

Abstract
High-frequency stimulation of the subthalamic nucleus (HFS-STN) is an effective treatment for alleviating the motor symptoms of parkinsonian patients. However, the neurochemical basis of its effects remains unknown. We showed previously that 1 h of HFS-STN in normal rats increases extracellular glutamate (Glu) level in the output nuclei of the STN, the globus pallidus (GP), and the substantia nigra pars reticulata (SNr), consistent with an increase in the activity of STN neurons. HFS-STN also increases GABA levels in the SNr, but the origin of this increase is unclear. We investigated the effectiveness of HFS-STN for improving Parkinson's disease symptoms, using intracerebral microdialysis to determine the extracellular Glu and GABA levels of the GP and SNr in response to HFS-STN in anesthetized hemiparkinsonian rats [6-hydroxydopamine lesion of the substantia nigra pars compacta (SNc)]. Basal levels of Glu and GABA in the GP and SNr were significantly higher in hemiparkinsonian than in intact rats. HFS-STN did not affect extracellular Glu level in the SNr of hemiparkinsonian rats but doubled the level of GABA. Ibotenic acid lesion of the GP abolished the increase in GABA levels in the SNr induced by HFS-STN in SNc-lesioned rats. These results provide neurochemical confirmation of the hyperactivity of the STN after dopaminergic denervation and suggest that the therapeutic effects of HFS-STN may result partly from the stimulation of pallidonigral fibers, thereby revealing a potential role for pallidal GABA in the inhibition of basal ganglial output structures during HFS-STN.
Keywords: subthalamic nucleus, substantia nigra pars reticulata, glutamate, GABA, basal ganglia, high-frequency deep brain stimulation, Parkinson's disease, microdialysis
Introduction
The subthalamic nucleus (STN) is a major component of the basal ganglia. It is involved in the transmission of cortical signals to the output nuclei of the basal ganglia, the substantia nigra pars reticulata (SNr), and the internal segment of the globus pallidum (Kitai and Deniau, 1981; Joel and Weiner, 1997; Smith et al., 1998). The STN plays a key role in the control of movement, and abnormal hyperactivity of the STN consecutive to the loss of dopaminergic (DA) cells in the substantia nigra pars compacta (SNc) is thought to play a critical role in the expression of Parkinson's disease (PD) symptoms (Miller and DeLong, 1987; Hollerman and Grace, 1992; Bergman et al., 1994; Hassani et al., 1996; Bevan et al., 2002). Thus, the STN constitutes a strategic target for the neurosurgical treatment of advanced PD.
In the last decade, high-frequency stimulation of the STN (HFS-STN) has become an increasingly used method to treat the parkinsonian motor syndrome and has been reported to induce clinical improvement in PD in both experimental animals and humans and drastically reduce daily levodopa requirements and dyskinesia (Benazzouz et al., 1993; Pollak et al., 1993; Limousin et al., 1995; Krack et al., 1998, 2003; Krause et al., 2001). The effects of HFS-STN look equivalent to those of an STN lesion (Benazzouz et al., 1995), suggesting that it may act by silencing STN neurons (Benabid et al., 2002; Dostrovsky and Lozano, 2002). Indeed, in vitro electrophysiological recordings have shown that HFS-STN may silence STN neurons via the depolarization-induced blockade of their activity (Beurrier et al., 2001; Magarinos-Ascone et al., 2002; Garcia et al., 2003). However, in vivo studies have suggested that this type of stimulation may also produce its beneficial effects by activating the axons of STN cells, STN afferents, or fibers passing close to the stimulation site (Windels et al., 2000, 2003; Dostrovsky and Lozano, 2002; Salin et al., 2002; Vitek, 2002; McIntyre et al., 2004a,b)
We further investigated the mechanisms underlying the effects of STN stimulation by in vivo intracerebral microdialysis analysis of the effects of HFS-STN on the extracellular glutamate (Glu) and GABA levels of the globus pallidus (GP) and SNr in normal and hemiparkinsonian rats. We also tested the hypothesis that passing fibers are stimulated from pallidal neurons by assessing the effects of HFS-STN on Glu and GABA levels in the SNr in normal and hemiparkinsonian rats with a unilateral GP lesion.
Materials and Methods
Animals. Studies were performed on male Sprague Dawley rats (Iffa Credo, Les Oncins, France) weighing between 280 and 350 g and housed under standard laboratory conditions (12 h light/dark cycle) with food and water provided ad libitum. Protocols conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (publication 865-23) and French Ministry of Agriculture regulations (authorization number 03-441).
Lesion procedures. All animals were anesthetized with chloral hydrate (400 mg/kg, i.p.) and secured in a Kopf stereotaxic apparatus (Phymep, Paris, France). For SNc lesioning, 15 animals, treated previously with desipramine (25 mg/kg, s.c.) to protect noradrenergic neurons, received an injection into the left SNc of 12 μg of 6-hydroxydopamine (6-OHDA) (Sigma, St. Quentin-Fallavier, France), dissolved in 4 μl of sterile 0.9% NaCl and 0.2% ascorbic acid, at a flow rate of 0.5 μl/min. The stereotaxic coordinates of the injection site relative to the bregma were anteroposterior (AP), -5.3 mm, lateral (L), +2.35 mm, and dorsoventral (DV), 7.5 mm, with the incisor bar at 3.3 mm below the interaural plane, according to the stereotaxic atlas of Paxinos and Watson (1982). After injections, animals were kept warm and allowed to recover from anesthesia. They were returned to the animal facility for 3 weeks, by which time the degeneration of DA neurons induced by the neurotoxin had stabilized, and were processed for microdialysis experiments. For unilateral GP lesions, 15 rats received local injections of 0.5 μg of ibotenic acid (Research Biochemicals, Illkirch, France). Ibotenic acid was dissolved in sterile NaCl (1 mg/ml) and infused at a flow rate of 0.2 μl/min into the left GP. Injections were performed at two sites per GP (0.5 μl each) to achieve homogeneous lesions (Konitsiotis et al., 1998; Miwa et al., 1998). Injection coordinates relative to the bregma were as follows: (1) anterior site: AP, -1.9 mm; L, 2.5 mm; DV, 6.5 mm; and (2) posterior site: AP, -1.2 mm; L, 3 mm; DV, 6.8 mm. For unilateral combined lesions (SNc plus GP), 15 rats were used, and there was a 5 d interval between the nigral 6-OHDA and injections of pallidal ibotenic acid.
Implantation of microdialysis probes and stimulation electrode. Rats were anesthetized by inhalation (1 L/min) of a 3% halothane/air mixture (22% O2, 78% N2) and mounted in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). The dorsal skull was exposed, and holes were drilled to facilitate the lowering of the dialysis probes bilaterally into the GP or the SNr and implantation of the stimulation electrode (left side, i.e., the same side as the 6-OHDA injection) in the STN. Stereotaxic coordinates were chosen according to the atlas of Paxinos and Watson (1982) and were as follows, relative to the bregma: (1) GP: AP, -1 mm; L, 3 mm; DV, 7.2 mm; (2) SNr: AP, -5.5 mm; L, 2.5 mm; DV, 8.4 mm; and (3) STN: AP, -3.7 mm; L, 2.5 mm; and DV, 8.4 mm. During microdialysis experiments, body temperature was maintained at 37°C with a feedback-controlled heating pad (Harvard Apparatus, Edenbridge, UK), and anesthesia was maintained by allowing the animal to inhale a 1% halothane/air mixture (1 L/min).
Electrical stimulation. Concentric stimulating bipolar electrodes (outer diameter, 200 μm; model NEX-100; Rhodes Medical Instruments, Woodland Hills, CA) were used. Stimuli were delivered with a World Precision Instruments (Stevenage, UK) acupulser and a stimulus isolation unit. Stimulation parameters corresponded to those used in clinical practice [frequency, 130 Hz, 60 μs rectangular pulse width, and 500 μA (3 V) intensity]. At the end of each experiment, an electrical lesion of the STN was made to check the electrode localization postmortem.
