

Abstract
Mammalian central synapses commonly specialize in one fast neurotransmitter, matching the content of their presynaptic vesicles with the appropriate receptors in their postsynaptic membrane. Here, I show that hippocampal cultures contain autaptic glutamatergic synapses that contravene this rule: in addition to postsynaptic glutamate receptors, they also express clusters of functional postsynaptic GABAA receptors yet lack presynaptic GABA. Hence, these synapses are presynaptically silent with respect to GABA. They can be unsilenced by loading GABA into presynaptic vesicles by endocytosis, after which a postload IPSC appears. This IPSC is similar to native IPSCs recorded from GABAergic interneurons in the same cultures. Thus, these “mistargeted” GABAA receptors, which apparently lack a signal that confers synaptic specificity, function almost normally. After GABA loading, glutamatergic miniature postsynaptic currents acquire a slow tail that is mediated by GABAA receptors, showing that synaptic vesicles can accommodate both the usual concentration of native glutamate and a saturating concentration of loaded GABA. After brief Ca2+-dependent exocytosis, endocytosis of GABA can proceed in low-Ca2+ external solution. The amplitude of the postload IPSC declines exponentially with repetitive stimulation as the endocytosed GABA passes through the presynaptic vesicle cycle and is depleted. Hence, by using GABA as an exogenous but physiological tracer, the properties of these presynaptically silent synapses can provide novel insights into the content and cycling of vesicles in presynaptic terminals.
Keywords: autapse, culture, endocytosis, EPSC, IPSC, synaptic vesicle
Introduction
To function correctly, fast central synapses typically require precise apposition of presynaptic and postsynaptic elements, ensuring that a single type of neurotransmitter is focally released onto the appropriate subsynaptic receptors. Surprisingly, some central synapses deviate from this simple functional architecture. For example, a population of synapses in the spinal cord and hippocampus can release more than one fast neurotransmitter onto multiple types of subsynaptic receptor (Jonas et al., 1998; Jo and Schlichter, 1999; Gutiérrez, 2000; Walker et al., 2001), and other types of synapse do not contain functional postsynaptic AMPA receptors until triggered insertion of the receptors occurs (“postsynaptically silent” synapses) (Malinow et al., 2000). These examples belong to a general class of synapses that lack a simple one-to-one correspondence between presynaptic and postsynaptic components. Here, I describe a new member of this class. This novel synapse, found in hippocampal cultures, expresses functional postsynaptic GABAA receptors, but its presynaptic vesicles do not normally contain GABA; it is thus “presynaptically silent” with respect to GABA.
The experiments presented here were motivated by the finding that some glutamatergic self-synapses (autapses) on cultured hippocampal pyramidal neurons are able to express, in addition to glutamate receptors, a high density of “mistargeted” GABAA receptors in their subsynaptic or perisynaptic membrane (Rao et al., 2000). Rao et al. (2000) used immunohistochemical techniques, so were unable to establish whether mistargeted GABAA receptors are physiologically functional. Functional GABAA receptors would confirm that certain aspects of receptor clustering operate correctly, even in the absence of a postulated signal that confers synaptic specificity. Moreover, functional GABAA receptors would make possible a new approach to studying the content and cycling of presynaptic vesicles.
Hippocampal pyramidal neurons express both subsynaptic and extrasynaptic GABAA receptors in their plasma membrane (Rao et al., 2000). To assay the electrophysiological properties of only subsynaptic GABAA receptors, a method must be found for exposing just those receptors to GABA. This can be accomplished by loading GABA into presynaptic vesicles and then triggering exocytosis of the vesicular contents into the synaptic cleft. The fluorescent stryl dyes [e.g., N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide (FM1-43)] are commonly loaded into vesicles by endocytosis from the bath solution (Betz et al., 1992). I reasoned that the same approach should also work for GABA.
In this paper, I show that exogenously applied GABA can, indeed, be endocytosed into presynaptic vesicles that normally contain only glutamate. Subsequent activity-dependent release then reveals a postload IPSC in the pyramidal neuron, confirming that mistargeted GABAA receptors function almost normally. Because the GABA is loaded briefly at one point in the presynaptic vesicle cycle, it can be used as an exogenous but physiological tracer to monitor the passage of vesicles through the cycle. Stryl dyes have been used extensively as synaptic vesicle tracers (Murthy, 1999), but, being lipophilic, they may report erroneous cycling kinetics under some circumstances (Pyle et al., 2000; Stevens and Williams, 2000; Aravanis et al., 2003). In contrast, GABA is itself a neurotransmitter and thus ideally suited to tracing the passage of native transmitter through the vesicle cycle.
Materials and Methods
Cell culture. Cultures were prepared according to standard methods (Bekkers and Stevens, 1991), with the following variations. Newborn Wistar rats were decapitated rapidly using methods approved by the Australian National University Animal Experimentation Ethics Committee. Hippocampal region CA1 was dissected out, dissociated, and then plated at a density of 8-10 × 104 cells/ml on 13-mm-diameter round glass coverslips that had been coated with agarose and then spotted with a mixture of calf skin collagen (0.5 mg/ml; Contact Technologies, Palo Alto, CA) and poly-d-lysine (0.1 mg/ml; Sigma, St. Louis, MO). At this plating density, after 3 weeks in culture, each coverslip contained two to six microislands (diameter, 100-200 μm), each occupied by a single neuron. The culture medium comprised the following (per 100 ml): 50 ml of DMEM with bicarbonate (D-0422; Sigma), 50 ml of DMEM with HEPES (D-1152; Sigma), 2 ml of B-27 supplement (Invitrogen, Mt. Waverley, Victoria, Australia), 5 ml of fetal bovine serum (Chemicon, Boronia, Victoria, Australia), and 1 ml of penicillin/streptomycin (100×; Invitrogen), pH 7.6 in a 5% CO2 incubator at 37°C. Cytosine β-d-arabinofuranoside (5 μm final concentration; Sigma) was added 3-5 d after plating to suppress proliferation of glia. Cultures were fed subsequently with fresh culture medium once per week.
