

Abstract
During rodent cortex development, cells born in the medial ganglionic eminence (MGE) of the basal telencephalon reach the embryonic cortex by tangential migration and differentiate as interneurons. Migrating MGE cells exhibit a saltatory progression of the nucleus and continuously extend and retract branches in their neuritic arbor. We have analyzed the migration cycle of these neurons using in vitro models.
We show that the nucleokinesis in MGE cells comprises two phases. First, cytoplasmic organelles migrate forward, and second, the nucleus translocates toward these organelles. During the first phase, a large swelling that contains the centrosome and the Golgi apparatus separates from the perinuclear compartment and moves rostrally into the leading neurite, up to 30 μm from the waiting nucleus. This long-distance migration is associated with a splitting of the centrioles that line up along a linear Golgi apparatus. It is followed by the second, dynamic phase of nuclear translocation toward the displaced centrosome and Golgi apparatus.
The forward movement of the nucleus is blocked by blebbistatin, a specific inhibitor of nonmuscle myosin II. Because myosin II accumulates at the rear of migrating MGE cells, actomyosin contraction likely plays a prominent role to drive forward translocations of the nucleus toward the centrosome. During this phase of nuclear translocation, the leading growth cone either stops migrating or divides, showing a tight correlation between leading edge movements and nuclear movements.
Keywords: migration, video microscopy, cortical interneurons, centrosome, Golgi apparatus, nonmuscle myosin II
Introduction
The cerebral cortex comprises two major functional populations of neurons born in different forebrain territories. Excitatory cortical neurons are generated in the dorsal telencephalon (Gorski et al., 2002), whereas a large population of inhibitory cortical interneurons originate in the medial ganglionic eminence (MGE) of the basal forebrain (Anderson et al., 1997; Lavdas et al., 1999; Wichterle et al., 2001) (for review, see Marin and Rubenstein, 2001). To reach the developing cortex, inhibitory interneurons migrate tangentially to the brain surface over long distances across various territories and cell substrates comprising progenitors in the subventricular zone, corticofugal axons in the intermediate zone, and postmitotic neurons in the developing cortex (Lavdas et al., 1999; Denaxa et al., 2001; Tanaka et al., 2003). A large variety of cues, neurotransmitters, growth factors, motogenic factors, or guidance molecules guide the journey of these neurons, in particular repulsive cues expressed in the basal telencephalon and attractive/permissive factors expressed in the dorsal cortex (Behar et al., 1996; Denaxa et al., 2001; Powell et al., 2001; Polleux et al., 2002; Marin et al., 2003; Wichterle et al., 2003).
During migration, future cortical interneurons exhibit saltatory nuclear movements and develop a branched leading process that undergoes complex morphological changes (Nadarajah et al., 2002; Polleux et al., 2002; Ang et al., 2003; Moya and Valdeolmillos, 2004). The present study was aimed at better analyzing these changes and the full migration cycle of cortical interneurons. In particular, we wanted to determine whether interneuron migration encompasses the three successive steps classically described in migrating cells: (1) extension of a leading process, (2) migration of the nucleus in this process, and (3) retraction of the trailing process (Morris et al., 1998; Walsh and Goffinet, 2000; Ridley et al., 2003).
It can be difficult to perform exact correlations of nuclear movements with transformations of branched neurites in explant or slice cultures because of their complex three-dimensional structure (Ang et al., 2003; Moya and Valdeolmillos, 2004). To perform a quantitative study of the nuclear and neuritic movements in cortical interneurons, we therefore allowed these cells to migrate in just one plane. MGE cells that give rise to a large population of cortical interneurons (Lavdas et al., 1999; Wichterle et al., 2001; Bellion and Métin, 2005) were thus cultured on a flat permissive migration substrate that consisted of either dissociated cortical cells or cortical axons.
Video microscopy analyses showed that forward nuclear movements in MGE cells are preceded by the long-distance migration of cytoplasmic organelles that form a swelling in the leading neurite. Using photonic and electronic microscopy, we show that the cytoplasmic swelling contains the centrosome and Golgi apparatus, which both undergo drastic morphological transformations when they migrate rostrally: the centrosome splits, and the Golgi apparatus acquires a linear conformation. A second unexpected result of our study was that myosin II transiently accumulates at the rear of the soma in migrating MGE cells. Blebbistatin, a selective nonmuscular myosin II inhibitor, inhibits long-distance nuclear translocations, showing that myosin contraction moves the nucleus forward. Therefore, in MGE cells, the actomyosin system periodically generates forces that push the nucleus toward the distant centrosome. The nucleokinesis in MGE cells comprises thus two phases that are in addition correlated with leading neurite outgrowth and splitting: (1) the forward migration of centrioles and Golgi apparatus at a long distance from the nucleus is associated with leading neurite elongation, and (2) the myosin II dependent forward migration of the nucleus is correlated with leading growth cone arrest or splitting.
Materials and Methods
Animals. Wild-type embryos and embryos ubiquitously expressing green fluorescent protein (GFP) were produced in the laboratory. GFP-expressing embryos resulted from crosses between OF1 females (IFFA Credo, Lyon, France) and GFPU transgenic males (The Jackson Laboratory, Bar Harbor, ME). The day of vaginal plug detection was considered embryonic day 0.5 (E0.5). Pregnant females were killed by cervical dislocation to collect embryos. The embryos were dissected in cold Leibovitz medium (L15; Invitrogen, San Diego, CA).
Grafts of MGE cells in forebrain slices. Brain slices were prepared from E12.5-E14.5 mouse embryos. Dissected brains were embedded in 4% low-melting-point agarose (type VII; Sigma, St. Louis, MO) in L15 medium (Invitrogen). Coronal sections (100 μm thick) were cut with a tissue slicer (Campden Instruments, Loughborough, UK) and deposited on polylysine/laminin-coated coverslips fixed to the bottom of 35 mm perforated Petri dishes with a paraffin/vaseline mixture (w/w, 90/10). Slices were covered with DMEM/F12 medium supplemented with 2 mm l-glutamine, 33 mm d-glucose, 3 mm sodium bicarbonate, 10 mm HEPES buffer, pH 7.4, 10 UI/ml penicillin/streptomycin, and 10% N2 and B27 complement (all from Invitrogen). Homochronic GFP-expressing MGE explants dissected as explained previously (Bellion and Métin, 2005) were grafted in the basal telencephalon of slices using glass micropipettes to transfer and inject the explants. Grafted slices were then transferred to a humidified incubator at 37°C with 5% CO2 and cultured for 1 d before imaging GFP-expressing MGE cells.
