

Abstract
Chronic opioid-induced drug dependence and withdrawal syndrome after opioid cessation remain a severe obstacle in clinical treatment of chronic pain and opioid drug addiction. One of the key symptoms during opioid withdrawal is a state of sensitized pain. The most significant molecular adaptation induced by chronic opioids in the brain is upregulation of the cAMP-signaling pathway. Although the cAMP system is known to have multiple effects on central neuron functions, how its upregulation mediates behavioral opioid dependence and withdrawal-induced pain in vivo remains unclear. In this study, we demonstrate that withdrawal from chronic morphine significantly upregulates the mRNA level of adenylyl cyclase (AC) VI and VIII isoforms and immunoreactivity of ACV/VI in the nucleus raphe magnus (NRM), a brainstem site critically involved in opioid modulation of pain. In cellular studies of NRM neurons containing μ-opioid receptors, we show that morphine withdrawal significantly increases glutamate synaptic transmission via a presynaptic mechanism mediated by an upregulated cAMP pathway. Morphine withdrawal also enhances the hyperpolarization-activated current in these neurons by increased intracellular cAMP. Both of the withdrawal-induced cAMP actions increase the excitability of these μ-receptor-containing neurons, which are thought to facilitate spinal pain transmission. Furthermore, in morphine-dependent rats in vivo, blocking the cAMP pathway significantly reduces withdrawal-induced pain sensitization. These results illustrate neurobiological mechanisms for the cAMP-mediated withdrawal pain and provide potential therapeutic targets for the treatment of opioid dependence and withdrawal-related problems.
Keywords: glutamate EPSCs, Ih, adenylyl cyclase, nucleus raphe magnus, hyperalgesia, opioid dependence
Introduction
Repeated use of opioid drugs, either for clinical treatment of chronic pain or as a result of drug abuse, induces neuroadaptive changes in the brain that form the neurobiological basis for drug dependence and addiction (Nestler, 2001; Williams et al., 2001; Chao and Nestler, 2004). Abrupt abstinence or withdrawal from opioid drugs causes a series of severe adverse symptoms, which keep drug-dependent individuals craving continued opioids. One of the core withdrawal symptoms is an increase in pain sensitivity (pain sensitization or hyperalgesia), which has been well documented both in animal studies (Mao et al., 1995; Nestler and Aghajanian, 1997) and in clinical reports, including those of opioid addicts (Doverty et al., 2001; Angst et al., 2003; Compton et al., 2003). Despite decades of research the underlying neurobiological mechanisms for opioid dependence and withdrawal remain unclear, leaving current treatment for opioid dependence and withdrawal mostly empirical with limited success. Early studies, using in vitro models of cell lines, showed that chronic morphine induces a compensatory increase in the adenylyl cyclase (AC) activity and intracellular cAMP concentration, and with morphine withdrawal the cAMP concentration overshoots to above premorphine levels (Sharma et al., 1975; Brandt et al., 1976). Subsequently, this chronic morphine-induced molecular adaptation of the cAMP cascade has been confirmed in many opioid addiction-related brain areas, and such a cAMP adaptation has been widely related to opioid dependence and withdrawal (for review, see Nestler and Aghajanian, 1997; Nestler, 2001; Williams et al., 2001; Watts, 2002; Chao and Nestler, 2004). Previous in vitro studies that used cAMP analogs have identified several direct cAMP-mediated actions on central neurons, including increasing synaptic transmission and augmenting the hyperpolarization-activated current (Ih) (for review, see Williams et al., 2001). It has been demonstrated recently that GABA synaptic transmission is augmented via an upregulated cAMP cascade in several brain areas of morphine-withdrawn rats (Bonci and Williams, 1997; Ingram et al., 1998; Jolas et al., 2000). The behavioral significance of this GABA synaptic adaptation remains to be demonstrated. Although the cAMP pathway has been implicated in the expression of many somatic symptoms of opioid withdrawal (Punch et al., 1997), the mechanisms by which this adaptive upregulation of the cAMP pathway mediates the behavioral syndrome of opioid dependence and withdrawal remain unclear. Especially lacking are mechanisms linking chronic opioid-induced cAMP upregulation in vivo to the behavioral symptom of pain sensitization during opioid withdrawal. In this study, we used a rat model of opioid dependence and withdrawal to investigate effects of chronic morphine on AC expression levels and, consequently, on cAMP-mediated synaptic actions and pain behaviors, focusing on neurons in the nucleus raphe magnus (NRM), a brainstem site critically involved in opioid modulation of pain transmission in the spinal cord (Scholz and Woolf, 2002; Fields, 2004).
Materials and Methods
Chronic morphine treatment and morphine withdrawal. Rats were treated with chronic morphine for 6 d by subcutaneous implantation of morphine pellets as described previously (Pan, 2003a). Placebo pellets were used as controls. For real-time PCR and Western blotting experiments, naloxone (1 mg/kg, i.p.) was injected on day 7 in both morphine- and placebo-treated rats 5-7 h before tissue samples were taken. For recordings, the morphine withdrawal was induced either by the application of naloxone (1 μm) to morphine-dependent brain slices maintained in 5 μm morphine in vitro or by the incubation of morphine-dependent slices in a morphine-free solution for at least 3 h (spontaneous withdrawal).
Relative quantitative real-time PCR and Western blotting. Tissue samples containing the NRM were taken from morphine-withdrawn or placebo control rats (n = 8). Total RNA was prepared by using an RNAqueous-4 PCR kit (Ambion, Austin, TX) and reverse transcribed by using a SYBR Green reverse transcription (RT)-PCR Reagents kit (Applied Biosystems, Foster City, CA). The sequences of primers designed with the Primer Express software (Applied Biosystems) included the following: for ACI, CCAGGGAAGAATTCAGGTGACT and GCCTCTGCACACAAACTGGTAT; for ACV, AACCAGGTGAACGCATGTCA and CTCTGGGAAGTTGCAGTTGGA; for ACVI, CTGCCTCAGCCTGCTTATGTG and GGAGTCCTGGCGGAAGCT; for ACVIII, TTGCCCTGACTGAAAGCATACA and CTGATGCCAATACGGAGTTCAAA; and for β-actin, CACCCACACTGTGCCCATCTA and ACGCTCGGTCAGGATCTTCAT.