Electrophysiology. Microislands occupied by a single neuron were identified in cultures that had been maintained 17-28 d in vitro. An array of flow pipes (400 μm diameter) was used to continuously superfuse an isolated neuron with external solution, ensuring that the entire neuron and microisland (diameter, <200 μm) were bathed in the desired solution. Solution changes were effected by lateral movement of the flow pipes, through which solution was flowing continuously. Exchanges were fast (<200 ms), as seen from the whole-cell response to GABA (see Fig. 1 B). Whole-cell patch-clamp recordings were obtained using a Multi-Clamp 700A amplifier (Molecular Devices, Union City, CA). Patch electrodes had resistances of 2-3 MΩ when filled with internal solution. Series resistance (3.5-7 MΩ) was monitored continuously for stability, and compensation was adjusted to 80-90% at a bandwidth of 10 kHz. Even with this high degree of compensation, significant errors probably remained for the larger EPSCs (>10 nA) (see Fig. 3A). Neurons were voltage clamped at -60 mV and autaptic currents elicited at 1-10 s intervals by a 2-ms-long step to 0 mV. Currents were filtered at 10 kHz and digitized at 50 kHz. The identity of each neuron (glutamatergic or GABAergic) was confirmed by both the kinetics of the postsynaptic current and its complete blockade by either 20 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) plus 50 μm d-2-amino-5-phosphonovaleric acid (d-APV) or 20 μm bicuculline. The electrode solution contained the following (in mm): 135 KCl, 7 NaCl, 1 EGTA, 2 MgCl2, 2 Na2ATP, 0.3 NaGTP, 10 HEPES, pH 7.2 with KOH, 50 sorbitol at 330 mOsm/kg. For 100 mm GABA internal solution, the KCl was reduced to 100 mm, and sorbitol was excluded. The standard bath solution contained the following (in mm): 125 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 25 HEPES, pH 7.4 with NaOH, 25 sorbitol at 310 mOsm/kg. The standard high-K+ GABA loading solution contained the following (in mm): 40 NaCl, 40 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 25 HEPES, pH 7.4, 100 GABA at 310 mOsm/kg. For loading solutions containing other concentrations of GABA (10-200 mm), the concentration of NaCl was adjusted to give an osmolarity of 310 mOsm/kg. Puffer applications of GABA (100 μm in bath solution for 50 ms) used a Picospritzer (General Valve, Fairfield, NJ). All chemicals were obtained from Sigma, except CNQX and d-APV (Tocris Cookson, Bristol, UK; Australian Laboratory Services, Sydney, New South Wales, Australia) and cyclosporin A (CsA) (Calbiochem, La Jolla, CA; Merck, Kilsyth, Victoria, Australia). Experiments were performed at room temperature (21-25°C).
Figure 1.
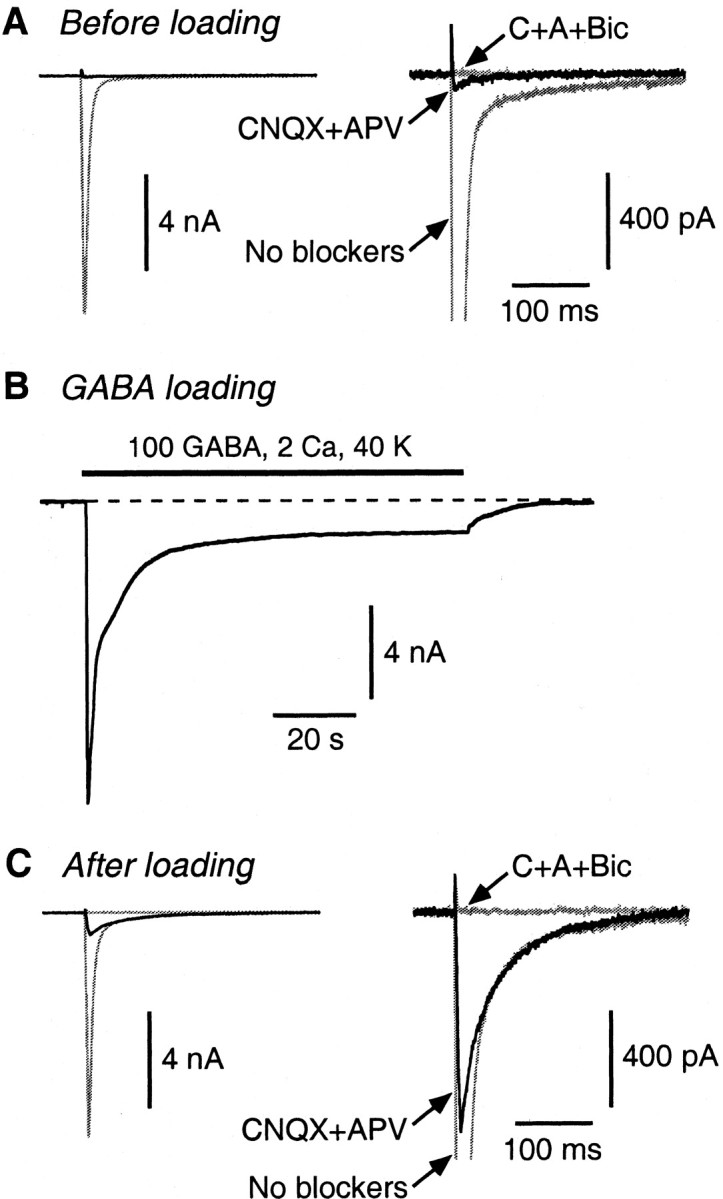
A postload IPSC appears after GABA loading. A, Before loading, the autaptic EPSC (large gray trace; No blockers) is almost fully blocked by 20 μm CNQX plus 50 μm d-APV (black trace). The addition of 20 μm bicuculline (flat gray trace; C+A+Bic) has little additional effect. The two panels show the same traces on different vertical scales. B, The cell was superfused for 90 s with a loading solution (in mm: 100 GABA, 2 CaCl2, and 40 KCl) that produced a large, desensitizing whole-cell GABA current. (The current is inward because the electrode solution contains a high chloride concentration.) C, After washing out the loading solution and resuming autaptic stimulation, the original EPSC reappears. Now, addition of CNQX plus d-APV reveals a large postload IPSC (black trace) that is blocked by bicuculline.
Figure 3.

Postload IPSCs behave presynaptically like glutamatergic synapses. A, Plot of native EPSC amplitude (Amp; measured at 2 s intervals) versus time. Bath-applied adenosine (100 μm) reversibly inhibits the EPSC. The right panel shows example traces at the numbered time points. B, Plot of native IPSC amplitude (measured at 10 s intervals) versus time. Adenosine (100 μm) has no effect. C, Plot of postload IPSC amplitude (2 s intervals; same cell as in A) versus time. Adenosine (100 μm) reversibly inhibits the postload IPSC. Bic, Bicuculline. D, Summary of Ado experiments. Both native EPSCs and postload IPSCs are significantly inhibited (*p < 0.05) by 100 μm adenosine, unlike native IPSCs. Error bars represent SEM.