Cocultures. MGE explants from E12.5-E14.5 GFP-expressing embryos were deposited either on wild-type dissociated cortical cells or cortical axons extending from wild-type cortical explants. Dissociated cells were prepared from E12.5-E14.5 embryos (most often from E13.5 embryos) and cultured for 1 d before receiving EGM explants. Long cortical axons were observed around E12.5 or E13.5 cortical explants cultured for 3-5 d. Cocultures were performed on polylysine/laminin-coated glass coverslips either placed on the bottom of culture wells or fixed to the bottom of perforated Petri dishes to image migrating MGE cells (see above). To prepare cortical explants and dissociated cortical cells, rostrocaudal stripes of dorsal cortex were dissected as explained by Bellion et al. (2003). Cells were mechanically dissociated by pipeting cortical tissue in DMEM/F12 medium containing 20% fetal calf serum (Invitrogen). A mean of 4000 cells/mm2 were deposited per polylysine/laminin-coated coverslip. Cortical explants and dissociated cells were cultured in DMEM/F12 medium with N2/B27 complement (see above). MGE explants were dissected in the rostral MGE (Bellion and Métin, 2005). Each explant was divided into smaller cubes (each side, 100-200 μm), which were deposited on the same coverslip, 1 mm apart from each other on average.
Pharmacology. Myosin II was inhibited with blebbistatin, a specific nonmuscle myosin II inhibitor (Straight et al., 2003). Three concentrations, 50, 70, and 100 μm, were tested in the cocultures. According to the published dose-response curves, the 70 and 100 μm concentrations were the most efficient and gave close results.
Time-lapse video microscopy. Before imaging, the grafted slices or the cocultures were cultured in Petri dishes equipped with glass coverslips, the culture medium was replaced with a HEPES-buffered MEM, which was phenol red free (Sigma), containing the following: 4 mm NaHCO3, 20 mm HEPES, 2 mm l-glutamine, 33 mm glucose, 1 mm pyruvate, 10 UI/ml penicillin/streptomycin, supplemented with N2 and B27 (Invitrogen). Two setups were successively used to acquire pictures. An inverted epifluorescence microscope (DM IRBE; Leica, Nussloch, Germany) equipped with a GFP emission/excitation filter, a humidified and thermo-regulated chamber, a mechanical shutter (Uniblitz; Vincent Associates, Rochester, NY), and a cooled CCD camera (CoolSnap HQ; PhotoMetrics, Huntington Beach, CA) was used at the beginning of this study. Pictures were captured every 3 or 5 min using a 25× oil immersion objective. Exposure time was 200 ms, and cocultures were imaged up to 10 h. We then imaged MGE cells with an inverted epifluorescence microscope (Axiovert; Carl Zeiss, Oberkochen, Germany) equipped with a monochromator, a motorized stage, a piezo z objective, and a humidified and thermo-regulated chamber. GFP-expressing cells were imaged through a 20× (Plan-Apochromat) or a 40× (Plan-Neofluar) objective every 3 or 5 min. Pictures were captured with a cooled CCD camera (Orca; Hamamatsu Photonics, Shizouka, Japan), and exposure time (500-800 ms) was controlled by the monochromator. Before pharmacological treatment, cocultures were imaged for 2-4 h. After drug application, cocultures were recorded from 6 to 12 h, depending on experiments. In both stations, image acquisition was performed using MetaMorph Software (Universal Imaging Corporation, West Chester, PA). Analyses were performed using MetaMorph and ImageJ (NIH Image; National Institutes of Health, Bethesda, MD) software.
Immunohistochemistry. After 2 d in vitro, the cocultures were fixed and processed for immunocytochemistry. The GFP was visualized using either rabbit serum (1:1000; Molecular Probes, Eugene, OR) or mouse monoclonal antibodies (clone 3E6; 1:100; Quantum Biotechnologies, Montreal, Quebec, Canada). Centrin-2 was revealed with a rabbit serum (1:2000) (Middendorp et al., 2000), ninein with a rabbit serum (1:1000) (Bouckson-Castaing et al., 1996), myosin II with mouse monoclonal antibodies (clone CC212; 1:1000) (Klotz et al., 1986), and the Golgi apparatus was labeled with mouse monoclonal antibodies directed against its medial compartment (clone CTR433; 1:100) (Jasmin et al., 1989). Filamentous actin (F-actin) was labeled with Texas Red phalloidin (1:40; Molecular Probes) and tubulin with class III β-tubulin (TUJ1) mouse monoclonal antibodies (IgG; 1:2000; Babco, Richmond, CA). Centrin-2, ninein, and myosin II were stained in cocultures fixed 10 min in -20°C methanol. To label GFP together with the Golgi apparatus, cocultures were sequentially fixed in 4% paraformaldehyde in 0.12 m phosphate buffer (PAF)/0.33 m sucrose at 4°C and in -20°C methanol. Actin and tubulin stainings were performed in cocultures fixed in PAF/0.33 m sucrose. After fixation, cocultures were rinsed in PBS and preincubated for 1 h in PBS with 10% normal goat serum (NGS)/0.2% Triton X-100. They were incubated overnight with primary antibodies diluted in PBS/2% NGS/0.2% Triton X-100. After rinses in PBS/0.2% Triton X-100 (PBT), cultures were incubated for 1 h in Alexa 488- and/or cyanine 3-coupled secondary antibodies (1:400; Jackson ImmunoResearch, West Grove, PA) diluted in PBS/2% NGS/0.2% Triton X-100. After rinses in PBS, cultures were incubated with 0.1% bis-benzimide in PBT, rinsed in PBS, and mounted in Mowiol 1,4-diazabicyclo-[2.2.2]octane. Labelings were analyzed on an epifluorescence microscope (DMRD; Leica) equipped with a CoolSnap camera (PhotoMetrics) and on a Leica TCS confocal microscope.
Electron microscopy. The ultrastructure of MGE cells was studied in cells cultured on cortical axons. Cocultures performed on polylysine laminin-coated plastic coverslips (Thermanox; Nalge Nunc, Rochester, NY) were fixed in 1% PAF/1% glutaraldehyde and postfixed in 2% OsO4. Before alcoholic dehydration and inclusion in Araldite, cultures were colored in 1% uranyl acetate in maleate buffer. Small blocks with isolated MGE cells migrating on cortical axons or with clustered MGE cells were separated from the rest of the culture. Ultrathin sections (70-80 nm) parallel to the plastic coverslips (that is, to the plane of migration of MGE cells) were collected on copper grids. They were colored with Reynolds solution and rinsed before observation with a Philips CM100 electron microscope.
Results
Coculture models
To perform quantitative analyses of the morphological changes that affect MGE cells during their migration cycle, we designed coculture models in which MGE cells migrated on a flat surface. GFP-expressing MGE explants were deposited either on a carpet of cortical axons or on a monolayer of dissociated cortical cells (Fig. 1B, C). MGE cells migrated on these permissive substrates, whereas they remained as large and dense aggregates on laminin. On both cortical axons and dissociated cortical cells, MGE cells were regularly spaced as observed in organotypic forebrain slices (Fig. 1A) and exhibited a characteristic polarized morphology with a frequently branched leading neurite (Fig. 1D). They became distributed homogeneously around MGE explants deposited on dissociated cortical cells (Fig. 1C), suggesting that the cues that delineate their migration pathway in vivo and in organotypic slices are missing or more randomly distributed in dissociated cells.
