

Abstract
Parkinson's disease (PD) is linked genetically to proteins that function in the management of cellular stress resulting from protein misfolding and oxidative damage. Overexpression or mutation of α-synuclein results in the formation of Lewy bodies and neurodegeneration of dopaminergic (DA) neurons. Human torsinA, mutations in which cause another movement disorder termed early-onset torsion dystonia, is highly expressed in DA neurons and is also a component of Lewy bodies. Previous work has established torsins as having molecular chaperone activity. Thus, we examined the ability of torsinA to manage cellular stress within DA neurons of the nematode Caenorhabditis elegans. Worm DA neurons undergo a reproducible pattern of neurodegeneration after treatment with 6-hydroxydopamine (6-OHDA), a neurotoxin commonly used to model PD. Overexpression of torsins in C. elegans DA neurons results in dramatic suppression of neurodegeneration after 6-OHDA treatment. In contrast, expression of either dystonia-associated mutant torsinA or combined overexpression of wild-type and mutant torsinA yielded greatly diminished neuroprotection against 6-OHDA. We further demonstrated that torsins seem to protect DA neurons from 6-OHDA through downregulating protein levels of the dopamine transporter (DAT-1) in vivo. Additionally, we determined that torsins protect robustly against DA neurodegeneration caused by overexpression of α-synuclein. Using mutant nematodes lacking DAT-1 function, we also showed that torsin neuroprotection from α-synuclein-induced degeneration occurs in a manner independent of this transporter. Together, these data have mechanistic implications for movement disorders, because our results demonstrate that torsin proteins have the capacity to manage sources of cellular stress within DA neurons.
Keywords: dopamine, dystonia, Parkinson, torsin, C. elegans, synuclein
Introduction
Parkinson's disease (PD) involves the progressive loss of dopaminergic (DA) neurons from the substantia nigra pars compacta (SNpc). Genetic forms of PD account for only 5-10% of known cases, and environmental factors seem pivotal to sporadic causality. Toxins that cause formation of excessive reactive oxygen species (ROS), such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), paraquat, rotenone, and 6-hydroxydopamine (6-OHDA), all induce DA neuron loss and parkinsonian phenotypes in animals (Simon et al., 1974; Langston et al., 1983; Tanner, 1992; Liou et al., 1997). Proteinaceous inclusions termed Lewy bodies are the pathological hallmark of PD and are found in brains from both familial and sporadic PD. Central to the formation of Lewy bodies is α-synuclein, a polypeptide with a propensity toward intracellular aggregation when mutated or present in multiple wild-type copies (Polymeropoulos et al., 1997; Conway et al., 2000, 2001; Singleton et al., 2003; Farrer et al., 2004). The specific metabolism of dopamine in neurons also contributes to additional vulnerability from cellular stress and degeneration (Fahn and Cohen, 1992; Jellinger et al., 1992; Jenner et al., 1992). In this context, familial PD has been linked to several genes in which many of the gene products function to manage protein degradation and oxidative stress (Dawson and Dawson, 2003).
TorsinA is a protein that is both highly expressed in DA neurons and a component of Lewy bodies, where it closely associates with α-synuclein (Shashidharan et al., 2000; Sharma et al., 2001; O'Farrell et al., 2004). TorsinA belongs to the functionally diverse AAA+ protein family (ATPases associated with cellular activities) that includes heat shock proteins (Hsp), proteases, and dynein (Neuwald et al., 1999). Others and we have demonstrated that torsins (torsinA and a Caenorhabditis elegans homolog, TOR-2) possess chaperone activity and can suppress intracellular protein aggregation in vivo (McLean et al., 2002; Caldwell et al., 2003). Dominant mutations in the DYT1 gene encoding torsinA cause early-onset torsion dystonia (EOTD) (Ozelius et al., 1997), another movement disorder in which only 30-40% of carriers display dystonic symptoms (Ozelius et al., 1997). Aberrant DA signaling has been suggested to play a role in EOTD (Augood et al., 2002, 2004; Maries et al., 2003). Moreover, torsinA was found recently to modulate cellular levels of the dopamine transporter (DAT) and other polytopic membrane-bound proteins (Torres et al., 2004).
The complexity of the human brain and limitations of cell culture preclude detailed analyses of genetic and environmental factors pertaining to movement disorders in vivo. Notably, C. elegans is ideal for such an analysis, because this anatomically and genetically defined transparent nematode has exactly 302 neurons, precisely 8 of which are DA (Sulston et al., 1975). This facilitates rapid scoring of DA neurodegeneration while animals age. Taking advantage of this system, we investigated the capacity of torsins to ameliorate the effects of α-synuclein overexpression and DA-specific toxicity. Here, we report that torsins can protect the DA neurons of C. elegans from defined cellular stresses linked to DA dysfunction. We further provide evidence toward elucidating potential mechanisms underlying torsin-mediated neuroprotection and explore the implications of these results for movement disorders.
Materials and Methods
C. elegans strains and protocols. Nematodes were maintained using standard procedures (Brenner, 1974). Transgenic lines were generated by transforming Pdat-1::GFP with Pdat-1::TOR-2 [strain UA13 (baEx13)], Pdat-1::TOR-2(K171T) [strain UA14 (baEx14)], Pdat-1::TOR-2(Δ367) [strain UA15 (baEx15)], Pdat-1::torsinA [strain UA16 (baEx16)], or Pdat-1::torsinA(ΔE302/303) [strain UA17(baEx17)] into wild-type C. elegans (N2 Bristol variety). Pdat-1::GFP alone [strain UA30 (baEx30)] and/or Pdat-1::TOR-2 [strain UA39 (baEx39)] were also injected into dat-1 knock-out worms [RM2702 (ok157), a functional null]. RM2702 (a gift from James Rand, Oklahoma Medical Research Foundation, Oklahoma City, OK) was also used for testing 6-OHDA specificity. For the construction of various α-synuclein-overexpressing lines, either Pdat-1::GFP and Pdat-1::α-synuclein alone [strain UA18 (baEx18)] or also with one of the Pdat-1::torsin constructs [Pdat-1::TOR-2, strain UA19 (baEx19); Pdat-1::TOR-2(Δ367), strain UA20 (baEx20); Pdat-1::torsinA, strain UA21 (baEx21); or Pdat-1::torsinA(ΔE302/303), strain UA22(baEx22)] were injected into N2 worms. The same combinations (except for mutant torsins) were injected into dat-1 worms to create Pdat-1::GFP and Pdat-1::α-synuclein alone [strain UA31 (baEx31)], Pdat-1::TOR-2 [strain UA32 (baEx32)], and Pdat-1::torsinA [strain UA33 (baEx33)]. For the generation of tyrosine hydroxylase (CAT-2)-overexpressing lines, either Pdat-1::GFP and Pdat-1::CAT-2 alone [strain UA23 (baEx23)] or also with one of the Pdat-1::torsin constructs [Pdat-1::TOR-2, strain UA24 (baEx24); Pdat-1::TOR-2(Δ367), strain UA25 (baEx25); Pdat-1::torsinA, strain UA26 (baEx26); or Pdat-1::torsinA(ΔE302/303), strain UA27(baEx27)] were injected. For the Hsp104-overexpressing line [strain UA28 (baEx28)], Pdat-1::GFP and Pdat-1::Hsp104 were coinjected. For the GFP::DAT-1 fusion lines, either Pdat-1::GFP::DAT-1 alone [strain UA34 (baEx34)] or this construct along with Pdat-1::TOR-2 [strain UA35 (baEx35)] or Pdat-1::TOR-2(Δ367) [strain UA36 (baEx36)] were injected into dat-1 worms. Each construct was injected at a concentration of 50 μg/ml, except for the Pdat-1::torsinA/torsinA(ΔE302/303) line [strain UA29 (baEx29)], in which only 25 μg/ml each of Pdat-1::torsinA and Pdat-1::torsinA(ΔE302/303) were used (along with Pdat-1::GFP). To construct Ptor-2::GFP transgenic worms [strain UA37 (baEx37)], 41 μg/ml Ptor-2::GFP-amplified DNA and 41 μg/ml rol-6 were coinjected. For each combination of plasmid constructs, multiple worm lines expressing stable extrachromosomal arrays were compared and three representative lines were used for experimental analysis, except for the 6-OHDA experiments and the analysis of green fluorescent protein (GFP) fluorescence in GFP::DAT-1 worms, in which single representative transgenic lines were used in repeated experiments after initial analysis on all stable lines.
Plasmid constructs and mutagenesis. Plasmids were constructed using Gateway technology (Invitrogen, San Diego, CA). Specifically, the unc-54 promoter region was excluded from pPD30.38 (a gift from Andrew Fire, Standford University, Stanford, CA) by double digestion using HindIII and KpnI and replaced by the dat-1 promoter region fragment amplified from pRN200 (Nass et al., 2002). The resulting novel vector was then converted into a Gateway destination vector, pDEST-DAT-1, using Gateway technology. Mutant versions of tor-2 cDNA were created using site-directed mutagenesis as described previously (Caldwell et al., 2003), except for modifications of internal primers specifically designed for each point mutation. Wild-type torsinA and torsinA(ΔE302/303) cDNAs were obtained from Ben Cravatt and Karen Kustedjo (Scripps Research Institute, La Jolla, CA). The GFP DNA sequence was amplified from pPD95.79 (a gift from A. Fire) using Gateway primers. The human α-synuclein cDNA plasmid was obtained from Philipp Kahle (Ludwig Maximilians University, Munich, Germany). The full-length cat-2 gene was amplified from C. elegans genomic DNA. Hsp104 cDNA was obtained from Rick Morimoto (Northwestern University, Chicago, IL). The GFP::DAT-1 fusion fragment was constructed by a PCR fusion approach (Hobert et al., 1999) using pME18S-FL3+dat-1 cDNA (a gift from Yugi Kohara, University of Tokyo, Tokyo, Japan) as the dat-1 cDNA template and pPD95.77 (a gift from A. Fire) as the GFP template. Gateway entry vectors were generated by BP reaction with pDONR201 or pDONR221 using PCR-amplified cDNA fragments encoding TOR-2, TOR-2(K171T), TOR-2(Δ367), torsinA, torsinA(ΔE302/303), α-synuclein, GFP, yeast Hsp104, a cat-2 genomic fragment, and GFP::DAT-1, Pdat-1::GFP, Pdat-1::TOR-2, Pdat-1::TOR-2(K171T), Pdat-1::TOR-2(Δ367), Pdat-1::torsinA, Pdat-1::torsinA(ΔE302/303), Pdat-1::α-synuclein, Pdat-1::CAT-2, Pdat-1::Hsp104, or Pdat-1::GFP::DAT-1. After this, all genes were cloned into the pDEST-DAT-1 vector via an LR reaction with the respective entry vector. To visualize native tor-2 expression, a PCR fusion-based approach was used to construct Ptor-2::GFP (Hobert, 2002). A genomic region consisting of 7070 bp upstream of the start codon of tor-2, as well as the entire genomic region of tor-2 (except for the last 16 bp and stop codon), was used to drive GFP expression. Information on specific primers used in these reactions is available on request.
