

Abstract
Striatal medium spiny neurons are an important site of convergence for signaling mediated by the neurotransmitters dopamine and glutamate. We report that in striatal neurons in primary culture, signaling through group I metabotropic glutamate receptors (mGluRs) 1/5 and the D1 class of dopamine receptors (DRs) 1/5 converges to increase phosphorylation of the mitogen-activated protein kinase ERK2 (extracellular signal-regulated kinase 2). Induction of mitogen-activated protein kinase kinase-dependent signaling cascades by either mGluR1/5 or DR1/5 gave rise to increases in phosphorylation of ERK2. Coactivation of mGluR1/5 and DR1/5 with (S)-3,5-dihydroxyphenylglycine and (+)-1-phenyl-2,3,4,5-tetrahydro-(1H)-3-benzazepine-7,8-diol hydrochloride enhanced the phosphorylation of ERK2. This interaction between mGluR1/5 and DR1/5 required protein kinase C (PKC), because the PKC inhibitors calphostin C, bisindolylmaleimide I, and Gö6976 blocked DR1/5-enhanced phosphorylation of ERK2. Use of the phosphatase inhibitors calyculin and okadaic acid indicated that inhibition of protein phosphatases 1 and 2A dramatically enhanced ERK2 phosphorylation by mGluR1/5. Coactivation of mGluR1/5 and DR1/5 also enhanced cAMP-response element binding protein (CREB) phosphorylation (compared with each receptor agonist alone) but did not enhance CREB-mediated transcriptional activity. Thus, signal transduction pathways activated by DR1/5 and mGluR5 interact to modify downstream events in striatal neurons while retaining numerous regulatory checkpoints.
Keywords: metabotropic glutamate receptor, dopamine receptor, ERK, PKC, striatum, signal transduction
Introduction
The striatum plays a critical role in motor control and several forms of implicit memory and is therefore involved in the pathophysiology of diverse illnesses, including Parkinson's disease and drug addiction. Medium spiny neurons are GABAergic projection neurons that constitute 95% of the neuronal types in the striatum (Kemp and Powell, 1971). Striatal function depends on the ability of these neurons to integrate inputs originating from dopaminergic midbrain neurons and glutamatergic cortical neurons. Abnormality in either of these inputs, or of their integration, is thought to play a role in disease.
Although medium spiny neurons in the adult striatum are characterized as expressing either the D1 or D2 class of dopamine receptors (DRs), they appear to express all classes of glutamate receptors. Attention has been focused on ionotropic glutamate receptor interactions with dopamine receptors in striatal neurons (Cepeda et al., 1993; Price et al., 1999), with fewer studies investigating dopamine receptor interactions with group I metabotropic glutamate receptors (mGluRs). Recent evidence suggests that interactions between metabotropic glutamate receptors and dopamine receptors are required for cocaine sensitization (Chiamulara et al., 2001). Here, we report the interaction of group I metabotropic glutamate receptors with the D1 class of dopamine receptors in striatal neurons. Group I mGluRs include mGluR1 and mGluR5 (mGluR1/5), which couple to Gq/11 to activate phospholipase Cβ (PLCβ). The D1 class of dopamine receptors include D1R and D5R (DR1/5) coupled to Golf to activate adenylyl cyclase (AC) (Zhuang et al., 2000; Corvol et al., 2001). Although convergence of PLC- and AC-mediated signaling pathways on downstream targets has been examined in glutamatergic neurons (Roberson et al., 1999), such convergence has not been tested in GABAergic striatal medium spiny neurons.
Few studies have dissected mGluR1/5 signal transduction in striatal neurons. Evidence from other neuronal types (Roberson et al., 1999; Hamilton and Nathanson, 2001) indicates that G-protein-coupled receptors can stimulate mitogen-activated protein kinases (MAPKs), extracellular signal-regulated kinase 1 (ERK1), and ERK2. We demonstrate here that mGluR1/5 stimulation increases ERK2 phosphorylation in cultured striatal neurons. Although forskolin stimulation of AC activates MAPKs in striatal neurons (Vincent et al., 1998; Zanassi et al., 2001), increased MAPK phosphorylation by selective DR1/5 activation has not been demonstrated in these neurons. Because protein kinase A (PKA) does not directly phosphorylate MAPKs, recruitment of Ras family members is the route hypothesized to mediate this effect in medium spiny neurons. Most interestingly, our data show that coactivation of DR1/5 and mGluR1/5 enhances ERK2 phosphorylation. Despite DR1/5-mGluR1/5 cooperation to elevate levels of ERK2 and cAMP-response element (CRE) binding protein (CREB) phosphorylation, we found that they do not cooperate in CREB-mediated transcriptional activity in medium spiny neurons. Although neither DR1/5 nor mGluR1/5 activation of ERK2 depends on protein kinase C (PKC), DR1/5-mGluR1/5-enhanced increases in ERK2 do require PKC. In addition, we observed that protein phosphatase 1 (PP1) and PP2A modulate mGluR1/5-mediated ERK2 phosphorylation. These findings, in conjunction with previous studies (Ferris et al., 1991a; Grewal et al., 2000; Lezcano et al., 2000), suggest mechanisms through which functional consequences of mGluR1/5 activation can be modulated by DR1/5 and open additional avenues for the pharmacological manipulation of striatal function.
Materials and Methods
Materials. Experiments were conducted according to the principles set forth in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. (RS)-2-chloro-5-hydroxyphenylglycine (CHPG), (S)-3,5-dihydroxyphenylglycine (DHPG), 2-methyl-6-(phenylethynyl)-pyridine hydrochloride (MPEP), (RS)-aminoindan-1,5-dicarboxylic acid (AIDA), (RS-4-carboxyphenylglycine (4CPG), (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid (LY367385), and bisindolylmaleimide I (GF109203X) were obtained from Tocris Cookson (Ellisville, MO). (+)-1-Phenyl-2,3,4,5-tetrahydro-(1H)-3-benzazepine-7,8-diol (SKF38393), dopamine, (R)-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine (SCH23390), calyculin A, calphostin C, and TTX were obtained from Sigma-RBI (St. Louis, MO). N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide dihydrochloride (H89), 12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo[2,3-a]pyrrolo[3,4-c]carbazole (Gö6976), forskolin, and okadaic acid (OA) were purchased from Calbiochem (La Jolla, CA). The mitogen-activated protein kinase kinase (MEK) inhibitor 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene (U0126) was obtained from Promega (Madison, WI). Concentrations of agents used in this study were determined by using published EC50/IC50 values by obtaining comparable published analyses done in cultured cells or by independent analysis performed by each author (or all three authors). Receptor antagonists were used at concentrations that were receptor selective. Transfections for CREB transcriptional assays were performed using the PathDetect system (Stratagene, La Jolla, CA). Analyses of CRE-mediated transcriptional activity were performed using the Tropix (Bedford, MA) Dual-Light luciferase assay kit.
Cell culture. Culture dishes were coated for 12-18 h with polyethylenimine (0.2% in 50 mm sodium borate, pH 8) and washed and treated with DMEM/10% fetal calf serum for 60 min before plating of cells. Striata were isolated from the pups of Sprague Dawley embryonic day 18 (E18)-timed pregnant females (Taconic, Germantown, NY) and dissociated in media by trituration with a fire-polished Pasteur pipette. Cells were plated in Neurobasal media (Invitrogen, Rockville, MD) at a density of 1.7 × 106 per 35 mm dish, to which glial cell line-derived neurotrophic factor (33 ng/ml), BDNF (50 ng/ml), B27 (1×), glutamine (0.5 mm), and pen/strep (50 U per 50 μg/ml) were added. No mitotic inhibitors were used. Cultures were maintained for 7-9 d before use.
Calcium imaging. Cells grown on polyornithine-coated coverslips were loaded with fluo-4 AM (1 mm in 20% Pluronic; Molecular Probes, Eugene, OR) for 30 min at room temperature. Coverslips were mounted in a Leiden perfusion chamber and positioned on the stage of an inverted microscope fitted with a Nikon (Tokyo, Japan) 20× fluorescent 0.75 numerical aperture lens. Cells were perfused with a solution containing the following (in mm): 130 NaCl, 5.36 KCl, 0.8 MgSO4, 1 Na2HPO4, 25 glucose, 20 HEPES, 1 Na-pyruvate, 1.50 CaCl2, 1 ascorbic acid, pH 7.3. Drugs were bath applied using a nine-barrel fast perfusion system (ALA Scientific Instruments, Westbrook, NY) connected to individual three-way valves (Lee Biomolecular Research Laboratories, Essex, CT).
Fluorescence images were acquired at 495 nm excitation and 510 nm emission through a microchannel plate intensifier with a CCD camera. Images were acquired every 500 ms, and data were extracted using the Synapse IP software environment (Synergy Research Systems, Silver Spring, MD). For analysis, fluorescence intensities were normalized (ΔF/Fo * 100). ΔF was calculated as the difference between the peak fluorescence (F) and the mean value of 20 data points before stimulation (Fo). Data were plotted using KaleidaGraph (Synergy Software, Reading, PA).