Figure 1.

Experimental models to study the migratory behavior of MGE cells. A, To image MGE cells migrating in cortical slices, MGE explants dissected from GFP-expressing embryos were grafted homotopically in wild-type forebrain slices. The experimental model is schematized on the left. The green dot represents the GFP-expressing MGE explant. The dotted frame indicates the limits of the picture shown on the right. B and C illustrate the two coculture models used in the present study. B, GFP-expressing MGE explants were placed at the tip of cortical axons growing on a polylysine/laminin-coated substrate. In the scheme on the left, the MGE explant is a green dot; cortical explant and cortical axons are shown in red. MGE cells migrate on cortical axons. C, GFP-expressing MGE explants were placed on a monolayer of wild-type dissociated cortical cells (in pink in the scheme on the left). MGE cells migrate randomly on cortical cells. D, MGE cells migrating on cortical cells show bifurcated or branched leading processes. In some cases, a long thin neurite is observed at the trailing side (white arrowhead). GFP-positive MGE cells were immunostained using a green fluorescent secondary antibody (A-D). Cortical axons were immunostained with TUJ1 antibodies using a red fluorescent secondary antibody (B). CX, Cortex; GE, ganglionic eminence. Scale bars: A, C, 100 μm; D, 20 μm.
In MGE cells, nuclear translocation is preceded by the long-distance migration of cytoplasmic organelles
Using GFP antibodies and bis-benzimide staining, we verified that the large swelling at the rear of migrating MGE cells was the nucleus (see Fig. 4A1,C1-C3). In MGE cells migrating on cortical axons or dissociated cortical cells, the nucleus alternated between resting phases and dynamic phases of translocation toward the leading edge. During the resting phase of the nucleus, cytoplasmic organelles initially located in the perinuclear compartment migrated forward and formed a swelling that increased in size while moving rostrally in the leading neurite (Fig. 2A). The swelling stopped its rostral migration at 15-30 μm from the center of the nucleus. This distance showed little variation among cells or among experiments (19.3 ± 2.3 μm; 92 measures in 43 cells) (Fig. 2B), suggesting a tight regulation. In a second phase, the nucleus translocated to the swelling. The duration of this second phase was most often between 3 and 9 min and rarely exceeded 12 min. This cyclically repeated two-step behavior generated a saltatory progression of the cell soma (supplemental movie 2A, available at www.jneurosci.org as supplemental material).
Figure 4.
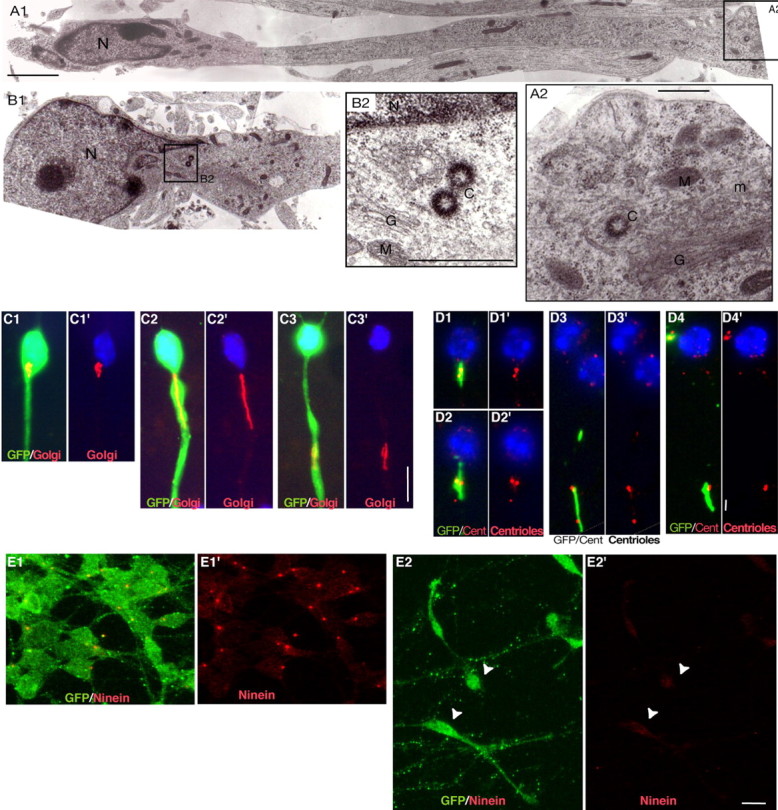
Localization and morphology of the centrosome and Golgi apparatus in MGE cells migrating on cortical cells. A1-B2, Ultrastructural analysis of MGE cells migrating on cortical axons reveals the presence of the centriole(s) and Golgi apparatus in a swelling of the leading process that can be located distant from (15 μm; A1) or close to (B1) the nucleus. The nucleus (N) occupies one pole of the cell and shows an indentation oriented toward the leading process (Gregory et al., 1988). Swellings contain one or two centrioles (C), depending on the plane of section (A2, B2). Centrioles were always located near the Golgi apparatus (G). M, Mitochondria; ER, rough endoplasmic reticulum; m, microtubules. Scale bars: A1, 2 μm; A2, 500 nm; B2, 200 nm. C1-C3′, Immunostainings of GFP (green; C1-C3) and the medial compartment of the Golgi apparatus (CTR433 in red) in migrating MGE cells. Nuclei are labeled with bis-benzimide (blue). The Golgi apparatus is located either close to the nucleus (C1′) or rostrally in a swelling of the neuritic trunk (C2′, C3′). It shows a compact (C1′, C3′) or a linear (C2′) conformation. Scale bar, 10 μm. D1-D4′, Immunostainings of the Golgi apparatus (CTR433 in green; D1-D4) and centrioles [centrin-2 (Cent) in red]. The two centrioles are associated with the Golgi apparatus (GA). In cells with a compact GA, the two centrioles appear as two close dots (D1′, D2′) or a single larger dot (D4′). In cells with a linear GA, the two centrioles are widely separated and line up along the GA (D3′). Scale bar, 2 μm. E1-E2′, In GFP-positive MGE cells aggregated on laminin (E1, E1′; GFP in green), ninein antibodies (red) strongly label a single dot in the centrosome. In MGE cells migrating on cortical axons (E2, E2′; GFP in green), the ninein-positive dot is no longer visible, whereas a faint labeling of the whole soma can be observed (E2′; white arrowheads). Scale bar, 10 μm.
Figure 2.