Preparation of C. elegans extracts for immunoblotting. Extracts were prepared after growth of each transgenic line to near confluence on two 100 mm nematode growth media (NGM) plates. Worms were collected by washing with M9 buffer (0.3% KH2PO4, 0.6% Na2HPO4, 0.5% NaCl, 1 mm MgSO4) and concentrated by centrifugation in a 1.5 ml microcentrifuge tube at 5000 × g for 1 min. The worm pellet was resuspended and lysed in 0.5 ml of worm lysis buffer (100 mm Tris, pH 6.8, 2% SDS, and 15% glycerol) by boiling for 5 min. This lysate was centrifuged again at 13,200 × g for 10 min, and the supernatant was collected and concentrated using a Centricon YM-10 column (Millipore, Bedford, MA) at 14,000 × g for 30 min. Protein concentration was determined using the bicinchoninic acid protein assay (Sigma, St. Louis, MO).
SDS-PAGE and Western blotting. SDS-PAGE and Western blotting were performed as described previously (Caldwell et al., 2003), unless indicated otherwise. For TOR-2 detection, a 1:800 dilution of rabbit anti-TOR-2 primary antibody (Caldwell et al., 2003) and a 1:10,000 dilution of horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (Amersham Biosciences, Piscataway, NJ) were used. For actin detection, a 1:8000 dilution of mouse anti-actin antibody (ICN Biochemicals, Costa Mesa, CA) and a 1:10,000 dilution of horseradish peroxidase-conjugated goat anti-mouse IgG secondary antibody (Amersham Biosciences) were used. For GFP detection, 140 μg of total protein was loaded, and a 1:1000 dilution of rabbit anti-GFP primary antibody (Clontech, Palo Alto, CA) and a 1:10,000 dilution of horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (Amersham Biosciences) were used. For Hsp104 detection, 200 μg of total protein was loaded, and a 1:800 dilution of mouse anti-Hsp104 primary antibody (a gift from Susan Lindquist, Whitehead Institute, Cambridge, MA) and a 1:10,000 dilution of horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (Amersham Biosciences) were used.
6-OHDA exposure and quantitative analyses of neuronal degeneration. Age-synchronized worms were obtained by treating gravid adults with 2% sodium hypochlorite and 0.5 m NaOH to isolate embryos (Lewis and Fleming, 1995). These embryos were grown for 30 h at 25°C. At the L3 stage, larvae were incubated with 10 mm (or 50 mm) 6-OHDA and 2 mm (or 10 mm) ascorbic acid for 1 h with gentle agitation every 10 min (Nass et al., 2002). The worms were then washed and spread onto NGM plates seeded with bacteria (OP50) and scored at time points ranging from 2 to 72 h after 6-OHDA exposure.
Immediately after 6-OHDA treatment, worms containing the nonintegrated transgenes were selected under a fluorescence dissecting microscope, based on the presence of GFP, and transferred to a fresh NGM plate seeded with OP50. For each time point, 30-40 worms were applied to a 2% agarose pad and immobilized with 3 mm levamisole. Worms were examined under a Nikon Eclipse E800 epifluorescence microscope equipped with an Endow GFP filter cube (Chroma Technology, Rockingham, VT). For the ease of analysis, only the four CEP DA neurons in the head of the worm were scored. A worm was scored as “wild type” when all four CEP neurons were present and their neuronal processes were intact; a worm was scored as having “dendrite blebbing,” “cell body rounding,” or “cell body loss” when at least one of the four neuronal dendrites or cell bodies was defective as described. These experiments were repeated three times. Images were captured with a Cool Snap CCD camera (Photometrics, Tucson, AZ) driven by MetaMorph software (Universal Imaging, West Chester, PA).
α-Synuclein- or CAT-2-induced neurodegeneration analyses. To obtain 7-d-old animals of α-synuclein and CAT-2 transgenic lines, nonintegrated L1 and L2 worms with green fluorescence were selected and allowed to grow to the 4 d adult stage (∼7 d after hatching). Thirty to 40 worms at each chosen stage were analyzed for each nonintegrated line, and the average of at least three stable lines for each combination of transgenes was reported. A worm was scored as wild type when it still preserved all four CEP cell bodies regardless of the morphology of the dendrites.
Analysis of fluorescence in GFP::DAT-1 worms. GFP fluorescence intensity of DA neurons in GFP::DAT-1 worms (with and without torsin) was measured using MetaMorph software (Universal Imaging). Thirty worms were analyzed under the microscope for each line generated. Only worms with visible GFP at 630× magnification were measured for GFP fluorescence intensity. One to two of the visibly brightest CEP cell bodies in each worm were measured for fluorescence intensity, which was recorded in terms of pixel intensity and assigned arbitrary units (A.U.). The average from all measured cell bodies was calculated and compared between at least three independent stable lines for each transgenic line, and the most representative one was chosen for direct comparison and statistical analyses as shown in Table 1. Additionally, scoring of GFP::DAT-1 dendritic and axonal expression was performed by analyzing six DA neurons in the anterior of 30 worms for each line at 630× magnification. A worm was scored as having “dendritic and/or axonal GFP” when both visible cell bodies and either dendritic or axonal GFP were observed. A worm was scored as possessing “cell body GFP only” when GFP was observed exclusively in cell bodies. A worm was scored as having “no GFP” when none of the anterior six DA neurons could be visualized.
Table 1.
Effect of torsin overexpression on GFP::DAT-1 expression within DA neurons
|
Worm straina |
Pdat-1::GFP::DAT-1 |
Pdat-1::GFP::DAT-1/TOR-2 |
Pdat-1::GFP::DAT-1/TOR-2 (Δ367) |
|---|---|---|---|
| Mean pixel intensity of cell bodies ± SEM (A.U.) | 2127 ± 59 (n=57) | 1356 ± 77 (n=44)b | 1865 ± 65 (n=38)bc |
| Worms with dendritic and/or axonal GFP | 66.7% | 13.3% | 23.3% |
| Worms with cell body GFP only | 33.3% | 70% | 63.4% |
| No GFP |
0% |
16.7% |
13.3% |
n represents the number of cell bodies analyzed from 30 worms (see footnote a).
Thirty worms were analyzed from each strain in which one to two cell bodies from each worm were analyzed for pixel intensity.
Mean pixel intensity of cell bodies from Pdat-1::GFP::DAT-1 worms is significantly different from cell bodies of Pdat-1::GFP::DAT-1/TOR-2 or Pdat-1::GFP::DAT-1/TOR-2(Δ367) worms (p < 0.001 and p < 0.05, respectively; ANOVA).
Mean pixel intensity of cell bodies from Pdat-1::GFP::DAT-1/TOR-2 (Δ367) worms is significantly different from Pdat-1::GFP::DAT-1/TOR-2 worms (p < 0.001; ANOVA).
3-Iodotyrosine treatment. Age-synchronized Pdat-1::CAT-2 embryos were obtained by bleaching gravid adults as described previously (Lewis and Fleming, 1995) and grown in 2 ml of M9 buffer containing 1 mg/ml 3-iodotyrosine (3-IT) for 24 h. After incubation, worms were washed three times using M9 buffer. Worms were scored for neurodegeneration at 1 and 24 h after treatment. Control worms received the same treatment, except that 3-IT was not added to the M9 buffer during the incubation.
Formaldehyde-induced fluorescence. Pdat-1::CAT-2 worms were visualized for high levels of dopamine using formaldehyde fixation as described previously by Lints and Emmons (1999). Worms were examined with a Nikon E800 epifluorescence microscope using a cyan fluorescent protein filter cube and compared with N2 control animals.
Immunocytochemistry. Worms were fixed with formaldehyde and stained for torsinA as described previously (McIntire et al., 1992), with slight modifications (details available on request). Mouse anti-torsinA antibody DM2A8 was a gift from Xandra Breakefield (Massachusetts General Hospital, Charlestown, MA) and was used at a concentration of 1:100 in PBS containing 0.5% Triton X-100. Alexa Fluor 594 goat anti-mouse IgG secondary antibody (Molecular Probes, Eugene, OR) was diluted 1:100 in PBS containing 1% BSA and 0.5% Triton X-100. Worms were examined immediately after being rinsed with PBS containing 1% BSA and 0.5% Triton X-100. GFP fluorescence was maintained by this procedure and observed for confirmation of tissue-specific localization of torsinA within the DA neurons. A methanol-based fixation method was used for the immunostaining of α-synuclein (Duerr et al., 1999). GFP fluorescence is lost using this fixation method; therefore, a 1:100 dilution of mouse anti-α-synuclein (Zymed, San Francisco, CA) and a 1:100 dilution of rabbit anti-GFP (Abcam, Cambridge, MA) antibodies were used. Alexa Fluor 594 goat anti-mouse IgG primary antibody and Alexa Fluor 488 goat anti-rabbit IgG secondary antibody were used to visualize α-synuclein and GFP, respectively. Worms were examined with a Nikon E800 epifluorescence microscope using both an Endow GFP filter cube as well as a Texas Red filter cube (Chroma Technology). Images were captured with a Cool Snap CCD camera (Photometrics) driven by MetaMorph software (Universal Imaging).
Statistical analyses. Statistical analyses were performed using the ANOVA Bonferroni's test. Comparisons were performed between multiple transgenic lines at individual time points but not across different time points. ANOVA was used for all analyses, except the 3-IT experiment, in which a t test was used. Experiments were performed at least three times, and all statistics were assessed using the Systat program.
