

Abstract
Signaling complexes are essential for the modulation of excitability within restricted neuronal compartments. Adaptor proteins are the scaffold around which signaling complexes are organized. Here, we demonstrate that the Camguk (CMG)/CASK adaptor protein functionally modulates Drosophila Ether-á-go-go (EAG) potassium channels. Coexpression of CMG with EAG in Xenopus oocytes results in a more than twofold average increase in EAG whole-cell conductance. This effect depends on EAG-T787, the residue phosphorylated by calcium- and calmodulin-dependent protein kinase II (Wang et al., 2002). CMG coimmunoprecipitates with wild-type and EAG-T787A channels, indicating that T787, although necessary for the effect of CMG on EAG current, is not required for the formation of the EAG-CMG complex. Both CMG and phosphorylation of T787 increase the surface expression of EAG channels, and in COS-7 cells, EAG recruits CMG to the plasma membrane. The interaction of EAG with CMG requires a noncanonical Src homology 3-binding site beginning at position R1037 of the EAG sequence. Mutation of basic residues, but not neighboring prolines, prevents binding and prevents the increase in EAG conductance. Our findings demonstrate that membrane-associated guanylate kinase adaptor proteins can modulate ion channel function; in the case of CMG, this occurs via an increase in the surface expression and phosphorylation of the EAG channel.
Keywords: calcium, protein kinase II, trafficking, Drosophila, phosphorylation, neuromodulation
Introduction
Ether-á-go-go (EAG), the Drosophila ortholog of KCNH1, is the founding member of a family of potassium (K) channels identified by the presence of domains with homology to the PAS (Per, Arnt, Sim) and cyclic nucleotide-binding domains of other proteins (Drysdale et al., 1991; Guy et al., 1991; Warmke et al., 1991; Morais Cabral et al., 1998). eag larvae exhibit spontaneous action potentials in the motor nerve and excitatory junctional potentials in muscle that are broadened compared with those of wild-type larvae (Ganetzky and Wu, 1985). EAG is present in the axons and terminals innervating the larval body-wall musculature (Wang et al., 2002) and is localized with synaptobrevin in the CNS (Sun et al., 2004).
In addition to hyperexcitability at the neuromuscular junction, eag mutants also exhibit defects in associative learning (Griffith et al., 1994). Intriguingly, there is substantial similarity in the electrophysiological and learning phenotypes of eag mutants and transgenic Drosophila expressing an inhibitory peptide of calcium- and calmodulin-dependent protein kinase II (CaMKII) (Griffith et al., 1994), and phosphorylation of EAG by CaMKII results in a dramatic increase in current (Wang et al., 2002). Recent work has demonstrated that Drosophila CaMKII interacts with the Camguk (CMG) adaptor protein (Lu et al., 2003). CMG is a member of the membrane-associated guanylate kinase (MAGUK) family of adaptor proteins and the Drosophila ortholog of CASK and Lin-2 (Hata et al., 1996; Hoskins et al., 1996; Dimitratos et al., 1997; Butz et al., 1998). CMG/CASK/LIN-2 is unique in that the N terminal (NT) displays considerable homology to CaMKII. The kinase domain of CMG, although appearing nonfunctional as a kinase, associates with CaMKII in an ATP- and calcium/calmodulin-dependent manner (Lu et al., 2003). Once calcium levels decline, CMG downregulates cellular CaMKII activity by releasing the kinase in a form that must be dephosphorylated before it can be activated by subsequent increases in calcium.
Given the regulation of EAG function by CaMKII, we sought to determine whether CMG also associates with EAG. Intriguingly, the interaction of CMG with EAG promoted an increase in whole-cell conductance and alterations in inactivation kinetics that were reminiscent of the changes in current observed as a consequence of phosphorylation by CaMKII. Indeed, mutation of EAG-T787, the site phosphorylated by CaMKII, prevented the effects of CMG without affecting immunoprecipitation of the complex. Moreover, CMG increased EAG surface expression, whereas mutation of T787 decreased EAG surface expression, indicating that CMG and phosphorylation affect channel function by the same mechanism. One possible model that could account for these findings is that CMG may locally increase kinase efficiency by ensuring that only active kinase is in the vicinity of the channel.
Materials and Methods
Plasmids and construction. For protein purification, plasmids containing DNA fragments spanning the indicated amino acids were generated by PCR using primers containing restriction sites and bases to maintain reading frame during subcloning. Glutathione S-transferase (GST) and 6Histidine (6H) fusion constructs were generated using the pGEX-4T1 or pGEX-KG vectors (Amersham Biosciences, Piscataway, NJ) (Guan and Dixon, 1991) or the pQE30 vector series (Qiagen, Valencia, CA), respectively. Where indicated, sequence encoding dihydrofolate reductase (DHFR) was subcloned into the pQE-eag construct to aid in the purification and identification of the small EAG fragment. pQE32-eag869-1107 was a gift from B. Ganetzky (University of Wisconsin, Madison, WI). With the exception of the Src homology 3 (SH3) domains of CMG and Drosophila discs large (DLG), GST-SH3 domain fusion proteins were a gift from Brian Kay (Argonne National Laboratory, Argonne, IL). Site-directed mutagenesis was performed using the PCR-based QuikChange mutagenesis kit (Stratagene, La Jolla, CA).
For expression of EAG mutant constructs in Xenopus oocytes, SphI and NaeI sites flanking the EAG SH3 domain-binding site were used to subclone single-site mutations from pQE-eag constructs into pGH19-eag (Wilson et al., 1998). A Kozak sequence was added to pGEX-cmg using PCR, and cmg, together with the Kozak sequence, was shuttled into pBluescript-KSM (a gift from W. Joiner and L. K. Kaczmarek, Yale University, New Haven, CT), which had been modified to contain the untranslated regions of the Xenopus β-globin gene and a more flexible cloning site.
All constructs were verified by sequencing.
Protein purification. GST fusion proteins were purified according to manufacturer protocols (Amersham Biosciences). BL21 cells were freshly transformed with the construct of interest. Large-scale (1 L) cultures were grown in Luria broth with ampicillin for 6 h and then induced for 6-12 h with isopropyl-β-d-thiogalactopyranoside. Bacteria were harvested by centrifugation, lysed, and cellular protein solubilized in buffer containing 150 mm NaCl, 20 mm Tris, pH 7.4, and 1% Triton X-100 in the presence of protease inhibitors (0.5 mm phenylmethanesulfonyl fluoride and 0.004 mg/ml each of aprotonin, pepstatin A, and leupeptin). In some cases, GST-CMG was purified by incubation with 0.8% N-lauroylsarcosine before solubilization and then neutralized with twice the concentration of Triton X-100. This increased the yield of CMG protein without affecting binding activity. Protein suspensions were centrifuged to remove insoluble material, and the supernatant was incubated for 60-90 min with glutathione-Sepharose 4B beads (Amersham Biosciences). Bead-immobilized proteins were extensively washed with 150 mm NaCl and 20 mm Tris, pH 7.4, and stored as a 50% slurry at 4°C until use. 6H fusion proteins were purified similarly using nickel-nitrilotriacetic acid beads (Qiagen) and a solubilization buffer containing 500 mm NaCl, 20 mm HEPES, pH 7.8, and 1% Triton X-100 in the presence of protease inhibitors. 6H fusion proteins were fractionally eluted from beads using successively increasing concentrations of imidazole ranging from 8 to 400 mm. Eluted fractions were checked for the presence of the desired protein using SDS-PAGE, and fractions containing the protein were pooled and dialyzed overnight in PBS [consisting of the following (in mm): 136 NaCl, 2.7 KCl, 10 Na2HPO4, 1.8 KH2PO4, pH 7.4]. Dialyzed protein was stored at 4°C or frozen at -20°C before use. Protein concentrations were determined by comparison to known concentrations of bovine serum albumin (BSA).