Primer concentrations were optimized. Real-time PCRs were performed by using SYBR Green I Master Mix and the ABI 7000 Sequence Detection System (Applied Biosystems). No other products were amplified, as indicated by a single peak in the melting curves for each sample. The β-actin gene was used as an internal normalizer. The threshold cycle (Ct) values for each sample were determined with the amplification plots within the logarithmic phase. The efficiency of amplification for the primers was comparable, as indicated by the lines in plots of ΔCt values against diluted cDNA (slopes < 0.1). Data were analyzed by using the 2-ΔΔCt method described by Livak and Schmittgen (2001). The AC expression was normalized to β-actin expression for each AC isomer. Statistical data are presented as mean ± SD. For Western blotting, the NRM tissues from placebo- and morphine-treated rats (n = 6-8 rats in each group) were homogenized on ice for 10 min in a buffer containing 50 mm Tris-Cl, 150 mm NaCl, 0.02 mm NaN2, 100 μg/ml phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, and 1% Triton X-100. The lysates were centrifuged at 14,000 rpm for 10 min at 4°C, and the supernatant was used for SDS-PAGE. Protein concentrations were determined by using the Bio-Rad (Hercules, CA) protein assay kit. The samples were treated with SDS sample buffer at 95°C for 5 min before electrophoresis with a 10% SDS polyacrylamide gel. Approximately 20 μg of total proteins was loaded. Proteins then were separated and electrotransferred onto nitrocellulose membrane. Samples were incubated overnight at 4°C with a primary antibody for AC (ACI, 1:1000; ACV/VI, 1:2000; ACVIII, 1:2000; Santa Cruz Biotechnology, Santa Cruz, CA) and for β-actin (1:10,000; Sigma-Aldrich, St. Louis, MO). ACV and AVI isoforms have greater sequence homology than other AC types (Katsushika et al., 1992), and the antibody from Santa Cruz Biotechnology (H-130) binds both ACV and ACVI. After three 10 min washes with TBS containing 0.2% Tween, the blots were incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (1:10,000; Amersham Biosciences, Arlington Heights, IL) for 1 h at room temperature with constant agitation and then washed and stained with 3,3′-diaminobenzidine tetrahydrochloride. The membranes were developed by the enhanced chemiluminescence (ECL) Plus kit (Amersham Biosciences). The intensity of bands was captured digitally and analyzed quantitatively with Kodak (Rochester, NY) 1D software, version 3.5.4. The immunoreactivity of all AC isoforms was normalized to that of β-actin.
Whole-cell voltage-clamp recording. Approximately 192 rats were used for whole-cell recordings in NRM slices in vitro. The brain of a male neonatal (9-14 d old) Wistar rat was cut in a vibratome in cold (4°C) physiological saline to obtain brainstem slices (200 μm thick) containing the NRM. A single slice was submerged in a shallow recording chamber and perfused with preheated (35°C) physiological saline [containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 11 glucose, and 25 NaHCO3, saturated with 95% O2/5% CO2, pH 7.2-7.4]. Slices were maintained at ∼35°C throughout the recording experiment. Neonatal rats were used for better visualization of neurons in brain slices with an infrared Nomarski microscope. It has been demonstrated that the physiological and pharmacological properties of neurons from these young rats are indistinguishable from those of adult rats (Pan et al., 1997). Visualized whole-cell voltage-clamp recordings were made from identified neurons with a glass pipette (resistance, 3-5 MΩ) filled with a solution containing the following (in mm): 126 potassium gluconate, 10 NaCl, 1 MgCl2, 11 EGTA, 10 HEPES, 2 ATP, and 0.25 GTP, pH adjusted to 7.3 with KOH; osmolarity, 280-290 mOsm/L. An Axopatch 1-D amplifier and AxoGraph software 4.7 (Axon Instruments, Union City, CA) were used for data acquisition and on-line/off-line data analyses. A seal resistance of ≥2GΩ and an access resistance of ≤15 MΩ were considered acceptable. Series resistance was compensated optimally. The access resistance was monitored throughout the experiment. All NRM cells that were recorded were classified into either a cell type that contains the μ-opioid receptor (termed “secondary cell”) or the other cell type that lacks the μ-receptor (termed “primary cell”), according to the criteria described in our previous study (Pan et al., 1990). Electrical stimuli of constant current (0.25 ms, 0.2-0.4 mA) were used to evoke EPSCs with bipolar-stimulating electrodes placed lateral (200-400 μm) to the recording electrode within the NRM. A pair of EPSCs was evoked by two stimuli with an interval of 40 ms. Paired pulse ratio (PPR) was calculated by dividing the amplitude of the second EPSC by the first one. The PPR has been used widely to determine synaptic action site (Manabe et al., 1993; Perkel and Nicoll, 1993; Dobrunz and Stevens, 1997; Ungless et al., 2001; Bie and Pan, 2003). It has an inverse relationship with the probability of presynaptic neurotransmitter release. Thus a decrease in the PPR indicates an increase in presynaptic transmitter release and vice versa. Miniature EPSCs (mEPSCs) were obtained in 60 s epochs in the presence of tetrodotoxin (TTX; 1 μm). The AxoGraph software was used to detect and measure the amplitude and intervals of the synaptic events and to analyze their distribution data. The amplitude of the Ih was determined by the difference in the current values between the beginning and the end of a 2 s hyperpolarizing voltage step from a holding potential of -50 mV. Ih data were fit with the Boltzmann equation, and then the voltage value (V1/2) at which one-half of the maximum conductance is activated was determined from the Ih activation curve (Pan, 2003b). In some experiments, NRM slices from chronic morphine-treated rats were incubated in the AC inhibitor cis-N-(2-phenylcyclopentyl)azacyclotridec-1-en-2-amine (MDL12330a; 100 μm) or in the protein kinase A (PKA) inhibitor N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H89; 10 μm) for at least 1 h. Firing of action potentials was evoked by a 3 s depolarizing current step in control, in morphine-withdrawn slices, and in withdrawn slices treated with the Ih blocker 4-(N-ethyl-N-phenylamino)-1,2-dimethyl-6-(methylamino)pyridinium chloride (ZD7288; 10 μm for >1 h). ZD7288 at 25-100 μm has been shown to selectively block the Ih in many types of central neurons, including μ-receptor-lacking neurons in the NRM (Harris and Constanti, 1995; Khakh and Henderson, 1998; Takigawa et al., 1998; Larkman and Kelly, 2001; Mellor et al., 2002; Pan, 2003b). Statistical analyses of the mEPSCs were performed by using Statview software (SAS Institute, Cary, NC) with the Kolmogorov-Smirnov test. Other numeral data were analyzed statistically with Student's t tests and presented as mean ± SEM. Generally, only one neuron was recorded in one slice, and one to two slices from each rat were used for recording. Drugs generally were applied via the bath solution unless otherwise specified.
Microinjection and behavioral experiments. Male Wistar rats (250-300 g) were maintained lightly anesthetized in a stereotaxic apparatus with a constant intravenous infusion of methohexital (10 mg/ml at 0.8 ml/h). A 26-gauge double-guide cannula was aimed at the NRM (anteroposterior, -10.0 from the bregma; lateral, 0; ventral, -10.5 from dura with the skull leveled) (Paxinos and Watson, 1986). Control placement of the cannula was 1.5 mm dorsal to the NRM placement. Tail-flick latency to a radiant heat stimulus was measured every 2 min. The heat intensity was set to elicit stable baseline latencies with a cutoff time of 12 s. After six baseline trials, the withdrawal-induced pain sensitization (hyperalgesia) was produced by an injection of naloxone (3 μg/kg, i.v.) in chronic morphine-treated rats. Drugs were microinjected into the NRM through a 33-gauge double-injector cannula with an infusion pump at a rate of 0.5 μl/min. ZD7288 or AP-5 and CNQX was microinjected immediately before naloxone injection. MDL12330a, H89, or recombinant protein-cAMP (Rp-cAMP) was microinjected 40 min before naloxone. Drug effects were analyzed statistically by an ANOVA for repeated measures and the Tukey-Kramer test of post hoc analysis. For histological verification of cannula placements, 0.5 μl of methylene blue was injected through the double-injector cannula. The rat brain was fixed in 4% paraformaldehyde for 30 min, and then NRM slices (200 μm thick) were cut and examined for cannula placement under a microscope. Data from rats in which cannula placements were outside of the NRM area were excluded from the results. Morphine and placebo pellets were kindly supplied by the National Institute on Drug Abuse. All other drugs were purchased either from Sigma-Aldrich or from Tocris Cookson (Ellisville, MO).