Microdialysis. Homemade microdialysis probes were prepared and used as described previously (Windels et al., 2000, 2003; Bruet et al., 2003). These consisted of a concentric arrangement of a stainless-steel tube (outer diameter, 0.4 mm) (Hamilton, Bonaduz, Switzerland) and a piece of polyethylene tubing (outer diameter, 1.09 mm) (Merck, Darmstadt, Germany) into which we placed a piece of silica tubing (outer diameter, 150 μm) (Merck). The silica tubing extended beyond the distal end of the steel tube and was covered by a cuprophan tubular dialysis membrane (Hospal, Lyon, France) sealed at the bottom. The length of the dialysis membrane was adapted according to the size of the brain nuclei studied: 2 mm for the GP and 1 mm for the SNr. The perfusion liquid flowed out of the distal end of the steel tube, passing proximally between the tube and the membrane (Tossman and Ungerstedt, 1986). The probes were perfused with artificial CSF (in mm: 149 NaCl, 2.8 KCl, 1.2 MgCl2, 1.2 CaCl2, and 5.4 glucose, pH 7.3) at a flow rate of 1 μl/min. Before implantation, each probe was tested in vitro in a standard amino acid solution to determine the recovery of amino acids (Tossman and Ungerstedt, 1986). After implantation, dialysate fractions were collected at 8 min intervals. The first 16 fractions were discarded to avoid the effects of parenchymal disturbance so that an approximate steady-state level was reached. The following eight fractions (64 min) were then collected to determine basal values, and the next eight dialysate fractions were collected during HFS-STN. Poststimulation effects were evaluated for ∼2 h by collecting the next 16 fractions. Dialysates were automatically collected with a refrigerated autosampler (Univentor, Zejton, Malta) and stored at -80°C until analysis.
Glutamate and GABA assay. The levels of Glu and GABA in the dialysate fractions were determined by using HPLC with fluorimetric detection, as described previously (Donzanti and Yamamoto, 1998; Windels et al., 2000). Briefly, samples and standards were derivatized with o-phthaldialdehyde. The injection was automatically processed by a refrigerated autoinjector (SIL 10 AXL; Shimadzu, Tokyo, Japan) into a 3 μm C18 reversed-phase column (100 × 4.6 mm). The mobile phase consisted of 0.05 m NaH2PO4, pH 5.8, with 12% acetonitrile, flowing at 1.2 ml/min and delivered by a Shimadzu LC-10AT pump. Extracellular levels of amino acids were estimated by rationing peak areas of each amino acid and their respective external standard (analytical software class LC10; Shimadzu). The running time for each determination was 30 min.
Histology. At the end of microdialysis experiments, animals without 6-OHDA-induced lesions (n = 8) were killed by decapitation under deep anesthesia; their brains were quickly removed from the skull and frozen in isopentane at -30°C. All animals bearing 6-OHDA lesions (n = 30) were perfused transcardially under chloral hydrate anesthesia with 100 ml of 0.9% NaCl, pH 7.2, supplemented with heparin (5 × 104 IU/ml), followed by 350 ml of cold fixative consisting of 4% paraformaldehyde in 0.1 m PBS, pH 7.3 (in mm: 2.6 KCl, 1.4 KH2PO4, 136 NaCl, and 8 NaH2PO4). Brains were quickly removed and immersed overnight at 4°C in 25% sucrose in 0.1 m phosphate buffer, pH 7.5, frozen in cooled (-40°C) isopentane, and then stored at -30°C. Serial frontal sections (30 μm thick) were cut with a cryostat microtome (Microm HM 500; Microm, Francheville, France). The correct location of the microdialysis probes and stimulation electrode was checked by collecting several tissue sections at the pallidal, nigral, and STN levels, according to the atlas of Paxinos and Watson (1982). The tissue sections were processed for classical cresyl violet staining. These histological controls were systematically performed for all of the animals of each experimental group. All animals presenting internal bleeding around the dialysis probes or electrodes were excluded (to prevent contamination of the microdialysates) (n = 2), as were animals with misplaced microdialysis probes or stimulation electrodes (n = 3). To assess the dopaminergic denervation induced by the nigral 6-OHDA injection, several tissue sections at the striatal and nigral levels from fixed brains of lesioned animals were processed by tyrosine hydroxylase (TH) immunohistochemistry. In this case, free-floating sections were thoroughly washed with TBS and incubated for 1 h in 0.3% Triton X-100 in TBS (TBST) and 3% normal rabbit serum (Miles, Elkhart, IN). Sections were then incubated with primary antisera diluted in TBST containing 1% normal rabbit serum and 1% sodium azide for 48 h, with shaking, at 4°C. The antiserum was diluted 1:100 for TH (mouse monoclonal; Roche Diagnostics, Meylan, France). Antibody binding was detected using the avidin-biotin-peroxidase complex. Diaminobenzidine (0.05%; Sigma) was used as chromagen, and 0.01% H2O2 (Merck, Rahway, NJ) was applied for 2-5 min, as described previously (Gesdon et al., 1979). Sections were then dehydrated, mounted in Eukitt, and covered with a coverslip before microscopy. GP lesions were evaluated by light microscopy to assess the loss of cell bodies and to observe the intense gliosis reaction leading to a shrinkage of this structure (see Fig. 2 D).
Figure 2.
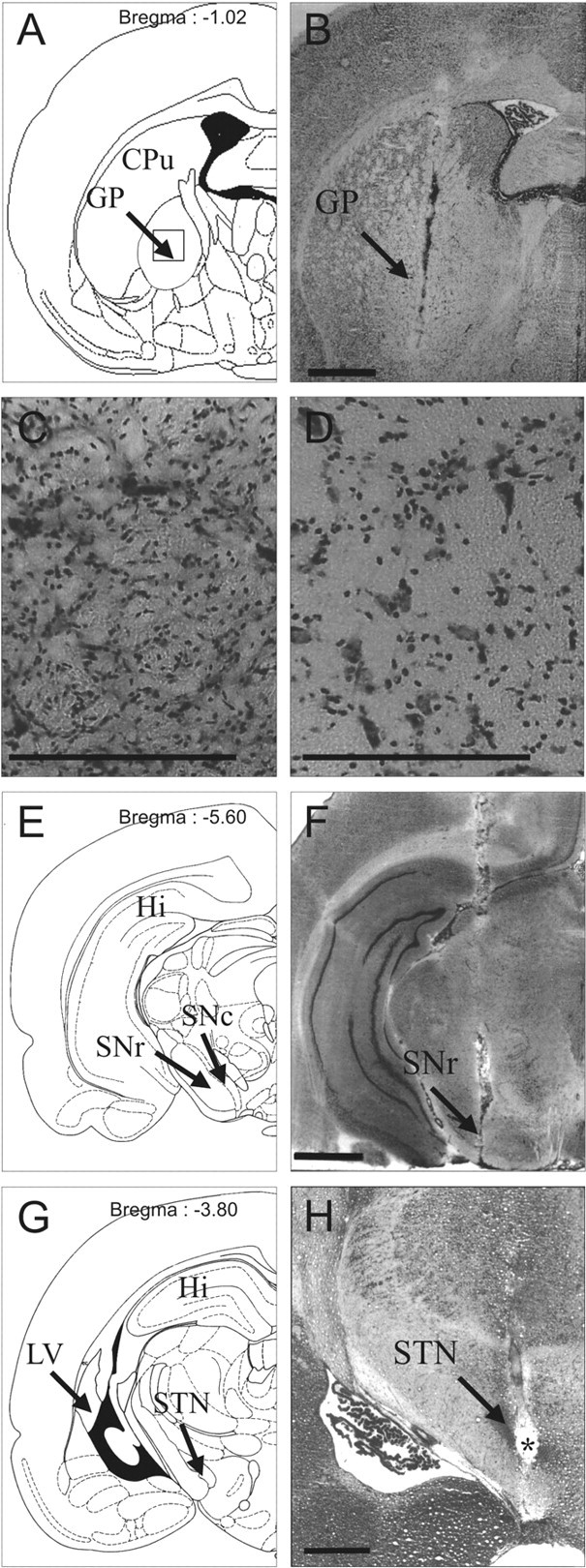
Photographs of cresyl violet-stained coronal rat-brain sections at pallidal (B-D), nigral (F), and subthalamic (H) levels and schematic diagrams (A, E, and G) adapted from the stereotaxic atlas of Paxinos and Watson (1982). B, F, H, Tissue sections at pallidal (B), nigral (F), and subthalamic (H) levels in hemiparkinsonian rats. Note the adequate implantation of microdialysis probes in the GP (B) and SNr (F) and the adequate localization of the stimulation electrode within the STN (H). H, Asterisk indicates stimulation point. Scale bar, 1 mm. C, D, Coronal sections at a higher magnification at the pallidal level (corresponding to the area delimited by a square in A) in GP-lesioned animals on the intact (C) or lesioned (D) side. Note the loss of neuronal cell bodies and the gliosis in the lesioned GP (D) after the two microinjections of 0.5 ml of ibotenic acid solution (1 mg/ml). Scale bar, 40 μm. CPu, Caudate-putamen nucleus; Hi, hippocampus; LV, lateral ventricle.