Peptides and antibodies. Peptides used for binding studies and immunization were synthesized by Open Biosystems (Huntsville, AL). An N-terminal peptide corresponding to amino acids 161-179 of the EAG sequence and a purified C-terminal (CT) fragment (6H-EAG1034-1174) were used for immunization of rabbits according to standard procedures (Open Biosystems). Sera were screened against the antigens in ELISAs and in Western blots of purified N- or C-terminal EAG fragments as appropriate. Purified GST-CMG152-897 was used for the immunization of guinea pigs as described previously (Dimitratos et al., 1997). Phosphospecific anti-EAG antibody was a gift from Leslie Griffith (Brandeis University, Waltham, MA).
In vitro binding assays. GST fusion proteins immobilized on Sepharose beads were mixed with 6H fusion proteins in PBS containing 0.5% Triton X-100 and protease inhibitors (EDTA-free Complete tablets; F. Hoffmann-La Roche, Basel, Switzerland) and incubated with shaking for 2-3 h at 4°C. Except where indicated otherwise, the protein concentrations used were 1-2 and 0.5-1 μm, respectively, in 100 μl of buffer containing a constant 10 μl bead bed volume. The beads were collected by centrifugation and then washed three times with PBS containing 0.1% Triton X-100, followed by three washes with 50 mm Tris, pH 8.0, 140 mm NaCl, and 0.1% Triton X-100 and a final wash with 50 mm Tris, pH 8.0. The pellets, including beads and associated proteins, were resuspended in sample buffer and analyzed by SDS-PAGE and Western blotting. Blots were probed with MRGS-6H antibody (Qiagen) followed by horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody and then visualized with ECL (Amersham Biosciences).
ELISAs. ELISAs were performed as described previously (Muller et al., 1996; Garcia et al., 1998) with minor modifications. GST-CMG fusion proteins were eluted from glutathione Sepharose according to manufacturer protocols (Amersham Biosciences). 6H-EAG869-1107, at a concentration of 0.16 μg/ml in BBS (125 mm borate, 75 mm NaCl, pH 8.5), was bound to the wells of Costar (Cambridge, MA) 96-well assay plates by overnight incubation (4°C). Plates were then washed extensively with BBS and blocked with BBS containing 1% BSA (BBS-BSA) before addition of serially diluted CMG protein at the indicated concentrations (50 μl per well). Each concentration was assayed in quadruplicate. After overnight incubation (4°C), plates were washed four times and then incubated with anti-GST antibody (1:1000 in BBS-BSA). Plates were washed again and incubated with alkaline phosphatase-conjugated anti-rabbit IgG (1:1000 in BBS-BSA). After a final set of washes, binding to 6H-EAG869-1107 was determined by treating wells with FAST p-nitrophenylphosphate (Sigma, St. Louis, MO) and measuring the colorometric reaction at 405 nm.
Immunocytochemistry. COS-7 cells were maintained at a subconfluent density at 37°C and 5% CO2 in DMEM supplemented with 10% fetal bovine serum (FBS). Twenty-four hours before transfection, cells were plated and grown to 80% confluence on 12 mm poly-l-lysine-coated glass coverslips (Becton Dickinson, Franklin Lakes, NJ). For transient transfection, each coverslip was washed twice with Opti-MEM (Invitrogen, Carlsbad, CA) and incubated for 10 h in 350 μl of Opti-MEM containing 0.4 μg of each of the indicated cDNAs and 1.5 μl of LipofectAMINE reagent (Invitrogen), followed by incubation in 1 ml of DMEM with 10% FBS for 12 h. Coverslips were washed with PBS and fixed with a 3:7 mixture of 50 mm glycine, pH 2.0, and absolute ethanol for 1 h at room temperature (RT). After washing three times, cells were permeabilized with 0.1% Triton X-100 in PBS for 20 min (RT) and blocked for 30 min in a blocking buffer consisting of 10% horse serum and 0.1% BSA in distilled deionized water. Cells were then incubated with a 1:500 dilution of CMG antisera in blocking buffer for 1 h at RT, washed with PBS, and incubated with a cyanine 2- or rhodamine-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA) for 30 min at 37°C. Coverslips were mounted on glass slides with Gelmount medium (Biomeda, Foster City, CA). Results were viewed with a Zeiss (Oberkochen, Germany) LSM 510 confocal microscope, and images were prepared for publication with Adobe Photoshop (version 7.0; Adobe Systems, San Jose, CA).
Immunoprecipitations. Body-wall muscle fibers and CNS tissue from third-instar larvae of the indicated genotypes were homogenized in radioimmunoprecipitation assay buffer (150 mm NaCl, 50 mm Tris, pH 8.0, 0.1% SDS, 1% IGEPAL CA-630, 0.5% sodium deoxycholate) containing Complete protease inhibitor on ice and solubilized for 20 min. Cellular debris was removed by centrifugation (3000 × g, 5 min, 4°C).
Fly head tissue was obtained from 15 ml of adult flies frozen in liquid nitrogen. Heads were collected by sieving and homogenized in ice-cold HM buffer (1 mm EDTA, 5 mm HEPES, pH 7.4, and Complete protease inhibitor). Cell debris was removed by centrifugation, and supernatant was further centrifuged to separate membranes (15,000 × g, 15 min). The heavy membrane pellet was solubilized in ice-cold buffer (HM buffer plus 100 mm NaCl and 1% Triton X-100) for 20 min, and supernatant was precleared with protein-A/G agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA).
For immunoprecipitations from Xenopus oocytes, 30 oocytes for each condition were homogenized in 1.5 ml of 100 mm NaCl, 1 mm DTT, 0.5% Triton X-100, 20 mm Tris, pH 7.4, and Complete protease inhibitor and then solubilized on ice for 15 min. Homogenate was centrifuged 20,000 × g, 4°C, for 10 min and the supernatant collected.
Protein concentrations of homogenates were determined using Bradford assay (Bio-Rad, Hercules, CA) and equilibrated across conditions by diluting homogenates to a concentration of 2 mg/ml. A volume of 600 μl of diluted extract was used for each immunoprecipitation reaction. CMG and EAG (NT) antisera were added to protein samples and incubated for 16 h at 4°C. Immunocomplexes were precipitated with protein-A/G beads for 2 h at 4°C and then washed three times in solubilization buffer and boiled for 10 min in sample loading buffer (250 mm Tris, pH 6.8, 12.5% glycerol, 0.125% bromophenol blue, 1% SDS, 3% β-mercaptoethanol). After addition of sample buffer, 5, 10, and 20 μl were loaded in each lane for extracts, immunoprecipitated proteins, and coimmunoprecipitated proteins, respectively.
Immunoprecipitations and cellular extracts were resolved via SDS-PAGE (8% acrylamide, 250 mm Tris, pH 6.8, 2.5% SDS) and transferred to polyvinylidene difluoride (PVDF) membrane for Western blotting. Blots were blocked (5% dry milk in TBS) and washed, probed with either CMG or EAG (CT) sera (1:1000 and 1:2000, respectively) followed by HRP-conjugated secondary antibody (1:2000), and visualized by ECL (Amersham Biosciences).
For experiments biotinylating surface membrane proteins, oocytes were washed in cold PBS and then incubated for 30 min in 2 mm sulfo-NHS-LC-biotin (Pierce, Rockford, IL) in PBS. The labeling reaction was quenched with three washes of 100 mm glycine in PBS. Fifty oocytes were homogenized in 1 ml of Buffer H (100 mm NaCl, 0.5% Triton X-100, 20 mm Tris, pH 7.4) supplemented with Complete protease inhibitor, 1 mm orthovanadate, 1 mm benzamidine, 1% phosphatase inhibitor cocktail 1 (Sigma), 2 μm microcystin-LR, and 5 mm 2-glycerol-phosphate.
Volume intensities of protein bands were quantified using Quantity One software (Bio-Rad). Experimental values (n ≥ 3) were corrected for background by subtracting values obtained for uninjected lanes and then were normalized to the value obtained for immunoprecipitated wild-type EAG in the absence of CMG.
Electrophysiology. For expression in Xenopus oocytes, plasmids were linearized and RNA transcribed using the appropriate RNA polymerase according to manufacturer instructions (Message Machine; Ambion, Austin, TX). RNA concentrations were quantified with spectrophotometric readings using an average reading for three or four dilutions of RNA.