Results
Torsins protect against 6-OHDA toxicity
The eight dopamine-producing cells of C. elegans consist of six neurons in the anterior (two pairs of CEPs, one pair of ADE neurons) and a pair in the posterior (PDE) of the nematode (Sulston et al., 1975). Previous studies have established that the CEP and ADE neurons undergo readily discernible neuronal degeneration after treatment with the dopamine-selective neurotoxin 6-OHDA (Nass et al., 2002). The toxicity of 6-OHDA is mediated through the formation of ROS by the generation of hydrogen peroxide and hydroxide radicals via a nonenzymatic auto-oxidation process (Kumar et al., 1995; Foley and Riederer, 2000). After exposure to 6-OHDA, C. elegans DA neurons exhibit a characteristic dose-dependent pattern of apoptotic cell death that was confirmed by ultrastructural analysis (Nass et al., 2002). This degeneration can be monitored in living animals by coexpression with GFP and categorized into three temporally and morphologically distinct stages, including neuronal process blebbing, cell body rounding with process loss, and cell body loss (Fig. 1A). These characteristic changes reproducibly appear in this order within a few hours, recapitulating observations in MPTP-treated monkeys and in 6-OHDA-treated rats, in which damage to striatal terminals leads to retrograde changes and precedes that of SNpc cell bodies (Berger et al., 1991; Herkenham et al., 1991).
Figure 1.
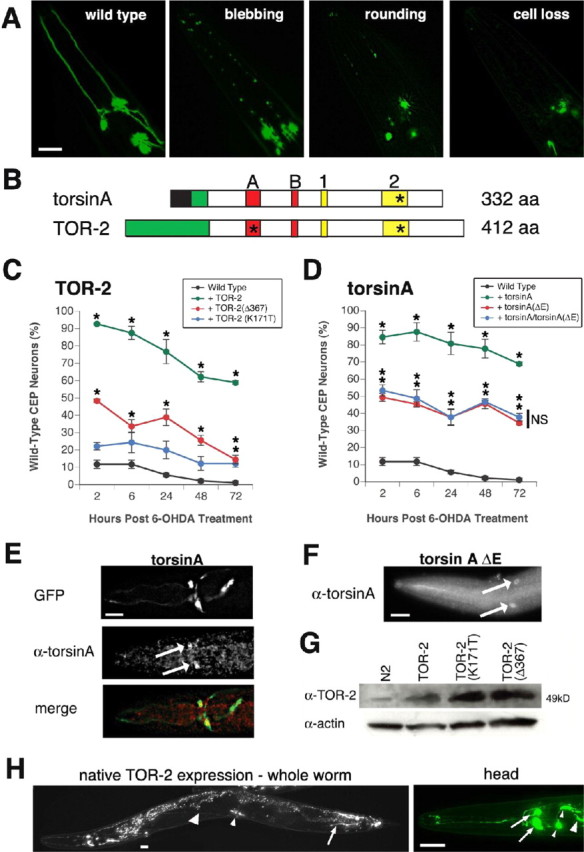
Torsin-mediated protection of C. elegans DA neurons against 6-OHDA. A, Different stages of DA neurodegeneration. From left to right, representative temporally staged images depicting CEP DA neuron morphology of wild type, neuronal process blebbing, process loss and cell body rounding, and cell body loss are shown. Scale bar, 20 μm. B, Schematic representation of torsin domains and mutations. Top, Human torsinA; bottom, C. elegans TOR-2; black region, signal peptide; green region, putative hydrophobic transmembrane domain; A, Walker A domain; B, Walker B domain; 1, sensor 1 domain; 2, sensor 2 domain. The asterisks mark the approximate positions of mutations that were analyzed. Mutations within sensor 2 correspond to the EOTD-associated mutation [torsinA(ΔE302/303)] and the structurally analogous change in worm TOR-2 [TOR-2(Δ367)]. The mutation within the Walker A domain corresponds to an alteration within a conserved ATP-binding site [TOR-2(K171T)]. C, TOR-2 protection against 6-OHDA. After 6-OHDA treatment, worms were examined for the presence of all four wild-type CEP neurons at specific time points: green, TOR-2; blue, TOR-2(K171T); red, TOR-2(Δ367); black, Pdat-1::GFP without TOR-2 overexpression. D, TorsinA protection against 6-OHDA. Green, TorsinA; blue, torsinA/torsinA (ΔE302/303) combination; red, torsinA(ΔE302/303); black, Pdat-1::GFP without torsinA overexpression. Statistical significance between Pdat-1::GFP and each of the six torsin-overexpressing lines was assessed at each time point between all transgenic lines by the ANOVA Bonferroni's test (*p < 0.05; NS, not significant). Error bars in C and D indicate SEM between independent experiments. E, Immunolocalization of torsinA in DA neurons in human torsin-overexpressing worms. Top, Two CEP neurons marked by GFP fluorescence; middle, torsinA antibody immunostaining in CEP cell bodies (arrows); bottom, merge of top and middle images. Scale bar, 20 μm. F, Immunolocalization of torsinA in torsinA(ΔE302/303)-overexpressing worms in CEP cell bodies (arrows). Scale bar, 20 μm. The GFP and merge images are not shown because they were reminiscent of those in Figure 1 E. G, Western analysis to verify TOR-2 overexpression in TOR-2-overexpressing worms. Actin was probed for each sample as a loading control. H, Native TOR-2 expression pattern in transgenic worms in which GFP is driven by a genomic region upstream and inclusive of the C. elegans tor-2 gene. Left, Entire worm depicting expression in the AVE neurons (large arrowhead), vulva muscle cells (small arrowhead), and PVW neurons (arrow); gut autofluorescence is also observed throughout the intestinal tract. Right, Close-up of the anterior head region, showing expression in the M1 pharyngeal neuron (large arrowhead), the AW class of neurons (2 small arrowheads), and the AVE interneurons (2 arrows). Expression in DA neurons is not observed. Scale bar, 20 μm.
To evaluate torsin activity in DA neuroprotection, transgenic lines of C. elegans overexpressing either human torsinA or worm TOR-2 under the control of a dopamine-specific neuronal promoter (Pdat-1, the C. elegans DAT promoter) were generated. Likewise, site-directed mutagenesis was used to generate specific changes in these proteins to evaluate their relative effects in the presence of oxidative stress (Fig. 1B). In all cases, animals were treated with various concentrations of 6-OHDA, and neurodegeneration was followed over time, as worms develop and age, by coexpression of a DA GFP marker in these transparent animals (Fig. 1C,D). While we documented all stages of neurodegeneration in the CEP neurons, we simplified these stages into two categories (wild type vs non-wild type) for ease of presentation. Protein expression was confirmed by in vivo immunolocalization and immunoblotting (Fig. 1E-G). Although the torsin transgenes expressed in our study do not cause overt morphological changes within the neurons at the compound microscope level, this does not eliminate the possibility that expression of either mutant or wild-type torsins could have more subtle morphological consequences as have been reported previously in cell culture studies (Kustedjo et al., 2000; Hewett et al., 2000; O'Farrell et al., 2002). It should be noted that the Pdat-1::torsinA and Pdat-1::TOR-2 transgenic animals (also expressing Pdat-1::GFP) exhibit a low background level of DA neuron loss comparable with Pdat-1::GFP alone (5-10%).
The overexpression of wild-type torsins (Pdat-1::torsinA and Pdat-1::TOR-2) in DA neurons significantly elevated the resistance of DA neurons to 6-OHDA at a concentration of 10 mm for at least 72 h compared with control worms (Fig. 1C,D). During this time course, the majority of the torsin-overexpressing worms still maintained wild-type DA neurons, with very few worms losing neuronal cell bodies (68.9 and 58.9% of wild-type neurons conferred by overexpression of torsinA and TOR-2, respectively, at 72 h; p < 0.01; ANOVA). In contrast, none of the DA neurons in control worms (Pdat-1::GFP) appeared to be wild type; nearly all of these animals lost at least one neuronal cell body. Expression of the EOTD-associated torsinA(ΔE302/303) protein exhibited significantly reduced neuroprotection from 6-OHDA toxicity with only 34.4% of the worms showing wild-type neurons at 72 h (Fig. 1D). Although this percentage of protection was statistically significant compared with control animals at each time point, it was also significantly different from that observed with wild-type torsin (68.9% wild type vs 34.4%) (Fig. 1D). Like-wise, a similar decrease in protection was seen in transgenic animals expressing mutant TOR-2 proteins in DA neurons. Site-directed mutagenesis designed to alter a conserved ATP-binding site [TOR-2(K171T)] showed only marginal neuroprotection (12.2% of worms with wild-type neurons at 72 h) (Fig. 1C); these results suggest that ATP binding is indispensable for torsin activity. This coincides with the assumption that torsins, as members of the AAA+ protein family, need ATP to function. Furthermore, a site-directed mutation that corresponds to a position that is structurally analogous to the ΔE302/303 deletion in human torsinA, TOR-2(Δ367), correspondingly showed only 14.5% of worms preserving wild-type neurons after 72 h (Fig. 1C). Western blotting was used to confirm the overexpression of TOR-2 and its mutant variants. The expression level of TOR-2 in Pdat-1::TOR-2, Pdat-1::TOR-2(K171T), or Pdat-1::TOR-2(Δ367) transgenic lines was much higher than in N2 control animals (Fig. 1G). Expression of human torsinA and torsinA(ΔE302/303) in DA neurons was verified by immunofluorescence using a torsinA-specific antibody (Fig. 1E,F).
Although distinctively lower than wild-type torsins, the protective effects resulting from the overexpression of any of these mutant gene products were still significant at most time points. This residual neuroprotection may suggest that critical residues retain partial activity in these specific torsin mutants. It is unlikely that the degree of neuroprotection observed was attributable to a nonspecific effect independent of the transgenes, because later results described herein demonstrate that overexpression of a related protein (Hsp104) does not provide any protection against 6-OHDA (see Fig. 7A). Regardless, the presence of either wild-type or mutant torsin proteins in these neurons affords at least moderate neuroprotection, because DA neurons of C. elegans do not contain the TOR-2 protein. Using a GFP reporter fusion, we have determined that native tor-2 gene expression is limited to just a few cells, including vulva muscles, a single cholinergic pharyngeal neuron (M1), two sensory neurons (AW class) and two interneurons (AVE) in the head, and a few neurons toward the tail, including a pair of PVW neurons (Fig. 1 H). Importantly for this study, tor-2 gene expression is unequivocally absent in DA neurons at any larval or adult stages. Thus, our functional assays for torsin activity in DA neurons is not complicated by endogenous torsin expression.
Figure 7.

