

Abstract
Tg(PrP-EGFP) mice express an enhanced green fluorescent protein (EGFP)-tagged version of the prion protein (PrP) that behaves like endogenous PrP in terms of its posttranslational processing, anatomical localization, and functional activity. In this study, we describe experiments in which Tg(PrP-EGFP) mice were inoculated intracerebrally with scrapie prions. Although PrP-EGFP was incapable of sustaining prion infection in Tg(PrP-EGFP)/Prn-p0/0 mice, it acted as a dominant-negative inhibitor that bound to, and fluorescently marked, deposits of PrPSc generated from endogenous PrP in Tg(PrP-EGFP)/Prn-p+/+ mice. Scrapie infection of these latter animals caused a progressive accumulation of fluorescent PrP-EGFP aggregates in neuropil, axons, and prominently in the Golgi apparatus of neurons. Our results provide an entirely new picture of PrPSc localization during the course of prion infection, and they identify for the first time intracellular sites of PrPSc formation that are not well visualized with conventional immunohistochemical techniques.
Keywords: prion, scrapie, transgenic, green fluorescent protein, neurodegeneration, Golgi
Introduction
Prions are infectious proteins of mammals, yeast, and fungi that propagate themselves by autocatalytic changes in conformational state (Aguzzi and Polymenidou, 2004; Wickner et al., 2004). PrPSc is a mammalian prion that is associated with several neurodegenerative disorders, including Creutzfeldt-Jakob disease, Gerstmann-Sträussler syndrome, and kuru in humans, as well as scrapie and bovine spongiform encephalopathy in animals (Prusiner, 2004). PrPSc is a conformationally altered isoform of PrPC, a surface glycoprotein of uncertain function that is expressed primarily on neurons and glia in the CNS (Prusiner, 1998).
PrPSc itself, or an intermediate generated as a result of the PrPC-PrPSc conversion process, is presumed to be responsible for the neurodegeneration seen in prion diseases (Chiesa and Harris, 2001). Therefore, determining the anatomical and subcellular localization of PrPSc is crucial for understanding the pathogenesis of these disorders. However, there is a serious technical difficulty in localizing PrPSc in tissue sections and cultured cells using traditional immunocytochemical staining techniques. Antibodies that recognize PrPC usually display weak affinity for PrPSc, attributable to epitope masking as a result of protein aggregation or conformational alterations (Serban et al., 1990; Peretz et al., 1997). Therefore, the detection of PrPSc in situ relies on antigen retrieval techniques that enhance the antigenicity of PrPSc by denaturing or hydrolyzing the protein (Kitamoto et al., 1986, 1987, 1992; Taraboulos et al., 1990). Unfortunately, these procedures also irreversibly damage cellular structures and can degrade or redistribute PrPSc. The requirement for antigen retrieval creates particular difficulties in localizing PrPSc at the subcellular level, and most PrPSc deposits visualized in brain sections by light or electron microscopic techniques are extracellular (Jeffrey et al., 1992, 1994b; DeArmond et al., 2004). Thus, we know very little about the intracellular compartments in which PrPSc accumulates and in which the initial events in PrPSc formation are likely to occur.
To circumvent these difficulties, we have constructed transgenic mice expressing PrP-EGFP, a fusion of wild-type PrP with enhanced green fluorescent protein (EGFP), under control of a PrP promoter. In a previous study, we showed that PrP-EGFP is posttranslationally processed and anatomically localized in neurons in the brains of transgenic mice in a way that is similar to that of endogenous PrP (Barmada et al., 2004). In that study, we also demonstrated that PrP-EGFP retains functional activity based on a genetic test in which the fusion protein was able to rescue the neurodegenerative phenotype induced by an N-terminally truncated form of PrP (Δ32-134). In the present study, we inoculated Tg(PrP-EGFP) mice with scrapie prions to directly visualize PrPSc in the brain without the need for antibody staining and antigen retrieval procedures. We demonstrate that, although PrP-EGFP is not itself a substrate for formation of PrPSc-EGFP, it acts as a ligand and inhibitor that binds specifically to PrPSc derived from endogenous PrPC, thus revealing sites where PrPSc is likely to be formed.
Materials and Methods
Transgenic mice. Construction of Tg(PrP-EGFP) mice has been described previously (Barmada et al., 2004). Transgene-positive animals were maintained on the Prn-p+/+ background by mating to C57BL/6J × CBA/J mice and on the Prn-p0/0 background by mating to Prn-p0/0 mice obtained from Charles Weissmann (Scripps Research Institute, Palm Beach, FL). The latter animals, which were originally created on a C57BL/6J/129 background (Büeler et al., 1992), have been maintained in our laboratory by crossing with C57BL/6J × CBA/J mice. Tg(PrP-EGFP+/+)/Prn-p0/0 mice were created by intercrossing Tg(PrP-)Prn-p0/0 mice. Tg(PrP-EGFP+/0)/tga20+/0/Prn-p0/0 animals were derived by crossing Tg(PrP-EGFP+/0)Prn-p0/0 mice with homozygous tga20 mice (Fischer et al., 1996) obtained from the European Mouse Mutant Archive (GSF National Research Center for Environment and Health, Institute of Experimental Genetics, Munich, Germany).
Scrapie infections. Inocula were prepared from the brains of terminally ill Tg(WT-E1+/+)/Prn-p0/0 mice (Chiesa et al., 1998) that express 3F4-tagged wild-type PrP and that had been infected with CD1-passaged Rocky Mountain Laboratory (RML) scrapie (obtained from Byron Caughey and Richard Race at Rocky Mountain Laboratories). Twenty-five percent (w/v) brain homogenates were prepared in sterile PBS using motorized tissue grinders. Homogenates were clarified by centrifugation for 5 min at 900 × g. Brain homogenates were diluted to 1% using sterile PBS, and 25 μl was injected intracerebrally into the right parietal lobes of 4- to 6-week-old recipient mice using a 25 gauge needle. Animals were monitored regularly for the appearance of clinical symptoms, including ataxia, weight loss, kyphosis, hyperexcitability, and hindlimb paralysis. Ataxia was assessed by observing the mice walking on a metal grid apparatus (Chiesa et al., 1998). Mice were scored as ill if they exhibited two or more clinical symptoms.
Protease-digestion assays. Twenty-five percent (w/v) brain homogenates were prepared as described above, and the protein concentrations were determined using a BCA assay kit (Pierce, Rockford, IL). The equivalent of 300 μg of protein was resuspended in 180 μl of BH buffer (0.5% sodium deoxycholate, 0.5% Triton X-100, 0.5% NP-40, 0.2% Sarkosyl, 150 mm NaCl, and 50 mm Tris-HCl, pH 7.5) and then incubated with proteinase K (PK) (final concentration, 20 μg/ml) for 30 min at 37°C. Digestion was terminated by addition of phenylmethylsulfonyl fluoride (PMSF) to a final concentration of 5 mm and incubation on ice for 10 min. Proteins were precipitated with methanol and then analyzed by SDS-PAGE and Western blotting using the following antibodies: anti-PrP monoclonal antibody 3F4 (Bolton et al., 1991), anti-PrP monoclonal antibody 8H4 (Zanusso et al., 1998), and anti-GFP rabbit polyclonal antibody (provided by Maurine Linder, Washington University).