Statistical analysis. The basal levels of the assayed substances were expressed as the concentrations found in the dialysates. Basal levels of the amino acids in the SNr and GP between the different experimental groups were analyzed with a Mann-Whitney U test. When data were expressed as percentages of controls, the average concentration of the eight samples preceding the HFS-STN was defined as 100%. The HFS-STN effects on extracellular Glu and GABA levels were analyzed with one-way ANOVA with repeated measures over time. Comparisons with prestimulation baseline were performed using a Dunnett's or Games-Howell test. A level of p < 0.05 was considered critical for statistical significance.
Results
Histological controls
Three weeks after injection, all animals receiving a unilateral injection of 6-OHDA (n = 30) showed a massive loss of TH immunolabeling in the SNc (Fig. 1A) and in the striatum (caudate-putamen nucleus) (Fig. 1B) on the lesioned side. Dense TH immunolabeling was detected throughout the SNc, the ventral tegmental area, the striatum, the nucleus accumbens, and the olfactory tubercles on the intact side in 6-OHDA-injected rats (Fig. 1A,B). Figure 2, B, F, and H, illustrates the correct implantation of the microdialysis probe in the parenchyma of the GP (Fig. 2B) and SNr (Fig. 2F) and of the stimulation electrode in the STN (Fig. 2H). The symmetric location of microdialysis probes made it possible to directly compare the bilateral data obtained from homologous anatomical areas. Figure 2D shows the massive loss of neuronal cell bodies and gliosis within the lesioned GP, with respect to the intact side (Fig. 2C).
Figure 1.
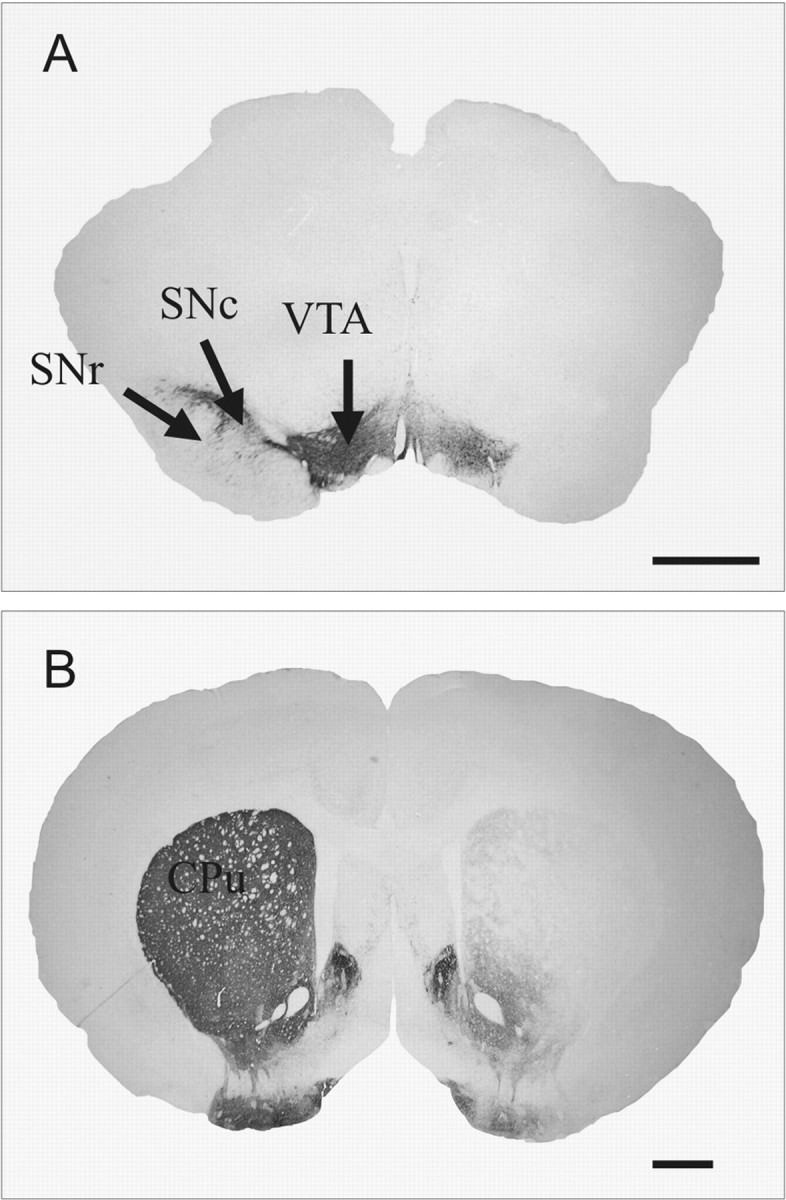
Photographs of TH-immunostained coronal rat-brain sections at the nigral (A) and striatal (B) levels in 6-OHDA-lesioned rats. Note the DA cell loss in the SNc (A) and the DA terminal loss in the striatum (B) on the lesioned side. CPu, Caudate-putamen nucleus; VTA, ventral tegmental area. Scale bar, 1 mm.
Basal levels of extracellular Glu and GABA in the SNr and GP are higher in hemiparkinsonian rats
Lesioning the SNc with 6-OHDA dramatically increased the basal level of Glu in output nuclei of the STN, the SNr, and the GP (Fig. 3A, Table 1), ipsilaterally to the lesion. In the SNr of SNc-lesioned rats, the mean Glu level was 4.21 ± 0.81 μm (n = 5), 9.5 times (p < 0.05) the basal levels reported previously for control intact animals (0.44 ± 0.06 μm; n = 7) (Windels et al., 2000). The Glu level in the GP was 16 times higher (p < 0.05) in SNc-lesioned animals (10.53 ± 1.36 μm; n = 6) than in controls (0.65 ± 0.04 μm; n = 6) (Windels et al., 2000). In contrast, lesioning of the GP did not result in significant changes in basal Glu levels in the SNr (0.27 ± 0.13 μm; n = 6, not significantly different from controls) (Fig. 3A, Table 1), and did not prevent the increase in Glu level induced by 6-OHDA treatment, with a level 2.6 and 3.4 times higher (1.15 ± 0.27 μm; n = 6; p < 0.05) in GP plus SNc-lesioned rats than in control and GP-lesioned animals, respectively. Although this increase in Glu levels remained detectable in GP plus SNc-lesioned rats, it was significantly lower (p < 0.05) (Fig. 3A) in GP plus SNc-lesioned rats than in SNc-lesioned animals. These high Glu levels in basal conditions (i.e., before HFS-STN) in the SNr and GP of hemiparkinsonian rats probably resulted from the hyperactivity of STN neurons induced by the SNc lesion (Hassani et al., 1996).
Figure 3.