Hsp104 does not protect against 6-OHDA-induced neurodegeneration. A, Comparison of the protective effects observed between torsins and yeast Hsp104 overexpressed in DA neurons. After 6-OHDA treatment, worms were examined for the presence of all four wild-type CEP neurons at specific time points. Filled circle, TOR-2; open circle, torsinA; open square, Hsp104; filled square, N2 background with only Pdat-1::GFP. Statistical significance between Pdat-1::GFP and each of the other transgenic lines was assessed by the ANOVA Bonferroni's test (*p < 0.05; NS, not significant). Error bars indicate SEM between independent experiments. B, Western analysis of Hsp104 expression in which Hsp104 was identified as a ∼100 kDa band present in extracts from Pdat-1::Hsp104 animals but not from N2 controls. Actin was probed as a loading control.
Because EOTD is a dominant-negative disease with limited penetrance, we also tested the effect of 6-OHDA treatment on animals coexpressing wild-type torsinA and mutant torsinA(ΔE302/303) together in DA neurons. These animals demonstrated much reduced protection from 6-OHDA when compared with wild type (37.7% at 72 h compared with 68.8% for torsinA; p = 0; ANOVA). Notably, the reduced levels of neuroprotection displayed by these worms was similar to that observed with mutant torsinA(ΔE302/303) alone (34.4% vs 37.7% at 72 h; p = 1; ANOVA) (Fig. 1D). Although these data are suggestive of a dominant-negative effect for torsinA(ΔE302/303), they are not conclusive because the precise wild-type/mutant torsinA ratio cannot be differentiated in transgenic worms.
Previous studies have determined that 6-OHDA is taken up specifically through the C. elegans dopamine transporter DAT-1 in wild-type worms; in contrast, dat-1 knock-out worms are resistant to this toxin (Nass et al., 2002). Our data corroborate these findings, because a high percentage of dat-1 mutant animals were not affected by 6-OHDA treatment (88.9% at 10 mm 6-OHDA) (Fig. 2). Interestingly, this percentage is similar to that of worms overexpressing wild-type TOR-2 (76.7%), raising the possibility that TOR-2 overexpression may result in a decrease in DAT-1 function. In line with this hypothesis, when TOR-2 was overexpressed in the dat-1 mutant background, the percentage of worms preserving all four CEPs was not significantly different (88.9% for Pdat-1::GFP in dat-1 vs 90% for Pdat-1::TOR-2 in dat-1). Additionally, similar results were obtained using 50 mm 6-OHDA, a concentration five times greater than the dose used in our previous trials (Fig. 2), indicating that the protective effects of either dat-1 knock-out or torsin overexpression were not 6-OHDA dosage dependent at these concentrations.
Figure 2.

Transgenic animals overexpressing TOR-2 exhibit neuroprotection from 6-OHDA at levels comparable with dat-1 mutant worms. Twenty-four hours after exposure to either 10 or 50 mm 6-OHDA, Pdat-1::GFP control worms (hatched bars), TOR-2-overexpressing worms (white bars), Pdat-1::GFP in dat-1 background worms (gray bars), and TOR-2-overexpressing worms in the dat-1 mutant background (black bars) were scored for evidence of DA neuron survival (n = 30-40 worms per line per independent experiment). Statistical significance was determined between control and other transgenic lines by the ANOVA Bonferroni's test (*p < 0.05). Error bars indicate SEM between independent experiments.
Torsins downregulate DAT-1 to protect against 6-OHDA
Torres et al. (2004) recently showed torsinA interacts with the C-terminal intracellular domain of DAT and that overexpression of wild-type torsinA impairs the cell surface localization of the DAT in cell culture. Together with our data demonstrating that TOR-2 overexpression results in greatly enhanced DA neuron resistance to 6-OHDA, we proceeded to evaluate whether torsin protection is partly mediated through downregulation of DAT-1. To examine this, we fused GFP to the N terminus of C. elegans DAT-1 and placed this construct under Pdat-1 control, thereby enabling us to monitor DAT-1 turnover by GFP fluorescence in transgenic worms, with and without torsins. When TOR-2 was co-overexpressed with Pdat-1::GFP::DAT-1, GFP fluorescence diminished greatly (Fig. 3A); this was evidenced by a decrease in fluorescence pixel intensity of DA neuronal cell bodies from 2127 to 1356 A.U. (p = 0) (Table 1). Furthermore, a higher percentage of worms with axonal and dendritic fluorescence (Fig. 3A, arrowheads and arrows, respectively) were observed in Pdat-1::GFP::DAT-1 animals (66.7%) compared with TOR-2 co-overexpressing worms (13.3%). Notably, 16.7% of TOR-2-overexpressing animals did not exhibit any visible fluorescence (Table 1). Because this latter group of worms was not included in our fluorescence intensity analysis, the 1356 A.U. observed in Pdat-1::GFP::DAT-1 with TOR-2 represents a skewed value. Overexpression of mutant TOR-2(Δ367) in DA neurons resulted in a significant reversal of GFP::DAT-1 pixel intensity, with a mean value of 1865 A.U. compared with wild-type TOR-2 (1356 A.U.). This moderate reversal, however, was still significantly less than wild-type neurons in the absence of any torsin expression (2127 A.U.). These results support the hypothesis that torsins can downregulate the expression of DAT-1. To ensure that GFP and DAT-1 are not cleaved in vivo, we confirmed the presence of this fusion protein within transgenic animals via Western blotting. These data demonstrated an ∼95 kDa band corresponding to GFP::DAT-1 in worm extracts probed with anti-GFP antibody, the intensity of which was much reduced in Pdat-1::GFP::DAT-1/Pdat-1::TOR-2 or Pdat-1::GFP::DAT-1/Pdat-1::TOR-2 (Δ367) compared with Pdat-1::GFP::DAT-1 alone (Fig. 3B). The overexpression of TOR-2 within these transgenic lines was confirmed by Western blotting (Fig. 3B).
Figure 3.

TOR-2 downregulates GFP::DAT-1. A, GFP fluorescence is greatly diminished within the axons (arrowheads) and dendrites (arrows) of DA neurons when TOR-2 is co-overexpressed and moderately diminished when TOR-2(Δ367) is co-overexpressed with GFP::DAT-1. Scale bar, 20 μm. B, Western analysis of GFP::DAT-1 demonstrating that there is a higher level of DAT-1 in worms that do not co-overexpress TOR-2. GFP antibody was used to detect the GFP::DAT-1 fusion protein band; TOR-2 antibody was used to detect the level of TOR-2 expression. Actin was probed for each line as a loading control.
Torsins do not significantly protect DA neurons against neurodegeneration induced by tyrosine hydroxylase overexpression
Dopaminergic neurons are particularly susceptible to oxidative stress as a result of DA metabolism, as well as the presence of other intracellular factors favoring the formation of ROS (Blum et al., 2001). Furthermore, it has been shown that single nucleotide polymorphisms in specific genes encoding enzymes in the DA metabolic pathway can also increase susceptibility to PD (Tan et al., 2000). Given these data, we set out to develop an in vivo model to study the effect of altered DA metabolism on neurodegeneration and investigate whether torsins have any impact on this process.
To increase intraneuronal DA production in vivo, we overexpressed the C. elegans cat-2 gene, encoding the nematode homolog of the DA biosynthetic enzyme tyrosine hydroxylase (TH). As expected, Pdat-1-driven overexpression of CAT-2 protein caused neuronal degeneration at all developmental stages in multiple stable lines, in which only 23.3% of 7-d-old animals maintained all four CEP neurons (Fig. 4A; supplemental Fig. S1, available at www.jneurosci.org as supplemental material). When either torsinA or TOR-2 was co-overexpressed with CAT-2, the percentage of wild-type CEP neurons was 39.0% (p = 0.65; ANOVA) and 37.5% (p = 1; ANOVA), respectively (Fig. 4A; supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Likewise, mutant torsinA(ΔE302/303) or TOR-2(Δ367) coexpression did not alter the percentage of worms with wild-type CEP neurons either [26.1% (p = 1) and 27.7% (p = 1), respectively; ANOVA] (Fig. 4A; supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Figure 4.

Torsin effect on TH/CAT-2 overexpression in DA neurons. A, A slight trend toward neuronal survival when torsinA or TOR-2 is coexpressed with CAT-2 is shown. The percentage of worms preserving all four wild-type CEPs at the 7-d-old stage was calculated for each of five CAT-2-overexpressing lines (n = 30-40 per stable line). Statistical significance between Pdat-1::CAT-2 and each of the torsin/CAT-2-overexpressing lines was assessed by the ANOVA Bonferroni's test; no significance was observed. B, 3-IT inhibition of CAT-2-induced neurodegeneration. Pdat-1::CAT-2 worms were incubated with or without 3-IT (black and white bars, respectively) in M9 buffer for 24 h, and neurodegeneration was analyzed 1 and 24 h after treatment. Statistical significance was assessed by t test (*p < 0.05; NS, not significant). Error bars in A and B indicate SEM between independent experiments. C, Strong FIF in CEP neuronal cell bodies (arrows) in Pdat-1::CAT-2 worms is indicative of high levels of dopamine. Scale bar, 20 μm. D, Western analysis of TOR-2 expression levels in TOR-2-overexpressing worms. Actin was probed as a loading control for each line. E, Immunolocalization of torsinA and torsinA(ΔE302/303) using anti-torsinA antibody in CEP cell bodies (arrows) of torsinA-overexpressing worms. Only torsinA immunostaining images are shown because GFP images were similar to those depicted in Figure 1 E. Scale bar, 20 μm.
To rule out the possibility that the neurodegeneration observed was attributable to nonspecific cytotoxicity unassociated with the normal enzyme activity of CAT-2/TH, we used a known TH inhibitor, 3-IT, to directly counteract the overexpression of CAT-2 and therefore the overproduction of dopamine (Ginns et al., 1988). 3-IT essentially suppressed the neurodegeneration in L1 worms (Fig. 4B), yielding 88.3% wild-type neuronal morphology in 3-IT-treated worms compared with 52.5% in nontreated animals (p = 0.002). This suppression is highly reversible and specific to 3-IT, because 24 h after the removal of 3-IT, previously treated worms exhibited essentially the same percentage of dopamine-specific degeneration (58.3%) as nontreated controls (56.6%; p = 0.64) (Fig. 4B). Furthermore, to verify that CAT-2 overexpression results in higher dopamine levels in DA neurons, worms were stained for dopamine using a formaldehyde-induced fluorescence (FIF) technique (Lints and Emmons, 1999). Pdat-1::CAT-2 worms showed strong FIF compared with N2 control animals, indicating much higher dopamine levels in the Pdat-1::CAT-2 line (Fig. 4C) (data not shown). Western blotting and immunofluorescent microscopy were used to confirm the overexpression of TOR-2, torsinA, and mutant forms (Fig. 4D,E). These data suggest that although wild-type torsins may slightly suppress the neurodegeneration associated with increased dopamine levels, this suppression is not statistically significant. This observed trend toward enhanced DA neuronal survival in the presence of torsin overexpression may reflect a more generalized role for protection from oxidative damage as reported previously in cell cultures exposed to hydrogen peroxide (Kuner et al., 2003; Shashidharan et al., 2004).