Immunoprecipitation of PrP-EGFP. Two hundred microliters of 25% brain homogenate were combined with 300 μl of PBS and 500 μl of 2% Triton X-100 (in PBS), and the mixture was incubated for 20 min at 4°C. Samples were precleared by incubation with 50 μl of protein G-Sepharose beads (Sigma, St. Louis, MO) for 20 min at 4°C, and the supernatants were transferred to siliconized microfuge tubes. For immunoprecipitation of PrP-EGFP, 10 μl of anti-GFP rabbit polyclonal antibody was added to each sample, together with protease inhibitors (1 μg/ml pepstatin, 1 μg/ml leupeptin, 2 mm EDTA, and 0.5 mm PMSF). As a control, some samples were also immunoprecipitated with 10 μl of nonspecific rabbit polyclonal IgG (Antibodies Incorporated, Davis, CA). Samples were mixed end-over-end at 4°C for 16 h, after which immune complexes were collected by addition of 500 μl of protein G-Sepharose beads. Beads were washed three times with 1% Triton X-100 (in PBS), washed twice with 150 mm NaCl and 50 mm Tris-HCl, pH 7.5, and then boiled in SDS-PAGE sample buffer before analysis by SDS-PAGE and Western blotting using 3F4, 8H4, or anti-GFP antibodies. In some cases, the beads were resuspended in 200 μl of 0.1% Sarkosyl and treated with PK (final concentration, 20 μg/ml) for 30 min at 37°C. Digestion was terminated by addition of PMSF to a final concentration of 5 mm, after which proteins were precipitated with methanol and analyzed by SDS-PAGE and Western blotting as above.
Sodium phosphotungstate precipitation. Two hundred microliters of 25% brain homogenate were combined with 300 μl of PBS and 500 μl of 2% Sarkosyl (in PBS), and the mixture was incubated for 10 min at 37°C. MgCl2 (final concentration, 1 mm) and 50 U of benzonase (Sigma, St. Louis, MO) were added, and samples were incubated for 30 min at 37°C. Sodium phosphotungstate (Sigma) from a 4% stock solution was added to reach a final concentration of 0.3%, and samples were incubated for 30 min at 37°C. Precipitates were then collected by centrifugation at 14,000 × g for 30 min at room temperature. Supernatants were removed, and the pellets were resuspended in 18 μl of 0.1% Sarkosyl. In some cases, resuspended proteins were treated with PK (final concentration, 50 μg/ml) for 1 h at 37°C. Ten microliters of SDS-PAGE sample buffer were added to each sample, after which proteins were separated by SDS-PAGE and then analyzed by Western blotting with 3F4, 8H4, or anti-GFP antibodies.
Fluorescence microscopy and immunostaining. Mice were perfused transcardially with 50 ml of 0.1 m phosphate buffer, pH 7.2, followed by 100 ml of 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.2. Brains were postfixed in the same fixative for 3 h at 4°C, transferred to 0.1 m phosphate buffer, pH 7.2, and stored at 4°C. Sagittal sections (50-100 μm thickness) were cut using a vibratome (The Vibratome Company, St. Louis, MO) and placed in sterile PBS containing 0.02% sodium azide. For visualization of intrinsic PrP-EGFP fluorescence, sections were mounted on glass slides using Gel/Mount solution (Biomeda, Foster City, CA).
To colocalize PrP-EGFP with other proteins, floating sections were rinsed three times in PBS and then blocked and permeabilized by incubation for 1 h at room temperature in PBS containing 0.3% Triton X-100 and 1% goat serum. Sections were then incubated for 16 h at 4°C using the following antibodies and dilutions: rat anti-mouse CD90.2 (Thy1.2) monoclonal antibody (1:50; BD Biosciences Pharmingen, San Diego, CA), rabbit anti-giantin polyclonal antibody (1:1000; Covance, Berkeley, CA), rabbit anti-translocon-associated protein α (TRAPα) polyclonal antibody (1:1000; Upstate Biotechnology, Lake Placid, NY), rat anti-lysosome-associated membrane protein 1 (LAMP1) monoclonal antibody (1D4B; 1:500; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), 3F4 (1:1000), 8H4 (1:1000), and anti-GFP antibody (1:1000). Sections were rinsed three times in PBS before incubation for 1 h at room temperature with secondary antibodies (Alexa 594-coupled goat anti-mouse, anti-rabbit, or anti-rat IgG from Molecular Probes, Eugene, OR) diluted 1:500 in PBS. After rinsing in PBS, sections were mounted on glass slides as above.
For PrPSc immunostaining, floating sections were pretreated with 96% formic acid for 15 min at room temperature. After rinsing in PBS, sections were then blocked and permeabilized in PBS containing 0.3% Triton X-100 and 1% goat serum. Sections were incubated with 8H4 antibody (1:250) for 16 h at 4°C and then rinsed in PBS before incubation for 1 h at room temperature with secondary antibody (Alexa 488-coupled goat anti-mouse IgG). Sections were rinsed in PBS and then mounted on glass slides as above.
Sections were viewed with a Zeiss (Oberkochen, Germany) LSM 510 confocal microscope with an Axiovert 200 laser-scanning system. Processing of digital images was accomplished using Zeiss LSM WS 510 software and Adobe Photoshop 6.0 (Adobe Systems, San Jose, CA).
Results
PrP-EGFP is not converted to PrPSc-EGFP, but it inhibits formation of PrPSc from endogenous PrPC
To determine whether PrP-EGFP could be converted to PrPSc-EGFP during prion infection, we inoculated Tg(PrP-EGFP+/+)/Prn-p0/0 mice with the RML strain of scrapie. The scrapie inoculum had been passaged through Tg(WT) mice (Chiesa et al., 1998) to introduce the 3F4 epitope tag, which is also present in the PrP-EGFP substrate. Tg(PrP-EGFP+/+)/Prn-p0/0 mice, which are homozygous for the transgene array, do not express endogenous PrP, and they express PrP-EGFP at approximately twice the level of endogenous PrP found in wild-type mice. These animals showed no signs of illness as late as 550 d postinoculation (dpi), similar to nontransgenic Prn-p0/0 mice inoculated with the same preparation of RML scrapie (Table 1, Fig. 1). These results indicate that prion infection cannot be sustained efficiently in Tg(PrP-EGFP+/+) mice in the absence of endogenous PrP, suggesting that PrP-EGFP may not be a substrate for formation of PrPSc-EGFP.
Table 1.
Clinical illness in mice inoculated with RML scrapie
|
Genotype |
Onset (dpi) |
Death (dpi) |
n |
|---|---|---|---|
| PrP-EGFP+/+/Prn-p0/0 | >550a | N/A | 20 |
| Prn-p0/0 | >550a | N/A | 4 |
| PrP-EGFP+/0/Prn-p+/+ | 206 ± 17 | 206 ± 17 | 19 |
| Prn-p+/+ | 157 ± 10 | 190 ± 22 | 21 |
| PrP-EGFP+/0/tga20+/0/Prn-p0/0 | 109 ± 7 | 112 ± 9 | 12 |
| tga20+/0/Prn-p0/0
|
92 ± 17 |
106 ± 11 |
20 |
For mice that remained healthy, the time after inoculation at which the animals were killed to terminate the experiment is given. n, Number of animals; N/A, not applicable.