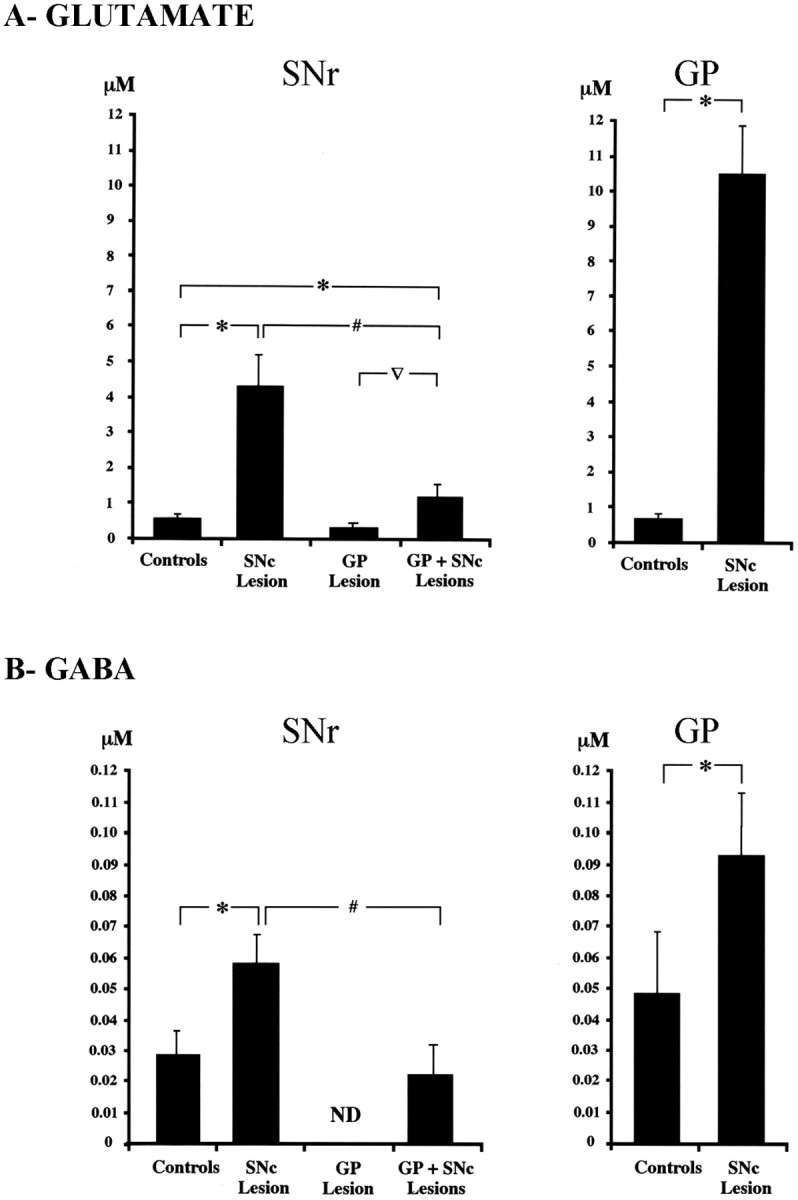
Basal levels of extracellular nigral and pallidal glutamate (A) and GABA (B) in controls and SNc-lesioned, GP-lesioned, and GP plus SNc-lesioned rats. *p < 0.05 compared with controls; #p < 0.05, comparison between SNc-lesioned and GP plus SNc-lesioned rats; ▿p < 0.05, comparison between GP-lesioned and GP plus SNc-lesioned rats. Values presented for the control group correspond to those obtained in a previous study (Windels et al., 2000). Error bars represent SEM.
Table 1.
Extracellular concentrations of glutamate and GABA (expressed as a micromolar concentration) measured in the SNr (n = 5-6) and GP (n = 6-7) on the stimulated side of intact, SNc-lesioned, GP-lesioned, and GP plus SNc-lesioned rats in basal conditions and under HFS-STN
|
|
|
Intacta |
SNc lesion |
GP lesion |
GP plus SNc lesions |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
Nuclei |
Basal |
HFS-STN |
Basal |
HFS-STN |
Basal |
HFS-STN |
Basal |
HFS-STN |
||||
| Glutamate (μm) | SNr | 0.44 ± 0.06 | 1.17 ± 0.16 (+166%; p < 0.01) | 4.21 ± 0.81 | 3.87 ± 0.46 (no effect) | 0.27 ± 0.13 | 0.45 ± 0.04 (+70%; p < 0.05) | 1.15 ± 0.27 | 1.03 ± 0.15 (no effect) | ||||
| GP | 0.65 ± 0.04 | 1.76 ± 0.11 (+172%; p < 0.01) | 10.5 ± 1.36 | 9.65 ± 0.58 (no effect) | |||||||||
| GABA (μm) | SNr | 0.028 ± 0.009 | 0.049 ± 0.015 (+76%; p < 0.05) | 0.056 ± 0.013 | 0.118 ± 0.029 (+112%; p < 0.05) | Not detectable | Not detectable | 0.02 ± 0.01 | 0.022 ± 0.01 (no effect) | ||||
|
|
GP |
0.048 ± 0.03 |
No effect |
0.094 ± 0.02 |
0.086 ± 0.01 (no effect) |
|
|
|
|
||||
Values presented for the intact rats correspond to those obtained in our previous study (Windels et al., 2000).
Interestingly, 6-OHDA treatment also doubled the mean basal GABA level in both the SNr, from 0.028 ± 0.09 μm in intact animals (n = 6) to 0.056 ± 0.03 μm in SNc-lesioned rats (n = 5; p < 0.05), and the GP, from 0.048 ± 0.002 μm in control animals (n = 6) to 0.094 ± 0.02 μm in SNc-lesioned rats (n = 6; p < 0.05) (Fig. 3B, Table 1). Basal GABA in the SNr may be released mostly by GABAergic fibers originating from the GP, because GABA was not detected in the SNr after lesioning of the GP. However, lesioning of the SNc with 6-OHDA increased the basal level of GABA in the SNr, with a level of 0.02 ± 0.01 μm (n = 6) recorded in GP plus SNc-lesioned rats (Fig. 3B, Table 1).
HFS-STN in hemiparkinsonian rats does not affect SNr extracellular Glu levels but increases GABA levels
In SNc-lesioned rats, HFS-STN induced no change in the Glu levels of both the SNr (n = 5) and the GP (n = 6) on the stimulated (ipsilateral to the SNc lesion) or the unstimulated (contralateral) side (Fig. 4, Table 1). Glu levels remained stable at around baseline values throughout the microdialysis experiment. This effect is in contrast to that of HFS-STN in intact rats, which strongly increases Glu levels in the SNr and GP (Table 1) (Windels et al., 2000). Thus, STN stimulation in SNc-lesioned rats fails to affect extracellular Glu levels in the output nuclei, possibly because the basal Glu level was already increased considerably by the dopaminergic lesion.
Figure 4.

Extracellular glutamate levels determined at 8 min intervals in the SNr (A) and GP (B) ipsilateral or contralateral to the stimulation in 6-0HDA SNc-lesioned rats. The prestimulation period (PRE-STIM; fractions 1-8) gave eight dialysates, whereas the stimulation period (STIM; fractions 9-16) and poststimulation period (POST-STIM; fractions 17-36) gave eight and 20 dialysates, respectively. The mean ± SEM of the eight dialysates collected before HFS-STN was used as the baseline. Results are expressed as a percentage of variation of this baseline value. Each percentage represents the mean variations ± SEM calculated from six animals. Note that Glu levels were not affected significantly by HFS-STN on either side in the GP and SNr. Error bars represent SEM.
The picture regarding the effect of HFS-STN on GABA levels was very different. In contrast to what was observed for Glu levels, HFS-STN induced a gradual increase in the extracellular GABA level in 6-OHDA-treated rats in the ipsilateral SNr during the 1 h HFS period (fractions 8-16). This increase was maximal in the last 16 min of the stimulation period (+112 ± 25%; n = 5; p < 0.05) (Fig. 5A). GABA level remained high during the poststimulation period through fractions 17-24 and then gradually decreased. This effect was specific to the ispilateral side, because the HFS-STN did not affect extracellular GABA level in the contralateral SNr (n = 5) (Fig. 5A). This effect is similar to that reported previously in intact rats (Table 1) (Windels et al., 2000). In contrast, HFS-STN did not affect the GABA level in the GP in either 6-OHDA-treated rats (n = 6) (Fig. 5B) or intact animals (Windels et al., 2000).
