

Abstract
Three of seven recently identified genes mutated in nonsyndromic mental retardation are involved in Rho family signaling. Two of the gene products, α-p-21-activated kinase (PAK) interacting exchange factor (αPIX) and PAK3, form a complex with the synaptic adaptor protein G-protein-coupled receptor kinase-interacting protein 1 (GIT1). Using an RNA interference approach, we show that GIT1 is critical for spine and synapse formation. We also show that Rac is locally activated in dendritic spines using fluorescence resonance energy transfer. This local activation of Rac is regulated by PIX, a Rac guanine nucleotide exchange factor. PAK1 and PAK3 serve as downstream effectors of Rac in regulating spine and synapse formation. Active PAK promotes the formation of spines and dendritic protrusions, which correlates with an increase in the number of excitatory synapses. These effects are dependent on the kinase activity of PAK, and PAK functions through phosphorylating myosin II regulatory light chain (MLC). Activated MLC causes an increase in dendritic spine and synapse formation, whereas inhibiting myosin ATPase activity results in decreased spine and synapse formation. Finally, both activated PAK and activated MLC can rescue the defects of GIT1 knockdown, suggesting that PAK and MLC are downstream of GIT1 in regulating spine and synapse formation. Our results point to a signaling complex, consisting of GIT1, PIX, Rac, and PAK, that plays an essential role in the regulation of dendritic spine and synapse formation and provides a potential mechanism by which αPIX and PAK3 mutations affect cognitive functions in mental retardation.
Keywords: cytoskeleton, synaptogenesis, dendritic spines, Rac, GIT1, PAK, PIX, MLC
Introduction
Dendritic spines are small, actin-rich protrusions that receive most of the excitatory synaptic inputs in the CNS. Spines are believed to mediate the synaptic plasticity that underlies cognitive functions such as learning and memory. In accordance with this, pathological studies have revealed dendritic spine abnormalities in patients with mental retardation (MR), further emphasizing the central role of spines in integrating information flow in the brain (Fiala et al., 2002). However, the molecular mechanism by which dendritic spines are involved in neuronal plasticity has only begun to be addressed. Recent studies showed that dendritic spines are highly dynamic structures that undergo actin-based motility both in vitro and in vivo (Fischer et al., 1998; Dunaevsky et al., 1999; Lendvai et al., 2000). This provides a structural basis by which synaptic plasticity could take place.
Dynamic rearrangements of the actin cytoskeleton are mediated by the Rho family of small GTPases, including Rho, Rac, and Cdc42 (Raftopoulou and Hall, 2004). Previously, we showed that Rac is involved in regulating spine morphogenesis and synapse formation in hippocampal neurons. Our work has led to the hypothesis that locally regulated Rac activation is central for synapse formation (Zhang et al., 2003). This localized activation of Rac could be achieved by the synaptic targeting of G-protein-coupled receptor kinase-interacting protein 1 (GIT1) and its binding partner p-21-activated kinase (PAK) interacting exchange factor (PIX), a guanine nucleotide exchange factor (GEF) for Rac (Zhang et al., 2003).
Once activated, Rac binds to a variety of downstream effector proteins. Among the most well characterized effectors are PAKs, which are a family of highly conserved serine/threonine kinases. PAKs are particularly interesting, because, in addition to binding active Rac, they also directly bind PIX, a Rac GEF (Manser et al., 1998). In fibroblasts, PIX and PAK, together with GIT1, coexist in a signaling module that shuttles between different subcellular compartments (Manabe et al., 2002). Interestingly, both αPIX and PAK3 mutations have been found in patients with nonsyndromic mental retardation, further pointing to an essential role of this signaling module in cognitive functions. However, the precise mechanism by which this signaling module participates in spine morphogenesis remains to be elucidated, and the downstream effectors of this complex are unknown.
In this study, we show that knockdown of GIT1 in hippocampal neurons affects spine morphology, confirming a central role for GIT1 in this process. We then addressed our hypothesis that Rac is locally activated in synapses by using fluorescence resonance energy transfer (FRET). Using this approach, local activation of Rac was observed at the synapses. To determine the downstream effectors, we examined the function of PAK1 and PAK3 in spine and synapse formation. We show that phosphorylated PAK localizes to synapses. Both PAK1 and PAK3 induce the formation of dendritic spines as well as the long, thin, dendritic protrusions that are not associated with presynaptic terminals. These effects are dependent on the kinase activity of PAK. We also show that the effects of PAK are mediated by phosphorylation of myosin II regulatory light chain (MLC). Finally, we show that activated PAK or MLC rescues the defects in GIT1 knockdown neurons, suggesting that GIT1 functions through PAK and MLC. Together, our results point to a signaling complex consisting of GIT1, PIX, Rac, and PAK that functions in regulating dendritic spine and synapse formation through modulating MLC activity. These results also provide a potential mechanism by which PIX and PAK mutations in mental retardation patients affect cognitive functions.
Materials and Methods
Antibodies and reagents. The myc monoclonal antibody (9E10) and polyclonal antibody (A14) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and were used at a ratio of 1:200 for immunostaining. The synaptic vesicle protein 2 (SV2) monoclonal antibody was obtained from Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA) and used at a ratio of 1:100. Polyclonal synapsin1 antibody was obtained from Chemicon (Temecula, CA) and was used at a ratio of 1:2000. Postsynaptic density-95 (PSD-95) antibodies used include the monoclonal antibody 6G6 from Affinity Bioreagents (Golden, CO) (1:200) and monoclonal antibody K28/43 (1: 100) from Upstate Biotechnology (Lake Placid, NY). The phospho-MLC antibody was a generous gift from Dr. Fumio Matsumura (Rutgers University, Piscataway, NJ) and has been described previously (Matsumura et al., 1998). The phospho-PAK antibody was kindly provided by Dr. Jonathan Chernoff (Fox Chase Cancer Center, Philadelphia, PA). The specificity of the phospho-PAK antibody has been described previously (Sells et al., 2000). The phospho-PAK antibody was raised against the activation loop of PAK, which is highly conserved. Therefore, it recognizes several different isoforms of PAK, including PAK1 and PAK3. Blebbistatin was purchased from Calbiochem (La Jolla, CA) and was reconstituted in DMSO. A final concentration of 50 μm blebbistatin was used in all experiments.
Plasmids. For the generation of the GIT1 small interfering RNA (siRNA) construct, the oligonucleotides 5′-CGGGATCCCCGCACTCAGCAACCGGCTCTTTCAAGAGAAGAGCCGGTTGCTGAGTGCTTTTTGGAAAAGCTTGGG-3′ and 5′-CCCAAGCTTTTCCAAAAAGCACTCAGCAACCGGCTCTTCTCTTGAAAGAGCCGGTTGCTGAGTGCGGGGATCCCG-3′ were annealed, digested with BamHI and HindIII, and subcloned into the BglII and HindIII sites of pSUPER. The siRNA sequence corresponds to nucleotides 887-905 of rat GIT1.