Results
Morphine withdrawal increases mRNA level and immunoreactivity of AC
We first used the real-time RT-PCR technique to examine whether withdrawal from chronic morphine changed the expression of AC at the mRNA level in the NRM. We focused on ACI, ACV, ACVI, and ACVIII, the four AC isoforms that are inhibited by acute opioids (Avidor-Reiss et al., 1997). The amplification plots demonstrated that the cycle thresholds for ACVI and ACVIII mRNAs in morphine-withdrawn rats were lower than those in placebo control rats. The average difference, or ΔCt, was -2.1 for ACVI and -1.9 for ACVIII, indicating that ACVI and ACVIII mRNA levels were upregulated in the NRM area during morphine withdrawal (Fig. 1A, B). When normalized to internal reference β-actin, the mRNA level of both ACVI and ACVIII was increased significantly in withdrawn rats (Fig. 1C). There was a small increase in ACV mRNA, but the difference did not reach statistical significance. ACI mRNA was not changed. To determine changes in these AC isoforms at the protein level, we used Western blotting for AC immunoreactivity. After being normalized to β-actin, the immunoreactivity of ACV/VI was enhanced significantly in the NRM from morphine-withdrawn rats, whereas ACI and ACVIII immunoreactivity was unchanged (Fig. 1D, E). Thus it appears that morphine withdrawal increases ACVI and ACVIII mRNA levels and ACV/VI protein levels in the NRM.
Figure 1.

Morphine withdrawal upregulates AC isoforms in the NRM area. A, B, Representative amplification plots of real-time PCRs for ACVI (A) and ACVIII (B). Con, Control. C, Fold changes in the mRNA of four AC isomers during morphine withdrawal. Error bars indicate SDs (n = 4 rats in placebo control and in withdrawal groups). D, Representative lanes of Western blots for ACI, ACV/VI, ACVIII, and corresponding β-actin from control and withdrawn rats. E, Group data of Western blots showing the percentage of changes in withdrawn rats from controls after normalization to β-actin. *p < 0.05; **p < 0.01. Error bars indicate SEs (n = 6-8 rats in each group).
Morphine withdrawal increases glutamate synaptic transmission through upregulated cAMP cascade
We next determined whether cAMP analogs could modulate glutamate synaptic transmission in normal NRM slices from morphine-naive rats, focusing on a class of NRM cells (termed secondary cells) that express μ-opioid receptors (Pan et al., 1997). In these cells methionine-enkephalin (10 μm), an endogenous opioid peptide acting on the μ-opioid receptor, produced a membrane hyperpolarization of 24 ± 4.6 mV (n = 14), an effect similar to what we have reported previously (Pan et al., 1997, 2000). Forskolin (10 μm), an AC activator, significantly increased the amplitude of glutamate-mediated evoked EPSCs in these cells sensitive to μ-receptor agonists (control, 95.1 ± 7.3 pA; forskolin, 144.1 ± 14.2 pA; p < 0.01; n = 12) (Fig. 2A). The membrane-permeable cAMP analog 8-bromo-cAMP (8-br-cAMP; 1 mm) also increased the EPSC amplitude (control, 110.9 ± 14.8 pA; 8-br-cAMP, 166.6 ± 19.2 pA; p < 0.01; n = 4). To determine the synaptic site of this cAMP action, we first used the paradigm of PPR. Forskolin significantly decreased the PPR of glutamate EPSCs (control, 1.73 ± 0.21; forskolin, 1.03 ± 0.14; p < 0.01; n = 5) (Fig. 2B) and so did 8-br-cAMP (control, 1.70 ± 0.08; 8-br-cAMP, 1.10 ± 0.07; p < 0.01; n = 4) (Fig. 2C). This indicates that a presynaptic site is involved in the cAMP action on glutamate release. Forskolin also produced a significant increase in the frequency, but not the amplitude, of the mEPSCs (for frequency: control, 5.70 ± 1.03 Hz; forskolin, 8.46 ± 1.07 Hz; p < 0.05; for amplitude: control, 20.76 ± 2.04 pA; forskolin, 20.64 ± 2.40 pA; p > 0.05; n = 5) (Fig. 2D). The opioid antagonist naloxone (1 μm) had no effect on the frequency or amplitude of the mEPSCs in these control cells (Fig. 2D). Similar effects of forskolin and 8-br-cAMP on EPSCs and PPR were observed in the other type of NRM cells that lack μ-opioid receptors (data not shown). These results indicate that activation of the cAMP pathway enhances glutamate synaptic transmission presynaptically by increasing glutamate release in NRM cells.
Figure 2.

Activation of cAMP pathway presynaptically increases glutamate synaptic transmission in μ-receptor-containing NRM cells from control rats. A-C, Superimposed glutamatergic EPSCs (A) and EPSC pairs normalized to the first EPSC (B, C) in control and in forskolin (10 μm) from the same cell (B) or recorded with an 8-br-cAMP-filled (1 mm) pipette (C). D, Cumulative frequency distribution of glutamate mEPSCs in control, in naloxone (1 μm), and in naloxone plus forskolin. cAMP, 8-Br-cAMP; Con, control; Forsk, forskolin; Nal, naloxone.
Because, as shown above, morphine withdrawal likely upregulates the cAMP cascade and cAMP analogs enhance glutamate synaptic transmission in NRM cells, morphine withdrawal should increase glutamate synaptic transmission in these cells. We then tested this hypothesis by using naloxone-precipitated and morphine-abstinent withdrawn slices from chronic morphine-treated rats. In morphine-dependent slices, naloxone (1 μm) significantly increased the amplitude of glutamate EPSCs in the μ-receptor-containing cells (dependent, 138.3 ± 20.1 pA; plus naloxone, 191.5 ± 29.6 pA; p < 0.01; n = 11) (Fig. 3A). It also reduced the PPR in these cells (dependent, 1.37 ± 0.13; plus naloxone, 1.08 ± 0.09; p < 0.01; n = 11) (Fig. 3C). In control slices from placebo-treated rats, naloxone was ineffective on the EPSC or the PPR in these cells (control, 1.94 ± 0.29; plus naloxone, 2.02 ± 0.36; p > 0.05; n = 8). In contrast to the effect of naloxone in these μ-receptor-containing cells, naloxone had no effect in the other type of cells lacking the μ-receptor from morphine-treated rats (treated, 128.6 ± 16.7 pA; plus naloxone, 123.4 ± 17.1 pA; p > 0.05; n = 8) (Fig. 3B). Furthermore, naloxone significantly increased the frequency, but not the amplitude, of the mEPSCs in the μ-receptor-containing cells (for frequency: dependent, 5.13 ± 1.3 Hz; plus naloxone, 9.47 ± 1.56 Hz; p < 0.05; for amplitude: dependent, 20.51 ± 0.7 pA; plus naloxone, 21.52 ± 1.01 pA; p > 0.05; n = 8) (Fig. 3E,F). Naloxone was without effect either on the PPR or on the mEPSCs in the NRM cells lacking μ-receptors. These results suggest that naloxone-precipitated withdrawal enhances glutamate synaptic transmission presynaptically by increasing glutamate release only in μ-receptor-containing NRM cells.