Analysis. Data acquisition and analysis used Axograph 4.9 (Molecular Devices). Stimulus artifacts were removed by subtracting an average of episodes recorded for each cell in CNQX plus d-APV plus bicuculline. In some cases, a small residual artifact was blanked for display (see Figs. 3, 5, 6). Some postload IPSCs were contaminated by a spontaneous, activity-independent GABA current that appeared after superfusion of the microisland with high GABA. This current had a slow rise time (∼20-50 ms) and decay time (∼200-500 ms) and was blocked by zero-Ca2+ bath solution but not by tetrodotoxin. It was much less common in neurons on microislands that did not contain glia, suggesting that it was caused by the action potential-independent release of GABA from underlying glial cells. Postload IPSCs that were contaminated by this current were excluded from the analysis. The weighted decay time constant of native and postload IPSCs was calculated as the following: (a1τ1 + a2τ2)/(a1 + a2), where a1 and a2 are the amplitudes and τ1 and τ2 the decay time constants obtained from a double-exponential fit to the IPSC decay. Miniature synaptic currents (minis) were captured using the template scaling algorithm in Axograph (Clements and Bekkers, 1997) and then selected to exclude overlapping or distorted events. After GABA loading, the prolonged tails on miniature synaptic currents (see Fig. 4 B), together with the large number of autaptic contacts in these older cultures, increased the baseline noise, making it impractical to quantify the frequency of minis with and without a GABA component. Errors are given as ±SEM. Unless stated otherwise, statistical comparisons used the two-tailed t test with significance at p = 0.05 and n equal to the number of cells.
Figure 5.

GABA can be endocytosed in the absence of Ca2+, provided Ca2+-dependent exocytosis has occurred previously. In this figure, the black traces are bicuculline-sensitive autaptic currents recorded in CNQX plus d-APV, and the gray traces are whole-cell currents elicited by bath application of the indicated solutions. A, After superfusion of a high-K+ GABA loading solution that lacks added Ca2+, there is little change in the autaptic current (middle black trace). Next, the same cell was superfused briefly (5 s) with a solution containing 2 mm Ca2+ and 40 mm K+ but no GABA to trigger exocytosis. The bath solution was then immediately switched to one containing 100 mm GABA and normal K+ but no added Ca2+. After endocytosis for 60 s in this Ca2+-free loading solution, a large postload IPSC emerged (right black trace). B, Exocytosis was triggered by a train of 300 autaptic stimuli at 20 Hz in normal Ca2+-containing bath solution. The inset (gray trace) shows the beginning and end of this train. Starting 1.5 s after the end of the train, the cell was superfused for 60 s with nominally Ca2+-free GABA loading solution. After this, a large postload IPSC emerged (right black trace).
Figure 6.

The amplitude of the postload IPSC irreversibly runs down with time, reflecting the passage of loaded GABA through the synaptic vesicle cycle. A, Plot of amplitude versus time for a typical experiment. The native EPSC amplitude was initially ∼25 nA [points near “1;” stimulus (Stim) rate 0.1 Hz]. After loading (40 mm K+, 100 mm GABA for 30 s), the cell was bathed in normal external solution containing CNQX plus d-APV to isolate the postload IPSC and then stimulated at 1 Hz (0-10 min). The amplitude of the IPSC declined exponentially [superimposed fit of a single exponential plus an offset; τ = 152 s; baseline confirmed by addition of bicuculline (+Bic)]. The stimulus rate was then reduced to 0.1 Hz (10-15 min) to confirm that the reduction in IPSC amplitude was irreversible and not simply caused by temporary exhaustion of the readily releasable pool of synaptic vesicles at the higher stimulation rate. Finally, the CNQX plus d-APV was washed out, confirming that the native EPSC was unchanged in amplitude (points near “2”). B, Single synaptic currents recorded near the numbered time points in A.
Figure 4.

Presynaptic vesicles at glutamatergic autapses can accommodate both loaded GABA and the normal concentration of glutamate. A, Individual miniature synaptic currents recorded in an isolated pyramidal neuron before GABA loading. The right panel shows normalized averaged minis recorded before (black; n = 42 minis) and during (gray, obscured; n = 56) application of 20 μm bicuculline. B, After loading, minis with a slow component (arrows) appear. The averages (right) show that the slow component is blocked by 20 μm bicuculline (Bic) (n = 48 minis before bicuculline; n = 41 after bicuculline). C, Dose-response plot of the amplitude of the evoked postload IPSC versus the concentration of GABA in the high-K+ loading solution. For each cell, the postload IPSC amplitude was expressed as a percentage of the native EPSC amplitude measured in that cell. This was done to normalize to the number of autapses on different cells. Each point is an average from five to seven cells. The superimposed logistic equation gives a half-maximal GABA concentration of 48 mm. Error bars represent SEM. D, Amplitude (Amp) histograms of minis recorded in bicuculline in the same cell before and after loading with 100 mm GABA. The amplitude distributions are not significantly different (p = 0.45; Kolmogorov-Smirnov test), suggesting that the presence of GABA in some vesicles does not affect the amount of glutamate that can be accommodated.
Results
A postload IPSC at glutamatergic autapses
Brief electrical stimulation of an isolated CA1 pyramidal neuron (24 d in vitro) elicited an autaptic current with the kinetics of an AMPA receptor-mediated EPSC (Fig. 1A, “No blockers”). This current was almost completely blocked by the glutamate receptor antagonists CNQX (20 μm) and d-APV (50 μm) (Fig. 1A, vertically expanded at right), and addition of the GABAA receptor antagonist bicuculline (20 μm) had little additional effect (Fig. 1A, “C+A+Bic”). This confirmed that the current was glutamatergic. The neuron was then superfused for 90 s with a high-K+/high-GABA loading solution containing 40 mm KCl, 2 mm CaCl2, and 100 mm GABA to encourage synaptic vesicle cycling and endocytosis of GABA from the superfusate (Fig. 1B). (The loading solution elicited an inward whole-cell current because the electrode solution contained a high concentration of chloride.) Finally, the GABA loading solution was washed away, and, after 60 s, the EPSC had recovered (Fig. 1C, left). Perfusion of CNQX plus d-APV now unmasked a postload autaptic IPSC that could be completely blocked by bicuculline (Fig. 1C, right). Similar results were obtained in 93 of 109 cells; in 16 cells, a postload IPSC did not appear.