All FRET probes were kindly provided by Dr. Michiyuki Matsuda (Osaka University, Osaka, Japan) and have been described previously (Itoh et al., 2002). Hemagglutinin (HA)-tagged PIX and dominant-negative (DN)-PIX (L238R, L239S) have been described previously (Zhang et al., 2003). All myc-tagged PAK1 constructs were generously provided by Dr. Jonathan Chernoff (Fox Chase Cancer Center). The mouse PAK3 cDNA in the pJ3H vector was a gift from Dr. Richard Cerione (Cornell University, Ithaca, NY). The myc-tagged PAK3 construct was generated by subcloning the PAK3 cDNA as a BamHI-BamHI fragment to pCMVmyc vector. All of the PAK3 mutants were generated with the QuikChange kit (Stratagene, La Jolla, CA) using myc-PAK3 as the template. The mutations introduced to PAK3 include K297R [kinase-dead (KD)-PAK3], T421E [constitutively active (CA)-PAK3], H78,81L, and K297R (DN-PAK3). MLC-green fluorescent protein (GFP) and MLC 18,19D-GFP were kindly provided by Dr. Kathleen Kelly (National Cancer Institute, Bethesda, MD).
Neuronal culture and transfection. Hippocampal low-density cultures were prepared from E19 rat embryos as described previously (Goslin et al., 1998). Neurons were plated on glass coverslips coated with 1 mg/ml poly-l-lysine at an approximate density of 70 cells/mm2 and were transfected using a modified calcium phosphate precipitation method as described previously (Zhang et al., 2003).
FRET imaging and analysis. Neurons were transfected with the FRET probes and imaged after 48 h on an Olympus (Melville, NY) IX70 inverted microscope with a 60× objective. Images were acquired using a cooled CCD camera (CH250; Photometrics, Tucson, AZ). For dual imaging of the FRET probes, a U-MF2 FRET cube (Olympus) and two emission filters, 480/40 for cyan fluorescent protein (CFP) and 545/30 for yellow fluorescent protein (YFP), were used. The ratio image of YFP/CFP was generated using ImageJ after background subtraction and was used to represent FRET efficiency. For photobleaching, cells were illuminated for 10 min with no neutral density filters (YFP excitation, 500/20).
Immunocytochemistry and image quantification. Neurons were fixed in PBS containing 4% paraformaldehyde with 4% sucrose for 15 min at room temperature and permeabilized with 0.2% Triton X-100 for 5 min. Alternatively, for PSD-95 staining, neurons were simultaneously fixed and permeabilized with cold methanol for 20 min. For simultaneous visualization of GFP and PSD-95, neurons were fixed in 1% formaldehyde with 4% sucrose for 3 min and then with cold methanol for 10 min. After blocking with 20% goat serum/PBS, the neurons were incubated with the appropriate antibodies in 5% goat serum/PBS. Coverslips were mounted with Vectashield mounting media (Vector Laboratories, Burlingame, CA). Images were acquired using a cooled CCD camera (Orca II; Hamamatsu, Shizuoka, Japan) attached to a Nikon (Tokyo, Japan) TE-300 inverted microscope with a 60× objective (numerical aperture, 1.4; Nikon). The linear densities of dendritic spines, protrusions, and PSD-95 clusters were quantified using NIH Image software. Spines are defined as stubby or mushroom-shaped protrusions with associated presynaptic terminals. Dendritic protrusions are defined as protrusions on the dendrites without associated synapses. A total of 80-100 primary and secondary dendrites from 15-20 neurons were quantified with each construct. For each dendrite, a 70-100 μm segment was analyzed. For quantification of the spine/shaft ratio, the fluorescent intensity of spines and shaft was measured, and the ratio was derived. More than 100 spines were measured for each construct. Each experiment was repeated at least three times with independent neuronal cultures.
Results
GIT1 knockdown affects spine morphology
We showed previously that the adaptor protein GIT1 localizes to synapses, and its mislocalization perturbs spine morphogenesis and synapse formation (Zhang et al., 2003). To establish more directly a role for GIT1 in spine and synapse formation, we used an RNA interference (RNAi) approach. A 19-nucleotide sequence corresponding to nucleotides 887-905 of the rat GIT1 sequence dramatically reduced the expression of endogenous GIT1 in hippocampal neurons, whereas pSUPER vector alone did not affect expression of endogenous GIT1 (Fig. 1A). When the fluorescent intensity of GIT1 immunostaining in siRNA transfected neurons versus nearby untransfected neurons was quantified, GIT1 siRNA reduced expression of endogenous GIT1 to 7.9 ± 2.3% of control levels (Fig. 1B). GIT1 siRNA has no effect on the expression of other proteins such as microtubule-associated protein 2 or actin (data not shown).
Figure 1.

Knockdown of GIT1 expression affects spine and synapse formation. A, Hippocampal neurons were cotransfected with GFP and either pSUPER vector alone or GIT1 siRNA at 6 d in culture and were fixed and immunostained for endogenous GIT1 at 12 d. GIT1 siRNA, but not pSUPER alone, caused a dramatic decrease in endogenous GIT1 expression. The neurons expressing the siRNA constructs were >95% viable as determined by trypan blue staining. B, Quantification of fluorescent intensity of GIT1 immunostaining in GIT1 siRNA transfected neurons as assayed by the mean fluorescent intensity in the soma. Fluorescent intensity of nearby untransfected neurons was measured, and these neurons served as controls. Values are normalized to control neurons. C, Expression of GIT1 siRNA in hippocampal neurons causes a decrease in spine and synaptic density. These defects are rescued by coexpressing GFP-tagged human GIT1. Scale bar, 10 μm. D, Quantification of the effects of GIT1 siRNA on the number of dendritic spines and protrusions. The effects are rescued by coexpressing human GIT1 (rescue). Asterisks indicate p < 0.0001. E, Quantification of the effects of GIT1 siRNA on synaptic density. The effects are rescued by coexpressing human GIT1 (rescue). For each condition, 85-95 dendrites of 18-20 neurons from three different cultures were analyzed. *p < 0.0001. Error bars represent SEM from three experiments.
Along with the knockdown of GIT1 expression, we observed a significant decrease in the number of spines as well as dendritic protrusions that are not associated with presynaptic terminals (Fig. 1C,D), demonstrating that GIT1 plays an important role in spine formation. In addition to the loss of spines, there was also a significant decrease in the number of synapses as visualized by SV2 immunostaining (Fig. 1C,E). To determine whether the effects of GIT1 siRNA were specifically attributable to a loss of GIT1 expression, we performed rescue experiments in which we expressed human GIT1 in the siRNA transfected neurons. Because the siRNA specifically targets the rat GIT1 sequence and does not significantly affect human GIT1 expression, we were able to re-express human GIT1 in the siRNA knock-down neurons. Expression of human GIT1 almost completely rescued the defects of the siRNA-expressing neurons (Fig. 1C-E). These results show that the effects of the siRNA are specifically attributable to loss of GIT1 expression, and GIT1 plays an important role in dendritic spine and synapse formation.