Torsins protect against α-synuclein-induced neurodegeneration
Mounting evidence for a potentially critical balance of α-synuclein expression, folding, and degradation in maintaining DA neuron survival points to the importance of potential mediators of these processes. Moreover, overexpression of α-synuclein can induce neurodegeneration in the DA neurons of worms and other systems (Feany and Bender, 2000; Masliah et al., 2000; Ostrerova-Golts et al., 2000; Lasko et al. 2003; Singleton et al., 2003; Farrer et al., 2004). Although all other known proteins involved with DA metabolism are conserved in C. elegans, there is no α-synuclein homolog in worms. Therefore, we sought to determine whether both human and worm torsins can protect DA neurons from the neurodegenerative effects of ectopic human α-synuclein overexpression, because immunolocalization and fluorescence resonance energy transfer studies have shown that α-synuclein colocalizes with torsinA within Lewy bodies (Shashidharan et al., 2000; Sharma et al., 2001; O'Farrell et al., 2004).
Transgenic worms overexpressing human α-synuclein in DA neurons exhibited significant neurodegeneration. Interestingly, neurodegeneration was more pronounced as animals aged; young larval-staged animals had very little degeneration, whereas 7-d-old Pdat-1::α-synuclein animals exhibited striking neuronal loss (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). This marked delay in α-synuclein-induced degeneration may reflect a lag time between the onset of α-synuclein insult and the complete loss of DA neurons as animals age. Notably, co-overexpression of α-synuclein with either torsinA or TOR-2 yielded a considerable enhancement in DA neuron survival in 7-d-old animals. The percentage of worms maintaining all four CEPs increased significantly for torsinA and TOR-2 from 26.1% (Pdat-1::α-synuclein alone) to 57.3 and 57.8% (p < 0.01 for both; ANOVA), respectively, at 7 d (Fig. 5A; supplemental Fig. S2, available at www.jneurosci.org as supplemental material). However, in transgenic animals co-overexpressing either mutant torsinA(ΔE302/303) or TOR-2(Δ367) with α-synuclein, the percentage of wild-type neurons was not significantly different from Pdat-1::α-synuclein alone (42.2 and 38.1%, respectively; p = 0.162 and 0.342; ANOVA) (Fig. 5A; supplemental Fig. S2, available at www.jneurosci.org as supplemental material), although there appeared to be some residual protection exhibited by these mutant torsins.
Figure 5.
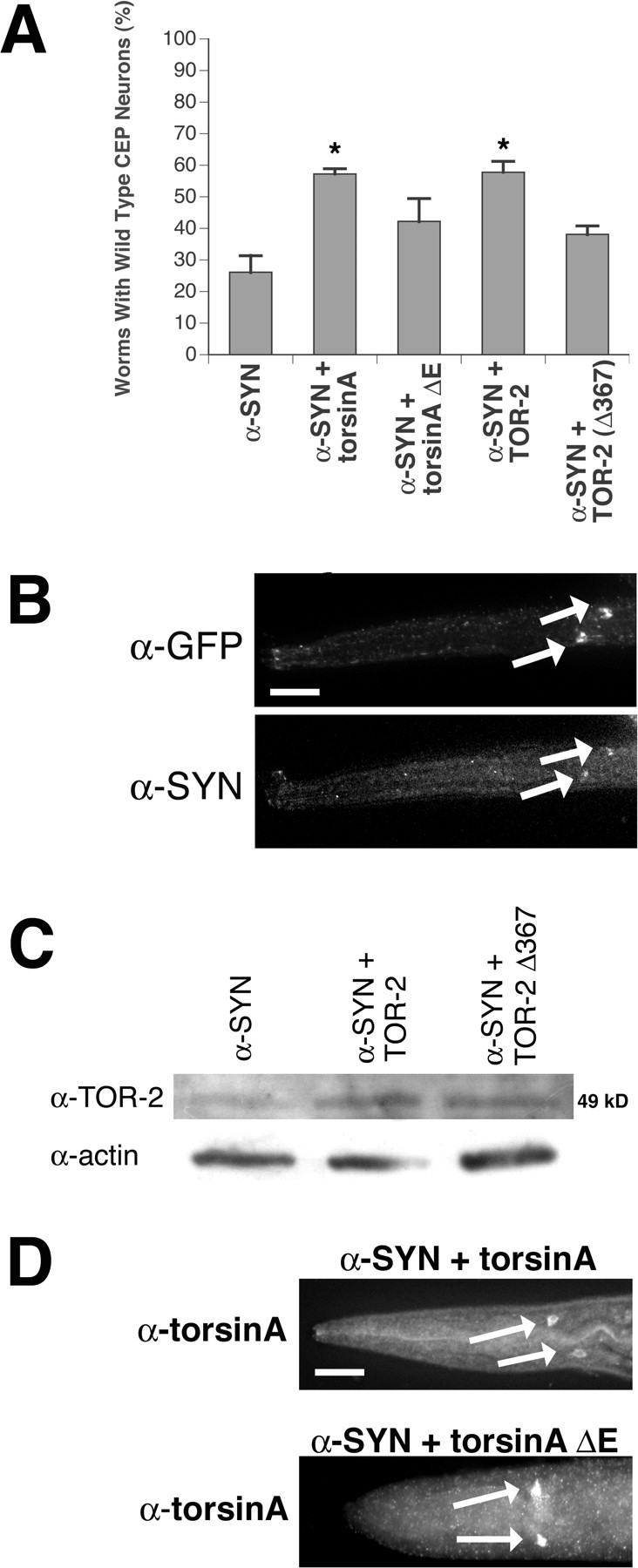
Torsin-mediated protection of DA neurons against α-synuclein overexpression. A, Overexpression of torsinA or TOR-2 protects against α-synuclein overexpression. The percentage of worms preserving all four wild-type CEP neurons at the 7-d-old stage was calculated for each of five α-synuclein-overexpressing lines (n = 30-40 per stable line). Statistical significance between Pdat-1::α-synuclein and each of four torsin/α-synuclein-overexpressing lines was assessed by the ANOVA Bonferroni's test (*p < 0.05). Error bars indicate SEM between independent experiments. B, Immunolocalization of α-synuclein within CEP cell bodies (arrows) of α-synuclein-overexpressing worms by α-synuclein antibody. The position of CEP cell bodies is depicted with α-GFP antibody. This worm is representative of all α-synuclein-overexpressing lines. Merged images were similar to those in Figure 1E. Scale bar, 20 μm. C, Western analysis of TOR-2 expression levels in TOR-2-overexpressing worms. Actin was probed for each line as a loading control. D, Immunolocalization of torsinA and torsinA(ΔE302/303) by torsinA antibody in CEP cell bodies (arrows) of torsinA-overexpressing worms. Only torsinA immunostaining images are shown because GFP images were similar to those depicted in Figure 1E. Scale bar, 20 μm.
Because multiple copies of the α-synuclein gene are sufficient to cause PD without additional mutations (Singleton et al., 2003; Farrer et al., 2004), we set out to determine whether the higher percentage of worms exhibiting neuroprotection in torsin-overexpressing lines was attributable to a protective effect or simply attributable to lower α-synuclein expression levels in our transgenic animals. A necessary limitation imposed by DNA microinjection of C. elegans is that transgene copy number is random. A commonly accepted method for circumventing this issue is the analysis of multiple stable lines. Thus, we analyzed three lines for each transgenic set generated. Indeed, in all torsin-overexpressing lines analyzed, wild-type neurons were maintained to a greater extent. Our analyses demonstrated that the average percentage of worms preserving all four CEP neurons in torsin-overexpressing lines was much higher than Pdat-1::α-synuclein lines. In fact, even the torsin-overexpressing lines exhibiting the lowest level of neuroprotection (54.5 and 51.7% for torsinA and TOR-2, respectively) still had higher percentages of DA neuron survival than the highest Pdat-1::α-synuclein line (36.7%). We demonstrated that α-synuclein was expressed in all α-synuclein-overexpressing lines by immunostaining (Fig. 5B). As before, the overexpression of human and worm torsin variants was confirmed by Western blotting and immunostaining (Fig. 5C,D). These data indicate torsins can mitigate the DA neurodegeneration induced by overexpression of α-synuclein, whereas the dystonia-associated mutant and its C. elegans counterpart retain partial, but greatly diminished, protective capacities.
Torsin protection against α-synuclein overexpression is independent of DAT-1
Torsins appear to provide robust protection against 6-OHDA via DAT-1 downregulation. Therefore, we proceeded to test whether the protection observed against α-synuclein overexpression is DAT-1 dependent. We also examined the possibility that α-synuclein-induced degeneration was itself dependent on the presence of this transporter. For this purpose, we created transgenic lines of worms overexpressing either Pdat-1::α-synuclein or Pdat-1::α-synuclein/Pdat-1::TOR-2 or Pdat-1::α-synuclein/Pdat-1::torsinA in a dat-1 knock-out background (Nass et al., 2002). Overexpression of α-synuclein still caused DA neurodegeneration in the absence of DAT-1 (46.5%), although to a lesser extent than when it is overexpressed in the wild-type N2 background (Figs. 5A, 6D). Thus, the presence of DAT-1 likely represents a source of additional dopamine for putative interactions with α-synuclein; this suggests an interplay between DAT-1 or dopamine with α-synuclein, either directly or indirectly, that leads to DA neuron loss in C. elegans. Regardless of the precise mechanism underlying the observed degeneration, torsins still provided significant protection as 75.0% of Pdat-1::α-synuclein/Pdat-1::TOR-2 (p = 0.011; ANOVA) and 68.7% (p = 0.044; ANOVA) of Pdat-1::α-synuclein/Pdat-1::torsinA worms preserved all four CEP neurons in the absence of dat-1 (Fig. 6A,D). α-Synuclein expression was confirmed by immunostaining using human α-synuclein antibody (Fig. 6B). TOR-2 overexpression was verified by Western blotting (Fig. 6C), and torsinA expression was confirmed by immunostaining (data not shown). In summary, these data indicate that α-synuclein-induced DA neurodegeneration is partially DAT-1 dependent and that torsins can protect DA neurons against α-synuclein overexpression in a manner independent of DAT-1 activity, as depicted in a comparison with wild-type animals retaining DAT-1 function (Fig. 6D).