Figure 3.

Naloxone-precipitated morphine withdrawal presynaptically increases glutamate synaptic transmission by cAMP upregulation. A, B, Glutamate EPSCs in control, in naloxone, and in naloxone plus forskolin from a μ-receptor-containing cell (A) and from a μ-receptor-lacking cell (B) of a morphine-dependent rat. Abbreviations are as in Figure 2. The following results were obtained from μ-receptor-containing cells. C, Normalized EPSC pairs in control and in naloxone in a morphine-dependent cell. D, Percentage increase by forskolin of the EPSC amplitudes in control (open circles; n = 12) and in withdrawn cells (filled circles; n = 8). *p < 0.05; **p < 0.01. E, F, Frequency (E) and amplitude (F) distributions of the mEPSCs in control, in naloxone, and in naloxone plus forskolin from the same morphine-dependent cell. G-I, Glutamate EPSCs (G, superimposed) and normalized EPSC pairs (H, I, staggered) in control and in naloxone from morphine-dependent cells treated with MDL12330a (MDL; 100 μm) or H89 (10 μm).
Next we examined whether this naloxone withdrawal-induced enhancement of glutamate neurotransmission was mediated by an upregulated cAMP cascade. We found that the EPSC-enhancing effect of forskolin in the μ-receptor-expressing cells was significantly larger in withdrawn slices than that in control slices (Fig. 3A,D), likely indicating a sensitized or upregulated AC system (Bonci and Williams, 1997; Ingram et al., 1998). Forskolin also increased the mEPSC frequency, but not the amplitude, in the withdrawn cells (Fig. 3E,F). In morphine-dependent slices treated with the AC inhibitor MDL12330a (100 μm), naloxone was no longer effective on the evoked EPSC amplitude (dependent/MDL12330a, 129.4 ± 17.3 pA; plus naloxone, 126.9 ± 20.2 pA; p > 0.05; n = 6) (Fig. 3G) or on the PPR (dependent/MDL12330a, 1.53 ± 0.07; plus naloxone, 1.50 ± 0.11; p > 0.05; n = 6) (Fig. 3H). Similar treatment with the PKA inhibitor H89 (10 μm) also abolished the effect of naloxone (for EPSC: dependent/H89, 123.2 ± 13.5 pA; plus naloxone, 128.4 ± 9.9 pA; p > 0.05; n = 8; for PPR: dependent/H89, 1.46 ± 0.11; plus naloxone, 1.39 ± 0.13; p > 0.05; n = 7) (Fig. 3I). In addition, consistent results were obtained in NRM spontaneous withdrawal slices. In these withdrawn slices, μ-receptor-containing cells displayed a significantly larger EPSC (control, 111.5 ± 7.8 pA; n = 76; withdrawn, 140.5 ± 8.2 pA; n = 27; p < 0.01) (Fig. 4A, B). They also exhibited significantly smaller PPRs than those in control slices (control, 1.89 ± 0.13, n = 33; withdrawn, 1.12 ± 0.04, n = 15; p < 0.01), indicating enhanced synaptic release of glutamate in these spontaneously withdrawn cells. Treatment of these withdrawn slices with MDL12330a (100 μm) significantly reduced the withdrawal effect on the PPR (plus MDL12330a, 1.59 ± 0.08; p < 0.01; n = 16) (Fig. 4C,D). These results obtained from both naloxone-induced and spontaneous withdrawal slices indicate the involvement of the cAMP pathway in the increased glutamate neurotransmission.
Figure 4.

Morphine abstinence-induced spontaneous withdrawal increases glutamate synaptic transmission by cAMP upregulation in μ-receptor-containing cells. A, Representative EPSCs in neurons from a control slice and from a spontaneously withdrawn slice. B, Group data of EPSC amplitudes from the same two groups as in A. Numbers in the columns indicate the cell numbers. C, Normalized EPSC pairs in neurons from a control slice, a withdrawn slice, and a withdrawn slice treated with MDL12330a (MDL). D, Group data of PPRs from the same three groups as in C. *p < 0.05; **p < 0.01. Withdr., Withdrawal. Error bars represent SEM.
Morphine withdrawal enhances hyperpolarization-activated current through upregulated cAMP cascade
cAMP also modulates the voltage dependence of the Ih or hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels (Ingram and Williams, 1994; Luthi and McCormick, 1999; Wainger et al., 2001; Mellor et al., 2002). These μ-receptor-expressing NRM cells, which display a large Ih (Pan, 2003b), had typical Ih properties, including blockade by cesium and by the Ih channel blocker ZD7288, but not by barium (data not shown). In control cells, the inclusion of 8-br-cAMP (1 mm) in the recording pipette increased the average amplitude of the Ih and Ih tails at submaximal levels, but not the maximum (Fig. 5A, B). Analysis with the Boltzmann equation revealed that the cAMP analog produced a positive shift of 8.7 mV, on average, in the Ih voltage dependence measured by V1/2 at the half-maximal current without affecting its maximum conductance (control, -76.1 ± 0.9 mV, n = 12; 8-br-cAMP, -67.4 ± 1.7 mV, n = 15; p < 0.001) (Fig. 5C). This is consistent with the Ih-modulating mechanism by cAMP described in previous studies (Ingram and Williams, 1994; Luthi and McCormick, 1999; Wainger et al., 2001; Pan, 2003b). Forskolin produced a smaller effect with a positive shift of 2.1 mV, which did not reach statistical significance (control, -75.1 ± 1.1 mV; forskolin, -73.0 ± 2.2 mV; p > 0.05; n = 9). Because we have shown above that the cAMP pathway in the NRM likely is upregulated during morphine withdrawal, one would expect that morphine withdrawal would modulate the Ih similarly in these μ-receptor-containing cells. Indeed, when compared with control slices, μ-receptor-containing cells in spontaneously withdrawn slices showed a positive shift of 3.5 mV in V1/2 (control, -76.1 ± 0.9 mV, n = 12; withdrawn, -72.6 ± 0.6 mV, n = 14; p < 0.01) (Fig. 5D). Treatment with MDL12330a (100 μm) mostly blocked this withdrawal-induced V1/2 shift (withdrawn/MDL12330a, -75.8 ± 0.8 mV; n = 18; p < 0.01 when compared with V1/2 in withdrawal and p > 0.05 when compared with V1/2 in control) (Fig. 5E). These data indicate that morphine withdrawal-induced Ih enhancement likely is mediated by the upregulated cAMP cascade.
Figure 5.