Localized Rac activation visualized by FRET
We hypothesized previously that GIT1 regulates synapse formation through local modulation of Rac activity (Zhang et al., 2003). To determine whether Rac is locally activated in the synapses, we used a FRET approach. A single-chain FRET probe, which has Rac, p21-binding domain (PBD), YFP (acceptor), and CFP (donor) encoded on a single cDNA, was developed to detect activated Rac in living cells (Itoh et al., 2002). Activation of Rac leads to the binding of the effector domain PBD, which brings YFP into close proximity of CFP and induces FRET. A FRET signal is observed as an increase in YFP emission and a decrease in CFP emission. Therefore, the emission ratio of YFP/CFP is used to represent FRET efficiency.
The FRET probe Raichu-wild-type (WT) Rac was transfected into hippocampal neurons at 8 d in culture and imaged at 10 d in culture. A FRET signal was observed in dendritic spines (Fig. 2A, arrows) and primarily at the base of dendritic protrusions (Fig. 2A). The dendritic spines are contacted by presynaptic terminals as determined by SV2 coimmunostaining (Fig. 2A, bottom, arrows). To confirm that there was FRET, photobleaching experiments were performed. As expected, photobleaching of YFP resulted in a 108% increase in CFP intensity. To further verify the validity of the FRET probe, we used a control FRET construct, in which WT Rac was replaced by the dominant-negative mutant, N17Rac. Raichu-N17Rac showed a decrease in FRET efficiency (Fig. 2B), further showing that the FRET signal observed with Raichu-WT Rac reflects Rac activation in these cells. Together, these results suggest that Rac is locally activated at the synapses in hippocampal neurons.
Figure 2.
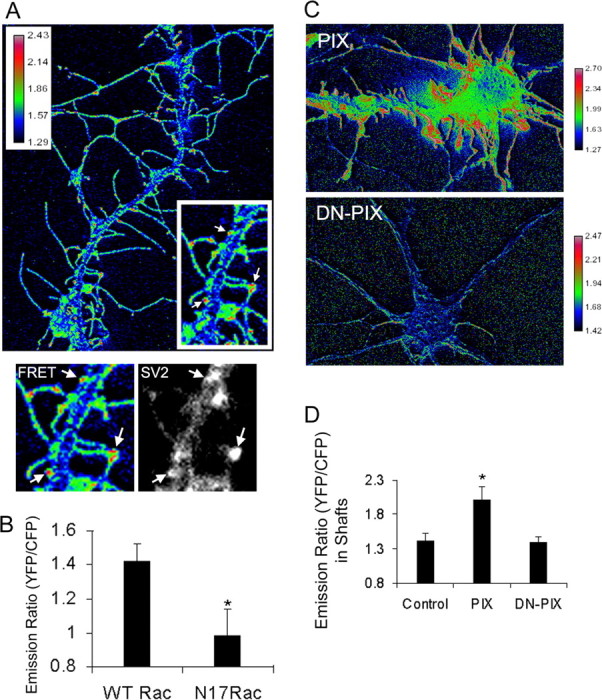
Localized activation of Rac visualized by FRET. A, Hippocampal neurons were transfected with Raichu-WT Rac at 8 d in culture and imaged at 10 d in culture. The ratio image of YFP and CFP was used to represent FRET efficiency. Note the low FRET efficiency in the dendritic shaft. Arrows point to active Rac localization in dendritic spines. These spines are in contact with presynaptic terminals as determined by SV2 coimmunostaining (bottom panels). B, Raichu-WT Rac and Raichu-N17Rac were transfected into hippocampal neurons, and FRET analyses were performed. Emission ratios are shown in the bar graph. *p < 0.001. C, HA-tagged PIX or DN-PIX (L238R, L239S; GEF deficient) were cotransfected with Raichu-WT Rac into the hippocampal neurons. FRET analyses were performed, and ratio images of YFP and CFP are shown to represent FRET efficiency. Ectopic expression of PIX results in high-FRET efficiency throughout the dendritic protrusions and increased FRET efficiency in the dendritic shafts. Expression of DN-PIX results in an overall reduction in FRET efficiency. D, Quantification of the emission ratios in the dendritic shafts of the neurons transfected with the PIX mutants. Note that in the dendritic shafts, the FRET efficiency is not significantly different between DN-PIX-expressing neurons and control neurons, indicating that under basal conditions, Rac is activated locally in dendritic spines. *p < 0.001. Error bars represent SEM from three experiments.
Effects of PIX on Rac activation
Our previous work led to the hypothesis that mislocalization of the GIT1/PIX complex away from the synapses results in mislocalized activation of Rac and the formation of multiple dendritic protrusions (Zhang et al., 2003). The mislocalization of PIX could be achieved by overexpressing PIX, which increases the nonsynaptic distribution of PIX. To examine the effects of PIX overexpression on Rac activation, we coexpressed either wild-type PIX or dominant-negative PIX (L238R, L239S; GEF deficient) with Raichu-WT Rac in hippocampal neurons at a ratio of 5:1. FRET analyses were performed on the transfected neurons. As shown in Figure 2C, coexpression of wild-type PIX resulted in an overall increase in FRET efficiency. Specifically, the highest FRET efficiency was observed throughout the dendritic protrusions as opposed to just the base of the protrusions in control neurons. In addition, significantly higher FRET efficiency was observed in the dendritic shafts (Fig. 2C,D), indicating mislocalized Rac activation in the shafts. This shows that overexpression of PIX results in an overall increase in Rac activation as well as mislocalization of Rac activation. When the dominant-negative PIX mutant was expressed in the neurons, a significant decrease in FRET efficiency was observed, which was accompanied by a decrease in the number of spines (Fig. 2C,D). This is consistent with our previous results that DN-PIX causes a decrease in spine and synaptic density (Zhang et al., 2003). Note that although there was an overall decrease in FRET efficiency in DN-PIX-expressing neurons, the FRET efficiency in the dendritic shafts was not significantly different from the control neurons. This further suggests that under basal conditions, Rac is locally activated in dendritic spines. Together, these results suggest that PIX regulates Rac activation, and locally regulated Rac activity is necessary for spine formation.