Western blot analysis and immunofluorescence. Cells were stimulated with various agents (each treatment in duplicate), washed with PBS, and solubilized in 1% SDS. Samples were boiled and aliquots taken for quantitation of protein using the Pierce (Rockford, IL) BCA protein assay kit. Ten micrograms of each sample were run on Novex (Wadsworth, OH) 4-20% SDS-PAGE and transferred to nitrocellulose. Membranes were stained posttransfer with Ponceau S to identify and rerun blots with uneven lane loading. Membranes were blocked with BSA and probed with either anti-active MAPK (1:5000 dilution; Promega) or anti-phospho-CREB (1:2000 dilution; Upstate Biotechnology, Lake Placid, NY). For all Western blots in which duplicate lanes are shown, data represent results from separately stimulated samples. Immunoreactivity was detected using enhanced chemiluminescence (Amersham Biosciences, Arlington Heights, IL). Immunofluorescence was performed on cells cultured 8 d in vitro (DIV). Cells were washed with PBS and fixed with 4% paraformaldehyde. Rabbit polyclonal antibody against mGluR1 or mGluR5 (Upstate Biotechnology) was used at a 1:1000 dilution for Western blot analysis and 1:300 dilution for immunofluorescence. The rat monoclonal anti-D1R antibody was a generous gift from Dr. Alan Levey (Emory University, Atlanta, GA) and was used at 1:500 for immunofluorescence. Rabbit polyclonal anti-phospho-p44/42 MAPK (Thr202/Tyr204; Cell Signaling Technology, Beverly, MA) was used at 1/500 for double-label immunofluorescence with mouse anti-neuronal-specific nuclear protein (NeuN) (1:500; Chemicon, Temecula, CA). Immunoreactivity was detected using either Alexa 568-conjugated or Alexa 488-conjugated goat anti-rabbit or anti-mouse antibody. Images were collected using a Micromax 5 MHz cooled CCD camera (Princeton Instruments, Trenton, NJ) and MetaMorph 4.0 image analysis software (Universal Imaging, West Chester, PA). Duplicate treatments were averaged and comprised an “N” of one.
Transcription/luciferase assays. At 5 d in vitro, cells were transfected with two vectors using the calcium phosphate method. One vector consisted of a galactosidase 4 (Gal4)-luciferase construct (pFR-Luc). The other construct contained a Gal4 DNA binding domain expressed either without CREB (pFC2-dbd; negative control) or with CREB (pFA2-CREB) fused to it. Twelve to 16 h after transfection, cells were stimulated with DHPG, SKF38393, or both together. Forskolin stimulation was used as a positive control for this system. After stimulation, cells were washed with PBS and lysed in Dual-Light lysis buffer. Extracts were assayed for luciferase activity using a luminometer (LB 9507 Lumat; Berthold, Bad Wildbad, Germany). All treatments were performed in duplicate; extracts were assayed in triplicate. Note that transfection with a lacZ construct was included as a control for transfection efficiency. There was no significant difference in β-galactosidase activity between dishes or between experiments. These data were not included in the final figure.
Data analysis. Results from Western blot analyses were collected with a Sierra Scientific (Sunnyvale, CA) video CCD camera and quantitated using ImageQuant (Molecular Dynamics, Sunnyvale, CA). Statistical analysis was conducted with Statview using ANOVA with post hoc Fisher's test. A probability of 0.05 was selected as the level of statistical significance. For calcium imaging experiments, data were analyzed using KaleidaGraph (Synergy Software). Statistical analysis was done using InStat (Statistical Services Centre, University of Reading, Reading, UK). All data are expressed as mean ± SEM.
Results
Group I metabotropic receptors are present and functional in primary striatal neurons
Before initiating biochemical experiments examining regulation of ERK phosphorylation in primary striatal neurons, we sought to establish that group 1 mGluRs were expressed by, and functional in the majority of, neurons at 7-10 DIV under our culture conditions. We chose to assay calcium elevation after mGluR1/5 activation. Stimulation with the nonselective group I mGluR agonist DHPG elicited an immediate increase in intracellular calcium levels, which persisted as long as agonist was present, and declined after its removal (Fig. 1A). To ensure that neuronal, not glial, responses to mGluR1/5 agents were quantitated, we marked cells that increased calcium in response to NMDA and collected data using mGluR1/5 selective agents only from NMDA responsive cells. Approximately 80% of cells in a field increased calcium in response to DHPG; of these, ∼90% also responded to NMDA. A concentration-response curve generated with DHPG was sigmoidal between 0.1 and 100 μm, and the EC50 value was calculated as 2.72 ± 0.34 μm. The concentration-response relationship for the highly selective mGluR5 agonist CHPG also was sigmoidal, and the EC50 value was 291 ± 20.75 μm (Fig. 1B). These values are in agreement with previously published data for CHPG and DHPG (Doherty et al., 1997; Toms and Roberts 1999). mGluR1 antagonists AIDA (10 μm) and LY367385 (10 μm) decreased the DHPG response by 28.5 ± 2.5% (n = 286) and 64.7 ± 1.6% (n = 322), respectively. The mGluR5 inverse agonist MPEP (20 μm) produced the greatest block of DHPG-mediated calcium elevation, inhibiting the DHPG response by 95.3 ± 1.1% (n = 312) (Fig. 1C1). We concluded that mGluR5 was the principal mediator of calcium flux elicited by DHPG. Involvement of NMDA receptors in the DHPG response was also observed, as reported previously (Pisani et al., 1997); 20 μm d-APV decreased the DHPG response by 41.8 ± 2.8% (n = 268). Calcium elevation by DHPG was dependent on external calcium, because brief removal of calcium while adding EGTA to the external solution reduced calcium rise by 92.5 ± 0.5% (n = 302) (Fig. 1C2). Blockade of calcium channels with cadmium also inhibited the DHPG response by 79.9 ± 1.6% (n = 132), which supports the importance of calcium influx in the DHPG effect (Fig. 1C3). The increase in calcium given by DHPG extended throughout the entire neuronal cell body and processes (Fig. 1D).
Figure 1.
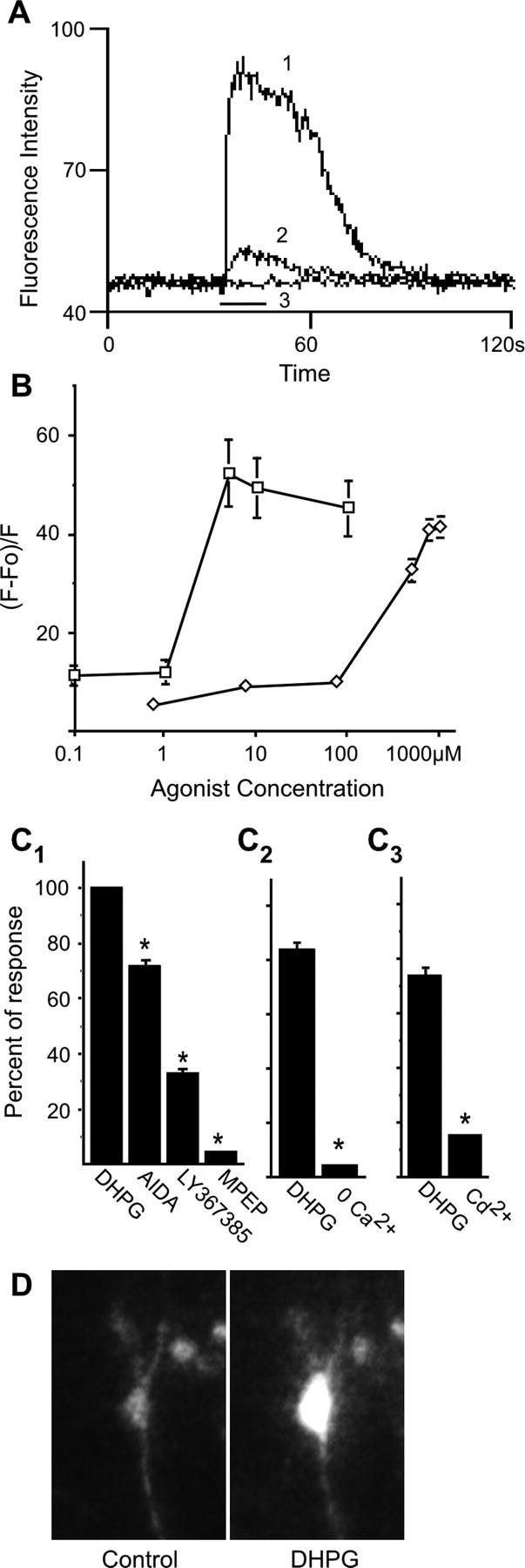
Metabotropic glutamate receptor agonists DHPG and CHPG evoke a cytoplasmic calcium rise in E18 striatal neurons. Agonists were applied for 20 s (black bar on x-axis), and antagonists were applied 3 min before stimulation with agonist. A, Traces show DHPG-evoked intracellular calcium signals. Trace 1, 10 μm; trace 2, 1 μm; trace 3, 4 μm DHPG in the presence of 20 μm MPEP. B, Concentration-response curves for DHPG (open squares; n = 45) and CHPG (open diamonds; n = 100). C1, Effect of the mGluR antagonists AIDA (10 μm; n = 286), LY367385 (10 μm; n = 322), and MPEP (20 μm; n = 312) on the response to 4 μm DHPG. C2, Removal of extracellular calcium blocks calcium rise by 4 μm DHPG (n = 302). C3, Blocking calcium channels with cadmium inhibits calcium rise by DHPG (n = 132). Data are presented as a percentage of the DHPG response remaining in the presence of the antagonist. *p < 0.001. D, An example of a calcium response in a single striatal neuron before application of DHPG and during the response to the agonist (4 μm).
mGluR5 activates ERK2 in striatal neurons
To ensure that experiments were performed at times of stable receptor expression in the cultures, we used Western blot analysis to examine levels of both mGluR1 and mGluR5 protein in cultured E18 striatal neurons as a function of DIV (Fig. 2A). The levels of mGluR5 were consistently higher than mGluR1 from 3 to 13 DIV. The expression of mGluR1 in E18 neurons varied with the number of DIV; peak levels occurred at 7 DIV. Experiments were performed at 7-9 DIV, during which period mGluR5 protein levels were relatively stable. The typical pattern of mGluR immunoreactivity in these neurons is shown for cells at 8 DIV. mGluR1 staining was visible primarily in the somata of these neurons, whereas mGluR5 immunoreactivity was apparent in both the somata and processes (Fig. 2B).