The follicular membrane of stage V-VI oocytes was removed by incubation in calcium-free OR2 solution [containing (in mm): 82.5 NaCl, 2.5 KCl, 1 MgCl2, 5 HEPES, pH to 7.6 with NaOH] containing collagenase (2 mg/ml; Type 1A; Sigma) for ∼2 h at room temperature with gentle agitation. Oocytes were injected with premixed stocks of eag and cmg RNA. Individual oocytes were injected with 0.1-0.2 ng of eag RNA and an excess of cmg RNA. In some oocyte batches (oocytes from the same frog), the capacity of oocytes to translate the eag constructs was verified by performing parallel injections of a larger volume of the same RNA mix and observing a linear increase in eag current amplitude. After RNA injections, oocytes were maintained in L-15 media (containing the following: 50% L-15, 15 mm HEPES, 50 mg/ml gentamycin, and 5 mg/ml BSA, pH to 7.4 with NaOH) at 18°C for 3-5 d. Recordings were performed using a Turbo TEC-10C amplifier (NPI Electronics, Tamm, Germany) and pClamp8 software (Molecular Devices, Union City, CA). The extracellular recording solution contained the following (in mm): 140 NaCl, 2 KCl, 1 MgCl2, 10 HEPES, pH 7.1, with NaOH. Pipettes were filled with 2 m KCl and had resistances of 0.3-0.6 MΩ. Experiments were performed at RT.
Amplitude measurements refer to the peak currents observed during test pulses to the indicated voltages. Activation and inactivation time constants were determined by fitting traces (excluding capacitative transients) with two exponentials and a steady state. Unless otherwise noted, measurements were statistically compared using a two-way ANOVA. Data are presented as the mean ± SEM.
Results
CMG modulates EAG current
To determine whether the CMG adaptor protein might regulate EAG function, we examined EAG channels expressed in Xenopus oocytes either alone or together with an excess of CMG. Figure 1, A and B, shows examples of EAG current traces and average current-voltage relationships obtained from a representative batch of oocytes (batch refers to oocytes obtained from the same frog). In response to test pulses to +60 mV (holding potential, -80 mV), coexpression with CMG resulted in peak currents that were increased 2.7-fold over control values (Fig. 1B). CMG produced increases in current in 19 of 21 batches of oocytes examined. The increase averaged 2.3-fold above EAG controls and was significant (p < 0.0001) when tested using a two-way ANOVA with oocyte batch and CMG as variables. When CMG was coexpressed with Shaker, another Drosophila potassium channel, no increase in Shaker current was observed (data not shown). The average peak Shaker currents in response to test pulses to +60 mV (holding potential, -100 mV) were 12.5 ± 0.6 μA (n = 8) and 10.7 ± 0.8 μA (n = 10) in the absence and presence of CMG, respectively.
Figure 1.

CMG increases EAG current and conductance. A, Coexpression with CMG increases EAG current amplitudes in Xenopus oocytes. Macroscopic currents recorded in two-electrode voltage clamp from representative oocytes in response to depolarizing voltage steps from -100 to 80 mV in 20 mV increments (holding potential, -80 mV). Linear leakage and capacitative components have been subtracted. Calibration bars apply to both sets of traces. B-G, Dark symbols and bars represent measurements obtained in the presence of CMG. B, Comparison of the average current-voltage relationships obtained for one batch of oocytes (batch refers to oocytes obtained from the same frog) injected with RNA encoding eag (n = 7) or eag together with an excess of cmg (n = 10), as indicated. Currents were elicited by a series of voltage steps from -100 to 80 mV (holding potential, -80 mV). Leak subtraction was performed using a P/4 protocol with pulses of opposite polarity preceding each test pulse (holding potential, -80 mV). C, Normalized conductance-voltage (GV) relationships for wild-type EAG channels, alone (n = 9) or in the presence of CMG (n = 16). For each oocyte, conductance was determined using the relationship G = Itail/(Vtail - EK) and normalized to the maximum conductance. Measurements were made in an extracellular solution containing 25 mm KCl to enhance tail currents. EK was experimentally determined for each oocyte. Boltzmann functions fit to the averaged data had a midpoint and slope of -12.2 mV and 10.3 for EAG and a midpoint and slope of -13.6 mV and 9.2 for EAG in the presence of CMG. D, Top, Scaled representative traces obtained for EAG alone (Ipeak = 3.0 μA) or together with CMG (Ipeak = 4.8 μA). Currents were elicited by a 400 ms pulse to 40 mV from a holding potential of -80 mV. Bottom, Kinetics of EAG currents in the absence and presence of CMG. Three batches of oocytes with a mean fold increase in current amplitude that approximated the mean for all oocytes were selected for kinetic analysis. For each oocyte, activation (left) and inactivation (middle) taus were obtained by fitting two exponentials plus a constant to the first 200 ms of the response to a 400 ms test pulse to 40 mV (holding potential, -80 mV). The percentage of inactivation (inact) was determined by dividing the mean current obtained during the final 10 ms of the pulse by the peak current observed during the pulse. n = 26 and 28 for EAG and EAG with CMG, respectively. The effect of CMG was analyzed using a two-way ANOVA with oocyte batch and CMG as variables (*p < 0.05; ***p < 0.001; N.S., not significant). E, CMG fails to affect EAG-T787A. Experiments were performed as in B. n = 9 and 9 in the absence or presence of CMG, respectively. F, GV relationships for EAG channels expressed alone or together with CMG. Currents shown in B were converted to conductance using the relationship G = Itest/(Vtest - EK). EK was assumed to be -80 mV. G, CMG increases the average whole-cell conductance in oocytes expressing the wild-type, but not mutant, channel. Fold increase in the whole-cell conductance for the oocytes described in D and three batches of oocytes expressing EAG-T787A alone or with CMG. Conductances were determined as described in F in response to a test pulse to 60 mV (holding potential, -80 mV). To normalize for variation in channel expression across different batches of oocytes, the mean conductance in the presence of CMG was normalized to the mean conductance obtained for that batch of oocytes expressing EAG or EAG-T787A alone. The number of oocytes examined for each condition is indicated above each bar. The effect of CMG was statistically analyzed as described in D. Error bars represent SEM.
The primary effect of CMG was on EAG current amplitudes. Analysis of the voltage dependence of EAG currents in an extracellular solution containing 25 mm KCl to enhance tail currents revealed little change in either the slope or midpoint of activation when EAG was coexpressed with CMG (Fig. 1C). Scaled representative traces for the two conditions are shown in Figure 1D (top). Examination of activation and inactivation kinetics revealed only a modest increase in the inactivation time constant and a decrease in the percentage of inactivation (Fig. 1D, bottom). The CMG-mediated decrease in inactivation was observed even when comparing oocytes with similar current amplitudes, suggesting that the effect on kinetics was not caused by a decreased efficiency of the voltage clamp for larger currents.
Many adaptor proteins affect the function of associated proteins by colocalizing the proteins with other modulators. Previous work has shown that EAG is phosphorylated by CaMKII activity endogenous to the oocytes even at basal calcium levels (Wang et al., 2002). Both the increase in current amplitude and the changes in inactivation in the presence of CMG are reminiscent of the changes in current observed as a consequence of phosphorylation. Because CaMKII has been shown to associate with CMG in the presence of ATP and calcium/calmodulin (Lu et al., 2003), we sought to determine whether the effect of CMG on EAG currents could be mediated by phosphorylation. As shown in Figure 1E, the effect of CMG was dependent on EAG-T787, the residue previously shown to be phosphorylated by CaMKII in vitro and in vivo. When threonine at position 787 was replaced by alanine (EAG-T787A), CMG failed to increase current or affect inactivation.
The increase in current observed for wild-type channels in the presence of CMG is most likely a result of an increase in the number of active channels given the lack of an appreciable change in either voltage dependence or kinetics. Figure 1F displays the change in EAG activity in terms of the whole-cell conductance for the batch of oocytes examined in Figure 1B. Figure 1G summarizes and compares the effect of CMG on the whole-cell conductance of oocytes expressing EAG and EAG-T787A for three representative oocyte batches. If changes in open probability or single-channel conductance do not contribute to the effect of CMG on EAG current, the average increase in conductance suggests a more than twofold increase in the number of functional channels at the plasma membrane.