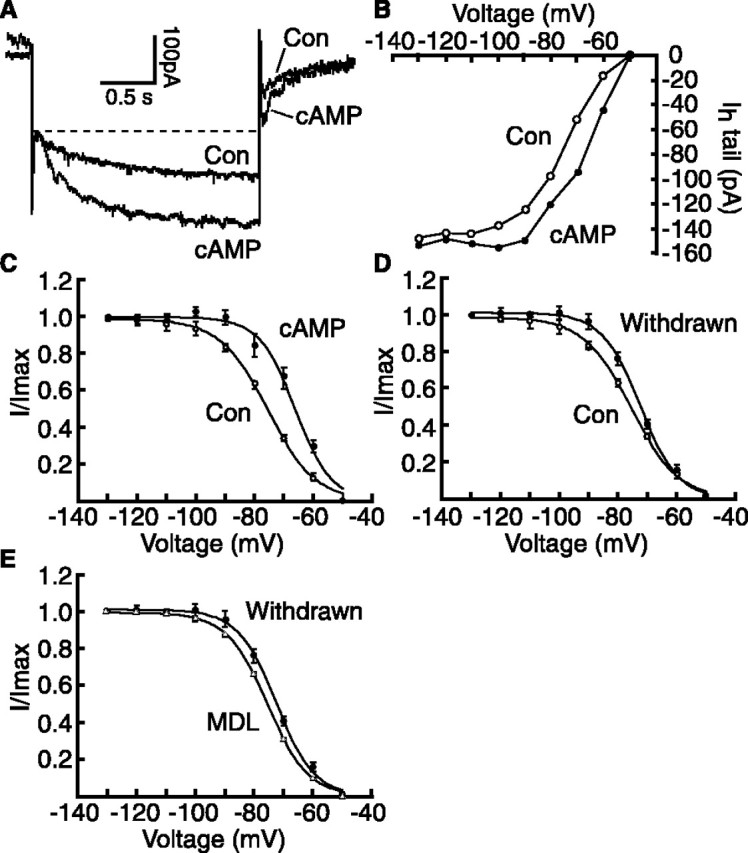
Morphine withdrawal enhances the Ih by cAMP upregulation in μ-receptor-containing cells. A, Current traces (aligned on the instantaneous current level) recorded with a control and an 8-br-cAMP-filled (1 mm) pipette during a voltage step (-50 to -80 mV). B, Averaged amplitudes of Ih tails recorded with control (n = 12) and 8-br-cAMP-filled pipettes (n = 15). C-E, Ih activation curves in control (V1/2 = -76.1 mV; n = 12) and in 8-br-cAMP (V1/2 = -67.4 mV; n = 15; p < 0.001) (C), in control (same as in C), in morphine-withdrawn cells (V1/2 = -72.6 mV; n = 14; p < 0.01) (D), in withdrawn cells (same as in D), and in another group of withdrawn cells treated with MDL12330a (MDL) (V1/2 = -75.8 mV; n = 18; p < 0.01) (E). Con, Control. Error bars represent SEM.
Inhibition of cAMP cascade attenuates withdrawal pain
Morphine withdrawal-induced increase in both glutamate synaptic transmission and the membrane-depolarizing Ih would increase the activity of these μ-receptor-expressing NRM cells. Consistently, these cells in spontaneously withdrawn slices displayed more-positive resting membrane potentials (control, -52.6 ± 0.8 mV, n = 22; withdrawn, -50.8 ± 0.7 mV, n = 23; p < 0.05) and a higher evoked firing rate (control, 17.6 ± 2.5 Hz, n = 21; withdrawn, 27.7 ± 1.7 Hz, n = 31; p < 0.01) (Fig. 6). Treatment of the withdrawn slices with a low concentration of the Ih blocker ZD7288 (10 μm) significantly reduced the firing rate (withdrawn/ZD7288, 18.2 ± 1.5 Hz; p < 0.01; n = 22) (Fig. 6), indicating an important Ih role in control of the cell excitability.
Figure 6.

Inhibition of Ih channels reduces evoked firing of μ-receptor-containing cells in withdrawn slices. Evoked firing of action potentials in cells from a control slice, a withdrawn slice, and a withdrawn slice (Withdr.) treated with ZD7288 (10 μm) is shown.
Previous studies, including ours, have shown that these μ-receptor-containing NRM cells may facilitate spinal pain transmission (Urban and Gebhart, 1999; Pan et al., 2000; Porreca et al., 2002; Bie and Pan, 2003; Fields, 2004). Therefore, we hypothesized that the cAMP-mediated mechanisms described above in these cells contributed to the symptom of pain sensitization (hyperalgesia) during opioid withdrawal. In behavioral experiments in rats in vivo, naloxone (3 μg/kg, i.v.) had no effect on pain behaviors in placebo-treated rats, but it significantly decreased the pain threshold measured by the tail-flick test in rats treated with chronic morphine, indicating morphine withdrawal-induced pain sensitization (Fig. 7A). Microinjection of the glutamate receptor antagonists AP-5 (197 ng in 1 μl) and CNQX (276 ng in 1 μl) into the NRM significantly attenuated the hyperalgesia (n = 4 rats) (Fig. 7A). This is consistent with our previous studies of acute morphine withdrawal, which suggest that glutamate-mediated activation of the μ-receptor-expressing cells, and not the other μ-receptor-lacking NRM cells, primarily mediates the withdrawal hyperalgesia (Pan et al., 1997, 2000; Bie and Pan, 2003). Supporting a role for Ih in the control of the firing of these cells shown above in vitro, microinjection of a low dose of ZD7288 (293 ng in 1 μl) also significantly reduced the withdrawal pain (n = 5 rats) (Fig. 7B). Pretreatment by microinjection of the AC inhibitor MDL12330a (377 ng in 1 μl) into the NRM significantly antagonized this withdrawal-induced pain sensitization (n = 6 rats) (Fig. 7C). Furthermore, NRM microinjection of the PKA inhibitor H89 (519 ng in 1 μl; n = 6 rats) or Rp-cAMP (8.9 μg in 1 μl; n = 5 rats) before the naloxone injection also suppressed the withdrawal pain (Fig. 7D, E). However, microinjection of the same dose of Rp-cAMP into a site directly dorsal to the NRM failed to alter the withdrawal-induced hyperalgesia (n = 3 rats) (Fig. 7F), indicating a local NRM-mediated effect for the reduction in withdrawal pain. Figure 8 shows representative cannula placements within the NRM and dorsal to the NRM.
Figure 7.