In cocultures, nuclear translocation is preceded by forward migration of perinuclear organelles. A, Time-lapse sequence of a GFP-expressing MGE cell migrating on wild-type dissociated cortical cells and imaged each 3 min using a 20× objective. Before the nuclear translocation (frames 01:39, 01:42), perinuclear material (white arrowhead) moves rostrally in the leading neurite and individualizes from the perinuclear compartment to which it remains linked by a thin bridge (open arrowhead). Elapsed time is indicated in hours and minutes on each frame. Scale bar, 10 μm. B, Graphs correlate the distance between the nucleus and rostral swelling (gray curves) with the successive nuclear displacements (black curves) in two cells. Nuclear translocations are represented as peaks in the black curve (black arrowheads). During waiting phases of the nucleus, the swelling moves away from the nucleus (ascending gray curve) until reaching a maximum distance (dmax; gray arrowheads). In the cell illustrated in the bottom graph, the nucleus reached the rostral swelling in two jumps.
We then verified that MGE cells exhibited the same saltatory nuclear movements in organotypic cortical slices as observed on dissociated cortical cells (Fig. 3 and supplemental movie 3A, available at www.jneurosci.org as supplemental material). In MGE cells migrating in E12.5-E14.5 cortical slices, long nuclear translocations were directed toward a swelling that previously differentiated in the leading neurite (Fig. 3A, blue squares). The mean amplitude of nuclear jumps was in the same range as that observed in cocultures (Fig. 3B1). In both cases, the speed of the nucleus could reach 130 μm/h during jumps. In slices, however, the dissociation between the swelling and the perinuclear compartment was quickly followed by a nuclear translocation, whereas the dissociation could be maintained up to 60 min in cocultures (Fig. 3B3). Nuclear translocations were thus significantlymore frequent in slices than in cocultures (Fig. 3B2), and the mean speed of MGE cells was twice as high as in the cocultures (34 ± 2.8 μm/h in E13.5 slices compared with 18 ± 0.4 μm/h in cocultures on E13.5 dissociated cortical cells) (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Figure 3.

MGE cells migrate faster in forebrain slices and show the same cycle of migration as on dissociated cortical cells. A, Time-lapse sequence shows the migration of a GFP-expressing MGE cell in an E14. 5 cortical slice. In a magnified view of the imaged slice (large picture on the left), the red circle locates the cell at the limit between the ventricular zone (VZ) and the intermediate zone (IZ). As in cocultures (Fig. 2), the nuclear migration is saltatory, and nuclear translocations end near swellings (blue squares) that form during waiting periods of the nucleus (black diamond). The leading growth cone division (red diamond) is correlated with a nuclear translocation (frames 30-35) and produces a bifurcation at the leading edge. One of the two paired branches collapses and retracts. Scale bar, 20 μm. CP, Cortical plate. B1-B3, Histograms show for MGE cells migrating on dissociated cortical cells (C; black bars) and for MGE cells migrating in E14.5 cortical slices (S14; white bars) that (1) the amplitude of nuclear translocations (B1) is the same, (2) the frequency of nuclear translocations (B2) is significantly increased in slices, and (3) swellings are observed for longer periods before nuclear translocation (B3) in cocultures (15-60 min) than in slices (<15 min). Error bars represent SE.
Our observations show that the nuclear migration of MGE cells is a two-step process. First, cytoplasmic organelles migrate forward at a long distance from the nucleus, and second, the nucleus translocates toward these organelles.
Forward migration and morphological changes of the Golgi apparatus and centrosome in migrating MGE cells
To identify the cytoplasmic organelles that perform a long-distance forward migration before each nuclear translocation, we performed an ultrastructural analysis of the proximal part of the neurite in MGE cells migrating on a substrate of cortical axons. Swellings could be observed either a long distance from (up to 15 μm) or close to the nucleus (Fig. 4A1,B1). The swelling comprised one or two centrioles if the plane of section passed through both centrioles, the Golgi apparatus, mitochondria, and rough endoplasmic reticulum (Fig. 4A2,B2). Golgi apparatus cisternas were always observed near centriole(s). In photonic microscopy, the Golgi apparatus presented two conformations, compact or linear, as a function of its distance from the nucleus (Fig. 4C1-C3′). The linear Golgi apparatus (Fig. 4C2) was observed in the leading process at a distance from the nucleus varying from 0 to 18 μm. The compact Golgi apparatus (Fig. 4C1,C3) was observed either far from the nucleus (18-25 μm) in the rostral swelling or, most often, near the nucleus. Intermediate shapes (half compact-half linear) were occasionally observed (data not shown). Double immunostainings confirmed that the two centrioles were closely associated with the Golgi apparatus (Fig. 4D1-D4′). In a proportion of cells exhibiting a linear Golgi apparatus, the two centrioles were dissociated from one another and distributed along the Golgi apparatus. In these cases, the intercentriolar distance could reach up to 6 μm (Fig. 4D3).
Therefore, in migrating MGE cells, the centrosome and the Golgi apparatus both migrate together away from the resting nucleus and undergo striking morphological changes during this rostral movement: the centrosome splits, the Golgi apparatus stretches along the long axis of the leading neurite, and the two centrioles line up along the Golgi apparatus. The Golgi apparatus seems to fold up again, and the centrioles to reassociate at a distance from the nucleus.
Myosin II controls the forward nuclear movements in MGE cells
In migrating neurons, microtubules of the perinuclear cage are anchored to the centrosome. They likely control nuclear movements by coupling the nucleus to the centrosome (Rivas and Hatten, 1995; Walsh and Goffinet, 2000; Xie et al., 2003; Bielas and Gleeson, 2004; Solecki et al., 2004). In migrating MGE cells, however, the anchorage of microtubules to the centrosome might be weak. Indeed, a centrosomal protein, ninein, which anchors microtubules to the centrosome (Bouckson-Castaing et al., 1996; Abal et al., 2002), was concentrated at the centrosome in nonmigrating MGE cells aggregated on laminin (Fig. 4 E1,E1′). This protein was no longer detected at the centrosome in MGE cells migrating on cortical axons (Fig. 4 E2,E2′). This suggests that microtubules might be redistributed in other locations and/or poorly anchored to the centrosome during migration. We therefore looked for an alternative system not involving microtubules that could produce the forces needed for nuclear translocations.
We thus investigated whether the actomyosin system could be involved in this process as already described in non-neuronal migrating cells (Sanchez-Madrid and del Pozo, 1999; Ridley et al., 2003). We observed that myosin II transiently accumulated at the rear of MGE cells during a certain phase of the migration cycle (Fig. 5A1,A2). When cocultures of MGE cells and cortical axons were treated with blebbistatin, a selective nonmuscle myosin II inhibitor, nuclear movements were inhibited in 60% of MGE cells within 30 min after application (n = 72 cells) (Fig. 5B1 and supplemental movie 5B1, available at www.jneurosci.org as supplemental material). Blebbistatin at 70 or 100 μm did not entirely abolish nuclear movements, and small amplitude shifts of the nucleus were still observed. In 71 imaged and responding cells, we observed a 68.6% decrease in the mean speed of nuclear migration (mean nuclear speed recorded 2 h before drug treatment, 35.1 ± 4.2 μm/h; mean nucleus speed recorded from 1-3 h after blebbistatin exposure, 11.1 ± 1.3 μm/h). Long and dynamic nuclear jumps were no longer observed, and the swelling stabilized at a short distance ahead of the nucleus (Fig. 5B2,B3).