The following experiments were designed to test the hypothesis that superfused GABA is endocytosed by presynaptic vesicles, passes through the vesicle cycle, and is released onto normal subsynaptic GABAA receptors, generating the postload IPSC.
GABA is released onto normal GABAA receptors
Because immunohistological experiments indicate that GABAA receptors are mistargeted to glutamatergic autapses (Rao et al., 2000), I first asked whether these receptors were dysfunctional as well as mistargeted. This was addressed by comparing postload IPSCs (measured in isolated pyramidal neurons) with native autaptic IPSCs (measured in isolated GABAergic interneurons).
The mean weighted decay time constant for postload IPSCs (82.3 ± 5.5 ms; n = 20) was not significantly different from that for native autaptic IPSCs (83.8 ± 12.1 ms; n = 7) (Fig. 2A,B, right), suggesting that the underlying GABAA receptors were identical. To check whether this result was sensitive to the identity of the postsynaptic neuron (pyramidal neuron for postload IPSCs, interneuron for native autaptic IPSCs), control recordings were made from synaptically connected interneuron-pyramidal neuron pairs. These confirmed that the decay kinetics of the simultaneously measured autaptic IPSC (from the presynaptic interneuron onto itself) and synaptic IPSC (from the interneuron onto the postsynaptic pyramidal neuron) were not significantly different (mean ratio of decays, 1.15 ± 0.21; n = 5). Thus, IPSC decay kinetics in this culture system are not affected by the identity of the postsynaptic neuron.
Figure 2.

Normal perisynaptic GABAA receptors mediate the postload IPSC. A, Examples of a native autaptic IPSC recorded in an isolated GABAergic interneuron (gray trace) and a postload IPSC recorded in an isolated pyramidal neuron (black trace) scaled so their peak amplitudes are equal. The rise time of the postload IPSC is slower than that of the native IPSC (left), but the decay is identical (right). B, Summary of kinetic measurements. The averaged 10-90% rise time (RT) for postload IPSCs (bar 2, left) is significantly greater than that for native IPSCs (bar 1; *p < 0.05). This is not caused by a systematic voltage-clamp error between interneurons and pyramidal neurons, because the rise time for native EPSCs (bar 3), measured in the same cells as the postload IPSCs, is as fast as that for native IPSCs. C, Pentobarbital (20 μm) reversibly slows the decay of both native (left traces) and postload (right traces) IPSCs. c, Control; PB, pentobarbital; w, washout. D, Summary of PB experiments. The weighted decay time constant of native and postload IPSCs is equally prolonged by PB. Error bars represent SEM.
In contrast to the decay kinetics, the mean 10-90% rise time of the postload and native IPSCs were significantly different (postload IPSC, 5.44 ± 0.38 ms, n = 19; native IPSC, 2.61 ± 0.43 ms, n = 8) (Fig. 2A,B, left). Two control experiments were done to test whether this difference was caused by measurement errors. First, it is possible that the voltage clamp is differentially effective in pyramidal neurons (expressing postload IPSCs) and interneurons (expressing native IPSCs). This was not the case, because the mean 10-90% rise time for native EPSCs, measured in the same pyramidal neurons that expressed the slowly rising postload IPSCs, was as fast as the rise time for native IPSCs (3.19 ± 0.18 ms) (Fig. 2B, left, bars 1 and 3). Second, it is possible that measurement errors are different for large and small currents; native EPSC and IPSC amplitudes are typically much larger than postload IPSC amplitudes. This was tested by partially blocking the native EPSC by addition of 2 μm CNQX, which reduced the mean amplitude to 817 ± 83 pA (n = 8), similar to the mean postload IPSC amplitude (912 pA; see below). This had no significant effect on the 10-90% rise time of the EPSC (blocked rise time, 107 ± 8% of control; n = 8). In summary, then, the slower rise for postload IPSCs is genuine and might indicate that GABAA receptors at glutamatergic terminals are not as tightly localized to the subsynaptic membrane as those at native GABA terminals, requiring the GABA to diffuse further to activate them.
The effects of GABAA modulators on the two kinds of IPSC were also examined. Bath perfusion of pentobarbital (20 μm) reversibly slowed the weighted decay time constant of both the native and postload IPSCs by the same factor (1.83 ± 0.10, n = 7; 1.87 ± 0.09, n = 6, respectively) (Fig. 2C,D). Similar results were obtained for diazepam (0.5 μm; data not shown). These results support the conclusion that postload IPSCs are generated by normal, fully functional GABAA receptors located at or near glutamatergic terminals.
Action potential-dependent release of GABA
If the loaded GABA is released phasically from glutamatergic terminals, the postload IPSC should be modulated by presynaptic manipulations that modify action potential-evoked release of neurotransmitter. This was tested in two ways. First, addition of tetrodotoxin (1 μm) or perfusion with Ca2+-free bath solution completely blocked the postload IPSCs (n = 3; data not shown), as expected for activity-dependent release. Second, the effect of the presynaptic modulator adenosine (Ado) was examined (Wu and Saggau, 1997). Bath application of Ado (100 μm) reversibly inhibited native autaptic EPSCs recorded in pyramidal neurons (to 0.42 ± 0.04 of control; n = 7) (Fig. 3A,D), whereas it had no effect on native autaptic IPSCs recorded in interneurons (1.02 ± 0.03; n = 8) (Fig. 3B,D). This confirms that the adenosine A1 receptor responsible for presynaptic inhibition is only present on glutamate-releasing terminals (Yoon and Rothman, 1991; Wu and Saggau, 1997). Like EPSCs, and unlike native IPSCs, postload IPSCs were strongly inhibited by Ado (0.17 ± 0.02; n = 7) (Fig. 3C,D). The greater apparent inhibition of postload IPSCs than native EPSCs (Fig. 3D) may be attributable to the incomplete (80-90%) compensation of series resistance, which would tend to give an underestimate of the control amplitudes of larger EPSCs (e.g., >10 nA) (Fig. 3A).
Together, these results suggest that the loaded GABA is, indeed, released from glutamatergic terminals by an activity-dependent process. Thus, postload IPSCs behave presynaptically like glutamate synapses (Fig. 3) but postsynaptically like GABA synapses (Fig. 2).