Phospho-PAK localizes to synapses
Because PAK is a major downstream effector of Rac, we determined whether PAK is also locally activated in the synapse. Activation of PAK is characterized by autophosphorylation of the kinase at several sites, including threonine 423. Autophosphorylation of this site correlates strongly with activation of PAK, and mutating this residue into glutamic acid yields a constitutively activated enzyme (Sells et al., 2000). To detect activated PAK in hippocampal neurons, a phospho-specific antibody that recognizes the phosphorylated form of threonine 423 was used. In cultured hippocampal neurons, phospho-PAK accumulates in puncta that colocalize with the excitatory synaptic marker PSD-95 (Fig. 3, arrows). Similar colocalization was also observed with SV2 (data not shown). These results suggest that PAK is locally activated in the synapses.
Figure 3.
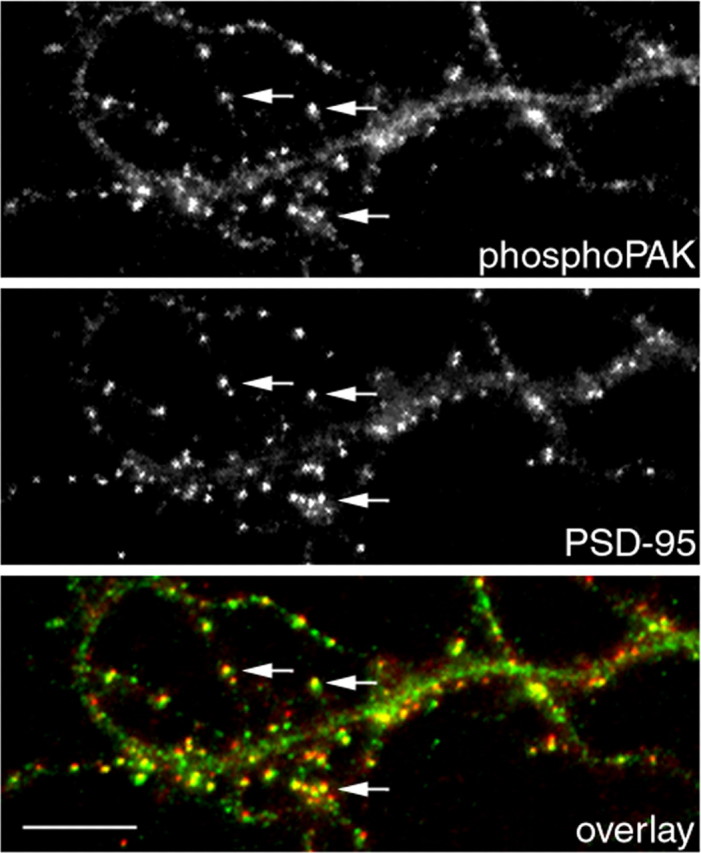
Active PAK localizes to synapses. Hippocampal neurons were fixed and double stained for phospho-PAK and PSD-95 at 14 d in culture. Phospho-PAK accumulates in clusters colocalizing with PSD-95 clusters (arrows). Scale bar, 10 μm.
Effects of PAK1 and PAK3 on spine morphogenesis and synapse formation
To examine the role of PAK in spine morphogenesis and synapse formation, various mutants of PAK1 were expressed in hippocampal neurons. We chose PAK1, because it is highly expressed in the brain and shows specific accumulation in a variety of brain regions, including the hippocampus (Manser et al., 1995). To analyze the effects on spine morphology, GFP was coexpressed with the PAK1 constructs at a 1:5 ratio to visualize spines. All constructs were introduced into the neurons at 7 d in culture, and the neurons were fixed and immunostained for synaptic markers at 14 d in culture. Overexpression of wild-type PAK1 resulted in an increase in the number of spines as well as long, thin, dendritic protrusions that are not associated with synapses (Fig. 4A). The number of dendritic spines in PAK1-expressing neurons increased by 39% compared with control neurons, whereas the number of dendritic protrusions increased by 176% (Fig. 4C). In addition, there is a concomitant increase in the number of PSD-95 clusters, suggesting an increase in excitatory synapse formation (Fig. 4B,D). Expression of DN-PAK1, which contains mutations in both the kinase domain (K299R) and the p21-binding domain (H83L, H86L), decreased dramatically the number of dendritic spines as well as the number of PSD-95 clusters (Fig. 4). Previous studies showed that this PAK1 mutant is able to interfere with the function of endogenous PAK (Sells et al., 1997; Tang et al., 1997). Because this mutant is also defective in the binding of GTPases, it should not sequester Rac or Cdc42 and therefore should not interfere with their interactions with other effectors. Thus, the dominant-negative effect should result from the association of the mutant with the effector proteins of PAK.
Figure 4.

PAK1 regulates dendritic spine and synapse formation. Different PAK1 constructs were transfected into hippocampal neurons at 7 d in culture and were fixed and immunostained at 14 d in culture. GFP was cotransfected with the PAK1 constructs to visualize the spines. Control neurons were cotransfected with GFP and an empty vector. Ectopic expression of WT-PAK1 or CA-PAK1 promotes the formation of dendritic spines and protrusions (A) as well as the formation of excitatory synapses as visualized by PSD-95 immunostaining (B). Expression of either DN-PAK1 or KD-PAK1 inhibits the formation of dendritic spines and excitatory synapses (A, B). C, Quantification of dendritic spines and protrusions in neurons transfected with the PAK1 constructs. *p < 0.001; **, ***p < 0.0005. D, Quantification of PSD-95 clusters in neurons transfected with the PAK1 constructs. *p < 0.001; **p < 0.0002. Scale bar, 10 μm. For each construct, 90-100 dendrites of 15-20 neurons from three independent cultures were analyzed. Error bars represent SEM from three experiments.
In non-neuronal cells, PAK has been shown to exert both kinase-dependent and kinase-independent effects. To determine whether the kinase activity of PAK is necessary for the formation of spines and protrusions, KD-PAK1, which has a single-point mutation in the kinase domain (K299R), was introduced into the neurons. Expression of this construct caused a dramatic decrease in the number of dendritic spines as well as PSD-95 clusters (Fig. 4), suggesting that the effect of PAK1 is kinase dependent. Furthermore, when CA-PAK1 (T423E) was expressed in the neurons, an increase in the density of both spines and dendritic protrusions was observed (Fig. 4A,C). There is also an increase in the number of PSD-95 clusters compared with control neurons (Fig. 4B,D). Together, these results show that the kinase activity of PAK1 is necessary for the formation of dendritic spines and excitatory synapses.
Another PAK isoform, PAK3, is expressed almost exclusively in the brain (Jaffer and Chernoff, 2002). Its expression is ubiquitous in most brain regions, including the cerebral cortex and the hippocampus (Allen et al., 1998). In addition, PAK3 mutations have been found in patients with nonsyndromic mental retardation. To investigate the role of PAK3 on spine morphogenesis and synapse formation, we made a series of PAK3 constructs including myc-tagged wild-type, constitutively active (T421E), kinase-dead (K297R), and dominant-negative (H78,81L and K297R) forms of PAK3. We observed similar effects with the PAK3 constructs as with PAK1 constructs (Fig. 5). These results show that, like PAK1, the kinase activity of PAK3 is necessary for dendritic spine morphogenesis and excitatory synapse formation.