Figure 2.
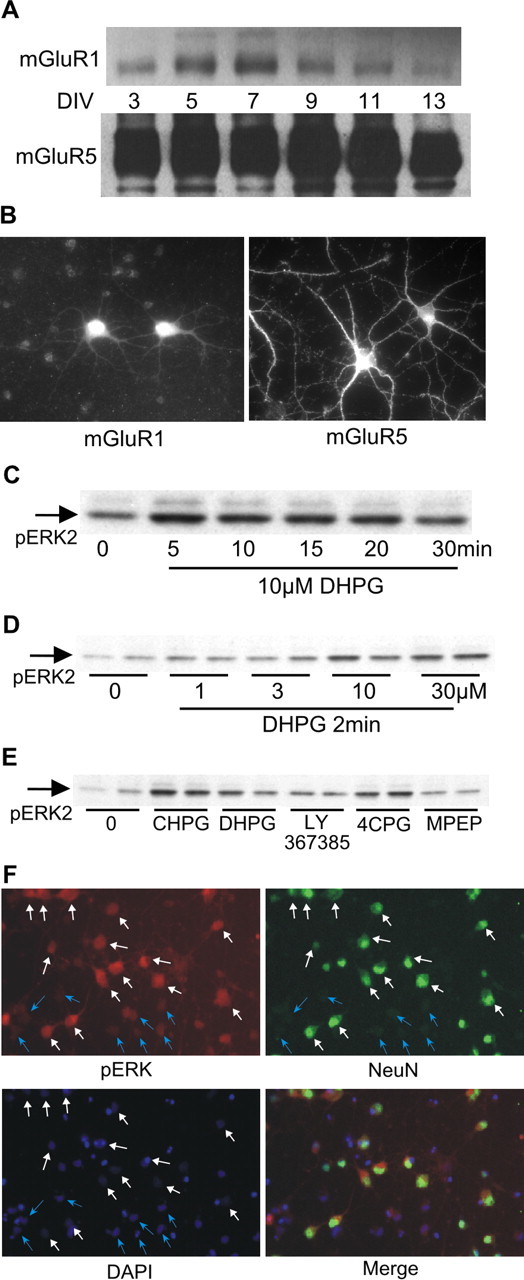
Group I metabotropic glutamate receptors stimulate ERK2 phosphorylation in E18 striatal neurons. A, Western blot analysis of mGluR1 and mGluR5 expression in cultured striatal neurons as a function of DIV. Protein content per lane for these blots was identical. Film exposure times were also identical to demonstrate the magnitude of the difference in protein levels of mGluR1 versus mGluR5. B, Immunofluorescence using subtype-specific antibodies shows that both mGluR1 and mGluR5 are localized in somata and processes, with mGluR5 more abundant than mGluR1. C, Time course of ERK2 phosphorylation by DHPG. D, Concentration-response of ERK2 phosphorylation by DHPG. E, Pharmacological characterization indicates that mGluR5 is the predominant group I mGluR mediating ERK2 phosphorylation in striatal neurons. mGluR agonist DHPG (10 μm) or CHPG (750 μm) was applied for 2 min. Receptor antagonist LY367385, 4CPG, or MPEP (see concentrations in Results) was applied 30 min before stimulation with DHPG. F, Cells were stimulated with DHPG and double labeled using anti-phosphoERK with anti-NeuN to mark neurons. White arrows indicate cells positive for NeuN; blue arrows indicate cells negative for NeuN. DAPI staining indicates the location of all cells.
Addition of DHPG (10 μm) to the media produced a rapid increase in ERK1 and ERK2 phosphorylation in E18 striatal neurons, peaking within 5 min and declining toward basal levels by 30 min (Fig. 2C). We focused our analyses on ERK2, because both the basal level and magnitude of DHPG-induced change were greater for ERK2 than ERK1. ERK2 phosphorylation significantly increased with 10 μm DHPG; 30 μm DHPG did not yield a greater increase (Fig. 2D). Based on data in Figures 1B and 2D, the concentration we selected for DHPG treatments was 10 μm. We further characterized mGluR1/5-mediated ERK2 phosphorylation with selective agonists and antagonists (Fig. 2E). The mGluR5 selective agonist CHPG (750 μm) (Doherty et al., 1997) increased ERK2 phosphorylation to a greater extent than 10 μm DHPG. Pretreatment with the mGluR5 inverse agonist MPEP (20 μm; 30 min) blocked DHPG-stimulated increases in phosphoERK2. Neither mGluR1 antagonist LY367385 (10 μm) nor 4CPG (100 μm) inhibited ERK2 phosphorylation by DHPG. These data led us to conclude that similar to the calcium imaging experiments, mGluR5 plays a more substantial role than mGluR1 in activating ERK2. Double-label immunofluorescence was used to visualize pERK in cultured cells, comparing neurons with glia. Cells were stimulated with DHPG and stained with rabbit anti-pERK and mouse anti-NeuN (neuronal-specific antigen). 4′,6′-Diamidino-2-phenylindole (DAPI) was used to visualize cells that were negative for both pERK and NeuN. The results indicated that cells with the greatest increase in pERK after DHPG treatment were predominantly neuronal in nature (Fig. 2F).
Because we observed that activation of mGluR5 in striatal neurons gave rise to a robust increase in intracellular calcium, we explored a few avenues whereby calcium rise could lead to ERK phosphorylation. To test whether calcium/calmodulin-dependent protein kinase II (CaMKII) could indirectly alter ERK2 phosphorylation, we preincubated cells with the selective CaMKII inhibitor 2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)amino-N-(4-chlorocinnamyl)-N-methylbenzylamine (1 and 10 μm; data not shown) but saw no suppression of DHPG-mediated ERK2 phosphorylation. We also determined whether calcium influx through voltage-dependent calcium channels could enhance ERK2 phosphorylation in these cells (Rosen et al., 1994; Farnsworth et al., 1995). The L-type calcium channel blocker nifedipine (10 μm) altered neither basal nor DHPG-mediated ERK2 phosphorylation (data not shown). N-, P-, and Q-type calcium channel blockade with 2 μm ω-conotoxin MVIIC had no effect on pERK2. Finally, exposure of cells to both nifedipine and ω-conotoxin MVIIC altered neither basal nor DHPG-stimulated pERK2 levels (data not shown). Nevertheless, pretreatment of cells with BAPTA AM blocked increased phosphoERK2 in response to DHPG (DHPG, 275 ± 27%; BAPTA plus DHPG, 127 ± 5; n = 3; p < 0.05). mGluR1/5 activation of ERK2 does not appear to depend on Src, because the Src-selective inhibitor SU6566 (1 μm) did not inhibit DHPG-mediated ERK2 phosphorylation (n = 4; data not shown).
DR1/5 enhances ERK2 phosphorylation by metabotropic glutamate receptors
Because of the hypothesized importance of dopamine-glutamate interactions for motor control and associative learning processes in the striatum (Alexander et al., 1990; Knowlton et al., 1996), we compared signaling cascades initiated by mGluR5 (which are PLCβ-coupled receptors) and DR1/5 (adenylyl cyclase linked). Although molecular details are lacking, a number of published reports provide evidence suggesting that significant interactions occur between pathways activated by mGluR and dopamine receptors in the striatum (Berke et al., 1998; Paolillo et al., 1998; Calabresi et al., 1999; Hanania and Johnson, 1999). In primary cultures derived from E18 striatum, we found that stimulation of neurons with 10 μm DHPG together with 10 μm R(+)SKF38393 enhanced ERK2 phosphorylation (see Fig. 4A). This effect was also observed in experiments in which DHPG was coapplied with either 10 μm SKF81297 or 100 μm dopamine (data not shown). In preliminary experiments, various combinations of DHPG and SKF38393 in concentrations ranging from 1 to 10 μm were tested for their ability to enhance ERK2 phosphorylation. For the quantitative and statistical analyses presented here, data from all experiments using 10 μm DHPG and 10 μm SKF38393 were included, without regard to the degree of enhancement exhibited.
Figure 4.

Pharmacology of DR1/5- and mGluR1/5-mediated ERK2 phosphorylation. A, ERK2 phosphorylation by DHPG (D) is attenuated by the mGluR5 antagonist MPEP (M) (20 μm; 30 min; n = 6). B, The D1 receptor antagonist SCH23390 (SCH) (20 μm; 30 min; n = 5) diminishes ERK2 phosphorylation by SKF38393 (SKF). C, The PKA inhibitor H89 (H) (5 μm; 60 min; n = 6) blocks DR1/5 phosphorylation of ERK2 without suppressing ERK2 phosphorylation by DHPG. D, Inhibition of MEK with 1 μm U0126 (60 min) suppresses basal as well as DHPG- and SKF38393 (S)-mediated ERK2 phosphorylation. The asterisk indicates a significant increase over control (controls are 0 for treatment with agonist alone or inhibitor for treatment with agonist in the presence of inhibitor).