Figure 1.

PrP-EGFP does not sustain scrapie propagation in Prn-p0/0 mice, and it delays the development of disease in Prn-p+/+ and tga20 mice. Mice were inoculated with scrapie and monitored for the development of clinical illness.
In contrast, Tg(PrP-EGFP+/0)/Prn-p+/+ mice, which express endogenous PrP as well as transgenically encoded PrP-EGFP, did become ill after inoculation with RML prions, with an incubation time of 206 ± 17 d (Table 1, Fig. 1). This incubation time was significantly longer (p < 0.0001; unpaired t test) than that for nontransgenic Prn-p+/+ mice, which became ill at 157 ± 10 d after RML inoculation. Thus, the presence of PrP—EGFP, in addition to wild-type PrP, delayed the onset of symptoms by ∼30%, suggesting that the fusion protein was inhibiting prion propagation in transgenic animals. A similar effect of PrP-EGFP was observed in tga20+/0/Prn-p0/0 mice that overexpress a wild-type PrP transgene by fivefold (Fischer et al., 1996) (Table 1, Fig. 1). In the absence of the PrP-EGFP transgene, these animals became ill at 92 ± 4 d after inoculation. In contrast, Tg(PrP-EGFP+/0)/tga20+/0/Prn-p0/0 mice did not exhibit symptoms until 109 ± 7 dpi. This 18% delay in incubation time was statistically significant (p < 0.0001; unpaired t test). Together, these data show that PrP-EGFP delays the development of prion disease sustained by wild-type PrP.
We tested for the presence of PrPSc in the brains of inoculated animals by Western blotting brain homogenates after protease digestion. PK treatment of PrPSc produces a protease-resistant core fragment of 27-30 kDa (PrP 27-30), whereas under the same conditions, PrPC is completely degraded. For these experiments, blots were developed with either of two anti-PrP antibodies: 8H4 (Zanusso et al., 1998), which recognizes both endogenous mouse PrP and transgenically encoded PrP-EGFP, or 3F4 (Bolton et al., 1991), which recognizes only PrP-EGFP because the appropriate epitope was engineered into the PrP moiety of the fusion protein. Brain homogenates from Tg(PrP-EGFP+/+)/Prn-p0/0 mice killed at 530 dpi showed no evidence of PrP 27-30, or other protease-resistant fragments, when blots were probed with either 8H4 or 3F4 antibodies (Fig. 2A,B, lanes 1, 2). This result demonstrates that PrP-EGFP is not converted into PrPSc-EGFP in these animals, consistent with the absence of clinical illness noted above. In contrast, significant amounts of PrP 27-30 were detected in the brains of Tg(PrP-EGFP+/0)/Prn-p+/+ mice by 200 dpi, correlating with the onset of symptoms in these animals (Fig. 2A, lanes 11-18). PrP 27-30 was detected in these samples with 8H4 antibody, but not with 3F4 antibody, indicating that the PrPSc produced was derived from endogenous PrP and not from PrP-EGFP (Fig. 2, compare A, B, lanes 11-18). Brain homogenates from nontransgenic Prn-p+/+ mice also demonstrated a significant accumulation of PK-resistant PrP when blotted with 8H4 antibody, but, in these animals, PrP 27-30 was detectable by 160 dpi, consistent with the earlier development of clinical symptoms in the nontransgenic mice (Fig. 2 A, lanes 7, 8). As expected, no PK-resistant PrP could be detected by immunoblotting samples from Prn-p+/+ mice with 3F4 antibody (Fig. 2 B, lanes 5-8) or by immunoblotting samples from Prn-p0/0 mice with either 8H4 or 3F4 antibodies (Fig. 2 A, B, lanes 3-4). In addition, uninoculated mice displayed no PK-resistant PrP (Fig. 2 A, B, lanes 5, 6, 9, 10). These results confirm that PrP-EGFP does not convert to PrPSc-EGFP and that it inhibits the formation of PrPSc from endogenous PrPC.
Figure 2.

PrP-EGFP does not convert to PrPSc-EGFP. Brain homogenates from scrapie-infected mice and uninfected mice (Un) of the indicated genotypes were incubated with PK (+ lanes) or left untreated (- lanes) before analysis by SDS-PAGE and Western blotting to detect PrP. A, Blots were probed with the anti-PrP antibody 8H4, which recognizes both PrP-EGFP (arrowhead at 60-70 kDa, to the right of lane 18) and endogenous PrP (30-36 kDa). B, Blots were probed with anti-PrP antibody 3F4, which recognizes only PrP-EGFP (arrowhead at 60-70 kDa, to the left of lane 9). Lanes representing PK-digested samples contain 10-fold more protein than those corresponding to undigested samples. The bands at 25 kDa in lanes 10, 16, and 18 are nonspecific and are not related to infection, because they appear in samples from uninoculated mice (lane 10). Molecular size markers are given in kilodaltons.
PrP-EGFP binds to PrPSc in the brains of infected animals
The inhibitory effect of PrP-EGFP on the conversion of endogenous PrPC to PrPSc suggested the possibility that PrP-EGFP interferes with some step in the conversion reaction by physically associating with PrPSc. To test for binding of PrP-EGFP to PrPSc, we immunoprecipitated PrP-EGFP from the brains of terminally ill Tg(PrP-EGFP+/0)/Prn-p+/+ mice using anti-GFP antibodies and analyzed the immunoprecipitates for the presence of PrPSc by PK digestion and immunoblotting with 8H4 antibody. Before PK digestion, the immunoprecipitated fraction contained PrP-EGFP (60-70 kDa), as well as endogenous PrP (30-35 kDa) (Fig. 3A, lane 2). After PK digestion, PrP-EGFP in the immunoprecipitate was degraded, but PrP 27-30 was clearly visible (Fig. 3A, lanes 3, 4). No PK-resistant protein was detected by probing the immunoprecipitated material with 3F4 antibody (data not shown), indicating again that PrP-EGFP does not adopt a PK-resistant form and that most of the PrP 27-30 visualized with 8H4 antibody was derived from endogenous PrPSc bound to PrP-EGFP. As specificity controls, immunoprecipitation of brain homogenate from terminally ill Tg(PrP-EGFP+/0)/Prn-p+/+ animals using nonspecific IgG failed to isolate either PrP-EGFP or endogenous PrP (Fig. 3A, lanes 5-7), and no PrPSc was immunoprecipitated from the brains of terminally ill Prn-p+/+ animals using anti-GFP antibodies (Fig. 3A, lanes 8-11). These data suggest that PrP-EGFP encoded by the transgene and PrPSc derived from endogenous PrPC specifically bind to one another in the brains of infected Tg(PrP-EGFP+/0)/Prn-p+/+ mice.
Figure 3.