Figure 6.
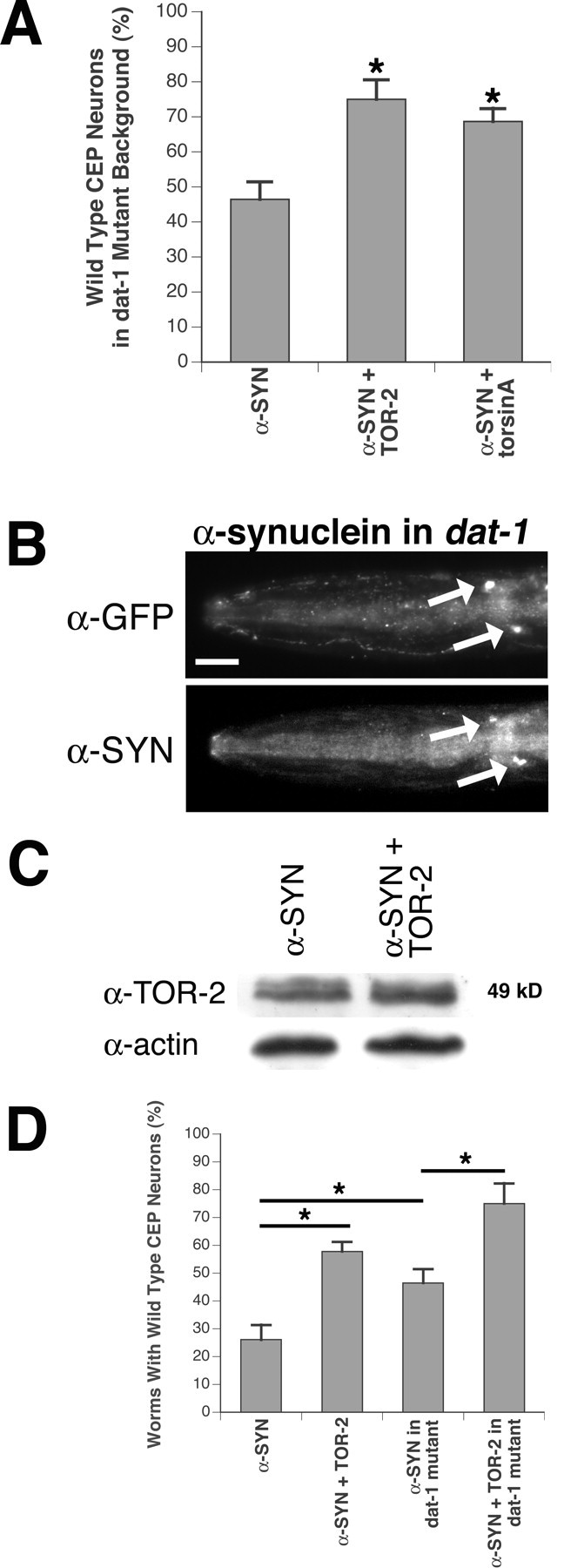
Torsin-mediated protection of DA neurons against α-synuclein overexpression in dat-1 knock-out animals. A, Overexpression of torsinA or TOR-2 protects against α-synuclein overexpression in dat-1 mutants. Percentage of 7-day-old worms preserving all four wild-type CEP neurons at each stage was calculated for each of three α-synuclein-overexpressing lines (n = 30-40 per stable line). Statistical significance between Pdat-1::α-synuclein and each of two α-synuclein/torsin-overexpressing lines was assessed by the ANOVA Bonferroni's test and is indicated with *p < 0.05. Error bars indicate SEM between independent experiments. B, Immunolocalization of α-synuclein within CEP cell bodies (arrows) of α-synuclein-overexpressing worms by α-synuclein antibody. The position of CEP cell bodies is depicted with GFP antibody. The worm depicted is representative of all α-synuclein-overexpressing lines. The merged images were similar to those in Figure 1 E. Scale bar, 20 μm. C, Western analysis of TOR-2 expression level in TOR-2-overexpressing worms. Actin was probed as a loading control. D, A comparison of DAT-1 depletion and torsin overexpression on α-synuclein-induced degeneration. Statistical significance was assessed by the ANOVA Bonferroni's test (*p < 0.05). Bars signify the worm strains compared in these analyses. Both overexpression of TOR-2 and loss of DAT-1 can contribute to neuroprotection against α-synuclein overexpression. Additionally, TOR-2 protection in DA neurons is independent of DAT-1. Error bars indicate SEM between independent experiments.
Hsp104 does not protect dopamine neurons from 6-OHDA toxicity
Phylogenetic analysis of torsin proteins indicates they share sequence similarity with members of the AAA+ family of ATPases, which includes molecular chaperones (Neuwald et al., 1999). We reported previously that torsins have the ability to function as molecular chaperones in ameliorating the formation of misfolded polyglutamine protein aggregates in C. elegans (Caldwell et al., 2003). Similarly, Satyal et al. (2000) demonstrated that another structurally similar ATPase, the yeast heat shock protein Hsp104, suppressed polyglutamine-induced toxicity and protein aggregation in worm body wall muscle cells. The marked cytoprotective nature of torsins in our studies led us to consider the distinctiveness of torsins in the context of other proteins within this same structural classification. Specifically, we wondered whether the functional relationship of torsin with DAT-1 is unique or common to other AAA+ family members. We therefore examined whether yeast Hsp104 could function to protect against 6-OHDA-induced neurodegeneration by overexpressing this AAA+ chaperone within C. elegans DA neurons. These experiments indicated that no protection was conferred by Hsp104 overexpression (1.1% with Hsp104 vs 0% wild-type neurons at 72 h; p = 1; ANOVA) (Fig. 7A). This is in contrast to overexpression of torsinA and TOR-2, in which 68.9 and 58.9% of DA neurons, respectively, were considered to be wild type at 72 h (p < 0.05; ANOVA) (Fig. 7A). The expression of Hsp104 was confirmed by Western blotting (Fig. 7B). Despite their functional equivalence in mediating protein folding, torsins, which are unique to metazoans, appear to differ mechanistically from structurally similar chaperones, possibly via their function in the turnover of polytopic proteins like DAT-1.
Discussion
Genetic predisposition to the effects of environmental perturbance may account for variance between affected and nonaffected individuals in several neurological diseases. Torsion dystonia and PD are two examples of movement disorders in which differences in mutational penetrance and sporadic occurrence play a significant role in onset and progression. In this regard, functional characterization of proteins that act in a neuroprotective capacity and are directly linked to DA dysfunction is significant. We have established a model system using known genetic components (α-synuclein, TH) in addition to a well established environmental trigger of DA neurodegeneration (6-OHDA) to investigate the neuroprotective capacity of torsins, an evolutionarily conserved protein family with functional and clinical significance for movement disorders, as well as DA neuron homeostasis.
How do torsins protect DA neurons against various cellular insults?
DA metabolism is believed to play a role in the selective toxicity of α-synuclein within neurons, because numerous lines of evidence have suggested that α-synuclein participates in regulating DA biosynthesis, vesicular storage, release, and reuptake (Sharon et al., 2001; Cabin et al., 2002; Lotharius and Brundin, 2002; Perez et al., 2002; Wersinger et al., 2003). Malfunction of α-synuclein, by virtue of mutation or overexpression, has been suggested as potentially disturbing a critical balance required for correct intracellular DA homeostasis. In this context, we investigated cellular conditions that may potentially impact DA neuron survival.
α-Synuclein toxicity is enhanced by the generation of ROS, and it has been shown that DA forms adducts with α-synuclein in vitro, stabilizing potentially toxic protofibrils (Conway et al., 2001; Xu et al., 2002). We have shown that α-synuclein-induced degeneration in C. elegans is at least partially DAT-1 dependent. In animals lacking functional DAT-1, dopamine reuptake is blocked, presumably limiting the intraneuronal interaction between α-synuclein and dopamine, which may have subsequent consequences for degeneration. It is also possible that the neurodegeneration observed is attributable to the misregulation of direct interactions between α-synuclein and DAT-1, because wild-type α-synuclein has been shown to modulate DAT activity (Sidhu et al., 2004).
Torsins exhibit potent neuroprotection from both 6-OHDA- and α-synuclein-induced degeneration. The mechanism by which torsins protect DA neurons from degeneration depends on how these specific insults exert their cellular effects. With respect to 6-OHDA-induced neurodegeneration, we initially hypothesized that torsins function in a capacity similar to other chaperone-like proteins to combat oxidative stress. For example, when Hsp27 is overexpressed, it responds to stressful oxidative stimuli by enhancing the degradation of ubiquitinated proteins by the 26S proteasome (Parcellier et al., 2003). The minor protection afforded by torsin overexpression in response to CAT-2-induced degeneration in vivo does not reflect a major role for torsins in directly combating the oxidative damage resulting from excess internal neuronal dopamine. Because dopamine and 6-OHDA are structurally related and induce cellular toxicity by similar means, an overt mechanistic difference between 6-OHDA treatment and CAT-2 overexpression is that 6-OHDA is applied exogenously and transported into DA neurons via the DAT, whereas CAT-2 overproduces dopamine intracellularly. Thus, we investigated the potential activity of torsins in modulating the DAT and demonstrated that TOR-2 overexpression downregulated DAT-1 levels in vivo, thereby providing an explanation for the dramatic neuroprotection observed after 6-OHDA exposure. An independent study performed by Torres et al. (2004) corroborates this finding by showing that torsinA overexpression downregulated the cell surface expression of the DAT in cell culture.