To determine whether transsynaptic events mediate enhancement, we repeated the stimulation in the presence of 0.5 μm TTX. Under these conditions, the enhancement was still observed when action potentials were blocked (Fig. 3B). To ensure that DR1/5-mGluR1/5 interactions were cell autonomous rather than transsynaptic, we conducted double-label immunofluorescence analysis on these neurons, staining for mGluR5 and DR1/5. At this developmental stage and under these culture conditions, DR1/5 appear to be expressed in approximately all neurons, as revealed by immunofluorescence using two different D1R antibodies (rat monoclonal shown here; similar results were obtained using rabbit polyclonal antibody; Research Diagnostics, Flanders, NJ). Results of staining using rat anti-D1R and rabbit anti-mGluR5 demonstrated that DR1/5 and mGluR5 are coexpressed in a majority of neurons in these cultures (Fig. 3C).
Figure 3.
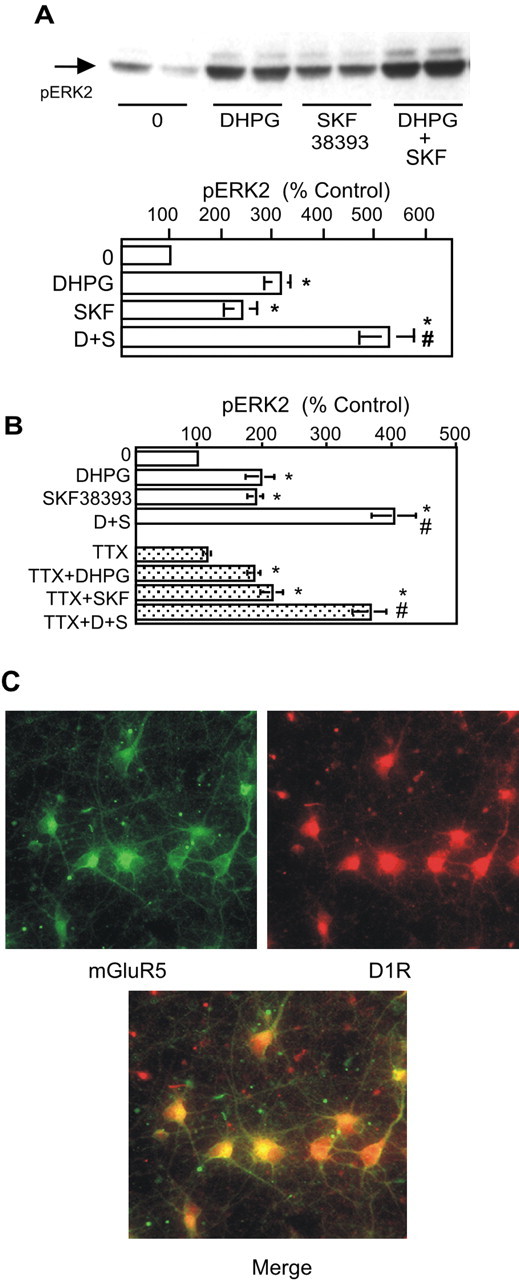
Coapplication of DR1/5 and mGluR1/5 agonists enhances phosphorylation of ERK2. A, The DR1/5 agonist SKF38393 (SKF) (10 μm; 2 min) significantly augments DHPG-mediated (10 μm; 2 min) phosphorylation of ERK2 (n = 16). The asterisk indicates a significant increase over control; # indicates significant difference from DHPG (D) or SKF38393 (S) treatment alone. B, DR1/5-mGluR1/5 interaction is cell autonomous, as demonstrated by the absence of an inhibitory effect in the presence of TTX. C, Double-label immunofluorescence reveals a high degree of coexpression of DR1/5 and mGluR5.
We quantified the contribution of mGluR5 to increased ERK2 phosphorylation by DHPG (Fig. 4A). Although MPEP pretreatment elevated basal pERK2, addition of DHPG with MPEP did not significantly increase ERK2 phosphorylation over MPEP alone. Preincubation of cells with the D1 receptor antagonist SCH23390 (20 μm) blocked SKF38393 increases in pERK2, confirming the role of DR1/5 in this effect (Fig. 4B).
Because DR1/5 are known to activate PKA, a role for PKA in agonist-mediated pERK2 elevation was examined. Basal levels of pERK2 were not altered when cells were incubated for 60 min with the PKA inhibitor H89 (5 μm; PKA-selective concentration) (Chijiwa et al., 1990). H89 did not block DHPG-evoked ERK2 phosphorylation but did prevent SKF38393 induction of pERK2 (Fig. 4C).
Activation of either Ras or Raf kinase is a prerequisite for stimulation of the protein kinase MEK, which phosphorylates ERK1 and ERK2. To confirm that the DR1/5-mGluR1/5 pathway convergence involved MEK, we pretreated cells with the MEK inhibitor U0126 at a concentration documented to selectively target this kinase (Duncia et al., 1998; Stanciu et al., 2000). At this concentration (1 μm), ERK2 phosphorylation was dramatically reduced compared with control (Fig. 4D). In the presence of U0126, receptor activation could not increase pERK2 levels over control. This confirmed that MEK is required for activation of ERK2 by mGluR1/5 and DR1/5 agonists. Similar results were observed when 50 μm 2′-amino-3′-methyoxyflavone was used to block MEK (data not shown).
The observation that the MEK inhibitor U0126 interfered with the ability of SKF38393 to increase ERK2 phosphorylation suggested that a potential point of interaction between the two signaling pathways occurred upstream of MEK. Based on published reports describing the ability of PKA to activate the MEK pathway in other cell types (Yarwood et al., 1996; Busca et al., 2000), we hypothesized that activation of a PKA pathway alone could increase ERK2 phosphorylation. Hence, we sought to define the concentration dependence of U0126 on signaling specifically through DR1/5 and generally through cAMP-dependent pathways. Cultures of striatal neurons were preincubated with U0126 ranging from 50 nm to 1 μm and followed by either SKF38393 or forskolin. ERK2 phosphorylation by forskolin was significantly reduced with 1 μm U0126 (Fig. 5A), whereas 0.1 μm U0126 blocked phosphorylation by SKF38393 (Fig. 5B). The data demonstrate that in striatal neurons, ERK phosphorylation is modulated in a significant and direct manner by DR1/5 activation.
Figure 5.
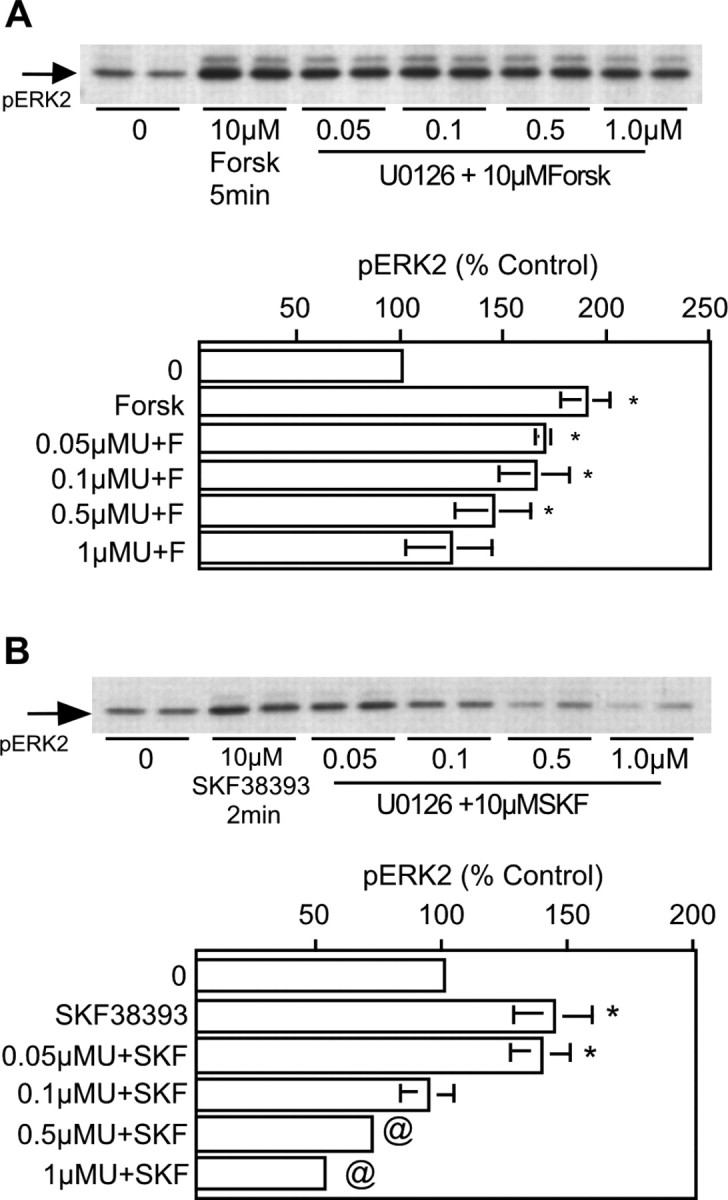
cAMP-mediated increases in ERK2 phosphorylation in striatal neurons are MEK dependent. A, Forskolin (Forsk) elicits a robust increase in ERK2 phosphorylation that is resistant to U0126 concentrations <1 μm (n = 3 for all conditions). B, ERK2 phosphorylation induced by SKF38393 (SKF) is radically reduced and driven to subbasal levels with 0.1 μm U0126 (n = 3 for all conditions). Cells were treated with U0126 for 60 min before agonist application. Asterisks indicate a significant increase over control (0); @ indicates a significant decrease compared with control.