PrP-EGFP binds to PrPSc derived from endogenous PrP in the brains of infected mice. A, PrP-EGFP was immunoprecipitated from brain homogenates prepared from scrapie-infected mice of the indicated genotypes using anti-GFP antibodies (αGFP, lanes 1-4, 8-11) or nonspecific IgG antibodies (IgG, lanes 5-7). The immunoprecipitates (IP) were incubated with PK (+ lanes) or left untreated (- lanes) before analysis by SDS-PAGE and immunoblotting with 8H4 antibody. Lanes 1 and 8 represent input (In) samples before immunoprecipitation. Lanes marked 10x represent 10-fold more protein than lanes marked 1x. PrP-EGFP migrates at 60-70 kDa, and endogenous PrP migrates at 30-36 kDa (arrowhead and bracket, respectively, to the left of lane 1). The band at 55 kDa (asterisk to the left of lane 1) is nonspecific. The position of PrP 27-30 is indicated by the bracket to the right of lane 4. B, PrPSc was precipitated from the brains of uninfected and scrapie-infected Tg(PrP-EGFP+/0)/Prn-p+/+ mice using NaPTA, separated by SDS-PAGE, and probed with antibodies 8H4 (lanes 1 and 2), 3F4 (lanes 3 and 4), or anti-GFP (αGFP, lanes 5 and 6). The arrowhead to the right of lane 6 indicates the position of PrP-EGFP. C, Same as B, but NaPTA pellets were digested with PK before SDS-PAGE and Western blotting. The 30 kDa band in lanes 5 and 6 corresponds to an intrinsically protease-resistant fragment GFP (Cody et al., 1993). D, The supernatants left after NaPTA precipitation of brain homogenates from uninfected (Un) and infected (Inf) Tg (PrP-EGFP+/0)/Prn-p+/+ mice were probed with anti-actin antibodies to demonstrate equal protein content.
As a second way of demonstrating a physical interaction between PrP-EGFP and PrPSc, we precipitated PrPSc from brain homogenates of infected and uninfected Tg(PrP-EGFP+/0)/Prn-p+/+ mice using sodium phosphotungstate (NaPTA) and analyzed the pellets for the presence of PrP-EGFP. NaPTA precipitation has been shown to selectively concentrate and purify PrPSc from tissue homogenates (Wadsworth et al., 2001). Western blots of NaPTA precipitates from the brains of Tg(PrP-EGFP+/0)/Prn-p+/+ animals were probed with either 8H4, 3F4, or anti-GFP antibodies. Although a small amount of PrP-EGFP could be found in NaPTA precipitates from the brains of uninfected animals (Fig. 3B, lanes 1, 3, 5), approximately fivefold more PrP-EGFP was found in precipitates from infected animals (Fig. 3B, lanes 2, 4, 6), indicating that PrP-EGFP was binding to precipitated PrPSc. Actin immunoblotting of the supernatants left after NaPTA precipitation demonstrated equivalent protein concentrations in each sample (Fig. 3D). After PK digestion of the NaPTA precipitates, PrP 27-30 was found only in samples from infected animals and was detectable only when the precipitates were probed with 8H4 but not with 3F4 or anti-GFP antibodies (Fig. 3C, lanes 2, 4, 6). This result indicates once again that the PrPSc found in infected Tg(PrP-EGFP+/0)/Prn-p+/+ mice was derived from endogenous PrPC and not from PrP-EGFP. Moreover, because NaPTA precipitation efficiently concentrates even small amounts of PrPSc from large volumes of tissue homogenates (Wadsworth et al., 2001), the absence of detectable PK-resistant PrP-EGFP in NaPTA precipitates from Tg(PrP-EGFP+/0)/Prn-p+/+ mice suggests that conversion of PrP-EGFP to PrPSc-EGFP must be minimal in these animals.
Together, the results obtained using anti-GFP immunoprecipitation (Fig. 3A) and NaPTA precipitation (Fig. 3B-D) indicate that PrP-EGFP binds specifically to endogenously derived PrPSc in the brains of Tg(PrP-EGFP+/0)/Prn-p+/+ mice.
Prion infection causes accumulation of fluorescent aggregates of PrP-EGFP
To determine whether PrPSc accumulation in infected mice altered the distribution of PrP-EGFP, brain sections from terminally ill Tg(PrP-EGFP+/0)/Prn-p+/+ animals and from age-matched, uninoculated control mice were examined by confocal fluorescent microscopy. Prominent fluorescent aggregates of PrP-EGFP were detected in the brains of the infected mice (Fig. 4A-D) but were absent from uninoculated control mice (Fig. 4E-H). The aggregates in the infected animals were seen in almost every brain region examined, although they were especially obvious in the granule cell layer of the cerebellum (Fig. 4A), layers II-V of the cerebral cortex (Fig. 4B), the thalamus (Fig. 4C), the corpus callosum (Fig. 4D), the dentate gyrus (see Fig. 6A-C), and the hippocampus (data not shown). Fluorescent aggregates were found in neuropil regions, in which they appeared as fine, granular deposits (Fig. 4A) as well as larger, plaque-like accumulations (Fig. 4C, arrowheads). Aggregates of PrP-EGFP were also frequently present within the cell bodies of neurons, where they often assumed a perinuclear distribution (Fig. 4B, arrows). These intracellular deposits were present in neurons throughout the brain but were most apparent in larger neurons, such as those in the interposed nuclei of the cerebellum (Fig. 5B-E), the hilus of the dentate gyrus (Fig. 6B,C), and the cerebral cortex (Fig. 6E,F). White matter tracts, such as those in the corpus callosum, exhibited linear, fluorescent deposits that appeared to fill dystrophic neurites and axonal swellings (Fig. 4D). PrP-EGFP aggregates were not observed in GFAP-positive astrocytes (data not shown). We observed similar kinds of PrP-EGFP aggregates in the brains of scrapie-infected Tg(PrP-EGFP+/0)/tga20+/0/Prn-p0/0 mice (not shown). The aggregates in these animals were less prominent, however, most likely reflecting the relatively low level of PrPSc that accumulates in the brains of infected tga20 mice (Fischer et al., 1996).
Figure 4.

Scrapie infection causes accumulation of fluorescent aggregates of PrP-EGFP. Brain sections from terminally ill Tg(PrP-EGFP+/0)/Prn-p+/+ mice (202-284 dpi; A-D) and from age-matched, uninoculated control mice (E-G) were viewed using confocal fluorescence microscopy. Sections are from the granule cell layer of the cerebellum (A, E), layers II-III of the cerebral cortex (B, F), the thalamus (C, G), and the corpus callosum (D, H). The red boxes over the anatomical models shown along the top indicate the brain regions from which sections were taken. The arrowheads in C indicate plaque-like PrP-EGFP aggregates in the neuropil, and the arrow in B indicates a perinuclear deposit of PrP-EGFP in a neuronal cell body. Scale bar: (in A) A-H, 20 μm.
Figure 6.