Figure 5.

Extracellular GABA levels determined at 8 min intervals in the SNr (A) and GP (B) ipsilateral or contralateral to stimulation in 6-OHDA SNc-lesioned rats (B). The prestimulation period (PRE-STIM; fractions 1-8) gave eight dialysates, whereas the stimulation period (STIM; fractions 9-16) and poststimulation period (POST-STIM; fractions 17-36) gave eight and 20 dialysates, respectively. The mean ± SEM of the eight dialysates collected before HFS-STN was used as the baseline. Results are expressed as a percentage of variation of this baseline value. Each percentage represents the mean variations ± SEM calculated from six animals. The data were analyzed using a one-way ANOVA test with repeated measures over time followed by a Dunnett's or Games-Howell test (n = 6 in each group). For SNr, F(2,24) = 21.68 and p < 0.05. Note the increase in extracellular GABA level induced by HFS-STN in the SNr ipsilateral to the stimulated side, with no significant change in GP on either side. *p < 0.05 compared with baseline values. Error bars represent SEM.
The HFS-STN-induced increase in SNr GABA level requires an intact GP
To check whether the source of GABA in the SNr could be the pallidonigral GABAergic fibers stimulated by HFS-STN, we measured the variations in Glu and GABA levels in the ipsilateral SNr of GP-lesioned and GP plus SNc-lesioned rats. GP lesions did not prevent the increase in extracellular Glu level in the SNr elicited by HFS-STN in nonhemiparkinsonian rats, with a significant 70 ± 10% maximum increase (n = 6; p < 0.05) obtained in the fifth fraction after the beginning of stimulation (Fig. 6A, Table 1). However, the increase in Glu level detected here, probably through the excitation of intact subthalamonigral neurons, is of lower amplitude in GP-lesioned animals than in controls. Extracellular Glu level remained significantly high and stable throughout the poststimulation period. In 6-OHDA-treated rats, lesioning of the GP did not change the absence of effect of HFS-STN on the SNr Glu level (n = 6) (Fig. 6B, Table 1).
Figure 6.

A, Extracellular Glu levels determined at 8 min intervals in the SNr ipsilateral to stimulation in ibotenic acid GP-lesioned rats. The prestimulation period (PRE-STIM; fractions 1-8) gave eight dialysates, whereas the stimulation period (STIM; fractions 9-16) and poststimulation period (POST-STIM; fractions 17-30) consisted of eight and 14 dialysates, respectively. The mean ± SEM of the eight dialysates collected before HFS-STN was used as the baseline. Results are expressed as a percentage of variation of this baseline value. Each percentage represents the mean variations ± SEM calculated from five animals. The data were analyzed using a one-way ANOVA test with repeated measures over time followed by a Dunnett's or Games-Howell test (n = 5). For SNr, F(2,18) = 23.456 and p < 0.05. Note the increase in extracellular Glu level induced by HFS-STN on the ipsilateral, stimulated side. *p < 0.05 compared with baseline values. B, C, Extracellular Glu (B) and GABA (C) levels, determined at 8 min intervals in the SNr ipsilateral to stimulation in GP plus SNc-lesioned rats. The prestimulation period (fractions 1-8) gave eight dialysates, where as the stimulation (fractions 9-16) and poststimulation (fractions 17-36) periods gave eight and 20 dialysates, respectively. The mean ± SEM of the eight dialysates collected before HFS-STN was used as the baseline. Results are expressed as a percentage of variation of this baseline value. Each percentage represents the mean variations ± SEM calculated from five animals. Note that HFS-STN did not affect extracellular Glu and GABA levels on the ipsilateral, stimulated side. Error bars represent SEM.
In contrast, the increase in the SNr GABA level induced by HFS-STN in intact rats was not observed in GP-lesioned rats (n = 6) (Fig. 6B, Table 1). GABA was undetectable before, during, and after HFS-STN. Similarly, the HFS-STN-induced increase in the SNr GABA level seen in rats with SNc lesions was abolished in rats bearing GP plus SNc lesions (Fig. 6B). Therefore, the increase in GABA level in the SNr induced by stimulation probably involved stimulation of the GABAergic fibers originating from the GP.
Discussion
SNc lesions induced a dramatic increase in extracellular Glu level in both the SNr and the GP. This extracellular Glu could come from STN axon terminals, glial cells, or both (Danbolt, 2001; Parpura et al., 2004). However, STN lesioning strongly decreases (by 80%) the basal Glu level in the SNr (Rosales et al., 1997) and its increase in response to HFS-STN (our unpublished observation). Therefore, most of the extracellular Glu detected in the output nuclei of the STN is probably linked to STN neuron activity. Such observations thus provide neurochemical confirmation of the hyperactivity of subthalamopallidal and subthalamonigral pathways induced by dopaminergic denervation. An increase in GABA level, smaller than the Glu level, was also measured in both SNr and GP in hemiparkinsonian rats. HFS-STN in hemiparkinsonian rats did not increase the already high level of Glu in the SNr but strongly increased the level of GABA. A lesion of the GP had little effect on the Glu level in the SNr, indicating an absence of hyperactivity of the STN not predicted by the “classic” model of basal ganglia functioning (Albin et al., 1989). Moreover, the GP lesion did not prevent the increase in basal levels of Glu in the SNr induced by the 6-OHDA treatment, suggesting that STN hyperactivity is not simply a consequence of the decrease in GP activity. Last, GP lesion completely abolished basal and stimulated GABA in the SNr. This suggests that GP GABAergic neurons constitute a major source of GABA in the SNr and that the stimulation of passing pallidonigral GABAergic fibers by HFS-STN is involved in its effects on basal ganglia output activity. These new data suggest that we should reevaluate the true contribution of the STN and GP to the effects of dopaminergic denervation and reconsider the mechanisms involved in HFS-STN effects.
Changes in basal Glu and GABA level induced by SNc lesioning
Many studies have shown that the loss of dopaminergic nigral neurons in animals and PD patients leads to hyperactivity of the subthalamopallidal, subthalamonigral, and corticostriatal pathways (Miller and DeLong, 1987; Bergman et al., 1990, 1994; DeLong, 1990; Hollerman and Grace, 1992; Calabresi et al., 1993; Bevan et al., 2002; Levy et al., 2002). Changes in STN firing probably lead to abnormal activity of basal ganglia output nuclei and to motor disturbances attributable to imbalance between the direct striatonigral and the trans-subthalamic pathways (Albin et al., 1989; Lannes and Micheletti, 1994; Chesselet and Delfs, 1996; Obeso et al., 1997). The large increase in Glu level in the SNr and GP in hemiparkinsonian rats under anesthesia is consistent with previous electrophysiological data showing that SNc lesion increases the discharge rate of STN neurons; the discharge pattern of these cells is also more “bursty” after SNc lesion (Hassani et al., 1996). This increase in Glu level in the SNr, which may reflect the hyperactivity of STN neurons, cannot solely be attributable to the removal of pallidal inhibition, because it persists in rats with GP plus SNc lesions. Similar conclusions were reached by Hassani et al. (1996), who evaluated the effects of pallidal and SNc lesions on electrophysiological activity in STN neurons. The change in SNr Glu levels after dopaminergic denervation may be attributable to an effect restricted to the STN or related structures. Many studies have reported direct dopaminergic innervation of the STN (Brown et al., 1979; Meibach and Katzman, 1979; Campbell et al., 1985; Hassani et al., 1997; Smith et al., 1998), which may control STN neuron activity directly or via presynaptic modulation of neurotransmitter release (Kreiss et al., 1997; Ni et al., 2001; Shen et al., 2003; Cragg et al., 2004).