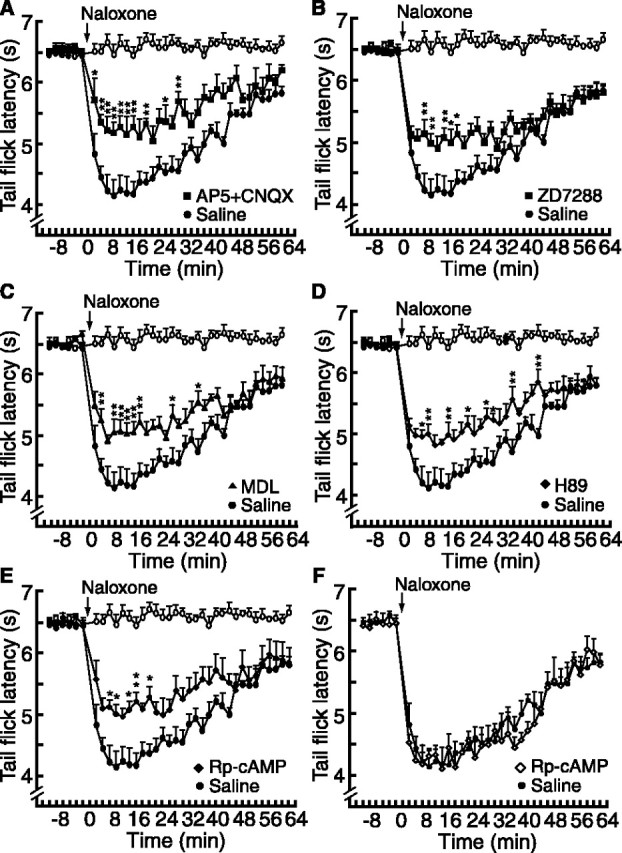
Inhibition of the cAMP pathway attenuates morphine withdrawal-induced pain sensitization. A-E, Plots of tail-flick latencies in placebo-treated rats (open circles) and in morphine-treated rats (filled symbols). Naloxone-induced withdrawal hyperalgesia (filled circles) was reduced by NRM microinjection of AP-5 and CNQX (A; filled squares; n = 4 rats), by ZD7288 (B; filled squares; n = 5), by MDL12330a (MDL) (C; filled triangles; n = 6), by H89 (D; filled diamonds; n = 6), and by Rp-cAMP (E; filled diamonds; n = 5). *p < 0.05; **p < 0.01 (ANOVA for repeated measures and the Tukey-Kramer test of post hoc analysis). F, Same microinjections of Rp-cAMP as in E, with cannula placement dorsal to the NRM (n = 3 rats). Error bars represent SEM.
Figure 8.
Placements of NRM microinjections. Photomicrographs show representative microinjection sites within the NRM area (A) and dorsal to the NRM (B). Scale bar, 1 mm.
Discussion
The current study has identified two neuroadaptations induced by in vivo administration of chronic morphine in rats: an increase in glutamate synaptic transmission and an enhancement of the Ih in the same population of μ-receptor-containing NRM neurons. Both adaptations appear to be mediated by chronic morphine-induced upregulation of the cAMP pathway in vivo. The current study also shows that both of the cAMP-mediated actions, which increase the activity of these presumably pain-facilitating neurons, contribute to the pain sensitization during morphine withdrawal.
Since its discovery in cell lines in vitro (Sharma et al., 1975; Brandt et al., 1976), chronic opioid-induced upregulation of the cAMP signaling pathway has been demonstrated in many brain areas after chronic morphine treatment (Nestler and Aghajanian, 1997; Jolas et al., 2000; Nestler, 2001; Williams et al., 2001; Chao and Nestler, 2004). These brain areas are critical either for the reinforcing effects of drugs of abuse including opioids in drug addiction, such as the ventral tegmental area (VTA) and the nucleus accumbens, or for the expression of somatic signs of opioid withdrawal, such as the noradrenergic locus ceruleus and the serotonergic dorsal raphe nucleus (Jolas et al., 2000; Chao and Nestler, 2004). The complete molecular mechanisms for the chronic opioid-induced cAMP upregulation remain to be demonstrated (Watts, 2002; Chao and Nestler, 2004). Recent research shows that AC has nine different isoforms for which the brain distributions and cellular locations are highly specific, indicating complex and multiple presynaptic and postsynaptic actions of the enzyme (Williams et al., 2001; Watts, 2002). Although ACI, ACIII, and ACVIII are stimulated by intracellular calcium (Ca2+), ACV and ACVI are inhibited by Ca2+ (Mons and Cooper, 1995). In COS-7 cells (African green monkey kidney cells) acute activation of μ-opioid receptors inhibits the activity of AC I, V, VI, and VIII isoforms, which are likely substrates for upregulation by chronic opioids (Avidor-Reiss et al., 1997). Indeed, a chronic morphine-induced increase in AC activity has been associated with increased levels of ACI and ACVIII mRNAs and immunoreactivity in the locus ceruleus and of ACVIII in the dorsal raphe nucleus (Nestler and Aghajanian, 1997; Jolas et al., 2000). Interestingly, increased immunoreactivity of ACVIII has been found in the hippocampus and amygdala of the human alcoholic brain (Yamamoto et al., 2001). Mice lacking both ACI and ACVIII display no synaptic plasticity of long-term potentiation in the hippocampus (Wong et al., 1999). Our current results indicating that withdrawal from chronic morphine increases the mRNA levels of ACVI and ACVIII and immunoreactivity of ACV/VI in the NRM are generally in agreement with previous reports indicating a role for NRM ACV/VI and ACVIII in chronic morphine-induced synaptic and behavioral adaptations.
The superactivated cAMP cascade and an overshoot in intracellular cAMP concentration after opioid withdrawal are a widely distributed molecular adaptation to chronic opioids (Maldonado et al., 1996; Nestler and Aghajanian, 1997; Williams et al., 2001). However, how such an adaptation in the cAMP pathway leads to behavioral dependence on opioids and withdrawal syndrome is still unclear, as are the underlying cellular mechanisms for the chronic opioid-induced behaviors. In cell preparations in vitro, the cAMP analogs or AC activators generally have two actions on central neurons. First, they increase neurotransmitter release in central glutamatergic and GABAergic synapses via both presynaptic and postsynaptic mechanisms (Greengard et al., 1991; Raman et al., 1996; Chavis et al., 1998; Ingram et al., 1998; Williams et al., 2001), including the enhancement of long-term potentiation, a well studied synaptic plasticity implicated in learning and memory (Salin et al., 1996). Second, they augment the Ih, which is important in the control of resting membrane potential, cell excitability, and firing patterns (Ingram and Williams, 1994; Luthi and McCormick, 1999; Williams et al., 2001; Mellor et al., 2002). The molecular and cellular mechanisms for cAMP modulation of the Ih channel (HCN channels) have been characterized in central neurons (Wainger et al., 2001; Williams et al., 2001).
However, in contrast to these cAMP analog-mediated mechanisms found in vitro, demonstration of the cellular effects of an upregulated endogenous cAMP cascade induced by in vivo administration of opioids in intact animals currently is limited only to GABAergic synapses. It has been shown recently that opioid withdrawal increases GABA synaptic transmission through an upregulated cAMP cascade in the VTA, the nucleus accumbens, and the periaqueductal gray (PAG) of chronic morphine-treated rats (Bonci and Williams, 1997; Ingram et al., 1998; Williams et al., 2001). Interestingly, this chronic opioid effect in all of these previous reports was observed exclusively on the inhibitory GABA synapses, with no effect on glutamate synapses. In fact, chronic morphine treatment has been reported to decrease NMDA EPSCs in the nucleus accumbens (Martin et al., 1999). In contrast, our results in the present study suggest that morphine withdrawal presynaptically increases glutamate synaptic transmission, likely via an upregulated cAMP cascade in μ-receptor-expressing NRM neurons. The sources of these glutamatergic terminals presently are unknown. Based on the current results, it appears that both the removal of the inhibitory effect of μ-receptors by naloxone or morphine abstinence and the cAMP-dependent activation of glutamate synaptic inputs increase the activity of these NRM cells during morphine withdrawal.