PrP-EGFP does not colocalize with Thy1 during scrapie infection. Sections from the cerebral cortex of terminally ill (267 dpi; A-C) and age-matched, uninfected (D, E) Tg(PrP-EGFP+/0)/Prn-p+/+ mice were immunostained for Thy1 and examined using confocal fluorescent microscopy. The intrinsic fluorescence of PrP-EGFP appears in green (A, D), and Thy1 immunofluorescence appears in red (B, E). Merged red and green images are shown in C and F. A, C, Arrowheads and arrows indicate, respectively, neuropil and intracellular aggregates of PrP-EGFP. Scale bar: (in A) A-F, 20 μm.
Figure 5.

Aggregation of PrP-EGFP occurs gradually during the course of scrapie infection. Brain sections from uninoculated Tg(PrP-EGFP+/0)Prn-p+/+ mice (A, F) or from scrapie-infected Tg(PrP-EGFP+/0)Prn-p+/+ mice at 100 dpi (B, G), 150 dpi (C, H), 200 dpi (D, I), 284 dpi (E), or 202 dpi (J) were viewed using confocal fluorescence microscopy. Sections were from the interposed nuclei of the cerebellum (A-E) and the thalamus (F-J). Scale bar: (in A) A-J, 20 μm.
We determined the time course of PrP-EGFP redistribution by examining brain sections from infected Tg(PrP-EGFP+/0)/Prn-p+/+ mice at various times after infection. At 100 dpi, the distribution of PrP-EGFP appeared no different from that in uninoculated animals (Fig. 5, compare B, G, with A, F). By 150 dpi, significant intracellular accumulations of PrP-EGFP were detectable (Fig. 5C,H). This initial aggregation of PrP-EGFP predates the development of clinical symptoms and astrocytosis, which were not apparent in these animals until 200 dpi (Fig. 1 and data not shown). By 200 dpi, intracellular deposits of PrP-EGFP were found in many neurons, and aggregates in the neuropil were also visible (Fig. 5D,I). In terminally ill animals (202-284 dpi), most neurons contained large intracellular accumulations of fluorescent protein, and extracellular deposits had increased in size and in number throughout the thalamus, cortex, and brainstem (Fig. 5E,J).
Together with the data demonstrating binding of PrP-EGFP to PrPSc, the fluorescence images suggest that PrP-EGFP acts as a specific ligand that marks the deposition of PrPSc in the brains of infected mice. However, it is also possible that the distribution of PrP-EGFP is altered non-specifically during the course of infection as a result of neuronal damage or dysfunction, which might cause changes in the cellular structures in which the fluorescent protein is normally localized. To rule out this possibility, we analyzed the distribution of Thy1, a neuronal protein that, like PrPC, is glycosylphosphatidylinositol (GPI) anchored and is localized to detergent-insoluble lipid rafts on the neuronal plasma membrane (Madore et al., 1999). In the cerebral cortex of uninfected Tg(PrP-EGFP+/0)/Prn-p+/+ mice, both PrP-EGFP and Thy1 were distributed throughout the neuropil, although Thy1 displayed a more punctate appearance that probably reflects its presence in distinct raft subdomains (Madore et al., 1999) (Fig. 6D-E). Both proteins were significantly redistributed in terminally ill animals, but the pattern of redistribution was unique for each protein. PrP-EGFP accumulated in large intracellular and extracellular deposits (Fig. 6A), whereas Thy1 was present in smaller, more punctate accumulations (Fig. 6B) that showed little overlap with PrP-EGFP (Fig. 6C). The fact that PrP-EGFP aggregates do not colocalize with Thy1, even at late stages of illness, indicates that the accumulations of PrP-EGFP represent an independent and specific feature of disease in transgenic animals. Moreover, the aggregation of PrP-EGFP preceded the redistribution of Thy1, as well as the onset of clinical symptoms and the development of astrocytosis and neuronal loss (data not shown). These data strongly suggest that the aggregation of PrP-EGFP observed in infected mice constitutes an early event in the pathogenesis of disease, rather than a secondary response to neurodegeneration.
PrP-EGFP accumulates in the Golgi apparatus of neurons beginning early in the course of scrapie infection
To more precisely determine the subcellular location of intraneuronal PrP-EGFP deposits in scrapie-infected Tg(PrP-EGFP+/0)/Prn-p+/+ mice, we colocalized these deposits with markers for several intracellular organelles. For these experiments, we focused on relatively large neurons found in the hilus of the dentate gyrus and in layers II-III of the cerebral cortex because of their easily identifiable organelles and prominent intracellular PrP-EGFP aggregates. By 150 d after infection, 50 d before the onset of clinical symptoms, animals displayed substantial intraneuronal accumulations of PrP-EGFP that colocalized with the resident Golgi protein giantin (Fig. 7B,E). PrP-EGFP continued to accumulate within the Golgi apparatus as the disease progressed, and, by the terminal stages of illness, PrP-EGFP aggregates completely filled the Golgi and were present in the adjacent neuropil as well (Fig. 7C,F). In contrast, PrP-EGFP aggregates in terminally ill mice did not colocalize with TRAPα, a marker for the endoplasmic reticulum (ER), or with LAMP1, a marker for lysosomes (Fig. 7G,H). In uninoculated, control mice, very little PrP-EGFP was visible within the cell bodies of neurons in the same brain regions (Fig. 7A,D). Thus, PrP-EGFP accumulates progressively in the Golgi apparatus of neurons beginning in the presymptomatic phase of the disease. Deposits are not visible in the ER or lysosomes, although our results do not rule out the possibility that these or other organelles might also be sites of PrP-EGFP deposition, particularly in the terminal stages of disease when the neuronal cell bodies are filled with fluorescent protein.
Figure 7.
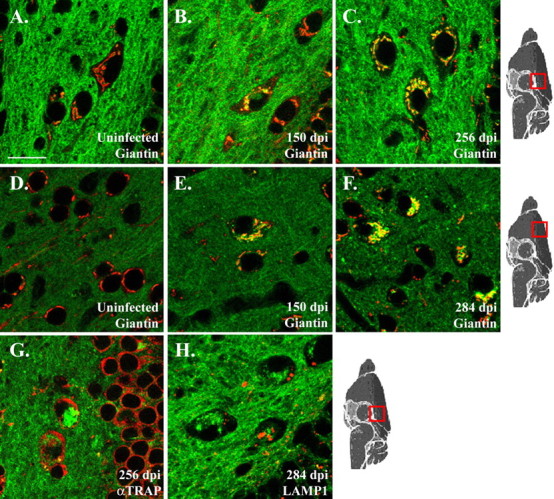
PrP-EGFP accumulates in the Golgi apparatus of neurons beginning early in the course of scrapie infection. Brain sections from the hilus of the dentate gyrus (A-C, G, H) and layers II and III of the cerebral cortex (D-F) of Tg(PrP-EGFP+/0)/Prn-p+/+ mice were immunostained for giantin (A-F), TRAPα (G), or LAMP1 (H) using Alexa 594-coupled secondary antibodies. Sections were then viewed by confocal fluorescence microscopy to reveal the intrinsic green fluorescence of PrP-EGFP and the red fluorescence from the immunostained marker proteins. All panels show merged green and red images. Mice were either uninoculated (A, D) or inoculated with scrapie (B, C, E-H; dpi are indicated on each panel). Scale bar: (in A) A-H, 20 μm.