The level of GABA in the SNr also increased in hemiparkinsonian rats. SNr neurons receive dense GABAergic inputs from the striatum, GP, and SNr neurons themselves, via axon collaterals playing a major role in the regulation of SNr neuron activity (Albin et al., 1989; Alexander and Crutcher, 1990; Mink, 1996; Celada et al., 1999). No basal GABA was detected in the SNr of rats with GP lesions, indicating that most of this GABA comes from the GP. This result is consistent with previous electrophysiological data suggesting that pallidonigral inputs predominate (Celada et al., 1999). GP lesion did not prevent the increase in basal GABA induced by dopaminergic denervation but may have decreased it. Thus, although part of the increase in nigral GABA may result from an increase in the activity of GP neurons (through the increase in Glu release from STN neuron terminals), other major sources of GABA, such as SNr neuron collaterals or other structures, appear to be involved.
Effects of HFS-STN
HFS-STN is an effective treatment for PD, but the mechanisms involved are unclear (Benabid et al., 2002; Dostrovsky and Lozano, 2002; Vitek, 2002; McIntyre et al., 2004a,b). In parkinsonian patients and animal models of PD, lesion (Bergman et al., 1990; Aziz et al., 1991; Guridi and Obeso, 2001) or pharmacological inactivation of the STN and HFS-STN (Benazzouz et al., 1993; Wichmann et al., 1994; Limousin et al., 1995, 1998; Krack et al., 2003) have similar beneficial effects on PD motor symptoms. Consequently, it has been suggested that HFS-STN may inhibit STN neuronal activity, reducing the excitatory drive of basal ganglia output nuclei. Such an effect has been observed in electrophysiological studies in vivo (Burbaud et al., 1994; Benazzouz et al., 1995, 2000; Tai et al., 2003) and in vitro (Beurrier et al., 2001; Magarinos-Ascone et al., 2002). However, recent electrophysiological data have shown that HFS-STN in monkeys increases the activity of basal ganglia output nuclei (Hashimoto et al., 2002, 2003) and that HFS may drive the STN activity of rat slices in vitro (Garcia et al., 2003). Consistent with these observations, we reported previously that extracellular Glu and GABA levels increase in the SNr during HFS-STN, suggesting the involvement of both excitatory and inhibitory fibers in the action of HFS-STN (Savasta et al., 2002).
We showed here that dopaminergic denervation prevented the increase in Glu level in both the GP and SNr normally triggered by HFS-STN in intact rats. This lack of effect on Glu level in STN target nuclei may be attributable to the hyperactivity of STN neurons in hemiparkisonian rats, with the level of Glu released already being too high for additional increase under stimulation. However, an increase in extracellular GABA level in the ipsilateral SNr was detected and was even greater in hemiparkinsonian rats. A recent in vivo electrophysiological study in intact rats also showed an increase in GABAergic synaptic transmission by HFS-STN (Maurice et al., 2003). This effect on GABA level was abolished by GP lesioning in intact and hemiparkinsonian rats. Thus, the stimulation of pallidonigral neurons by HFS-STN is sufficient to account for the increase in GABA release in the SNr, because GP neurons send axon collaterals to the SNr (Kita and Kitai, 1987, 1994).
In deep brain stimulation, electrodes are placed in a complex volume conductor in close proximity to somas, dendrites, and presynaptic and postsynaptic axons. It is unclear which elements are most susceptible to stimulation in our experiments (Ranck, 1975); however, in the cortex, axons have been reported to be more sensitive than neuronal somas (Nowak and Bullier, 1998a,b) and may be excited independently of soma depolarization (Rattay, 1999). It has also been suggested that, during deep brain stimulation, action potentials are initiated in the axons and may propagate antidromically, invading the soma and dendrites (Grill and McIntyre, 2001). GABAergic GP axons may be activated by synaptic stimulation of GP neurons via the activation of subthalamopallidal afferents or by direct stimulation of GABAergic pallidonigral fibers (Kita and Kitai, 1987, 1994). The former possibility is unlikely because HFS-STN did not increase Glu levels in the GP in hemiparkinsonian rats. It therefore seems more probable that HFS-STN activates GABAergic tracts passing close to the STN and that such a mechanism is responsible for the HFS-STN-induced increase in GABA level in the SNr. The role of this process in the clinical efficacy of HFS-STN is unclear, but it could be partly responsible for the inhibition of SNr neuron activity in vivo by HFS-STN in both intact and 6-OHDA-treated rats (Tai et al., 2003).
In conclusion, this study indicates that HFS-STN increases the level of GABA in the SNr, probably by an activation of the pallidonigral GABAergic pathway. The predominant effect of GABA in the SNr that has been demonstrated recently (Windels and Kiyatkin, 2004) supports the hypothesis that the increased GABA level observed may be actively involved in lessening the increased excitatory influence of the Glu subthalamic pathway on the SNr neurons in hemiparkinsonian rats. Through this action, HFS-STN may interfere with interactions involving direct and indirect corticostriatal output circuits by inhibiting subthalamic circuits. It will be of interest to determine to what extent this interference is involved in counteracting neuronal abnormal activity within the basal ganglia network caused by dopaminergic degeneration in PD and therefore the exact role of this process in the clinical effects of HFS-STN restoring motor functions in parkinsonian patients.
Footnotes
This work was supported by the Institut National de la Santé et de la Recherche Médicale, by European Community Grant QLK6-1999-02173 (Fifth Framework Programme for Research and Development), and by Action Concertée Incitative Grant 02 2 0409 from the Ministère de la Recherche et des Nouvelles Technologies. We thank Drs. Nicolas Hussy, Guy Chouvet, and Paul Krack for critical reading of this manuscript.
Correspondence should be addressed to Dr. Marc Savasta, Dynamique des Réseaux Neuronaux, Unité Mixte de Recherche, Institut National de la Santé et de la Recherche Médicale, Unité 704, Université Joseph Fourier, Unité de Formation par la Recherche en Biologie Bātiment B, Domaine Universitaire, 2280 rue de la Piscine, BP 53, 38041 Grenoble Cedex 09, France. E-mail: msavasta@ujf-grenoble.fr.