In addition, the present study provides evidence for another cellular effect of an upregulated cAMP cascade also induced by in vivo administration of morphine. It shows that morphine withdrawal augments the Ih, likely via an upregulated cAMP pathway in the same population of NRM cells for which the glutamate synaptic inputs are increased. An Ih involvement in the chronic morphine-induced increase of GABA tone via cAMP has been indicated in a previous study of the PAG (Jolas et al., 2000), but a direct effect of chronic opioids in vivo on Ih channels has yet to be demonstrated. Our results obtained from morphine-dependent rats show that in vivo administration of chronic morphine augments the membrane-depolarizing Ih by shifting its voltage dependence to a more depolarized potential without altering its maximum, which is consistent with the mechanisms for cAMP modulation of the Ih found in central neurons by the application of cAMP analogs in vitro (Ingram and Williams, 1994; Luthi and McCormick, 1999). Therefore, it appears that both an increase in glutamate synaptic inputs and augmentation of membrane-depolarizing Ih contribute to the increased excitability we observed in μ-receptor-containing NRM cells in withdrawn slices.
Some of the μ-receptor-expressing cells in the NRM project to spinal dorsal horn (Marinelli et al., 2002) and have been shown to have a facilitating action on spinal pain transmission (Fields, 2004). Growing evidence from recent pain research suggests that these μ-receptor-containing cells in the NRM contribute to many chronic pain conditions with pain sensitization (McNally, 1999; Millan, 2002; Porreca et al., 2002; Gebhart, 2004). These in vivo studies show that these cells and their descending pain-facilitating pathways commonly are activated in several forms of chronic and abnormal pain, including those induced by neuropathy, inflammation, illness-inducing agents, and chronic opioids. The mechanisms for their activation in those pain conditions mostly are unknown at present. Our previous studies have shown that these cells also play a critical role in the hyperalgesia induced by withdrawal from a single dose of acute morphine (Pan et al., 2000; Bie and Pan, 2003). The present study suggests that withdrawal from chronic morphine activates these cells by increasing their glutamate synaptic inputs and by augmenting the excitatory Ih, both likely via an upregulated cAMP pathway after chronic morphine treatment. Thus evidence from this study indicates that activation of these pain-facilitating cells contributes to the pain sensitization during morphine withdrawal. This is in line with previous substantial evidence demonstrating the involvement of glutamate receptors in the development of opioid dependence and the expression of withdrawal behaviors (Trujillo and Akil, 1991; Mao et al., 1995; Christie et al., 1997; Vekovischeva et al., 2001; Varney and Gereau, 2002; Inoue et al., 2003) as well as in withdrawal-induced hyperalgesia (Mao et al., 1995; Urban and Gebhart, 1999).
In summary, the present study shows two adaptive changes in pain-modulating brainstem neurons as a consequence of chronic morphine treatment. It provides possible cellular mechanisms for the behavioral pain sensitization during opioid withdrawal. Understanding the roles of these brainstem neurons and their activation mechanisms in abnormal pain conditions is beneficial to the development of improved therapies for opioid dependence and withdrawal.
Footnotes
This work was supported by National Institute on Drug Abuse Grant DA14524 and by an institutional fund of the M. D. Anderson Cancer Center.
Correspondence should be addressed to Dr. Zhizhong Z. Pan, Department of Anesthesiology, Box 110, University of Texas-M. D. Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, TX 77030. E-mail: zzpan@mdanderson.org.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253824-09$15.00/0
References
- Angst MS, Koppert W, Pahl I, Clark DJ, Schmelz M (2003) Short-term infusion of the mu-opioid agonist remifentanil in humans causes hyperalgesia during withdrawal. Pain 106: 49-57. [DOI] [PubMed] [Google Scholar]
- Avidor-Reiss T, Nevo I, Saya D, Bayewitch M, Vogel Z (1997) Opiate-induced adenylyl cyclase superactivation is isozyme-specific. J Biol Chem 272: 5040-5047. [DOI] [PubMed] [Google Scholar]
- Bie B, Pan ZZ (2003) Presynaptic mechanism for anti-analgesic and anti-hyperalgesic actions of κ-opioid receptors. J Neurosci 23: 7262-7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Williams JT (1997) Increased probability of GABA release during withdrawal from morphine. J Neurosci 17: 796-803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt M, Gullis RJ, Fischer K, Buchen C, Hamprecht B, Moroder L, Wunsch E (1976) Enkephalin regulates the levels of cyclic nucleotides in neuroblastoma X glioma hybrid cells. Nature 262: 311-313. [DOI] [PubMed] [Google Scholar]
- Chao J, Nestler EJ (2004) Molecular neurobiology of drug addiction. Annu Rev Med 55: 113-132. [DOI] [PubMed] [Google Scholar]
- Chavis P, Mollard P, Bockaert J, Manzoni O (1998) Visualization of cyclic AMP-regulated presynaptic activity at cerebellar granule cells. Neuron 20: 773-781. [DOI] [PubMed] [Google Scholar]
- Christie MJ, Williams JT, Osborne PB, Bellchambers CE (1997) Where is the locus in opioid withdrawal? Trends Pharmacol Sci 18: 134-140. [DOI] [PubMed] [Google Scholar]
- Compton P, Athanasos P, Elashoff D (2003) Withdrawal hyperalgesia after acute opioid physical dependence in nonaddicted humans: a preliminary study. J Pain 4: 511-519. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF (1997) Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron 18: 995-1008. [DOI] [PubMed] [Google Scholar]
- Doverty M, White JM, Somogyi AA, Bochner F, Ali R, Ling W (2001) Hyperalgesic responses in methadone maintenance patients. Pain 90: 91-96. [DOI] [PubMed] [Google Scholar]
- Fields HL (2004) State-dependent opioid control of pain. Nat Rev Neurosci 5: 565-575. [DOI] [PubMed] [Google Scholar]
- Gebhart GF (2004) Descending modulation of pain. Neurosci Biobehav Rev 27: 729-737. [DOI] [PubMed] [Google Scholar]
- Greengard P, Jen J, Nairn AC, Stevens CF (1991) Enhancement of the glutamate response by cAMP-dependent protein kinase in hippocampal neurons. Science 253: 1135-1138. [DOI] [PubMed] [Google Scholar]
- Harris NC, Constanti A (1995) Mechanism of block by ZD7288 of the hyperpolarization-activated inward rectifying current in guinea pig substantia nigra neurons in vitro J Neurophysiol 74: 2366-2378. [DOI] [PubMed] [Google Scholar]
- Ingram SL, Williams JT (1994) Opioid inhibition of Ih via adenylyl cyclase. Neuron 13: 179-186. [DOI] [PubMed] [Google Scholar]
- Ingram SL, Vaughan CW, Bagley EE, Connor M, Christie MJ (1998) Enhanced opioid efficacy in opioid dependence is caused by an altered signal transduction pathway. J Neurosci 18: 10269-10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Mishina M, Ueda H (2003) Locus-specific rescue of GluRϵ1 NMDA receptors in mutant mice identifies the brain regions important for morphine tolerance and dependence. J Neurosci 23: 6529-6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolas T, Nestler EJ, Aghajanian GK (2000) Chronic morphine increases GABA tone on serotonergic neurons of the dorsal raphe nucleus: association with an up-regulation of the cyclic AMP pathway. Neuroscience 95: 433-443. [DOI] [PubMed] [Google Scholar]