Comparison of PrP-EGFP fluorescence with conventional immunohistochemistry for visualization of PrPSc
We stained brain sections from scrapie-infected Tg(PrP-EGFP+/0)/Prn-p+/+ mice using anti-PrP antibodies to compare the distribution of PrPSc seen by immunohistochemistry with the pattern observed using the intrinsic fluorescence of PrP-EGFP. Sections were treated with 96% formic acid before immunostaining, which is a standard technique for enhancing the immunogenicity of PrPSc (Kitamoto et al., 1987). Sections from transgenic mice stained with 8H4 antibody showed PrP aggregates in the neuropil that had the same appearance as those in terminally ill, nontransgenic mice (Fig. 8B,C). Of note, these immunostained sections lacked the intracellular PrP deposits seen using EGFP fluorescence (Fig. 8A). Control sections from uninoculated transgenic mice showed a relatively uniform pattern of PrP-EGFP localization, with no visible aggregates, when viewed by either EGFP fluorescence (Fig. 8D) or immunostaining (Fig. 8E). Similar results were obtained in these experiments using 3F4 or anti-GFP antibodies for immunostaining (data not shown). These results suggest that the presence of EGFP-PrP facilitates the detection of intracellular aggregates of PrPSc that are not visualized by immunostaining after formic acid treatment.
Figure 8.

Comparison of PrP-EGFP fluorescence with conventional immunohistochemistry for visualization of PrPSc. Sections were taken from the cerebral cortex (layers II-III) of a terminally ill Tg(PrP-EGFP+/0)/Prn-p+/+ mouse (284 dpi; A, B), a terminally ill, nontransgenic Prn-p+/+ mouse (218 dpi; C), an uninfected Tg(PrP-EGFP+/0)/Prn-p+/+ mouse (D, E), and an uninfected Prn-p+/+ mouse (F). A, D, The intrinsic fluorescence of PrP-EGFP. B, C, E, F, PrPSc immunostaining using 8H4 antibody (with a red-coupled secondary antibody) after formic acid treatment of the sections. PrP immunostaining was relatively weak in sections from uninfected mice (E, F), resulting from loss of PrPC signal caused by formic acid pretreatment; staining was slightly more intense in transgenic animals (E) because of the presence of PrP-EGFP. Scale bar: (in A) A-F, 20 μm.
Discussion
Tg(PrP-EGFP) mice express an EGFP-tagged version of PrP that behaves like endogenous PrP in terms of its posttranslational processing, anatomical localization, and functional activity (Barmada et al., 2004). In this study, we describe experiments in which Tg(PrP-EGFP) mice were inoculated intracerebrally with scrapie prions. We found that, although PrP-EGFP was not itself converted to PrPSc-EGFP, the fusion protein served as a highly specific ligand that bound to PrPSc generated from endogenous PrPC. This feature allowed us to visualize the accumulation of PrPSc in situ by fluorescence microscopy without the need for immunocytochemical staining and the application of antigen retrieval techniques. Our results provide an entirely new picture of PrPSc localization during the course of prion infection, and they identify for the first time intracellular sites in which PrPSc formation is likely to occur.
Our results indicate that PrP-EGFP itself is incapable of sustaining a prion infection and of being converted to PrPSc-EGFP but that it acts as a dominant-negative inhibitor of prion propagation from endogenous PrP (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Tg(PrP-EGFP+/+)/Prn-p0/0 mice that express high levels of PrP-EGFP but lack endogenous PrP do not become ill after scrapie inoculation and do not accumulate protease-resistant PrP. In contrast, Tg(PrP-EGFP+/0)/Prn-p+/+ mice that coexpress PrP-EGFP and endogenous PrP do develop scrapie, but with a significant prolongation of incubation time and disease duration compared with Prn-p+/+ mice. Thus, the presence of PrP-EGFP appears to interfere with the conversion of endogenous PrPC to PrPSc. This inhibitory effect is likely to involve a physical interaction between PrP-EGFP and PrPSc, either direct or via a complex containing additional molecules. Consistent with this interpretation, PrPSc copurifies with PrP-EGFP isolated using anti-GFP antibodies, and conversely, PrP-EGFP copurifies with PrPSc that has been precipitated using NaPTA. In line with recent models for prion propagation (Caughey, 2001), we hypothesize that PrP-EGFP binds to PrPSc but that the presence of the bulky EGFP moiety prevents the subsequent conversion step that would generate PrPSc-EGFP. Additionally, we propose that PrP-EGFP competes with endogenous PrPC for access to binding sites on PrPSc, thereby slowing prion propagation in mice expressing both proteins. Interestingly, Meier et al. (2003) have observed an analogous phenomenon in transgenic mice expressing a soluble, dimeric form of PrP (PrP-Fc2) created by the fusion of two PrP moieties to an Ig Fc region. These results, coupled with our own, indicate that PrPC and PrPSc engage in a physical interaction during prion propagation and that genetically altered forms of PrPC can act as antagonists of this process.
Because of the specific affinity between PrP-EGFP and PrPSc, we envision that the fusion protein is incorporated into growing aggregates of PrPSc, thus marking the location of PrPSc and permitting its detection by fluorescence microscopy. Consistent with this proposal, we observed a striking and progressive accumulation of fluorescent aggregates in the brains of scrapie-inoculated Tg(PrP-EGFP+/0)/Prn-p+/+ mice during the course of infection. PrP-EGFP aggregates were observed in many brain areas and took several forms, including granular and plaque-like deposits in the neuropil as well as intracellular accumulations in neuronal somata and axon tracts. These aggregates were absent from uninoculated control mice. The redistribution of PrP-EGFP in infected animals preceded by ∼50 d the onset of clinical symptoms, the appearance of astrocytosis, and the detection of PrPSc on Western blots. At no point in the disease process did PrP-EGFP aggregates colocalize with Thy1, a GPI-anchored neuronal protein that, like PrPC, transits the secretory pathway and is present in lipid rafts on the plasma membrane (Madore et al., 1999). These observations, coupled with the fact that PrP-EGFP aggregates were first observed well before overt neuropathology developed, support the conclusion that changes in PrP-EGFP localization during infection are not secondary to pathological alterations in cellular organelles in which the fusion protein resides.