Synaptic vesicles contain glutamate and GABA
If loaded GABA is endocytosed by synaptic vesicles at glutamatergic terminals, the vesicles may contain both endocytosed GABA and the native glutamate. This was tested by comparing spontaneous minis in the same pyramidal neuron before and after GABA loading. Before loading, all minis had fast kinetics typical of AMPA receptor-mediated miniature EPSCs, were fully blocked by 20 μm CNQX (data not shown), and were unaffected by addition of 20 μm bicuculline (Fig. 4A, normalized averages with and without bicuculline superimposed at right). After GABA loading, some minis now had a slow component (Fig. 4B, arrow) that was blocked by 20 μm bicuculline (Fig. 4B, right, normalized averages). The slow component presumably reflects the release of loaded GABA from individual synaptic vesicles containing both glutamate and GABA. Note that some minis lacked a slow component (Fig. 4B, traces 1 and 4). These may have arisen from vesicles that did not endocytose GABA or from the ∼30% of autapses per pyramidal neuron that did not cluster GABAA receptors (Rao et al., 2000).
How much loaded GABA can be accommodated by glutamatergic vesicles? This was addressed by applying (for 90 s) high-K+ loading solutions containing a range of GABA concentrations (10-200 mm). The amplitude of the resultant postload IPSC was normalized to the amplitude of the native EPSC measured in the same pyramidal neuron, to correct for different numbers of autapses on different neurons. The dose-response plot obtained from these experiments saturated at GABA concentrations above ∼100 mm, with a half-maximal concentration of 48 mm and a saturating amplitude of 3.7% of the native EPSC amplitude (Fig. 4C). In absolute terms, the mean amplitude of all postload IPSCs (loading [GABA] ≥ 100 mm) was 912 ± 157 pA (n = 19). By comparison, the mean amplitude of native IPSCs recorded in autaptic interneurons in the same cultures was 10.7 ± 4.7 nA (n = 8), ∼10-fold larger.
The saturation in Figure 4C may be attributable to either a limitation on the concentration of GABA that can be accommodated in each vesicle or postsynaptic saturation of the GABAA receptors (Jones and Westbrook, 1995). If the latter is the case, the rise time of postload IPSCs might differ between subsaturating (10, 50 mm) and saturating (≥100 mm) GABA concentrations (Jones and Westbrook, 1995). However, the mean 10-90% rise time was not significantly different at all concentrations (5.89 ± 0.98, 5.25 ± 0.99, 5.54 ± 0.70, 5.60 ± 0.66, and 5.23 ± 0.72 ms for 10, 50, 100, 150, and 200 mm GABA, respectively). These rise times are slower than expected from models of GABAA receptor gating (Jones and Westbrook, 1995), perhaps because they are dominated by diffusional delay between the site of GABA release and the receptors (see Discussion).
Does the presence of a high concentration of exogenous GABA in a synaptic vesicle partially displace the native glutamate? If so, the mean amplitude of miniature EPSCs might be smaller after GABA loading, assuming that the size of miniature EPSCs reflects, at least in part, the amount of glutamate released from vesicles (Ishikawa et al., 2002). This was not the case: the amplitude distributions of miniature EPSCs (measured in the presence of bicuculline) were not significantly different before and after GABA loading (mean mini amplitude after loading, 99 ± 4% of mean amplitude before loading; n = 3) (Fig. 4 D). This suggests that glutamatergic vesicles are able to accommodate both loaded GABA and the usual concentration of glutamate.
Interestingly, some glutamatergic vesicles contained GABA before loading, indicated by the presence in some cells of a small bicuculline-sensitive autaptic current before exposure to the high-GABA loading solution (Figs. 1, 5). These “preload” IPSCs had a mean amplitude of 42 ± 6 pA (0.12 ± 0.02% of the native EPSC amplitude; n = 77), which is significantly smaller than the mean amplitude of the postload IPSCs (912 pA). The kinetics of the preload IPSCs (10-90% rise time, 5.51 ± 0.59 ms, n = 9; decay time constant, 87.7 ± 7.6 ms, n = 16) were not significantly different from those of the postload IPSCs. These results suggest that, even under quiescent conditions, the GABA component of transmission at glutamatergic autapses is not always entirely silent.
GABA loading requires endocytosis
The simplest model for GABA loading is that GABA enters presynaptic vesicles by the endocytosis that follows exocytosis, as occurs for stryl dyes (e.g., FM1-43). However, another possibility is that GABA is able to cross the plasmalemma into the cytoplasm of the presynaptic terminal, from which it is loaded into synaptic vesicles, perhaps by the nonselective action of glutamate vesicular transporters (Fon and Edwards, 2001; Otis, 2001). This was tested by recording from autaptic pyramidal neurons with 100 mm GABA in the intracellular solution; after waiting >30 min in whole-cell mode, the internal GABA should have diffused into the presynaptic terminals (Pan et al., 1993). After internal application of 100 mm GABA for 30-68 min, the IPSC amplitude (expressed as a percentage of the native EPSC amplitude in the same cell) was 0.27 ± 0.30% (n = 7). This was significantly smaller than the mean amplitude of postload IPSCs after external high-K+/100 mm GABA loading solution (3.7 ± 0.8%; n = 7), suggesting that little GABA enters via the cytoplasmic pathway.
If GABA enters presynaptic vesicles by endocytosis, the appearance of postload IPSCs should be blocked by an inhibitor of this process. After incubation of cultures in CsA (10 μm for 18-22 h) to block dynamin-dependent endocytosis (Engisch and Nowycky, 1998), the mean native EPSC amplitude was reduced from 24.4 ± 2.1 nA (n = 33) to 7.9 ± 1.0 nA (n = 4), presumably reflecting the depletion of spontaneously cycling vesicles by CsA. However, even when normalized to these smaller EPSCs, the postload IPSC amplitude in CsA-treated cells was only 0.62 ± 0.33% (n = 4), significantly smaller than control. Together, these results strongly suggest that loaded GABA is taken into presynaptic vesicles by endocytosis from the bath solution.
GABA endocytosis follows exocytosis
Given that external GABA can be endocytosed into synaptic vesicles, I next used this assay to examine some of the requirements for endocytosis. Superfusion of high-K+/high-GABA solution that lacked added Ca2+ was unable to cause endocytosis, as indicated by the lack of appearance of a postload IPSC (0.31 ± 0.10% of the native EPSC amplitude; n = 6; not significantly different from the preload amplitudes measured in the same cells) (Fig. 5A, left three traces). However, if exocytosis was first triggered by a 5-s-long application of high-K+ solution containing normal Ca2+ (but no GABA), endocytosis could subsequently proceed in a normal-K+ solution containing 100 mm GABA but no added Ca2+ (postload amplitude 2.64 ± 0.85% of native EPSC amplitude; n = 6; significantly greater than control) (Fig. 5A, right two traces). Thus, Ca2+-dependent exocytosis must precede endocytosis, but subsequent endocytosis does not require external Ca2+. Furthermore, exocytosis can be relatively brief (Fig. 5A, 5 s), but endocytosis, at least under these conditions, is relatively slow. A postload IPSC did not appear if, after exocytosis, GABA was present for <15-20 s.