DR1/5 enhancement of mGluR1/5 activation of ERK2 is PKC dependent
Along with generation of IP3 and increase in intracellular calcium concentration, DHPG stimulation of the PLC pathway leads to PKC activation. We therefore investigated the role of PKC in DHPG-mediated ERK2 stimulation and DR1/5-mGluR1/5 enhancement. For this analysis, we used three different PKC inhibitors. Calphostin C inhibits PKC by competing with DAG binding sites (Kobayashi et al., 1989). GF109203X is a potent inhibitor of PKCα and PKCβ1. Gö6976 selectively inhibits PKCα and PKCβ1 while having no effect on the atypical (λ and ζ) and novel (∂, ϵ, η, μ, and θ) PKC isozymes at micromolar concentrations (Martiny-Baron et al., 1993). Inhibitors were added 30-60 min before agonist application. Of the three inhibitors, only Gö6976 (1 μm) altered basal levels of pERK2 (data not shown); neither calphostin C (100 nm) nor GF109203X (500 nm) affected basal activity of ERK2 (Fig. 6A,B). None of the three inhibitors blocked the ability of DHPG to increase ERK2 phosphorylation. However, all three inhibitors prevented the enhanced activation of ERK2 by DR1/5-mGluR1/5, despite the slightly different profile of selectivity of these three commonly used inhibitors (Fig. 6A,B). These data provide the strongest evidence for the enhancement as a cell-autonomous event.
Figure 6.
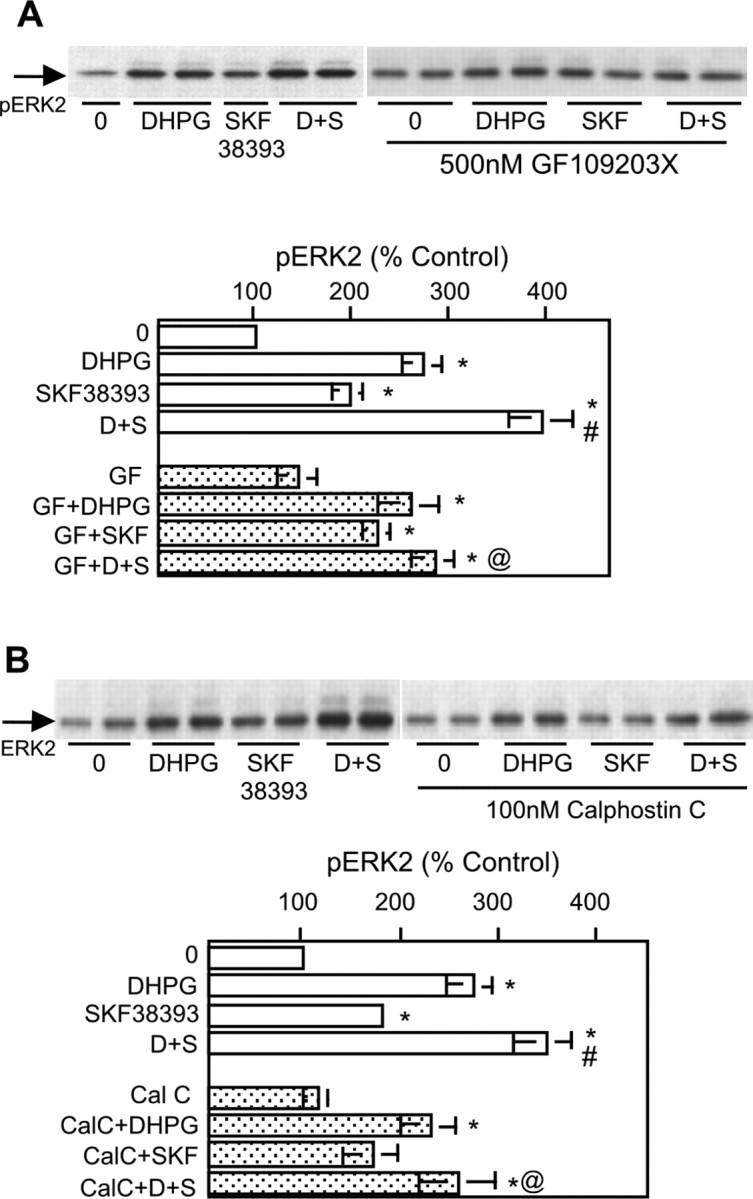
Protein kinase C mediates mGluR5-D1 receptor pathway interactions. The protein kinase C inhibitors GF109203X (GF) (A) (500 nm; n = 7) and calphostin C (CalC) (B) (100 nm; n = 5) block the DR1/5-enhanced activation of ERK2 by mGluR without affecting DHPG (D)-mediated ERK2 phosphorylation. Each inhibitor was applied 60 min before treatment with agonist. Graphical summary of results is shown below each panel. Asterisks indicate a significant increase over control (controls are 0 for treatment with agonist alone or inhibitor for treatment with agonist in the presence of inhibitor); # indicates significant difference from DHPG or SKF38393 (SKF; S) treatment alone; @ indicates significant difference from DHPG plus SKF38393 coapplication.
Protein phosphatases regulate mGluR1/5 signaling
In striatal medium spiny neurons, the dopamine and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32) is a substrate for PKA (Ouimet et al., 1984; Walaas and Greengard, 1984; Greengard et al., 1999). The culture conditions we used (serum-free Neurobasal medium supplemented with brain-derived neurotrophic factor) support DARPP32 expression (data not shown) (Ivkovic and Ehrlich, 1999). When phosphorylated by PKA, DARPP32 becomes an inhibitor of protein phosphatase PP1. We tested the hypothesis that PP1 inhibition by DR1/5 is a component of the pathway leading to DR1/5-mGluR1/5 enhancement. For this portion of the analysis, we used the phosphatase inhibitors okadaic acid and calyculin A. One limitation posed in using these agents is that they inhibit both PP1 and PP2A when used at nanomolar to low micromolar concentrations (Ishihara et al., 1989; Girault, 1993; Honkanan, 1994). We observed that these phosphatases play a significant role in the modulation of ERK2 phosphorylation (Fig. 7). Both calyculin A and okadaic acid elevated basal pERK2 but did not occlude DHPG- or SKF-mediated increases in pERK2 (data not shown). Both calyculin A and okadaic acid significantly enhanced ERK2 phosphorylation by DHPG. Okadaic acid treatment did occlude the DHPG-SKF interaction (5 nm OA, DHPG-SKF, 453 ± 129; DHPG plus OA, 382 ± 61; OA plus DHPG plus SKF, 502 ± 172; p > 0.05; n = 4). Calyculin A treatment yielded similar results (data not shown).
Figure 7.

Inhibition of protein phosphatases PP1 and PP2A before stimulation with DHPG enhances mGluR5-mediated ERK2 phosphorylation. Okadaic acid (A) or calyculin A (CalA) (B) pretreatment enhances ERK2 phosphorylation by DHPG (D) (10 μm; n = 5). Asterisks indicate a significant increase over control; # indicates a significant difference from DHPG treatment alone.
DR1/5-mGluR1/5 coactivation enhances CREB phosphorylation but not CREB-mediated transcription
One downstream event modulated by ERK2 kinase activity is phosphorylation of CREB. A minimal requirement for increasing CRE-mediated transcriptional activity via CREB is phosphorylation on Ser133. CREB phosphorylation was induced in striatal neurons with DHPG, SKF38393, or both and quantitated using an anti-phosphoCREB antibody that recognizes phosphorylated Ser133 (Fig. 8A). Treatment of neurons with either DHPG or SKF38393 elicited significant increases in phosphoCREB. The combined treatment of SKF with DHPG also increased CREB phosphorylation significantly over that seen with either DHPG or SKF alone. In summary, the DR1/5 enhancement of mGluR5 phosphorylation of ERK2 was apparently translated directly into similar changes in the magnitude of CREB phosphorylation.
Figure 8.

CREB phosphorylation, but not CREB-mediated transcriptional activity, demonstrates mGluR5/DR1/5 enhancement. A, mGluR5 and DR1/5 agonists each increase CREB phosphorylation; coactivation of both receptors significantly increases phosphoCREB levels beyond that for each agonist alone [10 μm DHPG (D); 10 μm SKF38393 (S); n = 11]. B, CREB-mediated transcriptional activity induced with DHPG, SKF38393, or forskolin was measured using the GAL-4/luciferase reporter system. Cells transfected with pFC2-dbd [dbd(-)CREB, no fusion with CREB] and pFR-Luc served as negative controls. All others were transfected with pFA2-CREB and pFR2-Luc and were either unstimulated [dbd(-) CREB and 0] or stimulated as indicated (n = 4). Asterisks indicates a significant increase over control (0); # indicates a significant difference from DHPG or SKF38393 treatment alone.