Figure 5.
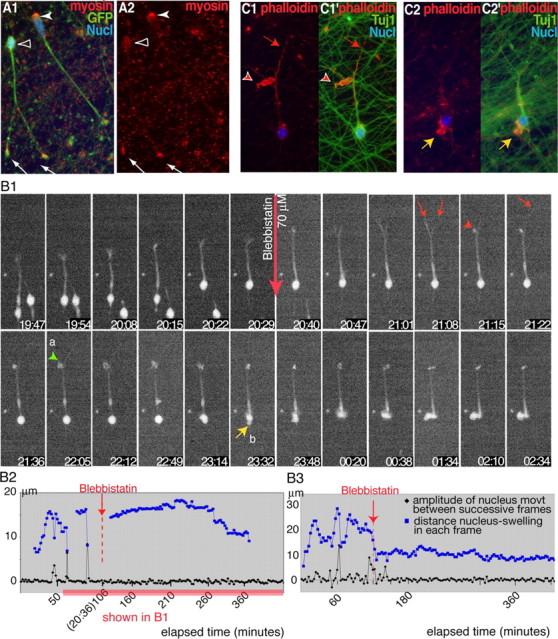
Blebbistatin, a nonmuscle myosin II inhibitor, inhibits nuclear movement in MGE cells. A1, A2, In GFP-positive MGE cells (A1; green) migrating on cortical axons, myosin II detected by CC212 antibodies (red) either concentrates at the rear of the nucleus (white arrowhead) or is distributed over the whole cell body (open arrowhead). Myosin II also accumulates in growth cones (white arrows). B1-B3, Time-lapse sequence (B1) shows the response of an MGE cell migrating on cortical axons to blebbistatin 70 μm (red arrow). Time is indicated in hours and minutes on each frame. A graphic representation of the nuclear and swelling movements is shown in B2 (same legend as Fig. 2 B). B3 illustrates similar movements in another treated MGE cell. Nuclear translocations occur during the control period (frames 19:54 and 20:22 in B1, peaks of the black curves in B2 and B3) and are no longer observed after drug treatment. Blebbistatin stabilizes the swelling (blue curve) ahead of the nucleus. In B1, 45 min after drug application, one of the two leading neurites retracts and forms a large protrusion (red arrowhead; frame 21:15), whereas the other neurite extends (red arrow; frame 21:22). Protrusions develop at the neuritic bifurcation (green arrowhead; a) and around the nucleus (yellow arrow; b). At the end of the sequence, the nucleus enters a lateral protrusion (frame 01:34). C1-C2′, MGE cells migrating on cortical axons and treated with blebbistatin for 6 h were stained with phalloidin (red) and TUJ1 antibodies (green; C1, C2). F-actin accumulates into large protrusions at previous branching points (red arrowhead; the red arrow shows the leading neurite) and in the perinuclear compartment (yellow arrow). Nucl, Nucleus.
Blebbistatin also induced striking morphological deformations in MGE cells. Large and active expansions strongly labeled with phalloidin developed at the tips of neurites, around the nucleus or the swelling (Fig. 5C1,C2). The nucleus could enter one of these lateral protrusions orienting itself perpendicularly to the long axis of the cell. In contrast, processes that did not produce ruffles became very thin, and their tips were difficult to localize in the movies. Some MGE cells extended several long and thin branches.
Thus, nonmuscle myosin II inhibition efficiently blocked nuclear translocation in a large proportion of migrating MGE cells and induced a drastic redistribution of actin within large and motile expansions. These results thus show myosin II to be a major actor in MGE cell migration.
The two steps of the nucleokinesis are correlated with specific transformations at the leading edge
At the leading edge of MGE cells, the growing rate of neurites showed large variations (45 ± 20 μm/h; n = 24 growth cones analyzed during 180 min). Phases of elongation alternated with growth cone pauses and/or leading growth cone splitting (Fig. 6A). Stops in neuritic elongation were correlated with nuclear translocations, whereas neurite(s) growth proceeded at the end of each nuclear translocation (Fig. 6B1, B2). In 21 cells imaged during at least 80 min, 29 of 36 (80%) divisions of a leading growth cone occurred during a nuclear translocation. The production of new branches by leading growth cone splitting were thus synchronized with forward nuclear movements. The same correlations were found in MGE cells that migrated in cortical slices (Fig. 3A and data not shown).
Figure 6.

Neuritic elongation and nuclear migration are correlated in MGE cells. A, Frames of the time-lapse sequence depicted in B1. A GFP-expressing MGE cell migrating on wild-type dissociated cortical cells was imaged each for 3 min using a 40× objective. Elapsed time is indicated in minutes on each frame. Three nuclear translocations (a,b,c) are illustrated. Between translocations a and b, the leading process elongates (red dotted curve). A growth cone splitting (white asterisk) is visible at the end of translocation c. B1, B2, Graphs depict the sequence of migration of two MGE cells. Graph B1 corresponds to the cell illustrated in A. The black curve shows the amplitude of nuclear displacements between successive frames. Peaks in the curve (black arrows) identify translocation phases (same letters in A and B1). Colored curves represent the distance from the rostral edge of the nucleus to growth cones on each frame (leading growth cone in red). Between two nuclear translocations (black arrows), the leading neurite elongates, and the red curve hence ascends. Nuclear translocations are associated with an arrest of neuritic elongation and a shrinkage of the leading neurite. C, Schematic representation of nucleokinesis in MGE cells. During the resting phase of the nucleus (white), the centrioles (yellow) and Golgi apparatus (red) migrate forward, and the leading neurite elongates. The centrioles split, and the Golgi apparatus elongates. Then, myosin II accumulates at the rear of the cell body (green) and pushes the nucleus toward the centrosome/Golgi apparatus.
Therefore, morphological changes of the neuritic arbor in MGE cells were correlated with the two phases of the nucleokinesis (Fig. 6C). Neuritic elongation occurred during the forward movement of the centrosome/Golgi apparatus, whereas leading growth cone arrest or splitting was associated with the myosin II-dependent phase of forward nuclear translocation.
Discussion
The present study shows that the migration cycle of MGE cells is remarkable in several aspects: (1) long nuclear displacements are preceded by movements of the same amplitude of the centrosome and Golgi apparatus; (2) the forward movement of the centrosome is associated with centriole splitting; (3) nuclear jumps toward the displaced centrosome are dependant on myosin II contractility; (4) transformations of the neuritic arbor at the leading edge are synchronized with phases of nuclear movement.