F. Windels' present address: National Institute on Drug Abuse/Intramural Research Program, 5500 Nathan Shock Drive, Baltimore, MD 21224.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255079-08$15.00/0
References
- Albin RL, Young AB, Penney JB (1989) The functional anatomy of basal ganglia disorders. Trends Neurosci 12: 366-375. [DOI] [PubMed] [Google Scholar]
- Alexander GE, Crutcher DM (1990) Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci 13: 266-271. [DOI] [PubMed] [Google Scholar]
- Aziz TZ, Peggs D, Sambrook MA, Crossman AR (1991) Lesion of the subthalamic nucleus for the alleviation of 1-methyl-4-phenyl-1,2,3,6-tethrahydropyridine (MPTP)-induced parkinsonism in the primate. Mov Disord 6: 288-292. [DOI] [PubMed] [Google Scholar]
- Benabid AL, Benazzouz A, Pollak P (2002) Mechanisms of deep brain stimulation. Mov Disord 17: S73-S74. [DOI] [PubMed] [Google Scholar]
- Benazzouz A, Gross C, Feger J, Boraud T, Bioulac B (1993) Reversal of rigidity and improvement in motor performance by subthalamic high-frequency stimulation in MPTP-treated monkeys. Eur J Neurosci 5: 382-389. [DOI] [PubMed] [Google Scholar]
- Benazzouz A, Piallat B, Pollak P, Benabid AL (1995) Responses of substantia nigra pars reticulata and globus pallidus complex to high-frequency stimulation of the subthalamic nucleus in rats: electrophysiological data. Neurosci Lett 189: 77-80. [DOI] [PubMed] [Google Scholar]
- Benazzouz A, Gao DM, Ni ZG, Piallat B, Bouali-Benazzouz R, Benzidine AL (2000) Effect of high-frequency stimulation of the subthalamic nucleus on the neuronal activities of substantia nigra pars reticulata and ventrolateral nucleus of the thalamus in the rat. Neuroscience 99: 289-295. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, DeLong MR (1990) Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science 249: 1436-1438. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, Karmon B, DeLong MR (1994) The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J Neurophysiol 72: 507-520. [DOI] [PubMed] [Google Scholar]
- Beurrier C, Bioulac B, Audin J, Hammond C (2001) High-frequency stimulation produces a transient blockade of voltage-gated currents in subthalamic neurons. J Neurophysiol 85: 1351-1356. [DOI] [PubMed] [Google Scholar]
- Bevan MD, Magill PJ, Terman D, Bolam JP, Wilson CJ (2002) Move to the rhythm: oscillations in the subthalamic nucleus-external globus pallidus network. Trends Neurosci 25: 525-531. [DOI] [PubMed] [Google Scholar]
- Brown LL, Makman MH, Wolfson LI, Dvorkin B, Warner C, Katzman R (1979) A direct role of dopamine in the rat subthalamic nucleus and intrapeduncular area. Science 206: 1416-1418. [DOI] [PubMed] [Google Scholar]
- Bruet N, Windels F, Carcenac C, Feuerstein C, Bertrand A, Poupard A, Savasta M (2003) Neurochemical mechanisms induced by high-frequency stimulation of the subthalamic nucleus: increase of extracellular striatal glutamate and GABA in normal and hemiparkinsonian rats. J Neuropathol Exp Neurol 62: 1228-1240. [DOI] [PubMed] [Google Scholar]
- Burbaud P, Gross C, Bioulac B (1994) Effect of subthalamic high frequency stimulation on substantia nigra pars reticulata and globus pallidus neurons in normal rats. J Physiol (Paris) 88: 359-361. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Pisani A, Mercuri NB, Bernardi G (1993) Electrophysiology of dopamine-denervated striatal neurons. Brain 116: 433-452. [PubMed] [Google Scholar]
- Campbell GA, Eckardt MJ, Weight FF (1985) Dopaminergic mechanisms in subthalamic nucleus of rat: analysis using horseradish peroxidase and microiontophoresis. Brain Res 333: 261-270. [DOI] [PubMed] [Google Scholar]
- Celada P, Paladini CA, Tepper JM (1999) GABAergic control of rat substantia nigra dopaminergic neurons: role of globus pallidus and substantia nigra pars reticulata. Neuroscience 89: 813-825. [DOI] [PubMed] [Google Scholar]
- Chesselet MF, Delfs JM (1996) Basal ganglia and movement disorders: an update. Trends Neurosci 19: 417-422. [DOI] [PubMed] [Google Scholar]
- Cragg SJ, Baufreton J, Xue Y, Bolam JP, Bevan MD (2004) Synaptic release of dopamine in the subthalamic nucleus. Eur J Neurosci 20: 1788-1802. [DOI] [PubMed] [Google Scholar]
- Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65: 1-105. [DOI] [PubMed] [Google Scholar]
- DeLong MR (1990) Primate models of movement disorders of basal ganglia origin. Trends Neurosci 13: 281-285. [DOI] [PubMed] [Google Scholar]
- Donzanti BA, Yamamoto BK (1998) An improved and rapid HPLC/EC method for the isocratic separation of amino acid neurotransmitters from brain tissues and microdialysis perfusate. Life Sci 43: 913-922. [DOI] [PubMed] [Google Scholar]
- Dostrovsky JO, Lozano AM (2002) Mechanisms of deep brain stimulation. Mov Disord 17 [Suppl 3]: S63-S68. [DOI] [PubMed] [Google Scholar]
- Garcia L, Audin J, D'Alessandro G, Bioulac B, Hammond C (2003) Dual effect of high-frequency stimulation on subthalamic neuron activity. J Neurosci 23: 8743-8751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesdon JL, Ternynck T, Avrameas S (1979) The use of avidin-biotin interaction in immunenzymatic techniques. J Neurochem 27: 1131-1139. [DOI] [PubMed] [Google Scholar]
- Grill WM, McIntyre CC (2001) Extracellular excitation of central neurons: implications for the mechanisms of deep brain stimulation. Thalamus Relat Syst 1: 269-277. [Google Scholar]
- Guridi J, Obeso JA (2001) The subthalamic nucleus, hemiballismus and Parkinson's disease: reappraisal of a neurosurgical dogma. Brain 124: 5-19. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Elder CM, Vitek JL (2002) A template subtraction method for stimulus artifact removal in high-frequency deep brain stimulation. J Neurosci Methods 113: 181-186. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Elder CM, Okun MS, Patrick SK, Vitek JL (2003) Stimulation of the subthalamic nucleus changes firing pattern of pallidal neurons. J Neurosci 20: 1916-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassani OK, Mouroux M, Feger J (1996) Increased subthalamic neuronal activity after nigral dopaminergic lesion independent of disinhibition via the globus pallidus. Neuroscience 72: 105-115. [DOI] [PubMed] [Google Scholar]
- Hassani OK, François C, Yelnik J, Feger J (1997) Evidence for a dopaminergic innervation of the subthalamic nucleus in the rat. Brain Res 749: 387-397. [DOI] [PubMed] [Google Scholar]
- Hollerman JR, Grace AA (1992) Subthalamic nucleus cell firing in the 6-OHDA-treated rat: basal activity and response to haloperidol. Brain Res 590: 291-299. [DOI] [PubMed] [Google Scholar]
- Joel D, Weiner I (1997) The connections of the primate subthalamic nucleus: indirect pathways and the open-interconnected scheme of basal ganglia-thalamocortical circuitry. Brain Res Brain Res Rev 23: 62-78. [DOI] [PubMed] [Google Scholar]
- Kita H, Kitai ST (1987) Efferent projections of the subthalamic nucleus in the rat: light and electron microscopic analysis with the PHA-L method. J Comp Neurol 260: 435-452. [DOI] [PubMed] [Google Scholar]
- Kita H, Kitai ST (1994) The morphology of globus pallidus projection neurons in the rat: an intracellular staining study. Brain Res 636: 308-319. [DOI] [PubMed] [Google Scholar]
- Kitai ST, Deniau JM (1981) Cortical inputs to the subthalamus: intracellular analysis. Brain Res 214: 411-415. [DOI] [PubMed] [Google Scholar]
- Konitsiotis S, Kafetzopoulos E, Anastasopoulos D, Blanchet PJ (1998) Opposite rotation induced by dopamine agonists in rats with unilateral lesions of the globus pallidus or substantia nigra. Behav Brain Res 92: 77-83. [DOI] [PubMed] [Google Scholar]
- Krack P, Benazzouz A, Pollak P, Limousin P, Piallat B, Hoffmann D, Xie J, Benabid AL (1998) Treatment of tremor in Parkinson's disease by subthalamic nucleus stimulation. Mov Disord 13: 907-914. [DOI] [PubMed] [Google Scholar]
- Krack P, Batir A, Van Blercom N, Chabardes S, Fraix V, Ardouin C, Koudsie A, Limousin PD, Benazzouz A, LeBas JF, Benabid AL, Pollak P (2003) Five-year follow-up of bilateral stimulation of the subthalamic nucleus in advanced Parkinson's disease. N Engl J Med 349: 1925-1934. [DOI] [PubMed] [Google Scholar]
- Krause M, Fogel W, Heck A, Hacke W, Bonsanto M, Trenkwalder C, Tronnier V (2001) Deep brain stimulation for the treatment of Parkinson's disease: subthalamic nucleus versus globus pallidus internus. J Neurol Neurosurg Psychiatry 70: 464-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiss DS, Mastropietro CW, Rawji SS, Walters JR (1997) The response of subthalamic nucleus neurons to dopamine receptor stimulation in a rodent model of Parkinson's disease. J Neurosci 17: 6807-6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannes B, Micheletti G (1994) Glutamate-dopamine balance in the striatum: pre- and post-synaptic interactions. In: The basal ganglia IV. New ideas and data on structure and functions (Percheron G, MacKenzie JS, Feger J, eds), pp 475-789. New York: Plenum.