- Katsushika S, Chen L, Kawabe JI, Nilakantan R, Halnon NJ, Homcy CJ, Ishikawa Y (1992) Cloning and characterization of a sixth adenylyl cyclase isoform: types V and VI constitute a subgroup within the mammalian adenylyl cyclase family. Proc Natl Acad Sci USA 89: 8774-8778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Henderson G (1998) Hyperpolarization-activated cationic currents (Ih) in neurons of the trigeminal mesencephalic nucleus of the rat. J Physiol (Lond) 510: 695-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkman PM, Kelly JS (2001) Modulation of the hyperpolarization-activated current, Ih, in rat facial motoneurons in vitro by ZD-7288. Neuropharmacology 40: 1058-1072. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25: 402-408. [DOI] [PubMed] [Google Scholar]
- Luthi A, McCormick DA (1999) Modulation of a pacemaker current through Ca2+-induced stimulation of cAMP production. Nat Neurosci 2: 634-641. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Blendy JA, Tzavara E, Gass P, Roques BP, Hanoune J, Schutz G (1996) Reduction of morphine abstinence in mice with a mutation in the gene encoding CREB. Science 273: 657-659. [DOI] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA (1993) Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol 70: 1451-1459. [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ (1995) Mechanisms of hyperalgesia and morphine tolerance: a current view of their possible interactions. Pain 62: 259-274. [DOI] [PubMed] [Google Scholar]
- Marinelli S, Vaughan CW, Schnell SA, Wessendorf MW, Christie MJ (2002) Rostral ventromedial medulla neurons that project to the spinal cord express multiple opioid receptor phenotypes. J Neurosci 22: 10847-10855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Ahmed SH, Blank T, Spiess J, Koob GF, Siggins GR (1999) Chronic morphine treatment alters NMDA receptor-mediated synaptic transmission in the nucleus accumbens. J Neurosci 19: 9081-9089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally GP (1999) Pain facilitatory circuits in the mammalian central nervous system: their behavioral significance and role in morphine analgesic tolerance. Neurosci Biobehav Rev 23: 1059-1078. [DOI] [PubMed] [Google Scholar]
- Mellor J, Nicoll RA, Schmitz D (2002) Mediation of hippocampal mossy fiber long-term potentiation by presynaptic Ih channels. Science 295: 143-147. [DOI] [PubMed] [Google Scholar]
- Millan MJ (2002) Descending control of pain. Prog Neurobiol 66: 355-474. [DOI] [PubMed] [Google Scholar]
- Mons N, Cooper DMF (1995) Adenylyl cyclases: critical foci in neuronal signaling. Trends Neurosci 18: 536-542. [DOI] [PubMed] [Google Scholar]
- Nestler EJ (2001) Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci 2: 119-128. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Aghajanian GK (1997) Molecular and cellular basis of addiction. Science 278: 58-63. [DOI] [PubMed] [Google Scholar]
- Pan ZZ (2003a) Opioid tolerance in adult and neonatal rats. In: Opioid research: methods and protocols (Pan ZZ, ed), pp 223-232. Totowa, NJ: Humana. [DOI] [PubMed]
- Pan ZZ (2003b) κ-Opioid enhancement of the hyperpolarization-activated current (Ih) through mobilization of intracellular calcium. J Physiol (Lond) 548: 765-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan ZZ, Williams JT, Osborne PB (1990) Opioid actions on single nucleus raphe magnus neurons from rat and guinea-pig in vitro J Physiol (Lond) 427: 519-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan ZZ, Tershner SA, Fields HL (1997) Cellular mechanism for anti-analgesic action of agonists of the kappa-opioid receptor. Nature 389: 382-385. [DOI] [PubMed] [Google Scholar]
- Pan ZZ, Hirakawa N, Fields HL (2000) A cellular mechanism for the bidirectional pain-modulating actions of orphanin FQ/nociceptin. Neuron 26: 515-522. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1986) The rat brain in stereotaxic coordinates, Ed 2. Sydney: Academic. [DOI] [PubMed]
- Perkel DJ, Nicoll RA (1993) Evidence for all-or-none regulation of neurotransmitter release: implications for long-term potentiation. J Physiol (Lond) 471: 481-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porreca F, Ossipov MH, Gebhart GF (2002) Chronic pain and medullary descending facilitation. Trends Neurosci 25: 319-325. [DOI] [PubMed] [Google Scholar]
- Punch LJ, Self DW, Nestler EJ, Taylor JR (1997) Opposite modulation of opiate withdrawal behaviors on microinfusion of a protein kinase A inhibitor versus activator into the locus coeruleus or periaqueductal gray. J Neurosci 17: 8520-8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Tong G, Jahr CE (1996) Beta-adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron 16: 415-421. [DOI] [PubMed] [Google Scholar]
- Salin PA, Malenka RC, Nicoll RA (1996) Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron 16: 797-803. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ (2002) Can we conquer pain? Nat Neurosci [Suppl] 5: 1062-1067. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Klee W, Nirenberg M (1975) Dual regulation of adenylate cyclase accounts for narcotic dependence and tolerance. Proc Natl Acad Sci USA 72: 3092-3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takigawa T, Alzheimer C, Quasthoff S, Grafe P (1998) A special blocker reveals the presence and function of the hyperpolarization-activated cation current Ih in peripheral mammalian nerve fibers. Neuroscience 82: 631-634. [DOI] [PubMed] [Google Scholar]
- Trujillo KA, Akil H (1991) Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science 251: 85-87. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A (2001) Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411: 583-587. [DOI] [PubMed] [Google Scholar]
- Urban MO, Gebhart GF (1999) Supraspinal contributions to hyperalgesia. Proc Natl Acad Sci USA 96: 7687-7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varney MA, Gereau RW (2002) Metabotropic glutamate receptor involvement in models of acute and persistent pain: prospects for the development of novel analgesics. Curr Drug Targets CNS Neurol Disord 1: 283-296. [DOI] [PubMed] [Google Scholar]
- Vekovischeva OY, Zamanillo D, Echenko O, Seppala T, Uusi-Oukari M, Honkanen A, Seeburg PH, Sprengel R, Korpi ER (2001) Morphine-induced dependence and sensitization are altered in mice deficient in AMPA-type glutamate receptor A subunits. J Neurosci 21: 4451-4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainger BJ, DeGennaro M, Santoro B, Siegelbaum SA, Tibbs GR (2001) Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 411: 805-810. [DOI] [PubMed] [Google Scholar]
- Watts VJ (2002) Molecular mechanisms for heterologous sensitization of adenylate cyclase. J Pharmacol Exp Ther 302: 1-7. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O (2001) Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev 81: 299-343. [DOI] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR (1999) Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron 23: 787-798. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Pohli S, Durany N, Ozawa H, Saito T, Boissl KW, Zochling R, Riederer P, Boning J, Gotz ME (2001) Increased levels of calcium-sensitive adenylyl cyclase subtypes in the limbic system of alcoholics: evidence for a specific role of cAMP signaling in the human addictive brain. Brain Res 895: 233-237. [DOI] [PubMed] [Google Scholar]