Figure 5.

PAK3 regulates dendritic spine and synapse formation. Different PAK3 constructs were transfected into hippocampal neurons at 7 d in culture and were fixed and immunostained at 14 d in culture. GFP was cotransfected with the PAK3 constructs to visualize the spines. Control neurons were cotransfected with GFP and an empty vector. Ectopic expression of WT-PAK3 or CA-PAK3 promotes the formation of dendritic spines and protrusions (A) as well as the formation of excitatory synapses as visualized by PSD-95 immunostaining (B). Expression of either DN-PAK3 or KD-PAK3 inhibits the formation of dendritic spines and excitatory synapses (A, B). Scale bar, 10 μm. C, Quantification of dendritic spines and protrusions in neurons transfected with the PAK3 constructs. *p < 0.001; **, ***p < 0.0005. D, Quantification of PSD-95 clusters in neurons transfected with the PAK3 constructs. *p < 0.001; **p < 0.0001. For each construct, 85-100 dendrites of 17-20 neurons from three independent cultures were analyzed. Error bars represent SEM from three experiments.
Active MLC localizes to dendritic spines
Because PAK directly phosphorylates MLC on serine 19 (Ramos et al., 1997; Chew et al., 1998; Van Eyk et al., 1998), we investigated the possibility that MLC acts downstream of PAK to mediate its effect on spine morphogenesis and excitatory synapse formation. We first examined the localization of activated MLC in hippocampal neurons. GFP-tagged wild-type MLC and activated MLC (T18D, S19D) were transfected into the neurons, and their localization was examined by immunocytochemistry. Wild-type MLC exhibited partial localization to the dendritic spines as shown by visualizing spines with rhodamine-phalloidin (Fig. 6A). In contrast, activated MLC showed a more specific accumulation in the spines (Fig. 6B). To quantify the localization of wild-type MLC versus activated MLC, the fluorescent intensity of the GFP-tagged MLC constructs in dendritic spines versus the shaft was measured, and a spine/shaft ratio was calculated. The wild-type MLC shows a spine/shaft ratio of 2.2 ± 0.4, whereas activated MLC shows a spine/shaft ratio of 4.1 ± 0.4 (Fig. 6C).
Figure 6.
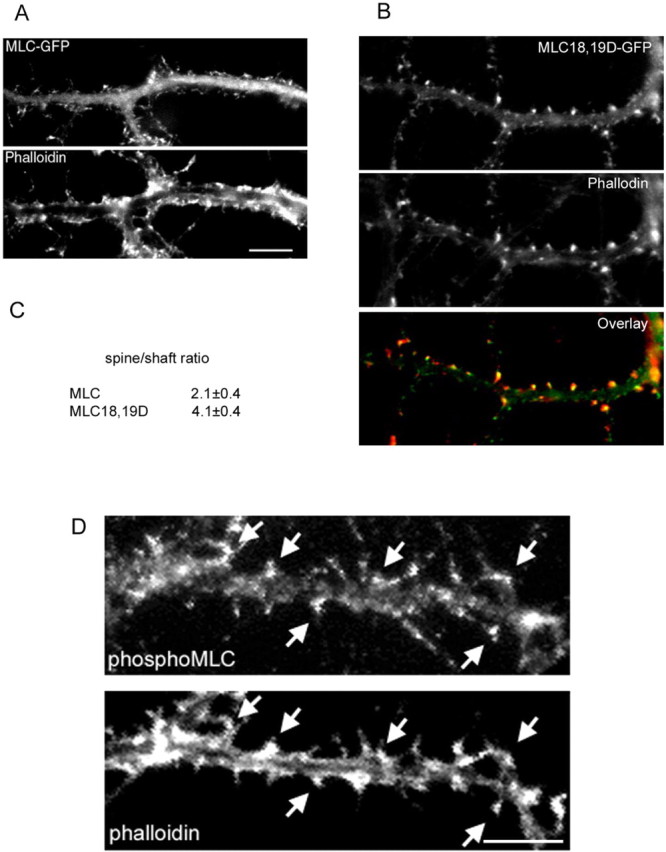
Active MLC localizes to dendritic spines. Hippocampal neurons were transfected with either GFP-tagged wild-type MLC (MLC-GFP) or GFP-tagged active MLC (MLC18,19D-GFP) at 7 d and were fixed and immunostained for rhodamine-phalloidin at 14 d in culture. A, MLC-GFP shows partial localization to spines. Scale bar, 10 μm. B, MLC18,19D-GFP localizes primarily to spines as determined by coimmunostaining with rhodamine-phalloidin. To quantify the localization of the MLC constructs, an average spine/shaft fluorescent intensity ratio was generated for each construct (C). MLC18,19D-GFP showed significantly higher accumulation in spines compared with MLC-GFP. D, Hippocampal neurons were coimmunostained for endogenous phosphoserine 19 MLC and rhodamine-phalloidin. Phospho-MLC localizes to dendritic spines (arrows). Scale bar, 10 μm.
To further show that MLC phosphorylated on serine 19 localizes to dendritic spines, we used a phospho-specific antibody against serine 19 of MLC (Matsumura et al., 1998). Phospho-Ser19 MLC localizes to dendritic spines as shown by visualizing spines with rhodamine-phalloidin (Fig. 6D, arrows). Together, these results show that activated MLC localizes to dendritic spines.
Effects of overexpressing MLC on spine and synapse formation
To investigate the role of MLC in spine morphogenesis and synapse formation, wild-type MLC and active MLC (MLC18,19D) were expressed in the neurons at 7 d in culture, and the number of spines and synapses were quantified at 14 d in culture. Ectopic expression of both wild-type and active MLC caused an over-abundance of spines as well as dendritic protrusions (Fig. 7A). The spine density in wild-type MLC-expressing neurons increased by 51% compared with GFP-expressing control neurons, whereas the spine density in MLC18,19D-expressing neurons increased by 88% (Fig. 7B). The density of dendritic protrusions increased by 82 and 93% in MLC and MLC18,19D-expressing neurons, respectively. In addition, a concomitant increase in the number of PSD-95 clusters was observed. The linear density of PSD-95 clusters increased by 62% (p < 0.002) and 85% (p < 0.0001) in MLC and MLC18,19D-expressing neurons, respectively (Fig. 7C). To determine whether the increase in the number of spines and PSD-95 clusters correlates with an increase in presynaptic boutons, immunostaining of SV2 was performed on neurons transfected with GFP or MLC constructs. Expression of either wild-type MLC or active MLC caused a moderate but significant increase in presynaptic terminals (Fig. 7C) (p < 0.0005 and p < 0.0001, respectively). These results show that expression of MLC promotes the formation of dendritic spines and synapses.