In contrast, torsin-mediated protection against α-synuclein overexpression may differ mechanistically from the aforementioned scenario, because it appears to be independent from DAT-1. Auluck et al. (2002) demonstrated that Hsp70 is able to suppress α-synuclein-induced neurotoxicity without changing the number, morphology, or distribution of the perinuclear protein inclusions in Drosophila, indicating that protofibrils, not the mature fibrils or aggregates, represent the neurotoxic species of α-synuclein. Because torsins have chaperone activity (McLean et al., 2002; Caldwell et al., 2003), these proteins can theoretically bind to prefibrillar α-synuclein intermediates, shielding them from aberrant interaction with cellular components, or engage in the clearance of fibrilized α-synuclein aggregates. Both of these scenarios are consistent with the finding that torsinA is physically associated with α-synuclein in Lewy bodies (Sharma et al., 2001; O'Farrell et al., 2004). The extremely small size of C. elegans DA neurons precludes our ability to visualize any putative inclusions, but regardless of whether prefibrillar α-synuclein intermediates or fibrilized α-synuclein aggregates are toxic, it is well accepted that the overexpression of α-synuclein is cytotoxic. In our study, torsins and α-synuclein were simultaneously co-overexpressed together in C. elegans DA neurons. Torsin activity is therefore concurrent with any supposed nucleation of α-synuclein protofibrils and thus may prevent oligomerization before it leads to neurodegeneration. The presence of DAT-1 in native conditions clearly is a source of DA that can contribute to cytotoxicity in the presence of α-synuclein; however, the ability of torsins to protect, even in a genetic null of dat-1, is indicative of functionality independent of the transporter. As discussed above, changes in dopamine metabolism may impact the ability of α-synuclein to induce degeneration. Thus, it is not surprising that torsins may also attenuate α-synuclein toxicity by decreasing the intraneuronal dopamine level through downregulation of DAT-1.
Consequences for EOTD
In patients with EOTD, disturbance of nigrostriatal DA function and changes in striatal DA turnover have been suggested as one potential causative factor underlying clinical symptoms associated with this movement disorder (Augood et al., 2002, 2004). A role for torsins in the folding, assembly, and/or trafficking of the DAT to the cell surface to ensure appropriate levels of this transporter may be significant in regulating neurotransmission. Our data showing that torsin-mediated protection from 6-OHDA-induced toxicity is similar between animals overexpressing TOR-2 and lacking DAT-1 lends support to this hypothesis. Thus, altered cell surface DAT levels may result in disturbed dopamine reuptake and signaling, which in turn may contribute to the clinical symptoms of dystonia. An association between EOTD mutant carriers and an increased risk of recurrent major depression has been reported recently (Heiman et al., 2004). It is intriguing to note, in the context of our observations, that the brains of patients with major depression exhibit a greater availability of DAT (Laasonen-Balk et al., 1999; Brunswick et al., 2003). Importantly, torsinA expression is not exclusive to DA neurons (Walker et al., 2001; Oberlin et al., 2004; Xiao et al., 2004). Therefore, the activity of torsins in modulating polytopic protein levels may have alternative consequences on different targets (i.e., transporters, channel proteins) within other neuronal subtypes.
EOTD-associated mutant torsinA(ΔE302/303) has been shown to bind wild-type torsinA, leading to its mislocalization to the nuclear envelope (NE) (Gonzalez-Alegre and Paulson, 2004; Goodchild and Dauer, 2004; Naismith et al., 2004; Torres et al., 2004). TorsinA may have undiscovered substrates at the NE, and the dominant nature of torsion dystonia may result from the loss of wild-type torsinA function or from its accumulation and aberrant activity when mislocalized, or both. These data correlate with our observed decrease in neuroprotection when mutant and wild-type torsins are coexpressed in C. elegans DA neurons, indicating a loss of torsin activity in the presence of torsinA(ΔE302/303).
Together, it is becoming readily apparent that the functional activity of torsin proteins as molecular chaperones has significant implications for both their native role in regulation of neurotransmission, as well as their aberrant activity in dystonia. To discern the mechanism of torsin-mediated protection, it is critical to evaluate the capacity of these proteins to act on a variety of cellular substrates. The development of functional assays for torsin activity in C. elegans allows for effector molecules to be systematically investigated by traditional genetic suppressor screens, genomic-scale knock-down of gene function by RNA interference and chemical library screening (Caldwell et al., 2004, 2005). The societal burden associated with movement disorders requires a comprehensive investigation of factors influencing neuroprotective proteins with the goal of understanding their contribution to disease etiology or for exploitation of their potential therapeutic value.
Footnotes
Worm strains used in this work were provided by the Caenorhabditis Genetics Center, which was funded by a grant from the National Institutes of Health (NIH). Sequencing was performed at The University of Alabama using an automated sequencer supported by a grant from the National Science Foundation. Movement disorders research in the Caldwell laboratory was supported by grants from the NIH, Bachmann-Strauss Dystonia and Parkinson Foundation, Dystonia Medical Research Foundation, Parkinson Disease Foundation, National Parkinson Foundation, Michael J. Fox Foundation, for Parkinson's Research, and by an Undergraduate Science Education Program grant from the Howard Hughes Medical Institute. We acknowledge the teamwork and cooperative spirit of all members of the Caldwell laboratory. We specifically thank Amber Clark, Iyare Izevbaye, and Shusei Hamamichi for assistance in vector and strain construction as well as Lucinda Carnell and Jing Yu for suggestions and insights. We are most grateful to Andy Fire, Karen Kustedjo, Ben Cravatt, Philipp Kahle, Jim Rand, Greg Mullen, Randy Blakely, Vijaya Ramesh, Xandra Breakefield, Yugi Kohara, and Susan Lindquist for generously providing strains, plasmids, and antibodies used in this work. We especially thank Gonzalo Torres and Marc Caron for sharing unpublished data, Janis O'Donnell for helpful discussions regarding 3-iodotyrosine methodologies, and Paul LeBlanc for assistance with formaldehyde-induced-fluorescence staining.
Correspondence should be addressed to Dr. Guy A. Caldwell, Department of Biological Sciences, The University of Alabama, Box 870344, Tuscaloosa, AL 35487-0344. E-mail: gcaldwel@bama.ua.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253801-12$15.00/0
References
- Augood SJ, Hollingworth Z, Albers DS, Yang L, Leung JC, Muller B, Klein C, Breakefield XO, Standaert DG (2002) Dopamine transmission in DYT1 dystonia: a biochemical and autoradiographical study. Neurology 59: 445-448. [DOI] [PubMed] [Google Scholar]
- Augood SJ, Hollingworth Z, Albers DS, Yang L, Leung J, Breakefield XO, Standaert DG (2004) Dopamine transmission in DYT1 dystonia. Adv Neurol 94: 53-60. [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM (2002) Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science 295: 809-810. [DOI] [PubMed] [Google Scholar]
- Berger K, Przedborski S, Cadet JL (1991) Retrograde degeneration of nigrostriatal neurons induced by intrastriatal 6-hydroxydopamine injection in rats. Brain Res Bull 26: 301-307. [DOI] [PubMed] [Google Scholar]
- Blum D, Torch S, Lambeng N, Nissou M, Benabid AL, Sadoul R, Verna JM (2001) Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson's disease. Prog Neurobiol 65: 135-172. [DOI] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabtidis elegans Genetics 77: 71-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunswick DJ, Amsterdam JD, Mozley PD, Newberg A (2003) Greater availability of brain dopamine transporter in major depression shown by [(99m)Tc] TRODAT-1 SPECT Imaging. Am J Psychiatry 160: 1836-1841. [DOI] [PubMed] [Google Scholar]
- Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, Lu B, Nussbaum RL (2002) Synaptic vesicle depletion correlates with attenuated synaptic response to prolonged repetitive stimulation in mice lacking alpha-synuclein. J Neurosci 22: 8797-8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell GA, Cao S, Sexton EG, Gelwix CC, Bevel JP, Caldwell KA (2003) Suppression of polyglutamine-induced protein aggregation in Caenorhabditis elegans by torsin proteins. Hum Mol Genet 12: 307-319. [DOI] [PubMed] [Google Scholar]
- Caldwell GA, Cao S, Gelwix CC, Sexton EG, Caldwell KA (2004) An animal model to discern torsin function: suppression of protein aggregation in C. elegans Adv Neurol 94: 79-85. [PubMed] [Google Scholar]
- Caldwell GA, Cao S, Izevbaye I, Caldwell KA (2005) Use of C. elegans to model human movement disorders. In: Animal models of movement disorders (LeDoux M, ed), pp 113-128. New York: Elsevier.
- Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury Jr PT (2000) Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc Natl Acad Sci USA 97: 571-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KA, Rochet JC, Bieganski RM, Lansbury Jr PT (2001) Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science 294: 1346-1349. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL (2003) Molecular pathways of neurodegeneration in Parkinson's disease. Science 302: 819-822. [DOI] [PubMed] [Google Scholar]
- Duerr JS, Frisby DL, Gaskin J, Duke A, Asermely K, Huddleston D, Eiden LE, Rand JB (1999) The cat-1 gene of Caenorhabditis elegans encodes a vesicular monoamine transporter required for specific monoamine-dependent behaviors. J Neurosci 19: 72-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S, Cohen G (1992) The oxidant stress hypothesis in Parkinson's disease: evidence supporting it. Ann Neurol 32: 804-812. [DOI] [PubMed] [Google Scholar]
- Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW (2004) Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol 55: 174-179. [DOI] [PubMed] [Google Scholar]
- Feany M, Bender WW (2000) A Drosophila model for Parkinson's disease. Nature 404: 394-398. [DOI] [PubMed] [Google Scholar]
- Foley P, Riederer R (2000) Influence of neurotoxins and oxidative stress on the onset and progression of Parkinson's disease. J Neurol 247 [Suppl 2]: II/82-II/94. [DOI] [PubMed] [Google Scholar]
- Ginns EI, Rehavi M, Martin BM, Weller M, O'Malley KL, LaMarca ME, McAllister CG, Paul SM (1988) Expression of human tyrosine hydroxylase cDNA in invertebrate cells using a baculovirus vector. J Biol Chem 263: 7406-7410. [PubMed] [Google Scholar]
- Gonzalez-Alegre P, Paulson HL (2004) Aberrant cellular behavior of mutant torsinA implicates nuclear envelope dysfunction in DYT1 dystonia. J Neurosci 24: 2593-2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild RE, Dauer WT (2004) Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc Natl Acad Sci USA 101: 847-852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman GA, Ottman R, Saunders-Pullman RJ, Ozelius LJ, Risch NJ, Bressman SB (2004) Increased risk for recurrent major depression in DYT1 dystonia mutation carrier. Neurology 63: 631-637. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Little MD, Bankiewicz K, Yang SC, Markey SP, Johannessen JN (1991) Selective retention of MPP+ within the monoaminergic systems of the primate brain following MPTP administration: an in vivo autoradiographic study. Neuroscience 40: 133-158. [DOI] [PubMed] [Google Scholar]
- Hewett J, Gonzalez-Agosti C, Slater D, Ziefer P, Li S, Bergeron D, Jacoby DJ, Ozelius LJ, Ramesh V, Breakefield XO (2000) Mutant torsinA, responsible for early-onset torsion dystonia, forms membrane inclusions in cultured neural cells. Hum Mol Genet 9: 1403-1413. [DOI] [PubMed] [Google Scholar]
- Hobert O (2002) PCR fusion-based approach to create reporter gene constructs for expression analysis in transgenic C. elegans Biotechniques 32: 728-730. [DOI] [PubMed] [Google Scholar]
- Hobert O, Moerman DG, Clark KA, Beckerle MC, Ruvkun G (1999) A conserved LIM protein that affects muscular adherens junction integrity and mechanosensory function in Caenorhabditis elegans J Cell Biol 144: 45-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger K, Kienzl E, Rumpelmair G, Riederer P, Stachelberger H, Ben-Shachar D, Youdim MB (1992) Iron-melanin complex in substantia nigra of parkinsonian brains: an X-ray microanalysis. J Neurochem 59: 1168-1171. [DOI] [PubMed] [Google Scholar]
- Jenner P, Dexter DT, Sian J, Schapira AH, Marsden CD (1992) Oxidative stress as a cause of nigral cell death in Parkinson's disease and incidental Lewy body disease. Ann Neurol [Suppl] 32: S82-S87. [DOI] [PubMed] [Google Scholar]
- Kumar R, Agarwal AK, Seth PK (1995) Free radical-generated neurotoxicity of 6-hydroxydopamine. J Neurochem 64: 1703-1707. [DOI] [PubMed] [Google Scholar]
- Kuner R, Teismann P, Trutzel A, Naim J, Richter A, Schmidt N, Bach A, Ferger B, Schneider A (2003) TorsinA protects against oxidative stress in COS-1 and PC12 cells. Neurosci Lett 350: 153-156. [DOI] [PubMed] [Google Scholar]
- Kustedjo K, Bracey MJ, Cravatt BF (2000) TorsinA and its torsion dystonia-associated mutant forms are lumen glycoproteins that exhibit distinct subcellular localizations. J Biol Chem 275: 27933-27939. [DOI] [PubMed] [Google Scholar]
- Laasonen-Balk T, Kuikka J, Viinamaki H, Husso-Saastamoinen M, Lehtonen J, Tiihonen J (1999) Striatal dopamine transporter density in major depression. Psychopharmacology (Berl) 144: 282-285. [DOI] [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I (1983) Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219: 979-980. [DOI] [PubMed] [Google Scholar]
- Lasko M, Vartiainen S, Moilanen A-M, Sirvio J, Thomas JH, Nass R, Blakley RD, Wong G (2003) Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human α-synuclein. J Neurochem 86: 165-172. [DOI] [PubMed] [Google Scholar]
- Lewis JA, Fleming JT (1995) Basic culture methods. Methods Cell Biol 48: 3-29. [PubMed] [Google Scholar]
- Lints R, Emmons S (1999) Patterning of dopaminergic neurotransmitter identity among Caenorhabditis elegans sensory neurons by a TGFβ family signaling pathway and a Hox gene. Development 126: 5819-5831. [DOI] [PubMed] [Google Scholar]
- Liou HH, Tsai MC, Chen CJ, Jeng JS, Chang YC, Chen SY, Chen RC (1997) Environmental risk factors and Parkinson's disease: a case-control study in Taiwan. Neurology 48: 1583-1588. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Brundin P (2002) Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci 3: 932-942. [DOI] [PubMed] [Google Scholar]
- Maries E, Dass B, Collier TJ, Kordower JH, Steece-Collier K (2003) The role of alpha-synuclein in Parkinson's disease: insights from animal models. Nat Rev Neurosci 4: 727-738. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L (2000) Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 287: 1265-1269. [DOI] [PubMed] [Google Scholar]
- McIntire SL, Garriga G, White J, Jacobson D, Horvitz HR (1992) Genes necessary for directed axonal elongation or fasciculation in C. elegans Neuron 8: 307-322. [DOI] [PubMed] [Google Scholar]
- McLean PJ, Kawamata H, Saadat S, Hewett J, Sharma N, Ueda K, Breakefield XO, Hyman BT (2002) TorsinA and heat shock proteins act as molecular chaperones: suppression of α-synuclein aggregation. J Neurochem 83: 846-854. [DOI] [PubMed] [Google Scholar]
- Naismith TV, Heuser JE, Breakefield XO, Hanson PI (2004) TorsinA in the nuclear envelope. Proc Natl Acad Sci USA 101: 7612-7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nass R, Hall DH, Miller DM, Blakely RD (2002) Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans Proc Natl Acad Sci USA 99: 3264-3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwald AF, Aravind L, Spouge JL, Koonin EV (1999) AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9: 27-43. [PubMed] [Google Scholar]
- Oberlin SR, Konakova M, Pulst S, Chesselet MF (2004) Development and anatomic localization of torsinA. Adv Neurol 94: 61-65. [PubMed] [Google Scholar]
- O'Farrell C, Hernandez DG, Evey C, Singleton AB, Cookson MR (2002) Normal localization of deltaF323-Y328 mutant torsinA in transfected human cells. Neurosci Lett 327: 75-78. [DOI] [PubMed] [Google Scholar]
- O'Farrell C, Lockhart PJ, Lincoln S, De Lucia M, Singleton AB, Dickson DW, Cookson MR (2004) Biochemical characterization of torsinB. Brain Res Mol Brain Res 127: 1-9. [DOI] [PubMed] [Google Scholar]
- Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farrer M, Wolozin B (2000) The A53T α-synuclein mutation increase iron-dependent aggregation and toxicity. J Neurosci 20: 6048-6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO (1997) The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet 17: 40-48. [DOI] [PubMed] [Google Scholar]
- Parcellier A, Schmitt E, Gurbuxani S, Seigneurin-Berny D, Pance A, Chantome A, Plenchette S, Khochbin S, Solary E, Garrido C (2003) HSP27 is a ubiquitin-binding protein involved in I-kappaBalpha proteasomal degradation. Mol Cell Biol 23: 5790-5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez RG, Waymire JC, Lin E, Liu JJ, Guo F, Zigmond MJ (2002) A role for α-synuclein in the regulation of dopamine biosynthesis. J Neurosci 22: 3090-3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276: 2045-2047. [DOI] [PubMed] [Google Scholar]
- Satyal SH, Schmidt E, Kitagawa K, Sondheimer N, Lindquist S, Kramer JM, Morimoto RI (2000) Polyglutamine aggregates alter protein folding homeostasis in C. elegans Proc Natl Acad Sci USA 97: 5750-5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, Hewett J, Ozelius LJ, Ramesh V, McLean PJ, Breakefield XO, Hyman BT (2001) A close association of torsinA and alpha-synuclein in Lewy bodies: a fluorescence resonance energy transfer study. Am J Pathol 159: 339-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon R, Goldberg MS, Bar-Josef I, Betensky RA, Shen J, Selkoe DJ (2001) Alpha-synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc Natl Acad Sci USA 98: 9110-9115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashidharan P, Good PF, Hsu A, Perl DP, Brin MF, Olanow CW (2000) TorsinA accumulation in Lewy bodies in sporadic Parkinson's disease. Brain Res 877: 379-381. [DOI] [PubMed] [Google Scholar]
- Shashidharan P, Paris N, Sandu D, Karthikeyan L, McNaught KS, Walker RH, Olanow CW (2004) Overexpression of torsinA in PC12 cells protects against toxicity. J Neurochem 88: 1019-1025. [DOI] [PubMed] [Google Scholar]
- Sidhu A, Wersinger C, Vernier P (2004) Alpha-synuclein regulation of the dopaminergic transporter: a possible role in the pathogenesis of Parkinson's disease. FEBS Lett 565: 1-5. [DOI] [PubMed] [Google Scholar]
- Simon H, LeMoal M, Galey D, Cardo B (1974) Selective degeneration of central dopaminergic systems after injection of 6-hydroxydopamine in the ventral mesencephalic tegmentum of the rat. Demonstration by the Fink-Heimer strain. Exp Brain Res 20: 375-384. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. (2003) Alpha-synuclein locus triplication causes Parkinson's disease. Science 302: 841. [DOI] [PubMed] [Google Scholar]
- Sulston J, Dew M, Brenner S (1975) Dopaminergic neurons in the nematode Caenorhabditis elegans J Comp Neurol 163: 215-226. [DOI] [PubMed] [Google Scholar]
- Tan EK, Khajavi M, Thornby JI, Nagamitsu S, Jankovic J, Ashizawa T (2000) Variability and validity of polymorphism association studies in Parkinson's disease. Neurology 55: 533-538. [DOI] [PubMed] [Google Scholar]
- Tanner CM (1992) Occupational and environmental causes of parkinsonism. Occup Med 7: 503-513. [PubMed] [Google Scholar]
- Torres GE, Sweeney AL, Beaulieu JM, Shashidharan P, Caron MG (2004) Effect of torsinA on membrane proteins reveals a loss of function and a dominant-negative phenotype of the dystonia-associated DeltaE-torsinA mutant. Proc Natl Acad Sci USA 101: 15650-15655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker RH, Brin MF, Sandu D, Gujjari P, Hof PR, Olanow WC, Shashidharan P (2001) Distribution and immunohistochemical characterization of torsinA immunoreactivity in rat brain. Brain Res 900: 348-354. [DOI] [PubMed] [Google Scholar]
- Wersinger C, Prou D, Vernier P, Sidhu A (2003) Modulation of dopamine transporter function by alpha-synuclein is altered by impairment of cell adhesion and by induction of oxidative stress. FASEB J 17: 2151-2153. [DOI] [PubMed] [Google Scholar]
- Xiao J, Gong S, Zhao Y, LeDoux MS (2004) Developmental expression of rat torsinA transcript and protein. Brain Res Dev Brain Res 152: 47-60. [DOI] [PubMed] [Google Scholar]
- Xu J, Kao SY, Lee FJ, Song W, Jin LW, Yanker BA (2002) Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson's disease. Nat Med 8: 600-606. [DOI] [PubMed] [Google Scholar]