Forward migration of the centrosome and Golgi apparatus in MGE cells
We show that nuclear jumps are preceded by the forward translocation of cytoplasmic organelles comprising in particular the centrosome and Golgi apparatus. These organelles segregate from the perinuclear compartment and reposition rostrally in the leading neurite in which they form a cytoplasmic swelling observable in numerous migrating neurons or glial progenitors (this study) (Kakita and Goldman, 1999; Nadarajah et al., 2002; Polleux et al., 2002; Ang et al., 2003; Moya and Valdeolmillos, 2004). The forward migration of the centrosome before nuclear translocation has also been observed in radially migrating cerebellar neurons (Solecki et al., 2004). Although the amplitude of centrosomal movement described by Solecki et al. (2004) is relatively short (1.4 μm on average, compared with 19.3 μm in this study), similar mechanisms might thus operate to control nucleokinesis in radially and tangentially migrating neurons. A specific property of MGE cells revealed by this study might therefore be their capacity to reposition the centrosome and Golgi apparatus at a large distance from the nucleus within the leading neurite. Interestingly, such a reorientation of the microtubule organizing center (MTOC) and Golgi apparatus has also been observed in cultured cortical interneurons during polarity reversal (Hayashi et al., 2003). In non-neuronal cells such as fibroblasts or astrocytes induced to migrate, nuclear movements are also preceded by a large rostral movement of the centrosome and Golgi apparatus (Etienne-Manneville and Hall, 2001; Palazzo et al., 2001).
In MGE cells, the maximum distance between the centrosome and the nucleus was quite constant, suggesting nevertheless a tight regulation. Indeed, in certain migrating cells, the cytoskeleton has been shown to establish a physical link between the nucleus and the centrosome (Walsh and Goffinet, 2000; Bielas and Gleeson, 2004). For example, microtubules form a cage around the nucleus of cerebellar neurons (Rivas and Hatten, 1995), and a fork anchored to the centrosome covers the nucleus in cortical neurons (Xie et al., 2003). In cerebellar neurons, the proteins LIS1 and doublecortin cooperate with microtubule-associated molecular motors such as dynein to control the coupling between the nucleus and the centrosome (Tanaka et al., 2004). In addition, proteins critical for neuronal migration belonging to the focal adhesion kinase/cyclin-dependent kinase 5 (FAK/cdk5) and mPar6α/atypical protein kinase C (PKCζ) signaling pathways (Gupta et al., 2003; Jossin et al., 2003) are enriched in the centrosome or in perinuclear microtubules (Xie et al., 2003; Solecki et al., 2004). It is thus important to determine the contribution of these proteins to the relative movements of the nucleus and centrosome in MGE cells, together with the role of Rho GTPases that control the polarity of the MTOC and Golgi apparatus during migration in non-neuronal cells (Hall, 1998; Ridley et al., 2003) and nucleokinesis in precerebellar neurons (Causeret et al., 2004).
A surprising result of our study is that a splitting of the centrosome occurs during its rostral repositioning. The two separated centrioles line up along the Golgi apparatus that forms a long tube parallel to the axis of movement of the cell. Such a linear morphology of the Golgi apparatus during migration has not been reported previously. It might result from the redistribution of proteins such as GMAP210, which stabilize microtubules and the Golgi apparatus around the centrosome (Rios et al., 2004). Centrosome splitting has not been observed in cerebellar neurons migrating in vitro along radial glial cells (Solecki et al., 2004; Tanaka et al., 2004). However, centriole dissociation has already been described in neutrophils induced to move by chemokinesis and in interphase fibroblasts (Schliwa et al., 1982; Piel et al., 2000). In both cases, this phenomenon was associated with the subcellular redistribution of microtubules possibly connected with the directionality of cell locomotion. Additional studies are thus needed to determine whether the dissociation of centrioles observed in MGE cells is related to the control of nuclear movement and/or to the control of the arrangement of microtubules bundles in the leading neuritic arbor. Indeed, as observed in neutrophils (Schliwa et al., 1982), centriole dissociation might permit the cell to control two bundles of microtubules independently of one another, for example when a bifurcation forms at the leading edge, or when one neuritic branch retracts while another one elongates.
Translocation of the nucleus toward the centrosome
A new result of our study is that nonmuscle myosin II controls nuclear movement in cortical interneurons. Recent genetic studies have revealed an important role of nonmuscle myosin II for controlling the migration of certain neuronal groups in the embryo (Ma et al., 2004). Here, we show that myosin II is important for the translocation of the nucleus toward the rostral centrosome in migrating MGE cells. We show that myosin II seems to accumulate periodically behind the nucleus, even in some cases forming a short tail at the rear of the cell. Patterns of distribution suggest that myosin II can contract the rear of the MGE cell to push the nucleus forward, as described in non-neuronal cells (Sanchez-Madrid and del Pozo, 1999; Ridley et al., 2003). The frequency of nuclear translocation might thus result from the transient accumulation of myosin II at the rear of MGE cells or from the highly regulated activation of myosin II (Matsumura et al., 1998). Indeed, intracellular Ca2+ transients observed just before or during nuclear movements in migrating MGE cells (Moya and Valdeolmillos, 2004) might play an important role in controlling the periodic activation of myosin II and the saltatory progression of the nucleus.
A spectacular consequence of myosin II inhibition by blebbistatin was the apparition of large and highly motile protrusions at the lateral border of the perinuclear compartment and at the tip of some neurites. These protrusions are enriched in F-actin and are reminiscent of the ruffles of migrating fibroblasts (Hall, 1998). We observed that in some cases, the nucleus rotates and enters a lateral expansion of the soma, showing that myosin II plays an important role in maintaining the nucleus aligned on the same axis with the MTOC and the leading growth cone. In Dictyostelium cells and fibroblasts, myosin II activity similarly restricts actin-dependant lateral membrane protrusions, thereby controlling cell polarity (Wessels and Soll, 1990; Ridley et al., 2003; Totsukawa et al., 2004). In addition to its motile function, myosin II is thus involved in maintaining a single axis of migration in MGE cells by inhibiting the formation of lateral protrusions.
Blebbistatin applied at concentrations that entirely block the ATPase activity of nonmuscle myosin II (Straight et al., 2003) did not entirely suppress nuclear movements. The smooth and residual nuclear movements might depend on microtubule-associated forces known to move the nucleus in granule cells (Rivas and Hatten, 1995; Solecki et al., 2004; Tanaka et al., 2004). Accordingly, nocodazole at low concentrations changes the dynamics of nucleokinesis in MGE cells (J.-P. Baudoin and C. Métin, unpublished data). Two sets of forces are likely therefore to control nuclear movements in tangentially migrating MGE cells: (1) microtubule-dependent pulling forces and (2) actomyosin-dependent pushing forces. Whether nonmuscle myosin II contributes to the radial migration of neurons is likely (Ma et al., 2004) but remains to be studied.