Figure 7.

Ectopic expression of MLC promotes spine and synapse formation. Hippocampal neurons were transfected with GFP, MLC-GFP, or MLC18,19D-GFP at 7 d in culture and were fixed and immunostained for synaptic markers at 14 d in culture. A, Expression of MLC or MLC-18,19D increases the number of dendritic spines and protrusions as well as SV2 and PSD-95 clusters. Scale bar, 20 μm. B, Quantification of dendritic spines and protrusions. *, **p < 0.001. C, Quantification of the number of SV2 clusters and PSD-95 clusters. A total of 95-100 dendrites of 20 neurons from three different cultures were analyzed for each construct. The differences between MLC constructs and GFP-expressing neurons are statistically significant (*p < 0.002; **p < 0.0001; ***p < 0.0005). Error bars represent SEM from three experiments.
Actomyosin contractility is necessary for spine and synapse formation
Phosphorylation of MLC leads to increased actomyosin contractility. To further investigate the role of actomyosin-based contractility in dendritic spine and synapse formation, we treated hippocampal neurons with blebbistatin, which inhibits myosin ATPase activity. Neurons were treated with 50 μm blebbistatin at 7 d in culture and were fixed and immunostained at 9 d in culture. Under these conditions, we did not observe a simplification of the neuronal network, suggesting that blebbistatin did not cause delayed neurite development, and any effect on spines is likely to be direct. Treatment of neurons with blebbistatin resulted in fewer dendritic spines as visualized by phalloidin staining (Fig. 8A,C). In addition, some neurons formed abnormally long, thin, dendritic protrusions (Fig. 8A, arrows). Along with the decrease in spine density, a decrease in the number of synapses was also observed. Immunostaining with a presynaptic marker, synapsin1, revealed that the number of presynaptic terminals decreased by 50% (Fig. 8B). Similar results were obtained with another presynaptic marker, SV2 (data not shown). In addition, a 71% decrease in the number of PSD-95 clusters was observed (Fig. 8B). Similar results were obtained when neurons were treated with 10 μm ML-7, which inhibits myosin light chain kinase activity and, thus, leads to decreased myosin light chain phosphorylation (data not shown). Together, these results suggest that actomyosin contractility is necessary for the formation of dendritic spines and synapses.
Figure 8.

Blebbistatin inhibits dendritic spine and synapse formation. A, Hippocampal neurons were treated with 50 μm blebbistatin at 7 d in culture and were fixed and immunostained for various synaptic markers at 9 d in culture. Control cultures were treated with DMSO. Arrows point to the abnormally long, thin, dendritic protrusions. Scale bar, 20 μm. B, Quantification of the number of synapses as visualized by both synapsin1 and PSD-95. *p < 0.0001. C, Quantification of the number of total protrusions, including dendritic spines and dendritic protrusions. *p < 0.0001. A total of 85-95 dendrites of 15-20 neurons from four independent cultures were analyzed for each condition. Error bars represent SEM from three experiments.
MLC functions downstream of PAK
If MLC functions downstream of PAK in regulating dendritic spine morphogenesis and synapse formation, inhibiting myosin ATPase activity should block the effects of constitutively active PAK. To address this, we transfected neurons with CA-PAK3 and treated these cells with 50 μm blebbistatin. Treatment of CA-PAK3-expressing neurons with blebbistatin almost completely inhibited the formation of dendritic spines and protrusions induced by CA-PAK3 (Fig. 9A,B). Similar results were obtained with CA-PAK1 (data not shown). This suggests that PAK functions through MLC in regulating spine and synapse formation.
Figure 9.

MLC functions downstream of PAK. A, Hippocampal neurons were cotransfected with GFP and CA-PAK3 at 6 d in culture and treated with either DMSO or 50 μm blebbistatin at 7 d in culture. After 48 h, neurons were fixed and immunostained for the myc epitope. Treatment of neurons with blebbistatin completely inhibited the protrusions induced by CA-PAK3. B, Quantification of the total number of protrusions, including dendritic spines and dendritic protrusions. *p < 0.0001. For each condition, 90-95 dendrites of 15-18 neurons from three independent cultures were analyzed. C, KD-PAK1 was cotransfected with either GFP or MLC18,19D-GFP at 7 d in culture and was fixed at 14 d in culture. Expression of KD-PAK1 was confirmed by immunostaining for the myc epitope. Coexpression of MLC18,19D-GFP rescues the defects in spine formation in KD-PAK1. Scale bars: top panels of each construct, 20 μm; enlarged pictures in the bottom panels, 10 μm. D, Quantification of the total number of protrusions in neurons expressing KD-PAK1 and GFP (control) or KD-PAK1 and MLC18,19D-GFP (MLC18,19D). The total number of protrusions includes dendritic spines and dendritic protrusions. *p < 0.0001. A total of 90-95 dendrites of 16-18 neurons from three different cultures were analyzed for each condition. E, Quantification of the relative fluorescent intensity of phospho-MLC in control neurons versus CA-PAK3-transfected neurons. Fluorescent intensity was normalized to the level of control neurons. *p < 0.0005. Error bars represent SEM from three experiments.
To further confirm the above results, we performed the reverse experiment. If MLC functions downstream of PAK, activated MLC should be able to rescue the effects of kinase-dead PAK constructs. To test this hypothesis, we cotransfected kinase-dead PAK constructs with MLC 18,19D-GFP. Coexpression of activated MLC rescued the defect in spine formation in KD-PAK1 (Fig. 9C,D). Similar results were obtained with KD-PAK3 (data not shown). This suggests that PAK mediates its effects by modulating the phosphorylation level of MLC.
To exclude the possibility that PAK and MLC are on parallel pathways, we examined whether PAK directly affects endogenous MLC phosphorylation. Constitutively active PAK constructs were transfected in hippocampal neurons, which were then immunostained for phospho-MLC. Staining intensity was then quantified. Expression of CA-PAK3 resulted in a 107% increase in the phospho-MLC staining (Fig. 9E), showing an increase in MLC phosphorylation. Similar results were obtained with CA-PAK1 (data not shown). Together, these results suggest that MLC functions downstream of PAK in regulating spine and synapse formation. However, other downstream effectors of PAK might also participate in this process.
PAK and MLC function downstream of GIT1
Finally, we examined whether GIT1 functions through PAK and MLC. To test this, we used RNAi to knock down endogenous GIT1 expression in the neurons. If PAK and MLC function downstream of GIT1, coexpression of activated forms of these molecules should be able to rescue the defects of GIT1 knockdown. When we coexpressed constitutively active PAK3 along with the GIT1 siRNA, the neurons showed a significantly increased number of spines as well as synapses compared with GIT1 siRNA-transfected neurons (Fig. 10) (p < 0.001). Similar results were obtained with constitutively active PAK1 (data not shown). A significant increase in spine and synaptic density was also observed when MLC 18,19D-GFP was coexpressed with GIT1 siRNA (Fig. 10) (p < 0.001). These results suggest that GIT1 functions through PAK and MLC in regulating dendritic spine and synapse formation.
