sokoloff L (1988) Measurement of local cerebral protein synthesis in vivo: influence of recycling of amino acids derived from protein degradation. Proc Natl Acad Sci USA 85: 9341-9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CB, Sun Y, Sokoloff L (1995) Effects of aging on regional rates of cerebral protein synthesis in the Sprague-Dawley rat: examination of the influence of recycling of amino acids derived from protein degradation into the precursor pool. Neurochem Int 27: 407-416. [DOI] [PubMed] [Google Scholar]
- Smith CB, Schmidt KC, Qin M, Burlin T, Cook M, Kang J, Saunders R, Bacher J, Carson RE, Channing M, Eckelman WC, Herscovitch P, Laverman P, Vuong B-K (2005) Measurement of regional rates of cerebral protein synthesis with l-[1-11C]leucine and PET with correction for recycling of tissue amino acids. II. Validation in rhesus monkeys. J Cereb Blood Flow Metab 25: 629-640. [DOI] [PubMed] [Google Scholar]
- Steward O, Schuman EM (2001) Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci 24: 299-325. [DOI] [PubMed] [Google Scholar]
- Sun Y, Deibler GE, Sokoloff L, Smith CB (1992) Determination of regional rates of cerebral protein synthesis in conscious adult rats adjusting for regional differences in recycling of leucine derived from protein degradation into the precursor pool. J Neurochem 59: 863-873. [DOI] [PubMed] [Google Scholar]
- Sun Y, Deibler GE, Jehle J, Macedonia J, Dumont I, Dang T, Smith CB (1995) Rates of local cerebral protein synthesis in the rat during normal postnatal development. Am J Physiol 268: R549-R561. [DOI] [PubMed] [Google Scholar]
- Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazleton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT (1997) Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci USA 94: 5395-5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Spangler CC, Klintsova AY, Grossman AW, Kim SH, Bertaina-Anglade V, Khaliq H, de Vries FE, Lambers FAE, Hatia F, Base CK, Greenough WT (2004) Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc Natl Acad Sci USA 101: 17504-17509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsh RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME (2001) Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA 98: 11024-11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisbeck JM, Huffman LC, Freund L, Gunnar M, Davis EP, Reiss AL (2000) Cortisol and social stressors in children with fragile X syndrome: a pilot study. J Dev Behav Pediatr 21: 278-282. [DOI] [PubMed] [Google Scholar]
- Zhang YQ, Bailey AM, Matthies HJG, Benden RB, Smith MA, Speese SD, Rubin GM, Broadie K (2001) Drosophila fragile X-related gene regulates the MAP1B homolog futsch to control synaptic structure and function. Cell 107: 591-602. [DOI] [PubMed] [Google Scholar]