Correlation between nucleokinesis and neuritogenesis
Graphic analyses revealed synchronies between nuclear movements and the change in morphology of neurites. In MGE cells, the start of a long nuclear jump always coincided with a transient arrest in neuritic elongation and, frequently, with leading growth cone splitting, revealing an unanticipated link between the process of branch formation and nuclear movement. Neuritic elongation quickly resumed thereafter and was pursued during the resting period of the nucleus. During this phase, the Golgi apparatus moved forward and took on a linear conformation. Whether this forward movement of the Golgi apparatus is causal to the increase in growth rate of the leading neurite remains to be determined. Genetic studies have identified ARFGEF2, a controller of trafficking from the trans-Golgi network, as an important regulator of neuronal migration in the developing human cortex (Sheen et al., 2004). In fibroblasts, it is also clear that anterograde vesicle trafficking from the Golgi apparatus toward the leading edge is required for migration (Schmoranzer et al., 2003; Prigozhina and Waterman-Storer, 2004).
The migration of MGE cells is thus an integrated and cyclic process as described previously for neurons moving radially along or independently from a radial glial support (Rivas and Hatten, 1995; Nadarajah et al., 2001). Although MGE cells migrated at a slower rate on dissociated cortical cells (10-15 μm/h) than in organotypic slices [37.5 μm/h in the present study; 50 μm/h in Nadarajah et al. (2002); 58 μm/h in Polleux et al. (2002); from 0 to 50 μm/h in Tanaka et al. (2003)], the fundamental properties of their migration were conserved in vitro. In addition, the formation of diverging branches at the leading edge used by tangentially migrating cells to change their direction in organotypic slices (Kakita and Goldman, 1999; Polleux et al., 2002) is conserved in vitro by MGE cells. It likely depends on intrinsic properties of the MGE cells rather on the polarized distribution of cues in the environment.
Footnotes
This work was supported by the Centre National de la Recherche Scientifique, Institut National de la Santé etde la Recherche Médicale, and grants from the Association pour la Recherche contre le Cancer (ARC; number 5934) and from the Fédération pour la Recherche sur le Cerveau to C.M. A.B. was funded by a fellowship from the ARC, and J.-P.B. was funded by a fellowship from the French Ministère de la Recherche et de la Technologie. We thank Antoine Triller for helping A.B. with video microscopy during initial stages of this work and Claude-Marie Bachelet and Jean-Paul Rio for helpful assistance with video microscopy and electron microscopy. Fiona Francis, Patricia Gaspar, Alain Prochiantz, Nicole Ropert, Antoine Triller, and Marion Wassef are acknowledged for their helpful comments at different stages of the preparation of this manuscript. Marion Wassef and Patricia Gaspar are acknowledged for their support during this work.
Correspondence should be addressed to Dr. C. Métin, Institut National de la Santé et de la Recherche Médicale, Unité 616, Développement Normal et Pathologique du Cerveau, Hôpital Pitié-Salpêtrière, 47 Boulevard de l'Hôpital, 75651 Paris Cédex 13, France. E-mail: metin@chups.jussieu.fr.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255691-09$15.00/0
A.B. and J.-P. B. contributed equally to this work.
References
- Abal M, Piel M, Bouckson-Castaing V, Mogensen M, Sibarita JB, Bornens M (2002) Microtubule release from the centrosome in migrating cells. J Cell Biol 159: 731-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL (1997) Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science 278: 474-476. [DOI] [PubMed] [Google Scholar]
- Ang Jr ES, Haydar TF, Gluncic V, Rakic P (2003) Four-dimensional migratory coordinates of GABAergic interneurons in the developing mouse cortex. J Neurosci 23: 5805-5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar TN, Li YX, Tran HT, Ma W, Dunlap V, Scott C, Barker JL (1996) GABA stimulates chemotaxis and chemokinesis of embryonic cortical neurons via calcium-dependent mechanisms. J Neurosci 16: 1808-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellion A, Métin C (2005) Early regionalisation of the neocortex and the medial ganglionic eminence. Brain Res Bull, in press. [DOI] [PubMed]
- Bellion A, Wassef M, Metin C (2003) Early differences in axonal outgrowth, cell migration and GABAergic differentiation properties between the dorsal and lateral cortex. Cereb Cortex 13: 203-214. [DOI] [PubMed] [Google Scholar]
- Bielas SL, Gleeson JG (2004) Cytoskeletal-associated proteins in the migration of cortical neurons. J Neurobiol 58: 149-159. [DOI] [PubMed] [Google Scholar]
- Bouckson-Castaing V, Moudjou M, Ferguson DJ, Mucklow S, Belkaid Y, Milon G, Crocker PR (1996) Molecular characterisation of ninein, a new coiled-coil protein of the centrosome. J Cell Sci 109: 179-190. [DOI] [PubMed] [Google Scholar]
- Causeret F, Hidalgo-Sanchez M, Fort P, Backer S, Popoff MR, Gauthier-Rouviere C, Bloch-Gallego E (2004) Distinct roles of Rac1/Cdc42 and Rho/Rock for axon outgrowth and nucleokinesis of precerebellar neurons toward netrin 1. Development 131: 2841-2852. [DOI] [PubMed] [Google Scholar]
- Denaxa M, Chan CH, Schachner M, Parnavelas JG, Karagogeos D (2001) The adhesion molecule TAG-1 mediates the migration of cortical interneurons from the ganglionic eminence along the corticofugal fiber system. Development 128: 4635-4644. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A (2001) Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell 106: 489-498. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR (2002) Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22: 6309-6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory WA, Edmondson JC, Hatten ME, Mason CA (1988) Cytology and neuron-glial apposition of migrating cerebellar granule cells in vitro J Neurosci 8: 1728-1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Sanada K, Miyamoto DT, Rovelstad Miyamoto, Nadarajah B, Pearlman AL, Brunstrom J, Tsai LH (2003) Layering defect in p35 deficiency is linked to improper neuronal-glial interaction in radial migration. Nat Neurosci 6: 1284-1291. [DOI] [PubMed] [Google Scholar]
- Hall A (1998) Rho GTPases and the actin cytoskeleton. Science 279: 509-514. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Kawai-Hirai R, Harada A, Takata K (2003) Inhibitory neurons from fetal rat cerebral cortex exert delayed axon formation and active migration in vitro. J Cell Sci 116: 4419-4428. [DOI] [PubMed] [Google Scholar]
- Jasmin BJ, Cartaud J, Bornens M, Changeux JP (1989) Golgi apparatus in chick skeletal muscle: changes in its distribution during end plate development and after denervation. Proc Natl Acad Sci USA 86: 7218-7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossin Y, Ogawa M, Metin C, Tissir F, Goffinet AM (2003) Inhibition of SRC family kinases and non-classical protein kinases C induce a reeler-like malformation of cortical plate development. J Neurosci 23: 9953-9959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakita A, Goldman JE (1999) Patterns and dynamics of SVZ cell migration in the postnatal forebrain: monitoring living progenitors in slice preparations. Neuron 23: 461-472. [DOI] [PubMed] [Google Scholar]