EAG and CMG coimmunoprecipitate from oocyte extracts
When oocyte extracts were probed with EAG (CT) antisera (Fig. 2A, top blot, left three lanes), two protein bands were clearly identified. The primary EAG band ranged between 180 and 200 kDa, depending on the protein markers used (Fig. 2A, top, lanes 2 and 3). The second, lower molecular weight, band was also reliably observed and is most likely an alternate modification of the protein or a degradation product. Both bands were absent in Western blots of extracts prepared from uninjected oocytes (Fig. 2A, top, lane 1).
Figure 2.

CMG associates with EAG and increases EAG surface expression and phosphorylation. A, Coimmunoprecipitation of EAG and CMG from Xenopus oocyte extracts. Lanes 1-3 show oocyte extracts for comparison; lane 4, immunoprecipitation with preimmune sera; lanes 5 and 6, immunoprecipitation with indicated antibody. CMG associates with both wild-type and EAG-T787A channels. Top (lanes 5, 6), Immunoprecipitation with CMG antisera. Bottom (lanes 5, 6), Reverse immunoprecipitation with EAG (NT) antisera. Similar results were observed in five experiments. Respective loads were 5, 10, and 20 μl for extracts, immunoprecipitated proteins, and coimmunoprecipitated proteins, respectively (see Materials and Methods). B, CMG and phosphorylation of EAG-T787 both increase EAG surface expression. Top, Oocytes were labeled with biotin for 30 min and then quenched with glycine as described in Materials and Methods before preparation of oocyte extracts. For each condition, 600 μl of extract obtained from 50 oocytes was used for immunoprecipitation with EAG (NT) antisera. Proteins were separated by SDS-PAGE, transferred to PVDF membranes, blotted with the indicated antisera, and processed with ECL. Gels were run in parallel with equal amounts of the precipitates. Bottom, Streptavidin-labeled bands were quantified by densitometry and normalized to the intensity of the EAG band in each experiment; data are presented as the mean ± SEM; n = 3 (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.005). C, CMG increases phosphorylation of EAG-T787. Top, EAG was immunoprecipitated from oocyte extracts with EAG (NT) antisera. Gels were run in parallel, proteins transferred to PVDF membranes, which were then probed with either EAG (CT) antisera (top) or antibody recognizing EAG phosphorylated at T787 (bottom) (Wang et al., 2002). Bottom, EAG labeled by phospho-specific antibody. Bands were quantified by densitometry and normalized to the EAG (CT)-labeled band and the corresponding region of the uninjected oocyte lane for each experiment; data are presented as the mean ± SEM; n = 3; p values as in C. IP, Immunoprecipitation.
Immunoprecipitation reactions indicated that, in oocytes, EAG and CMG are part of the same protein complex. Immunoprecipitations using anti-CMG, but not preimmune, sera coprecipitated the EAG protein (Fig. 2A, top, lanes 3 and 4). The reverse immunoprecipitation reaction is shown in the bottom blot (right three lanes). Immunoprecipitations using anti-EAG (NT), but not preimmune, sera coprecipitated CMG, which was observed as a single band near the predicted molecular weight of 103 kDa.
CMG and phosphorylation regulate EAG surface expression
One possible explanation of the failure of CMG to enhance EAG-T787A currents in our electrophysiological experiments is that phosphorylation of T787 may be required for the association of CMG with EAG. As shown in Figure 2A (top and bottom, lanes 6 and 9), there was no appreciable difference in the ability to immunoprecipitate the complex when EAG-T787A was expressed instead of the wild-type channel, indicating that phosphorylation is not a prerequisite for formation of the complex. Either phosphorylation is required for CMG to exert an effect on EAG currents or, alternatively, the increase in current observed in the presence of CMG is the result of an increase in the efficiency of phosphorylation.
To begin to distinguish between these alternatives, we further investigated the mechanism underlying the effect of CMG on EAG current. Oocytes were surface labeled with biotin, and, after biotin treatment, EAG was isolated by immunoprecipitation with EAG (NT) antisera (Fig. 2B, top blot). As shown in Figure 2B (bottom blot) for a parallel blot of the immunoprecipitate probed with streptavidin, only the lower EAG band was detectably biotinylated. More importantly, compared with EAG channels expressed alone, there was a clear increase in the biotinylation of EAG when CMG was coexpressed (bottom blot, lanes 2 and 3). Finally, biotinylated EAG was lowest when expressing EAG-T787A (compare lanes 2 and 4). Semiquantitative comparisons of band intensities normalized to the intensity observed for wild-type EAG indicated that the increases in biotinylation of EAG observed as a consequence of CMG and phosphorylation of EAG-T787 were significant (Fig. 2B, bottom). The average increase in biotinylation observed for EAG in the presence of CMG was comparable with the increase in whole-cell conductance observed in oocyte recordings (Fig. 1G). In addition, the decrease in biotinylation observed for EAG-T787A when compared with wild-type EAG closely approximated the previously reported decrease in current observed when oocytes are injected with approximately equal concentrations of RNA (Wang et al., 2002). A similar difference is observed in the experiments of Figure 1 (compare the current levels in B and E). These results suggest that CMG and phosphorylation of EAG-T787 increase EAG current by the same mechanism, namely by increasing the number of EAG channels in the plasma membrane.
CMG increases phosphorylation of EAG-T787
To determine whether the increase in current observed in the presence of CMG is the result of an increase in the efficiency of phosphorylation of EAG-T787, we immunoprecipitated EAG from oocyte extracts and probed the precipitate with an EAG antibody that specifically recognizes EAG phosphorylated at T787 (Wang et al., 2002). As shown in Figure 2C (left), whereas there was no detectable difference in the overall level of EAG as a function of CMG (top), phosphorylation of T787 was increased when CMG was coexpressed (bottom). Comparisons of the level of phosphorylation averaged across multiple experiments (Fig. 2C, right) indicated that the change in phosphorylation was significant. In summary, the enhancement of EAG current observed in the presence of CMG appears, at least in part, to be caused by an increase in the phosphorylation of T787. It remains unclear, however, whether phosphorylation is the sole mechanism underlying the increase in conductance and surface expression of EAG given the difference in the magnitudes of the effects. One possible explanation of this difference is that phosphorylation of EAG is more likely for channels in the membrane. Differences in the solubility of cytoplasmic versus membrane-associated channels may obscure differences in phosphorylation. Alternatively, the association with CMG and phosphorylation could have synergistic effects on the surface expression of EAG channels.
EAG recruits CMG to the plasma membrane in COS-7 cells
Additional indication of a functionally relevant interaction between EAG and CMG was obtained in COS-7 cells. Our ability to examine colocalization of EAG and CMG, as well as changes in the surface expression of EAG, was hampered by the fact that the primary epitope used by the only antibody, α-EAG(CT), that recognizes EAG in immunocytochemical experiments appears to be occluded when CMG is bound; in separate experiments, α-EAG(CT) was the only anti-EAG antibody examined that failed to coprecipitate CMG from oocyte extracts (D. D. Marble and G. F. Wilson, unpublished observations). Although we were unable to examine changes in the localization of EAG, experiments in COS-7 cells suggested that the EAG-CMG complex may play a role in localizing CMG, in addition to its role in the modulation of EAG current. Changes in the localization of CMG as a function of coexpression with EAG were assessed using another binding partner, Tbr-1, a T-box transcription factor known to associate with the mammalian CMG homolog CASK (Hsueh et al., 2000). As shown in Figure 3A, little or no background labeling was observed when cells were mock-transfected with empty vector (left top panel). CMG expressed alone (right top panel) was localized to the cytoplasm and displayed a pattern consistent with a distribution to the endoplasmic reticulum as has been observed in the case of CASK (Hsueh et al., 2000). Also consistent with previous experiments, cotransfection with Tbr-1 shifted CMG into the nucleus (right middle panel). Most importantly, when EAG was coexpressed with CMG and Tbr-1, EAG successfully recruited a significant fraction of the CMG protein away from the nucleus to the cytoplasm and the plasma membrane (indicated by the arrows in the inset of the left bottom panel). In contrast, coexpression with the Shaker K channel, another MAGUK-interacting protein (Kim et al., 1995), failed to compete with Tbr-1 to alter CMG localization (bottom right panel). Finally, plasma membrane localization of CMG was also observed in the presence of EAG alone (indicated by the arrows in the inset in the left middle panel). Differences in CMG localization as a function of coexpression with Tbr-1 and EAG were quantified using line scans of fluorescence intensity normalized to the maximum fluorescence observed in each image examined. Figure 3B shows representative line scans, taken across the segments indicated by the arrowheads, for the panels shown in Figure 3A. Figure 3C displays line scan data for the cytoplasm, nucleus, and membrane averaged across three separate experiments. In each case, EAG produced a significant change in the localization of CMG when compared with the localization of CMG in the presence of Tbr-1 (see also Fig. 6E).