A novel finding of our study is that the earliest accumulation of PrP-EGFP occurs intracellularly, within the Golgi apparatus of neurons, suggesting that PrPSc accumulates initially within this organelle. Because PrP-EGFP acts as an inhibitor of prion propagation, it may mark not only the location of existing PrPSc deposits but also sites of active PrPSc synthesis or places in which PrPC-PrPSc conversion intermediates accumulate. Thus, our results suggest that the production of PrPSc from PrPC may occur in the Golgi apparatus. As is the case for endogenous PrPC, a steady-state pool of PrP-EGFP can be visualized on the plasma membrane and within the Golgi complex of neurons in vitro (Ivanova et al., 2001) and in vivo (Barmada et al., 2004), reflecting transit of the protein through the Golgi apparatus toward the cell surface. In one scenario, extracellular PrPSc could gain access to the Golgi apparatus via an endocytic pathway and initiate conversion of PrPC within this organelle. This hypothesis is consistent with the existence of cellular trafficking routes that connect the endocytic pathway and the Golgi (Nichols et al., 2001) and with experiments demonstrating that the initial steps in PrPSc synthesis occur on the cell surface or after endocytosis of PrPC (Borchelt et al., 1992; Shyng et al., 1994). Alternatively, PrPSc may be formed initially in other cellular locations and later collect in the Golgi. In either case, once PrPSc accumulates in the Golgi it might then be trafficked to the cell surface or be released into the extracellular space when neurons degenerate. Previous studies have reported partial colocalization of PrPSc with markers for the Golgi apparatus (Taraboulos et al., 1990) and lysosomes (McKinley et al., 1991) in scrapie-infected cells in culture, but the applicability of these results to neurons in brain has been uncertain. Although we failed to detect accumulation of PrP-EGFP within lysosomes of infected neurons, it is possible that the acidic environment of these organelles disrupts the interaction between PrP-EGFP and PrPSc or quenches EGFP fluorescence (Kneen et al., 1998).
Visualization of PrPSc in Tg(PrP-EGFP) mice by fluorescence microscopy represents a significant advance over conventional immunohistochemical techniques because of the improved ability to detect intracellular deposits of PrPSc. Immunohistochemical studies with the light microscope typically describe several different patterns of PrPSc deposition in the brain, depending on prion strain and host, including dense plaques, diffuse plaques, granular deposits within the neuropil (“synaptic-like”), and peri-neuronal aggregates (Jeffrey et al., 1992, 1994b; DeArmond et al., 2004). Most of these deposits appear to be extracellular, a conclusion that is confirmed by electron microscopic studies that have identified both fibrillar and nonaggregated forms of PrPSc in spaces surrounding neurons and their processes (DeArmond et al., 1985; Jeffrey et al., 1994a, 1997). Published images showing intracellular accumulation of PrPSc in brain are rare (Piccardo et al., 1990; Laszlo et al., 1992; Arnold et al., 1995; Fournier et al., 2000; Kovacs et al., 2005). The difficulty in visualizing intracellular PrPSc deposits may be a consequence of the antigen retrieval techniques that are used to enhance the immunoreactivity of PrPSc. These procedures may cause loss or redistribution of PrPSc, particularly forms that are not strongly aggregated or protease resistant. In addition, these treatments can substantially alter cellular architecture and denature proteins used as markers for intracellular organelles. Consistent with published immunohistochemical studies of RML scrapie (Lloyd et al., 2004), we observed only diffuse neuropil deposits of PrP when formic acid-treated brain sections from infected Tg(PrP-EGFP+/0)/Prn-p+/+ mice were stained with antibodies to PrP or GFP (Fig. 8). Thus, imaging the intrinsic fluorescence of PrP-EGFP reveals additional intraneuronal deposits of PrPSc in the Golgi that are lost after the application of antigen retrieval techniques.
We do not believe that the novel features of PrPSc localization reported here are an artifact resulting from the expression of a foreign transgene. First, although Tg(PrP-EGFP+/0)/Prn-p+/+ mice display a delayed incubation time compared with Prn-p+/+ mice, the transgenic animals eventually show the same symptoms and neuropathological features as the nontransgenic ones, and they accumulate similar levels of PrPSc. Second, after immunohistochemical staining of formic acid-treated sections, the distribution of PrPSc in the neuropil was very similar in Tg(PrP-EGFP+/0)/Prn-p+/+ and nontransgenic animals (Fig. 8). This observation suggests that the expression of PrP-EGFP does not cause a major redistribution of PrPSc. However, visualization of PrPSc in Tg(PrP-EGFP+/0)/Prn-p+/+ mice is indirect, because it relies on binding of PrP-EGFP to PrPSc. Thus, it is possible that some deposits of PrPSc may not be evident by fluorescence microscopy because of the dissociation of PrPSc and PrP-EGFP. Conversely, some PrP-EGFP aggregates could form independently of PrPSc.
Tg(PrP-EGFP) mice represent a unique system for examining the pathogenesis and progression of prion disease in vivo. It may be possible to visualize PrPSc deposition in the brains or other organs of these animals while they are alive and to assess the effects of therapeutic agents. Moreover, the brains of scrapie-infected Tg(PrP-EGFP) mice can serve as a source of fluorescently tagged PrPSc to be used in cell biological experiments. Finally, PrP-EGFP, by virtue of its specific interaction with PrPSc, could be used as an affinity reagent for the isolation of proteins involved in prion replication.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS40975 (D.A.H.). S.J.B. was supported by the Medical Scientist Training Program at Washington University (NIH Grant T32GM07200). We are grateful to Bernardino Ghetti and Pedro Piccardo for their advice on this project, for commenting on this manuscript, and for performing initial PrPSc immunostaining experiments on paraffin-embedded sections. We also acknowledge Heather True-Krob, Michael Green, and members of the Harris laboratory for critical reading of this manuscript. We thank Charles Weissmann forPrn-p0/0 mice, Richard Kascsak for 3F4 antibody, and Man-Sun Sy for 8H4 antibody. We acknowledge Cheryl Adles and Michelle Kim for mouse colony maintenance and genotyping.