Endocytosis could also follow exocytosis that was triggered by a train of action potentials, instead of high K+. A train of 300 autaptic stimuli at 20 Hz was applied to the cell in normal bath solution (Fig. 5B, second trace from left; beginning and end of the train shown expanded in the inset). Immediately after the train, the cell was bathed for 60 s in normal-K+ solution containing 100 mm GABA but no added Ca2+. A large postload IPSC subsequently appeared, indicating robust endocytosis after the train (postload amplitude 6.55 ± 1.32%; n = 3; significantly greater than control) (Fig. 5B, right trace). These results strengthen the previous conclusion that GABA is endocytosed into synaptic vesicles from the bath, because the appearance of a postload IPSC strictly requires preceding vesicular exocytosis.
Loaded GABA passes through the vesicle cycle
Endocytosed GABA can be thought of as an exogenous tracer that is injected at one point in the synaptic vesicle cycle and expelled at a later point, where it is detected electrically as a postload IPSC. Because glutamatergic terminals are thought to lack mechanisms for synthesizing or transporting GABA (Takamori et al., 2000), the loaded GABA should pass inertly through the vesicle cycle and steadily be depleted with stimulation.
Before testing this prediction, control experiments were done to check the effect on neurons of a 60 s exposure to the high-K+/100 mm GABA loading solution. Native EPSC amplitudes recovered with a time constant of 13.6 ± 1.9 s (n = 8; data not shown), which presumably reflects recovery of the readily releasable pool of synaptic vesicles after strong K+ depolarization (Stevens and Tsujimoto, 1995; Stevens and Wesseling, 1999). In contrast, native IPSC amplitudes recovered much more slowly than EPSCs, with a time constant of 52.6 ± 6.6 s (n = 7; data not shown). This may reflect the slow recovery from desensitization of postsynaptic GABAA receptors after prolonged exposure to 100 mm GABA in the loading solution. To confirm this, the experiment was repeated using brief puffer applications of 100 μm GABA to pyramidal neurons after GABA loading. Puffer responses recovered with a mean time constant of 50.7 ± 10.2 s (n = 5; data not shown), which is not significantly different from the recovery time constant for native IPSCs. Together, these results suggest that postsynaptic GABAA receptors recover relatively slowly (τ = ∼50 s) from exposure to the high concentration of GABA used in the GABA loading solution. Therefore, in the experiments described below, the cell was allowed to rest without stimulation for >2 min after washout of the loading solution to avoid confounding GABA unloading and recovery from desensitization.
Figure 6 illustrates the basic unloading experiment in an isolated pyramidal neuron. The leftmost points (Fig. 6A, near “1”) show the amplitude of native EPSCs, evoked at 0.1 Hz, before loading. (Single synaptic currents near the numbered time points are shown in Fig. 6B.) After GABA loading (100 mm; arrow), the bath was switched to CNQX plus d-APV to isolate the postload IPSC, and the stimulation rate was increased to 1 Hz to accelerate unloading. The IPSC amplitude declined exponentially over 600 stimuli (Fig. 6A, points near “3” and “4”). Because part of this decline may have been caused by temporary depletion of the readily releasable pool of vesicles, after 600 stimuli, the stimulus rate was reduced to 0.1 Hz. The IPSC amplitude did not recover, confirming that the loaded GABA was irreversibly depleted (Fig. 6A, points near “5”). Finally, CNQX plus d-APV was washed out of the bath, revealing that the native EPSC remained near its original size (Fig. 6A, points near “2”). This confirms that the decline in postload IPSC amplitude was not caused by nonspecific rundown. Similar results were obtained in five cells. The mean decay time constant for postload IPSC amplitudes was 175 ± 39 s (n = 5; stimulation at 1 Hz).
Discussion
Here, I have described autaptic synapses that express functional postsynaptic GABAA receptors but that normally lack GABA in their presynaptic vesicles. Hence, these synapses are presynaptically silent with respect to GABA (but still function as normal glutamatergic synapses). It is possible to unsilence these synapses by loading bath-applied GABA into their presynaptic vesicles by endocytosis. The loaded GABA can subsequently be released onto perisynaptic GABAA receptors by additional rounds of normal, activity-dependent vesicular exocytosis.
What could be the functional significance of these synapses? They are not simply a curiosity of autaptic connections, because GABAA receptors can also cluster at glutamatergic terminals in mass hippocampal cultures that contain many more synapses than autapses (Kannenberg et al., 1999; Christie et al., 2002). It is unclear whether presynaptically silent GABA synapses also occur in vivo, although synapses that colocalize postsynaptic receptors for two fast neurotransmitters have been reported in slice preparations (Jonas et al., 1998; Jo and Schlichter, 1999; Walker et al., 2001).
If presynaptically silent GABA synapses are present in intact tissue, their unsilencing would require GABA to be loaded into presynaptic vesicles. How might this be achieved? One means would be through upregulation of GABA vesicular transporters (VGATs) (McIntire et al., 1997) and the GABA synthetic enzyme l-glutamic acid decarboxylase (GAD; possibly the GAD67 isoform) (Jin et al., 2003). Most excitatory terminals do not normally contain GAD or VGATs (Takamori et al., 2000), but GAD67 is found in hippocampal mossy fibers (Walker et al., 2001), and unknown isoforms of VGATs might exist. The finding that some autaptic EPSCs contained a small bicuculline-sensitive component before loading (Figs. 1A, 5) suggests that pyramidal neurons are sometimes able to package endogenous GABA in their vesicles, perhaps through the presence of ectopic synthetic enzymes and transporters for GABA.
Unsilencing might also be achieved by endocytosis of extracellular GABA, as was done artificially here (Kim et al., 2000). This seems unlikely under physiological conditions because of the GABA concentrations required. I have shown that >10 mm GABA must be present for significant loading and release to occur (Fig. 4C). However, the concentration of synaptically released GABA in the inhibitory cleft is thought to be only 300-500 μm (Jones and Westbrook, 1995; Perrais and Ropert, 1999), and the concentration that spills over to nearby glutamatergic synapses, and so is available for endocytosis, would be even lower (Rusakov et al., 1999).