Figure 3.

EAG-dependent translocation of CMG to the plasma membrane of COS-7 cells. COS-7 cells were transiently transfected with 0.4 μg of each of the cDNAs indicated below each panel. A, The subcellular distribution of CMG as a function of coexpressed proteins was assessed by indirect immunofluorescence using antisera directed against CMG, followed by rhodamine-conjugated anti-guinea pig Ig G. The left top panel shows the relative absence of CMG staining when cells were transfected with the empty pCDNA3 vector. The distribution of CMG shifts from the cytoplasm (right top panel) to the nucleus (left middle panel) when Tbr-1 is coexpressed as has been reported previously for the CMG homolog CASK (Hsueh et al., 2000). As shown in the bottom left panel, EAG competes with Tbr-1 to recruit a fraction of CMG to the plasma membrane, whereas Shaker fails to shift CMG from the nucleus. GW-CMV-Tbr-1 was a gift from M. Sheng (Massachusetts Institute of Technology, Cambridge, MA). Images were background subtracted for display using NIH Image software. Dashed boxes indicate the magnified areas displayed in the insets. Arrows within insets point to areas exhibiting particularly robust membrane staining. Scale bar, 10 μm. B, Representative line scans of fluorescence intensity for key panels in A. In each case, the scanned segment is the area between the two arrowheads. Intensity measurements were normalized to the highest intensity point of each image. C, Averaged, normalized line scan data from three separate experiments examining the distribution of CMG to the cytoplasm, nucleus, and membrane for three expression conditions: CMG (C), CMG and Tbr-1 (CT), and CMG, Tbr-1, and EAG (CTE). Note that the bottom left panel of A was excluded from the analysis, given our inability to distinguish between the nucleus and cytoplasm. One-way ANOVA comparisons of fluorescence intensity are between C versus CT and CT versus CTE conditions (*p < 0.05; **p < 0.01; ***p < 0.001; N.S., not significant). Error bars represent SEM.
Figure 6.
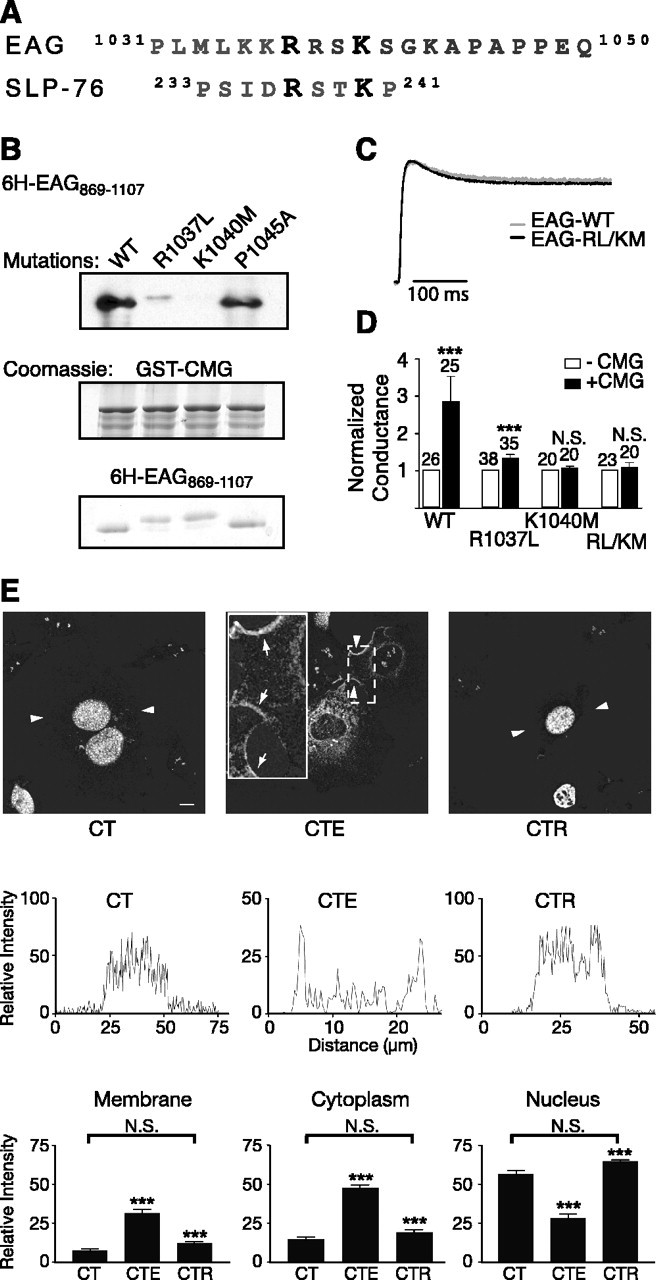
A noncanonical SH3-binding motif in EAG mediates the EAG-CMG interaction. A, Comparison of the amino acid sequence surrounding the fourth putative SH3-binding motif to the amino acid sequence of the noncanonical SH3-binding site in SLP-76, the Src homology 2 (SH2) domain containing a leukocyte protein of 76 kDa. Note that both proteins also contain prolines in the nearby sequence. B, In vitro binding assays comparing the association of 6H-EAG869-1107 and mutant EAG fragments (R1037L, K1040M, and R1037L/K1040M EAG) to GST-CMG, as indicated. Top, Immunoblotting with MRGS-6H antibody to detect interacting EAG fragments. Middle and bottom, Coomassie-stained gels showing the relative amounts of GST-CMG and EAG fragments present in each interaction experiment. C, Scaled representative traces obtained for wild-type and EAG-R1037L/K1049M channels. Currents were elicited by a 400 ms test pulse to +40 mV from a holding potential of -80 mV. Peak current amplitudes were 2.2 and 5.0 μA for EAG and EAG-R1037L/K1040M, respectively. D, Mutations in the EAG-binding site inhibit the effect of CMG on EAG conductance. Fold increase in the whole-cell conductance as a function of CMG coexpression for oocytes expressing wild-type, R1037L, K1040M, and R1037L/K1040M EAG channels. Conductances were determined as described in Figure 1G in response to a test pulse to 60 mV (holding potential, -80 mV). To normalize for normal variations in expression across different batches of oocytes, the mean conductance in the presence of CMG was normalized to the mean conductance obtained for oocytes expressing only EAG or the indicated EAG mutants. The SE and number of oocytes examined for each condition is indicated above each bar. The effect of CMG was statistically analyzed using a two-way ANOVA with oocyte batch and CMG as variables (***p < 0.001; N.S., not significant). WT, Wild type. E, Disruption of the CMG-binding site also disrupts the ability to compete with Tbr-1 and recruit CMG out of the nucleus of COS-7 cells. Top, Background-subtracted images for three expression conditions: CMG and Tbr-1 (CT), CMG, Tbr-1, and EAG (CTE), and CMG, Tbr-1, and EAG-RL/KM (CTR). Arrowheads indicate the segments used for the line scans presented in the middle panels. Scale bar, 10 μm. Middle, Representative line scans obtained as described in Figure 3C. Bottom, Averaged, normalized line scan data from three separate experiments examining the distribution of CMG to the cytoplasm, nucleus, and membrane for the three expression conditions. One-way ANOVA comparisons of fluorescence intensity between CT versus CTE and CTE versus CTR conditions were significant (***p < 0.001). In contrast, there was no significant difference (N.S.) between the CT and CTR conditions. Error bars represent SEM.