We also sought to determine whether activation of CREB by DHPG, SKF38393, or coapplied DHPG and SKF38393 would increase CREB-mediated transcriptional activity. To conduct these experiments, striatal neurons were transfected with two plasmids. One contained the GAL4 DNA-binding domain fused to CREB (pFA2-CREB), whereas the other possessed the GAL4 upstream activation sequence (UAS) fused to the luciferase gene (pFR-Luc). Induction of luciferase activity in transfected neurons therefore depended on the following: (1) successful cotransfection of both vectors and (2) a biological stimulus phosphorylating and activating CREB fused to the GAL4 DNA-binding domain. Under these circumstances, GAL4-phosphoCREB dimerizes and binds to the GAL4 UAS, transactivating luciferase expression. Control cells transfected with pFC2-dbd (negative control; construct lacking CREB) and pFR-Luc showed a decrease in basal luciferase activity compared with cells transfected with pFA2-CREB and pFR-Luc. This was reflective of basal levels of neuronal activity normally observed in these cells, which leads to CREB phosphorylation. Cells transfected with pFA2-CREB and pFR-Luc and stimulated with DHPG or SKF38393 alone increased CREB-mediated transcriptional activity to similar extents. In contrast to ERK2 and CREB phosphorylation, no enhancement of transcriptional activity was noted when both agonists were applied in combination. The possibility of a ceiling effect with regard to the ability of DHPG and SKF to activate CREB was examined in parallel samples treated with forskolin. Forskolin elevated CREB-mediated transcriptional activity twofold greater than that observed with any combination of DR1/5 or mGluR5 agonist (Fig. 8B). Therefore, the mGluR5-DR1/5 enhancement observed with ERK2 and CREB phosphorylation was not apparent at the level of CREB-mediated transcriptional activity.
Finally, by using results presented here in combination with data from related studies, we constructed a hypothetical model outlining the sequence of events after mGluR1/5 and DR1/5 activation (Fig. 9).
Figure 9.

Hypothetical model of elements involved in ERK2 phosphorylation by DR1/5 and mGluR5 activation in striatal neurons. Our data are consistent with the idea that G-protein-coupled phospholipase C activity increases intracellular calcium and elevates ERK2 phosphorylation by mGluR5. The increase in calcium triggers the Ras pathway and increases PKC activity required for DR1/5-mGluR5 pathway interactions. In parallel, DR1/5 activation may increase ERK2 phosphorylation via PKA-mediated increases in Rap 1 and B-Raf activity, also leading to MEK activation. The phosphatases PP1 and PP2A target MAPK for dephosphorylation.
Discussion
Striatal function depends on the integration of sensorimotor information from the cerebral cortex carried by glutamatergic neurons with information about the motivational state of the organism carried by dopamine neurons (Sacaan et al., 1992; Brown et al., 1999). At the cellular level, the concept of segregated signaling cascades extending from receptor to effector has given way to the recognition that interaction among biochemical pathways is a critical and highly regulated aspect of cellular function. Many examples have been documented in which signaling through one type of receptor can be modified by coactivation of a distinct type of receptor (Cepeda et al., 1993; Colwell and Levine, 1994; Pisani et al., 1997; Lezcano et al., 2000; Ciruela et al., 2001). The timing and pattern of receptor activation is very important, because it determines the overall excitability of a neuron. Integration of signals is reflected in patterns of neuronal firing, in modulation of phosphorylation of effector proteins, and in the regulation of gene expression. A complete understanding of the interactions between glutamate and dopamine receptor-mediated signaling events is a critical step in understanding striatal function, including the role of the striatum in implicit learning and memory.
mGluR5 is abundant and functional in striatal E18 neurons
Metabotropic receptors are expressed on striatal neurons (Testa et al., 1994; Ghasemzadeh et al., 1996); mGluR1 and mGluR5 are found on somata, dendritic shafts, and dendritic spines of DR1/5-expressing neurons. We used primary striatal cultures to minimize circuit-based interactions that might confound our analysis. Because the properties of some receptors can change dramatically under different culture conditions (Mao et al., 2004), we first established that mGluR1/5 activation was coupled to calcium mobilization in our cultures. Calcium concentrations increased throughout the cell in response to DHPG and exhibited oscillatory behavior typical of mGluR5 (Flint et al., 1999) (data not shown).
MGluR5-mediated signaling to the nucleus has not been explored extensively in striatal neurons. MAPK pathways are pivotal in carrying information from membrane to nucleus, resulting in extracellular signal-regulated gene expression. We therefore studied the effect of mGluR5 on ERK2 phosphorylation because of the likely significance of ERK2 in regulation of transcription factors (Sweatt, 2001; Yang et al., 2003). The availability of highly specific antibodies to phosphorylated ERK1/2 facilitated this analysis. Ras pathway activation leading to increased ERK2 phosphorylation has been characterized as a cellular event resulting from mGluR5 activation (Fiore et al., 1993; Ferraguti et al., 1999). We observed rapid increases in pERK2 with low concentrations of DHPG mediated primarily by mGluR5, the contribution from mGluR1 being much less apparent.
DR1/5 enhance mGluR5-mediated ERK2 phosphorylation
Roberson et al. (1999) and Watabe et al. (2000) reported the occurrence of interactions between PLC- and cAMP-mediated signal transduction pathways in hippocampal pyramidal cells that cooperated to increase ERK2 phosphorylation. Like glutamatergic hippocampal neurons, the GABAergic striatal neurons that we investigated demonstrated consistent, significant augmentation of pERK2 levels when mGluR1/5 and DR1/5 were stimulated simultaneously. Indeed, the finding that sensitization to cocaine is absent in mice lacking mGluR5 provides some of the strongest evidence to date that a critical interdependence exists between dopamine receptors and mGluR5 (Chiamulera et al., 2001). To establish an anatomic basis for the biochemical interactions between DR1/5 and mGluR5 pathways, we probed for DR1/5 and mGluR5 immunoreactivity in our cultures and found a high degree of coexpression of the two (Fig. 3C). Pharmacological analyses demonstrated that this interaction was dependent on PKA and MEK. Our data show striking, concentration-dependent inhibition of cAMP-mediated ERK2 phosphorylation with MEK inhibition.
There are at least four mechanisms by which DR1/5 and mGluR signals could interact. At the level of the cell membrane, DR1/5 can facilitate intracellular calcium release if the neuron is first “primed” by activation of a PLC-linked receptor, promoting DR1/5 binding to a protein termed calcyon (Lezcano et al., 2000). Calcyon is expressed in E18 striatal cultures (data not shown). At the level of cAMP generation, Paolillo et al. (1998) documented group 1 mGluR-mediated potentiation of dopamine-induced cAMP formation in striatal neurons. Subsequently, Hoffmann et al. (1999) found that ERK2 inhibited the cAMP phosphodiesterase phosphodiesterase 4D. The predominant striatal isoform PDE4B (Cherry and Davis, 1999) can also be phosphorylated by ERK (Lenhard et al., 1996). A third mechanism involves the Ras family member Rap1. In B-Raf-expressing cells, PKA activation of Rap1 leads to Rap1/B-Raf complex formation that can trigger the MEK-ERK signaling cascade (Vossler et al., 1997; Schmitt and Stork, 2000). Recent data support the idea that, in neurons, depolarization increases intracellular calcium levels, leading to activation of Rap1 and B-Raf in a PKA-dependent manner (Grewal et al., 2000; Morozov et al., 2003). We hypothesize that in striatal neurons, mGluR5-mediated calcium release can also activate B-Raf in a similar manner. Finally, PKA can phosphorylate IP3Rs and enhance their function (Ferris et al., 1991a,b). Because mGluR5 elicits calcium release via IP3 receptors, DR1/5-mGluR5 interactions may well involve PKA facilitation of IP3R function.
Although DR1/5 s and mGluR5 s acting in concert can substantially increase ERK2 phosphorylation, this enhanced effect does not recruit all possible pools of ERK2. Treatment of our neurons with low nanomolar phorbol 12-myristate 13-acetate (PMA) gave rise to even greater amounts of pERK2 than was achievable with DR1/5 and mGluR5 agonists (data not shown). Similarly, forskolin increased ERK2 more than SKF38393, DHPG or both together.
Interaction between signaling pathways is PKC dependent
The signaling pathway activated by mGluR5 includes PLCβ-mediated increases in the second messengers DAG and IP3. Our data demonstrate that activation of PKC is not required for ERK2 phosphorylation by DHPG or by SKF38393. The effects of DR1/5 and D2R stimulation differ in this regard, because D2R stimulation of pERK2 via PLC has been reported to be PKC dependent (Yan et al., 1999). Nevertheless, DR1/5-mGluR5 pathway interaction clearly depends on PKC. Although the predominant signaling pathway activated in striatal neurons by SKF38393 involves PKA (shown here in H89-mediated suppression of pERK2), DR1/5 activation of PKC has been described previously (Simpson and Morris, 1995; Gorelova and Yang, 2000) and could contribute to the observed interactions between DR1/5 and mGluR5. Overall, our data are consistent with the hypothesis that there is a shared substrate for PKA (DR1/5 activated) and PKC (mGluR5 activated) that modulates ERK2 phosphorylation.