- Levy R, Ashby P, Hutchinson WD, Lang AE, Lozano AM, Dostrovsky JO (2002) Dependence of subthalamic nucleus oscillations on movement and dopamine in Parkinson's disease. Brain 125: 1196-1209. [DOI] [PubMed] [Google Scholar]
- Limousin P, Pollak P, Benazzouz A, Hoffmann D, Le Bas JF, Broussolle E, Perret JE, Benabid AL (1995) Effect on parkinsonian signs and symptoms of bilateral subthalamic nucleus stimulation. Lancet 345: 91-95. [DOI] [PubMed] [Google Scholar]
- Limousin P, Krack P, Pollak P, Benazzouz A, Ardouin C, Hoffmann D, Benabid AL (1998) Electrical stimulation of the subthalamic nucleus in advanced Parkinson's disease. N Engl J Med 339: 1105-1111. [DOI] [PubMed] [Google Scholar]
- Magarinos-Ascone C, Pazo JH, Macadar O, Buno W (2002) High-frequency stimulation of the subthalamic nucleus silences subthalamic neurons: a possible cellular mechanism in Parkinson's disease. Neuroscience 115: 1109-1117. [DOI] [PubMed] [Google Scholar]
- Maurice N, Thierry AM, Glowinski J, Deniau JM (2003) Spontaneous and evoked activity of substantia nigra pars reticulata neurons during high-frequency stimulation of the subthalamic nucleus. J Neurosci 23: 9929-9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre CC, Savasta M, Walter BL, Vitek JL (2004a) How does deep brain stimulation work? Present understanding and future questions. J Clin Neurophysiol 21: 40-50. [DOI] [PubMed] [Google Scholar]
- McIntyre CC, Savasta M, Kerkerian-Le Goff L, Vitek JL (2004b) Uncovering the mechanism(s) of action of deep brain stimulation: activation, inhibition, or both. Clin Neurophysiol 115: 1239-1248. [DOI] [PubMed] [Google Scholar]
- Meibach MRC, Katzman R (1979) Catecholaminergic innervation of the subthalamic nucleus: evidence for a rostral continuation of A9 (substantia nigra) dopaminergic group. Brain Res 173: 364-368. [DOI] [PubMed] [Google Scholar]
- Miller WC, DeLong MR (1987) Altered tonic activity in the globus pallidus and subthalamic nucleus in the primate MPTP model parkinsonian. In: The basal ganglia II. Structure and function: current concepts (Carpenter MB, Jayaraman A, eds), pp 415-427. New York: Plenum.
- Mink JW (1996) The basal ganglia: focus selection and inhibition of competing motor programs. Prog Neurobiol 50: 381-425. [DOI] [PubMed] [Google Scholar]
- Miwa H, Nishi K, Fuwa T, Mizuno Y (1998) Globus pallidus lesions inhibit the induction of c-Fos by haloperidol in the basal ganglia output nuclei in rats. Neurosci Lett 205: 29-32. [DOI] [PubMed] [Google Scholar]
- Ni Z, Gao D, Bouali-Benazzouz R, Benabid AL, Benazzouz A (2001) Effect of microiontophoretic application of dopamine on subthalamic nucleus neuronal activity in normal rats and in rats with unilateral lesion of the nigrostriatal pathway. Eur J Neurosci 14: 373-381. [DOI] [PubMed] [Google Scholar]
- Nowak LG, Bullier J (1998a) Axons, but not cell bodies, are activated by electrical stimulation in cortical gray matter. I. Evidence from chronaxie measurements. Exp Brain Res 118: 477-488. [DOI] [PubMed] [Google Scholar]
- Nowak LG, Bullier J (1998b) Axons, but not cell bodies, are activated by electrical stimulation in cortical gray matter. II. Evidence from selective inactivation of cell bodies and axon initial segments. Exp Brain Res 118: 489-500. [DOI] [PubMed] [Google Scholar]
- Obeso J, Rodriguez MC, DeLong MR (1997) Basal ganglia pathophysiology. A critical review. Adv Neurol 74: 3-18. [PubMed] [Google Scholar]
- Parpura V, Scemes E, Spray DC (2004) Mechanisms of glutamate release from astrocytes: gap junction “hemichannels,” purinergic receptors and exocytic release. Neurochem Int 45: 259-264. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1982) The rat brain in stereotaxic coordinates. London: Academic. [DOI] [PubMed]
- Pollak P, Benabid AL, Gross C, Gao DM, Laurent A, Benazzouz A, Hoffmann D, Gentil M, Perret J (1993) Effects of the stimulation of the subthalamic nucleus in Parkinson disease. Rev Neurol (Paris) 149: 175-176. [PubMed] [Google Scholar]
- Ranck Jr JB (1975) Which elements are excited in electrical stimulation of mammalian central nervous system: a review. Brain Res 98: 417-440. [DOI] [PubMed] [Google Scholar]
- Rattay F (1999) The basic mechanism for the electrical stimulation of the nervous system. Neuroscience 89: 335-346. [DOI] [PubMed] [Google Scholar]
- Rosales MG, Martinez-Fong D, Morales R, Nunez A, Flores G, Gongora-Alfaro JL, Floran B, Aceves J (1997) Reciprocal interaction between glutamate and dopamine in the pars reticulata of the rat substantia nigra: a microdialysis study. Neuroscience 80: 803-810. [DOI] [PubMed] [Google Scholar]
- Salin P, Manrique C, Forni C, Kerkerian-Le Goff L (2002) High-frequency stimulation of the subthalamic nucleus selectively reverses dopamine denervation-induced cellular defects in the output structures of the basal ganglia in the rat. J Neurosci 22: 5137-5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savasta M, Windels F, Bruet N, Bertrand A, Poupard A (2002) Neurochemical modifications induced by high-frequency stimulation of subthalamic nucleus in rats. In: The basal ganglia VII (Nicholsson L, ed), pp 581-590. New York: Kluwer Academic/Plenum.
- Shen KZ, Zhu ZT, Munhall A, Johnson SW (2003) Dopamine receptor supersensitivity in rat subthalamus after 6-hydroxydopamine lesions. Eur J Neurosci 18: 2967-2978. [DOI] [PubMed] [Google Scholar]
- Smith Y, Shink E, Sidibe M (1998) Neuronal circuitry and synaptic connectivity of the basal ganglia. Neurosurg Clin N Am 9: 203-222. [PubMed] [Google Scholar]
- Tai CH, Boraud T, Bezard E, Bioulac B, Gross C, Benazzouz A (2003) Electrophysiological and metabolic evidence that high-frequency stimulation of the subthalamic nucleus bridles neuronal activity in the subthalamic nucleus and the substantia nigra reticulata. FASEB J 17: 1820-1830. [DOI] [PubMed] [Google Scholar]
- Tossman U, Ungerstedt U (1986) Microdialysis in the study of extracellular levels of amino acids in the rat brain. Acta Physiol Scand 128: 9-14. [DOI] [PubMed] [Google Scholar]
- Vitek JL (2002) Mechanisms of deep brain stimulation: excitation or inhibition. Mov Disord 17 [Suppl 3]: S69-S72. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Bergman H, DeLong MR (1994) The primate subthalamic nucleus. III. Changes in motor behavior and neuronal activity in the internal pallidum induced by subthalamic inactivation in the MPTP model of parkinsonism. J Neurophysiol 72: 521-530. [DOI] [PubMed] [Google Scholar]
- Windels F, Kiyatkin EA (2004) GABA, not glutamate, controls the activity of substantia nigra reticulata neurons in awake, unrestrained rats. J Neurosci 24: 6751-6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windels F, Bruet N, Poupard A, Urbain N, Chouvet G, Feuerstein C, Savasta M (2000) Effects of high-frequency stimulation of subthalamic nucleus on extracellular glutamate and GABA in substantia nigra and globus pallidus in the normal rat. Eur J Neurosci 12: 4141-4146. [DOI] [PubMed] [Google Scholar]
- Windels F, Bruet N, Poupard A, Feuerstein C, Bertrand A, Savasta M (2003) Influence of the frequency parameter on extracellular glutamate and gamma-aminobutyric acid in substantia nigra and globus pallidus during electrical stimulation of subthalamic nucleus in rats. J Neurosci Res 72: 259-267. [DOI] [PubMed] [Google Scholar]