- Klotz C, Bordes N, Laine MC, Sandoz D, Bornens M (1986) Myosin at the apical pole of ciliated epithelial cells as revealed by a monoclonal antibody. J Cell Biol 103: 613-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavdas AA, Grigoriou M, Pachnis V, Parnavelas JG (1999) The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci 19: 7881-7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Kawamoto S, Hara Y, Adelstein RS (2004) A point mutation in the motor domain of nonmuscle myosin II-B impairs migration of distinct groups of neurons. Mol Biol Cell 15: 2568-2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL (2001) A long, remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci 2: 780-790. [DOI] [PubMed] [Google Scholar]
- Marin O, Plump AS, Flames N, Sanchez-Camacho C, Tessier-Lavigne M, Rubenstein JL (2003) Directional guidance of interneuron migration to the cerebral cortex relies on subcortical Slit1/2-independent repulsion and cortical attraction. Development 130: 1889-1901. [DOI] [PubMed] [Google Scholar]
- Matsumura F, Ono S, Yamakita Y, Totsukawa G, Yamashiro S (1998) Specific localization of serine 19 phosphorylated myosin II during cell locomotion and mitosis of cultured cells. J Cell Biol 140: 119-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middendorp S, Kuntziger T, Abraham Y, Holmes Abraham, Bordes N, Paintrand M, Paoletti A, Bornens M (2000) A role for centrin 3 in centrosome reproduction. J Cell Biol 148: 405-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris NR, Efimov VP, Xiang X (1998) Nuclear migration, nucleokinesis and lissencephaly. Trends Cell Biol 8: 467-470. [DOI] [PubMed] [Google Scholar]
- Moya F, Valdeolmillos M (2004) Polarized increase of calcium and nucleokinesis in tangentially migrating neurons. Cereb Cortex 14: 610-618. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL (2001) Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci 4: 143-150. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Alifragis P, Wong RO, Parnavelas JG (2002) Ventricle-directed migration in the developing cerebral cortex. Nat Neurosci 5: 218-224. [DOI] [PubMed] [Google Scholar]
- Palazzo AF, Cook TA, Alberts AS, Gundersen GG (2001) mDia mediates Rho-regulated formation and orientation of stable microtubules. Nat Cell Biol 3: 723-729. [DOI] [PubMed] [Google Scholar]
- Piel M, Meyer P, Khodjakov A, Rieder CL, Bornens M (2000) The respective contributions of the mother and daughter centrioles to centrosome activity and behavior in vertebrate cells. J Cell Biol 149: 317-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polleux F, Whitford KL, Dijkhuizen PA, Vitalis T, Ghosh A (2002) Control of cortical interneuron migration by neurotrophins and PI3-kinase signaling. Development 129: 3147-3160. [DOI] [PubMed] [Google Scholar]
- Powell EM, Mars WM, Levitt P (2001) Hepatocyte growth factor/scatter factor is a motogen for interneurons migrating from the ventral to dorsal telencephalon. Neuron 30: 79-89. [DOI] [PubMed] [Google Scholar]
- Prigozhina NL, Waterman-Storer CM (2004) Protein kinase D-mediated anterograde membrane trafficking is required for fibroblast motility. Curr Biol 14: 88-98. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel Burridge, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR (2003) Cell migration: integrating signals from front to back. Science 302: 1704-1709. [DOI] [PubMed] [Google Scholar]
- Rios RM, Sanchis A, Tassin AM, Fedriani C, Bornens M (2004) GMAP-210 recruits gamma-tubulin complexes to cis-Golgi membranes and is required for Golgi ribbon formation. Cell 118: 323-335. [DOI] [PubMed] [Google Scholar]
- Rivas RJ, Hatten ME (1995) Motility and cytoskeletal organization of migrating cerebellar granule neurons. J Neurosci 15: 981-989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Madrid F, del Pozo MA (1999) Leukocyte polarization in cell migration and immune interactions. EMBO J 18: 501-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliwa M, Pryzwansky KB, Euteneuer U (1982) Centrosome splitting in neutrophils: an unusual phenomenon related to cell activation and motility. Cell 31: 705-717. [DOI] [PubMed] [Google Scholar]
- Schmoranzer J, Kreitzer G, Simon SM (2003) Migrating fibroblasts perform polarized, microtubule-dependent exocytosis towards the leading edge. J Cell Sci 116: 4513-4519. [DOI] [PubMed] [Google Scholar]
- Sheen VL, Ganesh VS, Topcu M, Sebire G, Bodell A, Hill RS, Grant PE, Shugart YY, Imitola J, Khoury SJ, Guerrini R, Walsh CA (2004) Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet 36: 69-76. [DOI] [PubMed] [Google Scholar]
- Solecki DJ, Model L, Gaetz J, Kapoor TM, Hatten ME (2004) Par6alpha signaling controls glial-guided neuronal migration. Nat Neurosci 7: 1195-1203. [DOI] [PubMed] [Google Scholar]
- Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ (2003) Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science 299: 1743-1747. [DOI] [PubMed] [Google Scholar]
- Tanaka D, Nakaya Y, Yanagawa Y, Obata K, Murakami F (2003) Multimodal tangential migration of neocortical GABAergic neurons independent of GPI-anchored proteins. Development 130: 5803-5813. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Serneo FF, Higgins C, Gambello MJ, Wynshaw-Boris A, Gleeson JG (2004) Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J Cell Biol 165: 709-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totsukawa G, Wu Y, Sasaki Y, Hartshorne DJ, Yamakita Y, Yamashiro S, Matsumura F (2004) Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J Cell Biol 164: 427-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CA, Goffinet AM (2000) Potential mechanisms of mutations that affect neuronal migration in man and mouse. Curr Opin Genet Dev 10: 270-274. [DOI] [PubMed] [Google Scholar]
- Wessels D, Soll DR (1990) Myosin II heavy chain null mutant of Dictyostelium exhibits defective intracellular particle movement. J Cell Biol 111: 1137-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Turnbull DH, Nery S, Fishell G, Alvarez-Buylla A (2001) In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development 128: 3759-3771. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Alvarez-Dolado M, Erskine L, Alvarez-Buylla A (2003) Permissive corridor and diffusible gradients direct medial ganglionic eminence cell migration to the neocortex. Proc Natl Acad Sci USA 100: 727-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Sanada K, Samuels BA, Shih H, Tsai LH (2003) Serine 732 phosphorylation of FAK by Cdk5 is important for microtubule organization, nuclear movement, and neuronal migration. Cell 114: 469-482. [DOI] [PubMed] [Google Scholar]