Signaling contributions from phosphatases
The phosphorylation state of a protein reflects the balance of kinase and phosphatase activity. DR1/5 activation in striatal neurons inhibits PP1 via DARPP-32, and PP2A directly dephosphorylates ERK1 and ERK2. As expected, inhibition of PP1 and PP2A increased mGluR5-mediated ERK2 phosphorylation (Braconi Quintaje et al., 1996; Keyse, 2000). This is consistent with a DR1/5-mediated decrease in PP1 activity that couples with DHPG actions to result in enhanced phosphoERK2. Lacking a fully selective inhibitor of PP1, interpretation of these data is limited by the effects of calyculin A and okadaic acid on PP2A. Identification of the mechanisms by which mGluR5 function is influenced by PP1 should provide important insights into signal integration within striatal neurons. An interesting observation by Yan et al. (1999) was that mice lacking DARPP-32 show a pronounced increase in basal MAPK (ERK1/2) phosphorylation, arising in part from disinhibition of PP1 by DARPP-32. Because PP1 has also been shown to physically interact with mGluR1/5, a suppression of PP1 by DARPP-32 activity could potentially enhance mGluR1/5 signaling (Croci et al., 2003). Future endeavors will be directed toward understanding the links between mGluR5, PP1, and ERK2, because only PP2A has been ascribed a role in regulating MAPK by dephosphorylation. Mao et al. (2005) have detailed PP2A activity in mGluR5 signaling via MEK/ERK.
Degree of CREB phosphorylation does not reflect the extent of CREB-mediated transcriptional activity
Neurotransmitter-induced changes in expression of specific transcripts are believed to contribute to the mechanism underlying synaptic plasticity. Activation of ERK1 and ERK2 can lead to phosphorylation of the transcription factor CREB via RSK2 (CREB kinase) (Xing et al., 1996); CREB phosphorylation is a requisite first step in the initiation of CRE-mediated transcriptional activity. CREB phosphorylation on Ser133 is mediated by a number of different kinases in response to diverse extracellular stimuli (for review, see Shaywitz and Greenberg, 1999). The regulation of transcription by CREB is complex, involving numerous kinases and proteins that stabilize CREB binding to DNA (such as the CREB-binding protein CBP and RNA helicase A). The biological relevance of CREB phosphorylation by multiple kinases is not well understood. We observed that amplification of signaling reflected by enhanced pERK2 and phosphoCREB after mGluR5 and DR1/5 activation was not mirrored by similar changes in the amount of CREB-mediated transcriptional activity. The failure of these neurons to demonstrate enhancement at the transcriptional level was not reflective of a ceiling effect, because in the same experiments, forskolin treatment increased luciferase activity to greater extents than agonist treatments. Rather, these data lend strong support to the idea that multiple checkpoints for regulation of transcription exist.
In conclusion, we have presented data supporting a very robust interaction between mGluR1/5 and D1R1/5. Although we have not yet discovered the precise components of this interaction, additional analysis of substrates for phosphorylation by PKC should reveal candidates for cross-talk between these signal transduction cascades.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke Intramural Research Program. We thank Drs. Hal Gainer, Leonard Lothstein, and Carol Torgan and members of the Molecular Plasticity Section for instructive discussions and critical reading of this manuscript. Dr. John Sarvey is remembered for his valuable contributions.
Correspondence should be addressed to Pamela J. Voulalas, Department of Physiology, University of Maryland School of Medicine, 655 West Baltimore Street, Baltimore, MD 21201. E-mail: pvoul001@umaryland.edu.
S. E. Hyman's present address: Office of the Provost, Harvard University, Cambridge, MA 02138.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253763-11$15.00/0
References
- Alexander GE, Crutcher MD, DeLong MR (1990) Basal ganglia-thalamocortical circuits: parallel substrates for motor, oculomotor, “prefrontal” and “limbic” functions. Prog Brain Res 85: 119-146. [PubMed] [Google Scholar]
- Berke JD, Paletzki RF, Aronson GJ, Hyman SE, Gerfen CR (1998) A complex program of striatal gene expression induced by dopaminergic stimulation. J Neurosci 18: 5301-5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braconi Quintaje SB, Church DJ, Rebsamen M, Valloton MB, Hemmings BA, Lang U (1996) Role of protein phosphatase 2A in the regulation of mitogen-activated protein kinase activity in ventricular cardiomyocytes. Biochem Biophys Res Commun 221: 539-547. [DOI] [PubMed] [Google Scholar]
- Brown J, Bullock D, Grossberg S (1999) How the basal ganglia use parallel excitatory and inhibitory learning pathways to selectively respond to unexpected rewarding cues. J Neurosci 19: 10502-10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busca R, Abbe P, Mantoux F, Aberdam E, Peyssonnaux C, Eychene A, Ortonne JP, Ballotti R (2000) Ras mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J 19: 2900-2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Marfia GA, Bernardi G (1999) Glutamate-triggered events inducing corticostriatal long-term depression. J Neurosci 19: 6102-6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Buchwald NA, Levine MS (1993) Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci USA 90: 9576-9580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JA, Davis RL (1999) Cyclic AMP phosphodiesterases are localized in regions of the mouse brain associated with reinforcement, movement, and affect. J Comp Neurol 407: 287-301. [PubMed] [Google Scholar]
- Chiamulera C, Epping-Jordan MP, Zocchi A, Marcon C, Cottiny C, Tacconi S, Corsi M, Orzi F, Conquet F (2001) Reinforcing and locomotor stimulant effects of cocaine are absent in mGluR5 null mutant mice. Nat Neurosci 4: 873-874. [DOI] [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H (1990) Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinoline-sulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem 265: 5267-5272. [PubMed] [Google Scholar]
- Ciruela F, Escriche M, Burgueno J, Angulo E, Casado V, Soloviev MM, Canela EI, Mallol J, Chan WY, Lluis C, McIlhinney RA, Franco R (2001) Metabotropic glutamate 1alpha and adenosine A1 receptors assemble into functionally interacting complexes. J Biol Chem 276: 18345-18351. [DOI] [PubMed] [Google Scholar]
- Colwell CS, Levine MS (1994) Metabotropic glutamate receptors modulate N-methyl-d-aspartate receptor function in neostriatal neurons. Neuroscience 61: 497-507. [DOI] [PubMed] [Google Scholar]
- Corvol JC, Studler JM, Schonn JS, Girault JA, Herve D (2001) Galpha(olf) is necessary for coupling D1 and A2a receptors to adenylyl cyclase in the striatum. J Neurochem 76: 1585-1588. [DOI] [PubMed] [Google Scholar]
- Croci C, Sticht H, Brandstatter JH, Enz R (2003) Group I metabotropic glutamate receptors bind to protein phosphatase 1C. Mapping and modeling of interacting sequences. J Biol Chem 278: 50682-50690. [DOI] [PubMed] [Google Scholar]
- Doherty AJ, Palmer MJ, Henley JM, Collingridge GL, Jane DE (1997) (RS)-2-chloro-5-hydroxyphenylglycine (CHPG) activates mGlu5, but no mGlu1, receptors expressed in CHO cells and potentiates NMDA responses in the hippocampus. Neuropharmacology 36: 265-267. [DOI] [PubMed] [Google Scholar]
- Duncia JV, Santella III JB, Higley CA, Pitts WJ, Wityak J, Frietze WE, Rankin FW, Sun JH, Earl RA, Tabaka AC, Teleha CA, Blom KF, Favata MF, Manos EJ, Daulerio AJ, Stradley DA, Horiuchi K, Copeland RA, Scherle PA, Trzaskos JM, et al. (1998) MEK inhibitors: the chemistry and biological activity of U0126, its analogs, and cyclization products. Bioorg Med Chem Lett 8: 2839-2844. [DOI] [PubMed] [Google Scholar]
- Farnsworth CL, Freshney NW, Rosen LB, Ghosh A, Greenberg ME, Feig LA (1995) Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature 376: 524-527. [DOI] [PubMed] [Google Scholar]
- Ferraguti F, Baldani-Guerra B, Corsi M, Nakanishi S, Corti C (1999) Activation of the extracellular signal-regulated kinase 2 by metabotropic glutamate receptors. Eur J Neurosci 11: 2073-2082. [DOI] [PubMed] [Google Scholar]
- Ferris CD, Huganir RL, Bredt DS, Cameron AM, Snyder SH (1991a) Inositol trisphosphate receptor: phosphorylation by protein kinase C and calcium calmodulin-dependent protein kinases in reconstituted lipid vesicles. Proc Natl Acad Sci USA 88: 2232-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris CD, Cameron AM, Bredt DS, Huganir RL, Snyder SH (1991b) Inositol 1,4,5-trisphosphate receptor is phosphorylated by cyclic AMP-dependent protein kinase at serines 1755 and 1589. Biochem Biophys Res Commun 175: 192-198. [DOI] [PubMed] [Google Scholar]
- Fiore RS, Murphy TH, Sanghera JS, Pelech SL, Baraban JM (1993) Activation of p42 mitogen-activated protein kinase by glutamate receptor stimulation in rat primary cortical cultures. J Neurochem 61: 1626-1633. [DOI] [PubMed] [Google Scholar]
- Flint AC, Dammerman RS, Kriegstein AR (1999) Endogenous activation of metabotropic glutamate receptors in neocortical development causes neuronal calcium oscillations. Proc Natl Acad Sci USA 96: 12144-12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemzadeh MB, Sharma S, Surmeier DJ, Eberwine JH, Chesselet MF (1996) Multiplicity of glutamate receptor subunits in single striatal neurons: an RNA amplification study. Mol Pharmacol 49: 852-859. [PubMed] [Google Scholar]