EAG and CMG coimmunoprecipitate from Drosophila extracts
Immunoprecipitation reactions using protein extracts from wandering third-instar larvae indicated that EAG and CMG associate in vivo. In the experiment of Figure 4, CMG was immunoprecipitated and the precipitates separated by SDS-PAGE. Blotting with CMG antisera identified a single band near the molecular weight predicted for CMG, 103 kDa (Fig. 4A), for immunoprecipitates from extracts of both wild-type and eag null mutants (eagsc29). More importantly, as shown in Figure 4B, EAG was observed in both extracts (left panel) and CMG immunoprecipitates (right panel) from wild-type larvae but was absent in eagsc29 larvae. As observed for oocytes, the primary EAG band was higher than the molecular weight predicted for EAG, but this band was identified by three antisera raised against different regions of the EAG protein (data not shown). The EAG-CMG complex also could be observed in immunoprecipitation reactions using membrane fractions prepared from adult fly heads (data not shown). In addition, reciprocal coimmunoprecipitation of CMG using α-EAG(NT) antisera was possible, but only when CMG was overexpressed using the Gal4-upstream activation sequence system coupled with the neuron-specific C155-elav driver; this is presumably because of an observed decrease in EAG precipitation efficiency when using α-EAG(NT) versus α-EAG(CT) antisera (data not shown). These results suggest that EAG and CMG are part of a protein complex in vivo; however, neither these results nor our electrophysiological experiments in oocytes address whether the interaction between EAG and CMG is direct.
Figure 4.

Native EAG and CMG coimmunoprecipitate from Drosophila extracts. Protein extracts were prepared from the nervous system, imaginal discs, and body muscle fibers of third-instar larvae. CMG antisera were used to immunopreciptate CMG and associating proteins from extracts prepared from the indicated genotypes. Precipitated proteins were resolved by SDS-PAGE, transferred to nitrocellulose, and then probed with either CMG (A) or EAG (NT) antisera (B, right), followed by HRP-conjugated anti-guinea pig or anti-rabbit IgG, respectively. For comparison, input extract probed with EAG (NT) antisera is shown in the left panel of B. Respective loads were 5, 10, and 20 μl for extracts, immunoprecipitated proteins, and coimmunoprecipitated proteins, respectively (see Materials and Methods). Similar results were obtained in five experiments. Bands were visualized using ECL. WT, Wild type; IP, immunoprecipitation.
In vitro binding assays identify a direct interaction between EAG and CMG
A direct association of EAG and CMG was suggested by in vitro binding assays (Fig. 5). In these assays, GST-tagged CMG or CMG domains were immobilized on glutathione Sepharose beads and the ability to “pull down” 6H-tagged EAG fragments assessed in Western blots using antibody directed against the MRSG-6H tag. The EAG and CMG fragments used to map the respective EAG- and CMG-binding sites are shown in Figure 5, A and B, together with a rating of their relative binding. As shown in Figure 5C for the EAG fragment 6H-EAG869-1107, nearly all of the EAG available associated with GST-CMG (compare lanes 1 and 3). There was minimal background binding to the bead-immobilized GST tag, indicating that the association of EAG with CMG was not a result of a nonspecific effect.
Figure 5.

A direct interaction between EAG and CMG adaptor protein in in vitro binding assays. A, Schematic representation and respective binding for the EAG protein constructs used in mapping experiments. For comparison, full-length EAG is shown at the top with the primary domains as indicated, including the six putative transmembrane domains, pore (P), and the region with homology to cyclic nucleotide-binding domains (CNBD). The amino acid residues contained in each EAG construct are noted by the subscripted numbers on the left, and the relative binding of each construct to full-length CMG is indicated on the right. Note that the EAG C-terminal cytoplasmic domain contains six SH3-binding motifs as defined by the PxxP consensus sequence. The locations of these potential binding sites are indicated by asterisks. B, Schematic representation and respective binding for the CMG constructs used in mapping experiments as described for A. CK, CaMKII-like domain. Binding to 6H-EAG869-1107 is shown in C-F. This fragment was chosen for display because there was minimal degradation during purification. C, 6H-EAG869-1107 binding to full-length CMG and CMG domains. GST-tagged CMG fragments immobilized on glutathione Sepharose beads were assayed for their ability to pull down 6H-EAG869-1107 as described in Materials and Methods. Top, Western blot probed with MRGS-6H antibody to detect interacting EAG fragments. Bottom, Bound 6H-EAG869-1107 quantified by densitometry; data are presented as the mean ± SEM; n = 3. D, 6H-EAG869-1107 binding to GST-SH3 domain fusion proteins. Top, The ability of purified GST-SH3 domains, derived from the proteins indicated above each lane, to pull down 6H-EAG869-1107 was determined in immunoblots by comparing the relative amount of associating 6H-EAG869-1107 to the amount available during the interaction experiment (left lane). As shown in lanes 2 and 3, there was little or no background binding to either glutathione Sepharose or immobilized GST tag. Bottom, Bound 6H-EAG869-1107 quantified by densitometry; data are presented as the mean ± SEM; n = 3. E, Dose-response curves comparing the binding of CMG and the CMG SH3 domains to 6H-EAG869-1107. Wells were coated overnight with 6H-EAG869-1107, washed, and then incubated with the indicated concentrations of GST fusion protein. Bound protein was detected using anti-GST antibody, followed by alkaline phosphatase-conjugated secondary antibody and treatment with p-nitrophenylphosphate. Binding was determined by colorimetric reaction at 405 nm. Each concentration was assayed in quadruplicate. Data are presented as the mean ± SEM; n = 4. O.D., Optical density. F, Effect of salt concentration on the 6H-EAG869-1107-CMG complex. Experiments examining the binding to GST-CMG were performed as described in Materials and Methods, with the exception that PBS buffer was modified to include the indicated NaCl concentrations. Complex formation was relatively unaffected in up to 1000 mm salt. Bottom, Bound 6H-EAG869-1107 quantified by densitometry; data are presented as the mean ± SEM; n = 3.
EAG C-terminal fragments associating with full-length CMG associated most robustly with the SH3 and guanylate kinase (GUK) domains of CMG (Fig. 5C). Although interactions with the GST-tagged CaMKII-like and PDZ [postsynaptic density-95 (PSD-95)/DLG/zona occludens-1] domains of CMG were also observed, the SH3 and GUK domains came closest to approximating the binding observed to the full-length CMG protein. Recent structural studies have suggested that the SH3 domain of PSD-95, another MAGUK protein, requires the GUK domain for proper orientation of the SH3 domain-binding pocket (McGee et al., 2001). However, in our experiments, there was no additional increase in the association of EAG when using a larger CMG fragment including both the SH3 and GUK domains (Fig. 5B, GST-CMG584-898).
Additional experiments further characterized the interaction of EAG with CMG in regard to its specificity, affinity, and mechanism. Figure 5D shows in vitro pull-down assays examining the binding of 6H-EAG869-1107 to a selection of SH3 domain fusion proteins immobilized on glutathione Sepharose beads. The association of EAG with the CMG SH3 domain was specific because there was minimal binding to the SH3 domains of src and the human DLG ortholog (Fig. 5D). Although EAG also bound to the SH3 domain of DLG, coexpression of EAG with DLG in oocytes failed to produce an increase in current (data not shown). In ELISAs (Fig. 5E), full-length CMG bound 6H-EAG869-1107 with a Kd of 1-2 nm, whereas the CMG SH3 domain displayed a Kd of ∼20 nm. Thus, interactions with other domains of CMG likely contribute to the affinity of the interaction. Finally, as shown in Figure 5F, the interaction between EAG and CMG was resistant to increases in salt concentration. Binding to CMG only decreased when the salt concentration was increased to above 1 m, suggesting that binding is not solely mediated by electrostatic interactions.