Correspondence should be addressed to David A. Harris, Department of Cell Biology and Physiology, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO 63110. E-mail: dharris@cellbiology.wustl.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255824-09$15.00/0
References
- Aguzzi A, Polymenidou M (2004) Mammalian prion biology: one century of evolving concepts. Cell 116: 313-327. [DOI] [PubMed] [Google Scholar]
- Arnold JE, Tipler C, Laszlo L, Hope J, Landon M, Mayer RJ (1995) The abnormal isoform of the prion protein accumulates in late-endosome-like organelles in scrapie-infected mouse brain. J Pathol 176: 403-411. [DOI] [PubMed] [Google Scholar]
- Barmada S, Piccardo P, Yamaguchi K, Ghetti B, Harris DA (2004) GFP-tagged prion protein is correctly localized and functionally active in the brains of transgenic mice. Neurobiol Dis 16: 527-537. [DOI] [PubMed] [Google Scholar]
- Bolton DC, Seligman SJ, Bablanian G, Windsor D, Scala LJ, Kim KS, Chen CM, Kascsak RJ, Bendheim PE (1991) Molecular location of a species-specific epitope on the hamster scrapie agent protein. J Virol 65: 3667-3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Taraboulos A, Prusiner SB (1992) Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J Biol Chem 267: 16188-16199. [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Fluethmann H, Lipp H-P, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C (1992) Normal development and behavior of mice lacking the neuronal cell-surface PrP protein. Nature 356: 577-582. [DOI] [PubMed] [Google Scholar]
- Caughey B (2001) Interactions between prion protein isoforms: the kiss of death? Trends Biochem Sci 26: 235-242. [DOI] [PubMed] [Google Scholar]
- Chiesa R, Harris DA (2001) Prion diseases: what is the neurotoxic molecule? Neurobiol Dis 8: 743-763. [DOI] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Ghetti B, Harris DA (1998) Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 21: 1339-1351. [DOI] [PubMed] [Google Scholar]
- Cody CW, Prasher DC, Westler WM, Prendergast FG, Ward WW (1993) Chemical structure of the hexapeptide chromophore of the Aequorea green-fluorescent protein. Biochemistry 32: 1212-1218. [DOI] [PubMed] [Google Scholar]
- DeArmond SJ, McKinley MP, Barry RA, Braunfeld MB, McColloch JR, Prusiner SB (1985) Identification of prion amyloid filaments in scrapie-infected brain. Cell 41: 221-235. [DOI] [PubMed] [Google Scholar]
- DeArmond SJ, Ironside JW, Bouzamondo-Bernstein E, Peretz D, Fraser JR (2004) Neuropathology of prion diseases. In: Prion biology and diseases, Ed 2 (Prusiner SB, ed), pp 777-856. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C (1996) Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 15: 1255-1264. [PMC free article] [PubMed] [Google Scholar]
- Fournier JG, Escaig-Haye F, Grigoriev V (2000) Ultrastructural localization of prion proteins: physiological and pathological implications. Microsc Res Tech 50: 76-88. [DOI] [PubMed] [Google Scholar]
- Ivanova L, Barmada S, Kummer T, Harris DA (2001) Mutant prion proteins are partially retained in the endoplasmic reticulum. J Biol Chem 276: 42409-42421. [DOI] [PubMed] [Google Scholar]
- Jeffrey M, Goodsir CM, Bruce ME, McBride PA, Scott JR, Halliday WG (1992) Infection specific prion protein (PrP) accumulates on neuronal plasmalemma in scrapie infected mice. Neurosci Lett 147: 106-109. [DOI] [PubMed] [Google Scholar]
- Jeffrey M, Goodsir CM, Bruce ME, McBride PA, Fowler N, Scott JR (1994a) Murine scrapie-infected neurons in vivo release excess prion protein into the extracellular space. Neurosci Lett 174: 39-42. [DOI] [PubMed] [Google Scholar]
- Jeffrey M, Goodsir CM, Bruce M, McBride PA, Scott JR, Halliday WG (1994b) Correlative light and electron microscopy studies of PrP localisation in 87V scrapie. Brain Res 656: 329-343. [DOI] [PubMed] [Google Scholar]
- Jeffrey M, Goodsir CM, Bruce ME, McBride PA, Fraser JR (1997) In vivo toxicity of prion protein in murine scrapie: ultrastructural and immunogold studies. Neuropathol Appl Neurobiol 23: 93-101. [PubMed] [Google Scholar]
- Kitamoto T, Tateishi J, Tashima T, Takeshita I, Barry RA, DeArmond SJ, Prusiner SB (1986) Amyloid plaques in Creutzfeldt-Jakob disease stain with prion protein antibodies. Ann Neurol 20: 204-208. [DOI] [PubMed] [Google Scholar]
- Kitamoto T, Ogomori K, Tateishi J, Prusiner SB (1987) Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest 57: 230-236. [PubMed] [Google Scholar]
- Kitamoto T, Shin R-W, Doh-ura K, Tomokane N, Miyazono M, Muramoto T, Tateishi J (1992) Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in patients with Creutzfeldt-Jakob disease. Am J Pathol 140: 1285-1294. [PMC free article] [PubMed] [Google Scholar]
- Kneen M, Farinas J, Li Y, Verkman AS (1998) Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys J 74: 1591-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Preusser M, Strohschneider M, Budka H (2005) Subcellular localization of disease-associated prion protein in the human brain. Am J Pathol 166: 287-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszlo L, Lowe J, Self T, Kenward N, Landon M, McBride T, Farquhar C, McConnell I, Brown J, Hope J, Mayer RJ (1992) Lysosomes as key organelles in the pathogenesis of prion encephalopathies. J Pathol 166: 333-341. [DOI] [PubMed] [Google Scholar]
- Lloyd SE, Thompson SR, Beck JA, Linehan JM, Wadsworth JD, Brandner S, Collinge J, Fisher EM (2004) Identification and characterization of a novel mouse prion gene allele. Mamm Genome 15: 383-389. [DOI] [PubMed] [Google Scholar]
- Madore N, Smith KL, Graham CH, Jen A, Brady K, Hall S, Morris R (1999) Functionally different GPI proteins are organized in different domains on the neuronal surface. EMBO J 18: 6917-6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley MP, Taraboulos A, Kenaga L, Serban D, Stieber A, DeArmond SJ, Prusiner SB, Gonatas N (1991) Ultrastructural localization of scrapie prion proteins in cytoplasmic vesicles of infected cultured cells. Lab Invest 65: 622-630. [PubMed] [Google Scholar]
- Meier P, Genoud N, Prinz M, Maissen M, Rulicke T, Zurbriggen A, Raeber AJ, Aguzzi A (2003) Soluble dimeric prion protein binds PrPSc in vivo and antagonizes prion disease. Cell 113: 49-60. [DOI] [PubMed] [Google Scholar]
- Nichols BJ, Kenworthy AK, Polishchuk RS, Lodge R, Roberts TH, Hirschberg K, Phair RD, Lippincott-Schwartz J (2001) Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J Cell Biol 153: 529-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz D, Williamson RA, Matsunaga Y, Serban H, Pinilla C, Bastidas RB, Rozenshteyn R, James TL, Houghten RA, Cohen FE, Prusiner SB, Burton DR (1997) A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J Mol Biol 273: 614-622. [DOI] [PubMed] [Google Scholar]
- Piccardo P, Safar J, Ceroni M, Gajdusek DC, Gibbs Jr CJ (1990) Immunohistochemical localization of prion protein in spongiform encephalopathies and normal brain tissue. Neurology 40: 518-522. [DOI] [PubMed] [Google Scholar]
- Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95: 13363-13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB (2004) Prion biology and diseases, Ed 2 (Prusiner SB, ed). Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Serban D, Taraboulos A, DeArmond SJ, Prusiner SB (1990) Rapid detection of Creutzfeldt-Jakob disease and scrapie prion proteins. Neurology 40: 110-117. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Heuser JE, Harris DA (1994) A glycolipid-anchored prion protein is endocytosed via clathrin-coated pits. J Cell Biol 125: 1239-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraboulos A, Serban D, Prusiner SB (1990) Scrapie prion proteins accumulate in the cytoplasm of persistently infected cultured cells. J Cell Biol 110: 2117-2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsworth JD, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J (2001) Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet 358: 171-180. [DOI] [PubMed] [Google Scholar]
- Wickner RB, Edskes HK, Ross ED, Pierce MM, Baxa U, Brachmann A, Shewmaker F (2004) Prion genetics: new rules for a new kind of gene. Annu Rev Genet 38: 681-707. [DOI] [PubMed] [Google Scholar]
- Zanusso G, Liu D, Ferrari S, Hegyi I, Yin X, Aguzzi A, Hornemann S, Liemann S, Glockshuber R, Manson JC, Brown P, Petersen RB, Gambetti P, Sy MS (1998) Prion protein expression in different species: analysis with a panel of new mAbs. Proc Natl Acad Sci USA 95: 8812-8816. [DOI] [PMC free article] [PubMed] [Google Scholar]