- Girault JA (1993) Protein phosphorylation and dephosphorylation in mammalian central nervous system. Neurochem Int 23: 1-25. [DOI] [PubMed] [Google Scholar]
- Gorelova NA, Yang CR (2000) Dopamine D1/D5 receptor activation modulates a persistent sodium current in rat prefrontal cortical neurons in vitro. J Neurophysiol 84: 75-87. [DOI] [PubMed] [Google Scholar]
- Greengard P, Allen PB, Nairn AC (1999) Beyond the dopamine receptor: the DARPP-32/protein phosphatase-1 cascade. Neuron 23: 435-447. [DOI] [PubMed] [Google Scholar]
- Grewal SS, Horgan AM, York RD, Withers GS, Banker GA, Stork PJ (2000) Neuronal calcium activates a Rap1 and B-Raf signaling pathway via the cyclic adenosine monophosphate-dependent protein kinase. J Biol Chem 275: 3722-3728. [DOI] [PubMed] [Google Scholar]
- Hamilton SE, Nathanson NM (2001) The M1 receptor is required for muscarinic activation of MAP kinase in murine cerebral cortical neurons. J Biol Chem 276: 15850-15853. [DOI] [PubMed] [Google Scholar]
- Hanania T, Johnson KM (1999) Regulation of NMDA-stimulated [14C]GABA and [3H]acetylcholine release by striatal glutamate and dopamine receptors. Brain Res 844: 106-117. [DOI] [PubMed] [Google Scholar]
- Hoffmann R, Baillie GS, MacKenzie SJ, Yarwood SJ, Houslay MD (1999) The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. EMBO J 18: 893-903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkanan RE, Codispoti BA, Tse K, Boynton AL (1994) Characterization of natural toxins with inhibitory activity against serine/threonine protein phosphatases. Toxicon 32: 339-350. [DOI] [PubMed] [Google Scholar]
- Ishihara H, Martin BL, Brautigan DL, Karaki H, Ozaki H, Kato Y, Fusetani N, Watabe S, Hashimoto K, Uemura D, Hartshorne DJ (1989) Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem Biophys Res Commun 159: 871-877. [DOI] [PubMed] [Google Scholar]
- Ivkovic S, Ehrlich ME (1999) Expression of the striatal DARPP-32/ARPP-21 phenotype in GABAergic neurons requires neurotrophins in vivo and in vitro. J Neurosci 19: 5409-5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp JM, Powell TP (1971) The synaptic organization of the caudate nucleus. Philos Trans R Soc Lond B Biol Sci 262: 403-404. [DOI] [PubMed] [Google Scholar]
- Keyse S (2000) Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol 12: 186-192. [DOI] [PubMed] [Google Scholar]
- Knowlton BJ, Mangels JA, Squire LR (1996) A neostriatal habit learning system in humans. Science 273: 1399-1402. [DOI] [PubMed] [Google Scholar]
- Kobayashi E, Ando K, Nakano H, Iida T, Ohno H, Morimoto M, Tamaoki T (1989) Calphostins (UCN-1028), novel and specific inhibitors of protein kinase C. I. Fermentation, isolation, physico-chemical properties and biological activities. J Antibiot (Tokyo) 42: 1470-1474. [DOI] [PubMed] [Google Scholar]
- Lenhard JM, Kassel DB, Rocque WJ, Hamacher L, Holmes WD, Patel I, Hoffman C, Luther M (1996) Phosphorylation of a cAMP-specific phosphodiesterase (HSPDE4B2B) by mitogen-activated protein kinase. Biochem J 316: 751-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lezcano N, Mrzljak L, Eubanks S, Levenson R, Goldman-Rakic P, Bergson C (2000) Dual signaling regulated by calcyon, a D1 dopamine receptor interacting protein. Science 287: 1660-1664. [DOI] [PubMed] [Google Scholar]
- Mao L, Tang Q, Samdani S, Liu Z, Wang JQ (2004) Regulation of MAPK/ERK phosphorylation via ionotropic glutamate receptors in cultured rat striatal neurons. Eur J Neurosci 19: 1207-1216. [DOI] [PubMed] [Google Scholar]
- Mao L, Yang L, Arora A, Choe ES, Zhang G, Liu Z, Fibuch EE, Wang JQ (2005) Role of protein phosphatase 2A in mGluR5-regulated MEK/ERK phosphorylation in neurons. J Biol Chem, in press. [DOI] [PubMed]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C (1993) Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J Biol Chem 268: 9194-9197. [PubMed] [Google Scholar]
- Morozov A, Muzzio IA, Bourtchouladze R, Van-Strien N, Lapidus K, Yin D, Winder DG, Adams JP, Sweatt JD, Kandel ER (2003) Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron 39: 309-325. [DOI] [PubMed] [Google Scholar]
- Ouimet CC, Miller PE, Hemmings Jr HC, Walaas SI, Greengard P (1984) DARPP-32, a dopamine- and adenosine 3′:5′-monophosphate-regulated phosphoprotein enriched in dopamine-innervated brain regions. III. Immunocytochemical localization. J Neurosci 4: 114-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolillo M, Montecucco A, Zanassi P, Schinelli S (1998) Potentiation of dopamine-induced cAMP formation by group I metabotropic glutamate receptors via protein kinase C in cultured striatal neurons. Eur J Neurosci 10: 1937-1945. [DOI] [PubMed] [Google Scholar]
- Pisani A, Calabresi P, Centonze D, Bernardi G (1997) Enhancement of NMDA responses by group I metabotropic glutamate receptor activation in striatal neurones. Br J Pharmacol 120: 1007-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price CJ, Kim P, Raymond LA (1999) D1 dopamine receptor-induced cyclic AMP-dependent protein kinase phosphorylation and potentiation of striatal glutamate receptors. J Neurochem 73: 2441-2446. [DOI] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD (1999) The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci 19: 4337-4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen LB, Ginty DD, Weber MJ, Greenberg ME (1994) Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron 12: 1207-1221. [DOI] [PubMed] [Google Scholar]
- Sacaan AI, Bymaster FP, Schoepp DD (1992) Metabotropic glutamate receptor activation produces extrapyramidal motor system activation that is mediated by striatal dopamine. J Neurochem 59: 245-251. [DOI] [PubMed] [Google Scholar]
- Schmitt JM, Stork PJ (2000) Beta 2-Adrenergic receptor activates extracellular signal-regulated kinases (ERKs) via the small G protein rap1 and the serine/threonine kinase B-Raf. J Biol Chem 275: 25342-25350. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68: 821-861. [DOI] [PubMed] [Google Scholar]
- Simpson CS, Morris BJ (1995) Induction of c-fos and zif/268 gene expression in rat striatal neurons, following stimulation of D1-like dopamine receptors, involves protein kinase A and protein kinase C. Neuroscience 68: 97-106. [DOI] [PubMed] [Google Scholar]
- Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW, DeFranco DB (2000) Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem 275: 12200-12206. [DOI] [PubMed] [Google Scholar]
- Sweatt JD (2001) The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem 76: 1-10. [DOI] [PubMed] [Google Scholar]
- Testa CM, Standaert DG, Young AB, Penney Jr JB (1994) Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci 14: 3005-3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toms NJ, Roberts PJ (1999) Group 1 mGlu receptors elevate [Ca2+]i in rat cultured cortical type 2 astrocytes: [Ca2+]i synergy with adenosine A1 receptors. Neuropharmacology 38: 1511-1517. [DOI] [PubMed] [Google Scholar]
- Vincent SR, Sebben M, Dumuis A, Bockaert J (1998) Neurotransmitter regulation of MAP kinase signaling in striatal neurons in primary culture. Synapse 29: 29-36. [DOI] [PubMed] [Google Scholar]
- Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ (1997) cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell 89: 73-82. [DOI] [PubMed] [Google Scholar]
- Walaas SI, Greengard P (1984) DARPP-32, a dopamine- and adenosine 3′: 5′-monophosphate-regulated phosphoprotein enriched in dopamine-innervated brain regions. I. Regional and cellular distribution in the rat brain. J Neurosci 4: 84-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe AM, Zaki PA, O'Dell TJ (2000) Coactivation of β-adrenergic and cholinergic receptors enhances the induction of long-term potentiation and synergistically activates mitogen-activated protein kinase in the hippocampal CA1 region. J Neurosci 20: 5924-5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Feng J, Fienberg AA, Greengard P (1999) D2 dopamine receptors induce mitogen-activated protein kinase and cAMP response element-binding protein phosphorylation in neurons. Proc Natl Acad Sci USA 96: 11607-11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Sharrocks AD, Whitmarsh AJ (2003) Transcriptional regulation by the MAP kinase signaling cascades. Gene 320: 3-21. [DOI] [PubMed] [Google Scholar]
- Yarwood SJ, Kilgour E, Anderson NG (1996) Cyclic AMP stimulates the phosphorylation and activation of p42 and p44 mitogen-activated protein kinases in 3T3-F442A preadipocytes. Biochem Biophys Res Commun 224: 734-739. [DOI] [PubMed] [Google Scholar]
- Xing J, Ginty DD, Greenberg ME (1996) Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 273: 959-963. [DOI] [PubMed] [Google Scholar]
- Zanassi P, Paolillo M, Feliciello A, Avvedimento EV, Gallo V, Schinelli S (2001) cAMP-dependent protein kinase induces cAMP-response element-binding protein phosphorylation via an intracellular calcium release/ERK-dependent pathway in striatal neurons. J Biol Chem 276: 11487-11495. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Belluscio L, Hen R (2000) GOLFα mediates dopamine D1 receptor signaling. J Neurosci 20: RC91(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]