Although postsynaptic GABAA receptors are unlikely to be activated by the small amount of GABA that is endocytosed and later released from synaptic vesicles, a similar mechanism might work for other agonist/receptor combinations. Neuromodulators or transmitters that are present at relatively high extracellular concentrations during periods of intense activity could be retained within synaptic vesicles and subsequently released onto high-affinity receptors (Nicoll et al., 1990). In this way, a memory of previous extracellular conditions could be stored in vesicles to be replayed at a later time.
Although the physiological role of presynaptically silent synapses remains speculative, these synapses have immediate value as a tool for studying synaptic function and development. First, they are informative about the targeting of postsynaptic receptors. Previous studies on postsynaptic targeting have suggested the existence of a synaptogenic signal common to both excitatory and inhibitory synapses, as well as an additional specificity-conferring signal that normally ensures the postsynaptic receptors are matched to the correct presynaptic transmitter phenotype (Rao et al., 2000). This specificity-conferring signal is presumably absent at some terminals in hippocampal cultures, allowing the mistargeting of GABAA receptors to glutamatergic autapses. Here, I have shown that mistargeted GABAA receptors on pyramidal neurons operate like normal subsynaptic GABAA receptors at inhibitory synapses, with identical decay kinetics and modulation (Fig. 2), despite the absence of the specificity-conferring signal. However, the postload and native IPSCs do differ in two important respects. First, the postload IPSCs have a slower rise time, a difference that cannot be explained by voltage-clamp errors (Fig. 2B). The most likely explanation is that postload IPSCs arise in GABAA receptors that are not as tightly localized under presynaptic terminals as are those at native GABAergic terminals, requiring the released GABA to diffuse further (Takamori et al., 2000). It is unlikely that this slight dispersal of GABAA receptors would be apparent using light microscopy (Rao et al., 2000). Note that these perisynaptic GABAA receptors still produce IPSCs with the same decay kinetics as native IPSCs (Fig. 2B), in contrast with truly extrasynaptic GABAA receptors, which have been reported to decay much more slowly (Banks and Pearce, 2000). The second difference between postload and native IPSCs is that the mean saturating amplitude of postload IPSCs is ∼10-fold smaller than the mean amplitude of native IPSCs (0.9 vs 10.7 nA). This may reflect the greater spatial dispersion of mistargeted receptors, so these receptors experience a lower GABA concentration. Alternatively, there may be a lower density of GABAA receptors at each contact or fewer contacts per pyramidal neuron, or the GABA concentration inside loaded vesicles may be less than that inside native vesicles (see below). In summary, the absence of the postulated specificity-conferring signal appears to produce a measurable deficiency in receptor clustering at the postsynaptic membrane, but the individual GABAA receptors otherwise function normally.
These synapses also provide a method for studying the contents of synaptic vesicles. I showed that the normalized postload IPSC amplitude saturates when the concentration of GABA in the loading solution is 100 mm (Fig. 4C). The GABA concentration inside the synaptic vesicle just before release will be the same as that in the loading solution provided (1) the vesicle interior equilibrates with the outside solution during endocytosis, and (2) the GABA concentration does not change as the vesicle passes through its cycle. Assumption 1 seems likely, because GABA is a freely diffusing small molecule, although the transient existence of a narrow fusion pore could limit its entry (Aravanis et al., 2003). Assumption 2 may also be true, because the synaptic vesicle retains its identity as it passes through the cycle (Murthy and Stevens, 1998), although neurotransmitter could spontaneously leak out of vesicles (Zhou et al., 2000). Assuming free diffusion and small leakage, however, we can state that the maximum effective GABA concentration in vesicles is ∼100 mm; above this, either the release mechanism or postsynaptic GABAA receptors are saturated. This supports modeling studies that estimate the intravesicular GABA concentration to be 80-100 mm (Busch and Sakmann, 1990; Axmacher et al., 2004).
GABA loading also provides a convenient assay for endocytosis at small central synapses. Endocytosis at these synapses has frequently been studied using either the fluorescent lipophilic FM dyes (Betz et al., 1996; Murthy, 1999) or the pH-sensitive fluorescent marker synaptophluorin (Sankaranarayanan et al., 2000; Gandhi and Stevens, 2003). However, some dyes may not enter and exit vesicles rapidly (Klingauf et al., 1998; Pyle et al., 2000; Stevens and Williams, 2000; Aravanis et al., 2003), and overexpression of a vesicle-associated protein, synaptophluorin, may perturb the system (Sankaranarayanan et al., 2000). GABA loading offers a complementary approach, in which the tracer is a small, readily diffusible, physiological molecule, GABA, which presumably enters synaptic vesicles as readily as the native neurotransmitter, glutamate, exits them. Here, I showed that, after Ca2+-dependent exocytosis, endocytosis of GABA can proceed in low-Ca2+ solution (Fig. 5). This supports previous results with FM1-43 (Ryan et al., 1993, 1996; Ryan and Smith, 1995; Wu and Betz, 1996) but does not address more recent suggestions that Ca2+ inhibits or accelerates endocytosis under some circumstances (Cousin and Robinson, 2000; Sankaranarayanan and Ryan, 2001; Wu, 2004).
Finally, GABA loading can provide information on vesicle cycling kinetics. The amplitude of the postload IPSC ran down irreversibly with a mean time constant of 175 s (1 Hz stimulation), equivalent to a loss of ∼0.6% of the GABA response with each action potential (Fig. 6). FM1-43 destaining is reported to occur at a similar rate (0.5% fluorescence lost per action potential; 2-20 Hz stimulation) (Reuter, 1995; Ryan and Smith, 1995; Isaacson and Hille, 1997). In general, GABA should be a better tracer for monitoring exocytosis than FM dyes, because its exit from vesicles does not depend on the rate at which dyes can departition from the membrane (Henkel and Betz, 1995; Klingauf et al., 1998). The activity-dependent decline of the postload IPSC should thus provide an accurate assay for the loss of neurotransmitter from synaptic vesicles, particularly under conditions in which fast, kiss-and-run-mediated exocytosis is expected to predominate.
In conclusion, by characterizing the properties of presynaptically silent GABA synapses in culture, I have given a striking demonstration that synaptic vesicles can indiscriminately endocytose extracellular solution, preserving a “snapshot” of previous conditions. Subsequent exocytosis then delivers an echo of the previous extracellular environment, potentially generating a novel form of short-term synaptic plasticity.