Figure 10.

PAK and MLC function downstream of GIT1. A, Hippocampal neurons were transfected with pSUPER-GIT1 siRNA along with GFP (control), CA-PAK3, or activated MLC (MLC18,19D-GFP) at 6 d in culture and were fixed and immunostained for synapsin1 at 12 d in culture. Both CA-PAK3 and MLC 18,19D-GFP were able to rescue the GIT1 siRNA-induced defects. B, Quantification of the number of synapses in neurons transfected with the indicated constructs. *p < 0.001. C, Quantification of the total number of protrusions, including dendritic spines and protrusions not associated with presynaptic terminals. For each condition, 85-95 dendrites of 15-20 neurons from independent cultures were analyzed. *p < 0.001. Error bars in represent SEM from three experiments.
Discussion
Increasing evidence implicates the formation, maintenance, and plasticity of dendritic spines in learning and memory. Spine changes that are associated with learning processes have been observed from invertebrates to rodents (Lendvai et al., 2000; Smart and Halpain, 2000). In addition, pathological changes in dendritic spines are often observed in brain disorders such as nonsyndromic mental retardation (Fiala et al., 2002). As the importance of spine morphogenesis in cognitive function has become clearer, there has been accelerating interest in understanding its molecular basis. The dynamic, actin-rich nature of dendritic spines has pointed to the Rho family as a central contributor, because they are key regulators of actin dynamics and organization (Matus, 2000; Hering and Sheng, 2001; Etienne-Manneville and Hall, 2002). Previously, we showed that the adaptor protein GIT1 functions to localize PIX, a Rac GEF, to dendritic spines and that mislocalization of GIT1 leads to alterations in spine morphogenesis and synapse formation. These observations lead to the hypothesis that GIT1 functions to locally regulate Rac activity in spines and synapses (Zhang et al., 2003). In the present study, we provided strong support for this hypothesis by showing that GIT1 is required for normal spine morphogenesis. In addition, we showed that Rac activation, which is regulated by PIX, is localized to spines. Furthermore, we have extended the observations by identifying a signaling pathway downstream of Rac, including PAK and MLC, which functions in the regulation of spine morphogenesis and synapse formation. Thus, GIT1 appears to organize a signaling module containing an activator and effector of Rac that localizes and regulates Rac activity in dendritic spines and synapses.
Our FRET experiments show localized Rac activation in dendritic spines and at the base of dendritic protrusions. In other cell types, Rac mediates the polymerization of actin through its interactions with regulators of actin assembly, resulting in the extension and retraction of protrusions. By analogy, these Rac-mediated activities in spines would likely function to regulate the formation of dendritic protrusions, activity-dependent spine remodeling, and perhaps spine stabilization.
The PAK family of kinases is a well characterized effector of Rac, and PAK3 is mutated in patients with nonsyndromic mental retardation. Here, we demonstrate an essential role of PAK in dendritic spine morphogenesis and excitatory synapse formation that is dependent on its kinase activity. Our results are consistent with previous reports that showed a PAK inhibitory peptide hinders spine formation in hippocampal neurons (Penzes et al., 2003) and the cerebral cortex (Hayashi et al., 2004). In addition, PAK3 has been shown recently to be involved in synapse formation and spine morphogenesis in hippocampal slices (Boda et al., 2004).
We further show that PAK exerts its effects by modulating the phosphorylation state of MLC, and that increased phospho-MLC levels correspond to increased spine and synapse formation. How might MLC phosphorylation regulate spine morphogenesis? In non-neuronal cells, phosphorylated MLC localizes to both the leading edge and the rear of a migrating cell (Matsumura et al., 1998). Phosphorylation of MLC at the leading edge presumably serves to stabilize lamellipodia, because kinase-dead PAK, which fails to promote MLC phosphorylation, causes fibroblasts to extend multiple lamellipodia without a predominant leading edge (Sells et al., 1999). Therefore, similar to its role in fibroblasts, MLC phosphorylation could promote dendritic spine morphogenesis by stabilizing the actin network at this site. Interestingly, wild-type MLC can also increase spine and synapse formation. This suggests that a significant fraction of wild-type MLC is in an active, phosphorylated form.
Intriguingly, the PAK mutants do not completely recapitulate the phenotype of the Rac mutants (Zhang et al., 2003). We previously showed that activated Rac promotes formation of dendritic protrusions but inhibits the formation of both dendritic spines and synapses. However, activated PAK promotes the formation of both dendritic spines and protrusions. Because both molecules induce the formation of dendritic protrusions, it is likely that the stabilization of protrusions is differentially regulated by these two molecules. Clearly, other downstream effectors of Rac must also be involved in this process.
Finally, as mentioned above, both PAK3 and αPIX mutations have been identified in patients with nonsyndromic mental retardation (Allen et al., 1998; Bienvenu et al., 2000; Kutsche et al., 2000). One of the PAK3 mutations, C419T, results in premature termination of the protein. This renders a mutant that retains the GTPase binding activity but not the kinase activity (Allen et al., 1998), corresponding to the KD-PAK3 construct that we used in this study. Our results indicate that KD-PAK3 causes a significant decrease in the number of dendritic spines and excitatory synapses. Although the dendritic spine pathology in the patients with the PAK3 mutation is not known, a decrease in spine density is one of the common forms of spine abnormality in mental retardation. The αPIX mutation found in patients with nonsyndromic MR leads to a C-terminal truncation mutant (Kutsche et al., 2000). We showed previously that PIX is targeted to synapses by the C-terminal GIT1-binding domain (Zhang et al., 2003). Thus, the PIX mutation could lead to mislocalization of PIX away from synapses, mislocalized Rac activation, and the formation of long, thin, dendritic protrusions, which is another very common form of spine pathology in MR patients.
Together, our results point to a signaling complex containing GIT1, PIX, active Rac, and PAK, which is involved in the regulation of spine morphogenesis and synapse formation. A mislocalized, active signaling complex leads to an overabundance of dendritic protrusions, whereas a loss-of-function mutation in the signaling complex leads to decreased spine density. Either of these would result in decreased synapse formation. Therefore, our results provide a potential mechanism by which mutations in Rho family signaling, such as PIX and PAK, lead to decreased neuronal connectivity and cognitive defects in nonsyndromic mental retardation.
Footnotes
This work was supported by National Institutes of Health Grant GM23244. We thank Drs. Kathleen Kelly, Michiyuki Matsuda, Fumio Matsumura, Jonathan Chernoff, Rick Cerione, Chris Turner, Lorraine Santy, and Jim Casanova for generously providing reagents and Dr. Claire Brown for her help with the FRET analyses.