Mapping the EAG interaction site identifies a noncanonical SH3-binding motif
Additional in vitro binding experiments were performed with the aim of mapping the primary interaction site in EAG. The C-terminal cytoplasmic domain of EAG contains six putative SH3 domain-binding sites, as identified by the presence of a PxxP consensus sequence (Sparks et al., 1998). Indeed, as indicated on the right of Figure 5A, any EAG C-terminal fragment that contained the fourth putative SH3-binding motif directly associated with GST-tagged CMG. To determine whether this region was sufficient for the interaction, 38 amino acids spanning the fourth SH3 motif (residues 1032-1070) were attached to a DHFR tag. Because DHFR-EAG1032-1070 associated with CMG (Fig. 5A), the fourth SH3 motif appears necessary and sufficient for the interaction.
Two strategies were used to definitively establish the fourth SH3-binding motif as the site of the interaction with CMG. First, a peptide containing the fourth PxxP motif together with additional flanking amino acids (EAG1039-1054) was synthesized, cross-linked to beads, and then examined for its ability to pull down CMG SH3 domain that had been cleaved from the GST tag using thrombin. No association was observed although a 16 amino acid peptide that has been used to investigate the specificity of binding to the Src SH3 domain (src peptide) (Sparks et al., 1998) associated with the Src SH3 domain under the same experimental conditions (data not shown). Second, rationalizing that the peptide may not have contained a sufficient number of flanking amino acids, we generated a point mutation in the first proline of the fourth SH3-binding motif in the 6H-EAG869-1107 fragment (P1045A). Little or no decrease in the association of this fragment with full-length CMG was observed (Fig. 6B, compare lanes 1 and 4).
Recent work has established that some SH3 domains interact with a noncanonical binding motif characterized by an RxxK consensus sequence (Harkiolaki et al., 2003; Liu et al., 2003). As shown in Figure 6A, this sequence also is present in EAG just four amino acids upstream of the fourth PxxP motif. Indeed, binding to CMG was substantially reduced when leucine was substituted for arginine at residue 1037 (Fig. 6B, lane 2, R1037L869-1107). Binding to the SH3 domain was eliminated when the other basic residue in this motif, lysine at position 1040, was replaced by methionine (Fig. 6B, lane 3, K1040M).
To determine whether the above mutations were sufficient to disrupt the effect of CMG on EAG current, we returned to the oocyte expression system. As shown in Figure 6C, which compares scaled representative traces for wild-type and EAG-R1037L/K1040M channels, channel function was essentially unaffected by mutations in the SH3 domain-binding site. Figure 6D shows the result of coexpressing CMG with wild-type EAG, EAG-R1037L, EAG-K1040M, or the double mutant, EAG-R1037L/K1040M. As observed in our previous experiments, CMG produced a more than twofold increase in the whole-cell conductance of wild-type channels. The CMG-mediated increase was drastically reduced for EAG-R1037L and eliminated in EAG-K1040M and the double mutant. Finally, the SH3-binding motif also was required for the effect of EAG on CMG localization in COS-7 cells. As shown in Figure 6E, although wild-type EAG produced the same changes in CMG localization observed in the experiments of Figure 3 (CTE-labeled panels), EAG-R1037L/K1040M failed to compete with Tbr-1 (CTR-labeled panels). When statistically analyzed using a one-way ANOVA, there was no significant difference in the distribution of CMG to the membrane, cytoplasm, or nucleus when comparing averaged line scan data for the CMG and Tbr-1 and CMG, Tbr-1, and EAG-R1037L/K1040M conditions. Together, these results indicate that a direct interaction between EAG and CMG is required for the effect of EAG on the localization of CMG, as well as the effect of CMG on EAG current.
Discussion
Our findings indicate that EAG is functionally regulated by an association with the CMG adaptor protein. The primary effect of CMG is to increase EAG current and whole-cell conductance, and this increase depends on a direct association between proteins, because mutations of the CMG-binding motif in EAG prevent the effect. In addition, EAG successfully competes with another CMG/CASK-interacting protein to alter the localization of CMG. Thus, another distinct function of the EAG-CMG complex may be to localize CMG to the plasma membrane.
The mechanism primarily responsible for the increase in current appears to be an increase in the surface expression of EAG. Intriguingly, effects opposite those produced by CMG, namely decreased EAG current, decreased surface expression of EAG, and decreased inactivation, are observed when phosphorylation of EAG-T787 is prevented either by mutation of the phosphorylation site or by inhibition of CaMKII (Wang et al., 2002; present study). These findings suggest that the effect of CMG is produced by an increase in the phosphorylation of EAG-T787. In agreement, CMG increased the phosphorylation of EAG-T787, and mutation of this site prevented the CMG-mediated increase in EAG current without disrupting formation of the EAG-CMG complex. These results suggest that the mechanism underlying the effect of CMG is indirect and point to a change in the localization or activity of either a kinase or phosphatase in the vicinity of the channel. In addition, however, the possibility that CMG also directly affects the surface expression of phosphorylated EAG has not been ruled out. Direct and indirect effects of CMG may additively affect the membrane association of EAG channels.
At present, the enzyme most likely responsible for mediating the effect of CMG is CaMKII. EAG-T787 has been shown previously to be phosphorylated by CaMKII, inhibition of CaMKII has been shown to decrease EAG current, and CaMKII directly associates with CMG (Wang et al., 2002; Lu et al., 2003). However, it is not known whether other kinases such as protein kinase A can also phosphorylate T787, nor has the phosphatase responsible for dephosphorylating T787 been identified. Our results do not rule out the possibility that CMG may change the localization, activity, or efficiency of another kinase or a protein phosphatase. Nonetheless, given that CMG interacts with CaMKII (Lu et al., 2003), and given that no interaction between CMG (or its orthologs) and other kinases or phosphatases has been identified, the most likely model accounting for the CMG effect on EAG is one in which a common association with CMG colocalizes EAG with CaMKII and increases the efficiency of phosphorylation.
Intriguingly, after decreases in calcium, the association with CMG promotes phosphorylation of T306 of CaMKII, rendering the kinase inactive and then releasing it (Lu et al., 2003). CaMKII cannot be activated by subsequent increases in calcium as long as phosphorylation of T306 persists. The creation of a pool of inactive kinase predicts that CMG should have produced a decrease in EAG current, rather than the observed increase at basal calcium levels. These observations can be reconciled, however, if the cytoplasm and the local membrane are considered as separate compartments. Because CMG only associates with active kinase, CMG, in addition to globally increasing the pool of inactive kinase, may ensure that only active kinase is in the vicinity of the channel. Alternatively, EAG may directly or indirectly change the effect of CMG on CaMKII. Assuming that the majority of EAG is normally localized at the membrane, either alternative would result in an increase in active kinase at the membrane versus the cytoplasm. The stability of the CMG/CaMKII complex may also be regulated by the activity history of the synapse. Calcium influx can cause T287 phosphorylation, which, in addition to making the kinase activity calcium independent, can decrease the off-rate of CaM by four orders of magnitude (Putkey and Waxham, 1996). CaMKII with “trapped” CaM would be resistant to deactivation by CMG even at low intracellular calcium. EAG that was localized at a synapse that had been previously active then would be expected to be susceptible to CMG regulation.
The common association of CMG with EAG and CaMKII suggests that CMG acts as the central scaffold for the complex. However, EAG also directly associates with activated CaMKII (Sun et al., 2004), suggesting that EAG and CMG play equally important roles. The association between EAG and CaMKII constitutively activates the kinase and CaMKII remains associated with EAG, even once calcium returns to resting levels (Sun et al., 2004). Thus, the presence of EAG in the complex may alter the net effect of CMG on CaMKII activity. Finally, although phosphorylation of EAG-T787 is likely to occur via the constitutively active kinase, this constitutive activity is considerably lower than that of the fully activated kinase (Sun et al., 2004). Indeed, in our experiments, the association with CMG further increased EAG currents and phosphorylation of the channel above and beyond the levels achieved by the CaMKII endogenous to oocytes. CMG may localize and then transfer CaMKII to EAG, or CaMKII holoenzyme, which has 12 subunits, may be able to bind to both EAG and CMG simultaneously. In addition, CMG may increase the number of CaMKII molecules in the vicinity of the channel, and interlocking all three proteins may further stabilize an EAG-CMG-CaMKII complex.