Correspondence should be addressed to A. F. Horwitz at the above address. E-mail: horwitz@virginia.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253379-10$15.00/0
References
- Allen KM, Gleeson JG, Bagrodia S, Partington MW, MacMillan JC, Cerione RA, Mulley JC, Walsh CA (1998) PAK3 mutation in nonsyndromic X-linked mental retardation. Nat Genet 20: 25-30. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, des Portes V, McDonell N, Carrie A, Zemni R, Couvert P, Ropers HH, Moraine C, van Bokhoven H, Fryns JP, Allen K, Walsh CA, Boue J, Kahn A, Chelly J, Beldjord C (2000) Missense mutation in PAK3, R67C, causes X-linked nonspecific mental retardation. Am J Med Genet 93: 294-298. [DOI] [PubMed] [Google Scholar]
- Boda B, Alberi S, Nikonenko I, Node-Langlois R, Jourdain P, Moosmayer M, Parisi-Jourdain L, Muller D (2004) The mental retardation protein PAK3 contributes to synapse formation and plasticity in hippocampus. J Neurosci 24: 10816-10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew TL, Masaracchia RA, Goeckeler ZM, Wysolmerski RB (1998) Phosphorylation of non-muscle myosin II regulatory light chain by p21-activated kinase (gamma-PAK). J Muscle Res Cell Motil 19: 839-854. [DOI] [PubMed] [Google Scholar]
- Dunaevsky A, Tashiro A, Majewska A, Mason C, Yuste R (1999) Developmental regulation of spine motility in the mammalian central nervous system. Proc Natl Acad Sci USA 96: 13438-13443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A (2002) Rho GTPases in cell biology. Nature 420: 629-635. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM (2002) Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev 39: 29-54. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Knutti D, Matus A (1998) Rapid actin-based plasticity in dendritic spines. Neuron 20: 847-854. [DOI] [PubMed] [Google Scholar]
- Goslin K, Asmussen H, Banker G (1998) Rat hippocampal neurons in low-density culture. Cambridge, MA: MIT.
- Hayashi ML, Choi SY, Rao BS, Jung HY, Lee HK, Zhang D, Chattarji S, Kirkwood A, Tonegawa S (2004) Altered cortical synaptic morphology and impaired memory consolidation in forebrain-specific dominant-negative PAK transgenic mice. Neuron 42: 773-787. [DOI] [PubMed] [Google Scholar]
- Hering H, Sheng M (2001) Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci 2: 880-888. [DOI] [PubMed] [Google Scholar]
- Itoh RE, Kurokawa K, Ohba Y, Yoshizaki H, Mochizuki N, Matsuda M (2002) Activation of Rac and cdc42 video imaged by fluorescent resonance energy transfer-based single-molecule probes in the membrane of living cells. Mol Cell Biol 22: 6582-6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffer ZM, Chernoff J (2002) p21-activated kinases: three more join the Pak. Int J Biochem Cell Biol 34: 713-717. [DOI] [PubMed] [Google Scholar]
- Kutsche K, Yntema H, Brandt A, Jantke I, Nothwang HG, Orth U, Boavida MG, David D, Chelly J, Fryns JP, Moraine C, Ropers HH, Hamel BC, van Bokhoven H, Gal A (2000) Mutations in ARHGEF6, encoding a guanine nucleotide exchange factor for Rho GTPases, in patients with X-linked mental retardation. Nat Genet 26: 247-250. [DOI] [PubMed] [Google Scholar]
- Lendvai B, Stern EA, Chen B, Svoboda K (2000) Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo Nature 404: 876-881. [DOI] [PubMed] [Google Scholar]
- Manabe R, Kovalenko M, Webb DJ, Horwitz AR (2002) GIT1 functions in a motile, multi-molecular signaling complex that regulates protrusive activity and cell migration. J Cell Sci 115: 1497-1510. [DOI] [PubMed] [Google Scholar]
- Manser E, Chong C, Zhao ZS, Leung T, Michael G, Hall C, Lim L (1995) Molecular cloning of a new member of the p21-Cdc42/Rac-activated kinase (PAK) family. J Biol Chem 270: 25070-25078. [DOI] [PubMed] [Google Scholar]
- Manser E, Loo TH, Koh CG, Zhao ZS, Chen XQ, Tan L, Tan I, Leung T, Lim L (1998) PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol Cell 1: 183-192. [DOI] [PubMed] [Google Scholar]
- Matsumura F, Ono S, Yamakita Y, Totsukawa G, Yamashiro S (1998) Specific localization of serine 19 phosphorylated myosin II during cell locomotion and mitosis of cultured cells. J Cell Biol 140: 119-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus A (2000) Actin-based plasticity in dendritic spines. Science 290: 754-758. [DOI] [PubMed] [Google Scholar]
- Penzes P, Beeser A, Chernoff J, Schiller MR, Eipper BA, Mains RE, Huganir RL (2003) Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron 37: 263-274. [DOI] [PubMed] [Google Scholar]
- Raftopoulou M, Hall A (2004) Cell migration: Rho GTPases lead the way. Dev Biol 265: 23-32. [DOI] [PubMed] [Google Scholar]
- Ramos E, Wysolmerski RB, Masaracchia RA (1997) Myosin phosphorylation by human cdc42-dependent S6/H4 kinase/gammaPAK from placenta and lymphoid cells. Recept Signal Transduct 7: 99-110. [PubMed] [Google Scholar]
- Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J (1997) Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol 7: 202-210. [DOI] [PubMed] [Google Scholar]
- Sells MA, Boyd JT, Chernoff J (1999) p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J Cell Biol 145: 837-849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sells MA, Pfaff A, Chernoff J (2000) Temporal and spatial distribution of activated Pak1 in fibroblasts. J Cell Biol 151: 1449-1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart FM, Halpain S (2000) Regulation of dendritic spine stability. Hippocampus 10: 542-554. [DOI] [PubMed] [Google Scholar]
- Tang Y, Chen Z, Ambrose D, Liu J, Gibbs JB, Chernoff J, Field J (1997) Kinase-deficient Pak1 mutants inhibit Ras transformation of Rat-1 fibroblasts. Mol Cell Biol 17: 4454-4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eyk JE, Arrell DK, Foster DB, Strauss JD, Heinonen TY, Furmaniak-Kazmierczak E, Cote GP, Mak AS (1998) Different molecular mechanisms for Rho family GTPase-dependent, Ca2+-independent contraction of smooth muscle. J Biol Chem 273: 23433-23439. [DOI] [PubMed] [Google Scholar]
- Zhang H, Webb DJ, Asmussen H, Horwitz AF (2003) Synapse formation is regulated by the signaling adaptor GIT1. J Cell Biol 161: 131-142. [DOI] [PMC free article] [PubMed] [Google Scholar]