The clearest demonstration of the physiological importance of CMG/CASK/LIN-2, to date, has been obtained in Caenorhabditis elegans, in which a complex of proteins including LIN-2, LIN-7, and LIN-10 localizes the receptor tyrosine kinase LET-23 to the basolateral surface of vulval precursor cells. The absence of LIN-2, LIN-7, or LIN-10 results in mislocalization of LET-23 receptors, causing a vulvaless phenotype (Kaech et al., 1998). The LIN-2-LIN-7-LIN-10 complex appears evolutionarily conserved because a similar complex, consisting of CASK, Velis, and the munc18-1-interacting protein Mint1, homologs of LIN-2, LIN-7, and LIN-10, respectively, has been observed in the mammalian CNS (Butz et al., 1998). These authors suggest that CASK is a scaffold for proteins involved in synaptic vesicle exocytosis and cell adhesion. The presynaptic complex may include not only Velis and Mint1, but also N-type calcium channels and neurexins (Hata et al., 1996; Maximov et al., 1999). When combined with the previously demonstrated presynaptic localization of EAG, CMG, and CaMKII (Wang et al., 2002; Lu et al., 2003; Sun et al., 2004), our findings provide additional support for a role of CMG/CASK in presynaptic function in vivo.
Footnotes
This work was supported by National Institutes of Health Grants MH62648 (G.F.W.) and 440860-29088 (P.J.B.) and by a University of Michigan Regent's Fellowship to D.D.M. We thank Leslie Griffith, Johannes Hell, Ed Chapman, Brian Kay, and Barry Ganetzky for advice and assistance at various stages of this project.
Correspondence should be addressed to Gisela F. Wilson, Department of Biology, 3057 Kraus Natural Science Building, 830 North University Avenue, University of Michigan, Ann Arbor, MI 48109-1048. E-mail: wilsongf@umich.edu.
S. Dimitratos's present address: Inscent, Inc., 17905 Sky Park Circle, Irvine, CA 92614.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254898-10$15.00/0
References
- Butz S, Okamoto M, Sudhof TC (1998) A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell 94: 773-782. [DOI] [PubMed] [Google Scholar]
- Dimitratos SD, Woods DF, Bryant PJ (1997) Camguk, Lin-2, and CASK: novel membrane-associated guanylate kinase homologs that also contain CaM kinase domains. Mech Dev 63: 127-130. [DOI] [PubMed] [Google Scholar]
- Drysdale R, Warmke J, Kreber R, Ganetzky B (1991) Molecular characterization of eag: a gene affecting potassium channels in Drosophila melanogaster Genetics 127: 497-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganetzky B, Wu C-F (1985) Genes and membrane excitability in Drosophila Trends Neurosci 8: 322-326. [Google Scholar]
- Garcia EP, Mehta S, Blair LA, Wells DG, Shang J, Fukushima T, Fallon JR, Garner CC, Marshall J (1998) SAP90 binds and clusters kainate receptors causing incomplete desensitization. Neuron 21: 727-739. [DOI] [PubMed] [Google Scholar]
- Griffith LC, Wang J, Zhong Y, Wu CF, Greenspan RJ (1994) Calcium/calmodulin-dependent protein kinase II and potassium channel subunit eag similarly affect plasticity in Drosophila Proc Natl Acad Sci USA 91: 10044-10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan KL, Dixon JE (1991) Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal Biochem 192: 262-267. [DOI] [PubMed] [Google Scholar]
- Guy HR, Durell SR, Warmke J, Drysdale R, Ganetzky B (1991) Similarities in amino acid sequences of Drosophila eag and cyclic nucleotide-gated channels. Science 254: 730. [DOI] [PubMed] [Google Scholar]
- Harkiolaki M, Lewitzky M, Gilbert RJ, Jones EY, Bourette RP, Mouchiroud G, Sondermann H, Moarefi I, Feller SM (2003) Structural basis for SH3 domain-mediated high-affinity binding between Mona/Gads and SLP-76. EMBO J 22: 2571-2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata Y, Butz S, Sudhof TC (1996) CASK: a novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J Neurosci 16: 2488-2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins R, Hajnal AF, Harp SA, Kim SK (1996) The C. elegans vulval induction gene lin-2 encodes a member of the MAGUK family of cell junction proteins. Development 122: 97-111. [DOI] [PubMed] [Google Scholar]
- Hsueh YP, Wang TF, Yang FC, Sheng M (2000) Nuclear translocation and transcription regulation by the membrane-associated guanylate kinase CASK/LIN-2. Nature 404: 298-302. [DOI] [PubMed] [Google Scholar]
- Kaech SM, Whitfield CW, Kim SK (1998) The LIN-2/LIN-7/LIN-10 complex mediates basolateral membrane localization of the C. elegans EGF receptor LET-23 in vulval epithelial cells. Cell 94: 761-771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Niethammer M, Rothschild A, Jan YN, Sheng M (1995) Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature 378: 85-88. [DOI] [PubMed] [Google Scholar]
- Liu Q, Berry D, Nash P, Pawson T, McGlade CJ, Li SS (2003) Structural basis for specific binding of the Gads SH3 domain to an RxxK motif-containing SLP-76 peptide: a novel mode of peptide recognition. Mol Cell 11: 471-481. [DOI] [PubMed] [Google Scholar]
- Lu CS, Hodge JJ, Mehren J, Sun XX, Griffith LC (2003) Regulation of the Ca2+/CaM-responsive pool of CaMKII by scaffold-dependent auto-phosphorylation. Neuron 40: 1185-1197. [DOI] [PubMed] [Google Scholar]
- Maximov A, Sudhof TC, Bezprozvanny I (1999) Association of neuronal calcium channels with modular adaptor proteins. J Biol Chem 274: 24453-24456. [DOI] [PubMed] [Google Scholar]
- McGee AW, Dakoji SR, Olsen O, Bredt DS, Lim WA, Prehoda KE (2001) Structure of the SH3-guanylate kinase module from PSD-95 suggests a mechanism for regulated assembly of MAGUK scaffolding proteins. Mol Cell 8: 1291-1301. [DOI] [PubMed] [Google Scholar]
- Morais Cabral JH, Lee A, Cohen SL, Chait BT, Li M, Mackinnon R (1998) Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell 95: 649-655. [DOI] [PubMed] [Google Scholar]
- Muller BM, Kistner U, Kindler S, Chung WJ, Kuhlendahl S, Fenster SD, Lau LF, Veh RW, Huganir RL, Gundelfinger ED, Garner CC (1996) SAP102, a novel postsynaptic protein that interacts with NMDA receptor complexes in vivo. Neuron 17: 255-265. [DOI] [PubMed] [Google Scholar]
- Putkey JA, Waxham MN (1996) A peptide model for calmodulin trapping by calcium/calmodulin-dependent protein kinase II. J Biol Chem 271: 29619-29623. [DOI] [PubMed] [Google Scholar]
- Sparks AB, Rider JE, Kay BK (1998) Mapping the specificity of SH3 domains with phage-displayed random-peptide libraries. Methods Mol Biol 84: 87-103. [DOI] [PubMed] [Google Scholar]
- Sun XX, Hodge JJ, Zhou Y, Nguyen M, Griffith LC (2004) The eag potassium channel binds and locally activates calcium/calmodulin-dependent protein kinase II. J Biol Chem 279: 10206-10214. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wilson GF, Griffith LC (2002) Calcium/calmodulin-dependent protein kinase II phosphorylates and regulates the Drosophila eag potassium channel. J Biol Chem 277: 24022-24029. [DOI] [PubMed] [Google Scholar]
- Warmke J, Drysdale R, Ganetzky B (1991) A distinct potassium channel polypeptide encoded by the Drosophila eag locus. Science 252: 1560-1562. [DOI] [PubMed] [Google Scholar]
- Wilson GF, Wang Z, Chouinard SW, Griffith LC, Ganetzky B (1998) Interaction of the K channel beta subunit, Hyperkinetic, with eag family members. J Biol Chem 273: 6389-6394. [DOI] [PubMed] [Google Scholar]