Footnotes
This work was supported by recurrent funding from The John Curtin School of Medical Research. I thank Ann Marie Craig, George Richerson, and Greg Stuart for helpful comments on this manuscript.
Correspondence should be addressed to Dr. John M. Bekkers, Division of Neuroscience, The John Curtin School of Medical Research, The Australian National University, Canberra, Australian Capital Territory 0200, Australia. E-mail: John.Bekkers@anu.edu.au.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254031-09$15.00/0
References
- Aravanis AM, Pyle JL, Tsien RW (2003) Single synaptic vesicles fusing transiently and successively without loss of identity. Nature 423: 643-647. [DOI] [PubMed] [Google Scholar]
- Axmacher N, Stemmler M, Engel D, Draguhn A, Ritz R (2004) Transmitter metabolism as a mechanism of synaptic plasticity: a modeling study. J Neurophysiol 91: 25-39. [DOI] [PubMed] [Google Scholar]
- Banks MI, Pearce RA (2000) Kinetic differences between synaptic and extrasynaptic GABAA receptors in CA1 pyramidal cells. J Neurosci 20: 937-948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF (1991) Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci USA 88: 7834-7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Mao F, Bewick GS (1992) Activity-dependent fluorescent staining and destaining of living vertebrate motor nerve terminals. J Neurosci 12: 363-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Mao F, Smith CB (1996) Imaging exocytosis and endocytosis. Curr Opin Neurobiol 6: 365-371. [DOI] [PubMed] [Google Scholar]
- Busch C, Sakmann B (1990) Synaptic transmission in hippocampal neurons: numerical reconstruction of quantal IPSCs. Cold Spring Harb Symp Quant Biol 55: 69-80. [DOI] [PubMed] [Google Scholar]
- Christie SB, Miralles CP, De Blas AL (2002) GABAergic innervation organizes synaptic and extrasynaptic GABAA receptor clustering in cultured hippocampal neurons. J Neurosci 22: 684-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM (1997) Detection of spontaneous synaptic events using an optimally scaled template. Biophys J 73: 220-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin MA, Robinson PJ (2000) Ca2+ influx inhibits dynamin and arrests synaptic vesicle endocytosis at the active zone. J Neurosci 20: 949-957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engisch KL, Nowycky MC (1998) Compensatory and excess retrieval: two types of endocytosis following single step depolarizations in bovine adrenal chromaffin cells. J Physiol (Lond) 506: 591-608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fon EA, Edwards RH (2001) Molecular mechanisms of neurotransmitter release. Muscle Nerve 24: 581-601. [DOI] [PubMed] [Google Scholar]
- Gandhi SP, Stevens CF (2003) Three modes of synaptic vesicular recycling revealed by single-vesicle imaging. Nature 423: 607-613. [DOI] [PubMed] [Google Scholar]
- Gutiérrez R (2000) Seizures induce simultaneous GABAergic and glutamatergic transmission in the dentate gyrus-CA3 system. J Neurophysiol 84: 3088-3090. [DOI] [PubMed] [Google Scholar]
- Henkel AW, Betz WJ (1995) Staurosporine blocks evoked release of FM1-43 but not acetylcholine from frog motor nerve terminals. J Neurosci 15: 8246-8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Hille B (1997) GABAB-mediated presynaptic inhibition of excitatory transmission and synaptic vesicle dynamics in cultured hippocampal neurons. Neuron 18: 143-152. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Sahara Y, Takahashi T (2002) A single packet of transmitter does not saturate postsynaptic glutamate receptors. Neuron 34: 613-621. [DOI] [PubMed] [Google Scholar]
- Jin H, Wu H, Osterhaus G, Wei J, Davis K, Sha D, Floor E, Hsu CC, Kopke RD, Wu JY (2003) Demonstration of functional coupling between γ-aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proc Natl Acad Sci USA 100: 4293-4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo Y-H, Schlichter R (1999) Synaptic corelease of ATP and GABA in cultured spinal neurons. Nat Neurosci 2: 241-245. [DOI] [PubMed] [Google Scholar]
- Jonas P, Bischofberger J, Sandkühler J (1998) Corelease of two fast neurotransmitters at a central synapse. Science 281: 419-424. [DOI] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL (1995) Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron 15: 181-191. [DOI] [PubMed] [Google Scholar]
- Kannenberg K, Sieghart W, Reuter H (1999) Clusters of GABAA receptors on cultured hippocampal cells correlate only partially with functional synapses. Eur J Neurosci 11: 1256-1264. [DOI] [PubMed] [Google Scholar]
- Kim K-T, Koh D-S, Hille B (2000) Loading of oxidizable transmitters into secretory vesicles permits carbon-fiber amperometry. J Neurosci 20: RC101(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingauf J, Kavalali ET, Tsien RW (1998) Kinetics and regulation of fast endocytosis at hippocampal synapses. Nature 394: 581-585. [DOI] [PubMed] [Google Scholar]
- Malinow R, Mainen ZF, Hayashi Y (2000) LTP mechanisms: from silence to four-lane traffic. Curr Opin Neurobiol 10: 352-357. [DOI] [PubMed] [Google Scholar]
- McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM (1997) Identification and characterization of the vesicular GABA transporter. Nature 389: 870-876. [DOI] [PubMed] [Google Scholar]
- Murthy VN (1999) Optical detection of synaptic vesicle exocytosis and endocytosis. Curr Opin Neurobiol 9: 314-320. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Stevens CF (1998) Synaptic vesicles retain their identity through the endocytic cycle. Nature 392: 497-501. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC, Kauer JA (1990) Functional comparison of neurotransmitter receptor subtypes in mammalian central nervous system. Physiol Rev 70: 513-565. [DOI] [PubMed] [Google Scholar]
- Otis TS (2001) Vesicular glutamate transporters in cognito. Neuron 29: 11-14. [DOI] [PubMed] [Google Scholar]
- Pan ZZ, Tong G, Jahr CE (1993) A false transmitter at excitatory synapses. Neuron 11: 85-91. [DOI] [PubMed] [Google Scholar]
- Perrais D, Ropert N (1999) Effect of zolpidem on miniature IPSCs and occupancy of postsynaptic GABAA receptors in central synapses. J Neurosci 19: 578-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle JL, Kavalali ET, Piedras-Renteria ES, Tsien RW (2000) Rapid reuse of readily releasable pool vesicles at hippocampal synapses. Neuron 28: 221-231. [DOI] [PubMed] [Google Scholar]
- Rao A, Cha EM, Craig AM (2000) Mismatched appositions of presynaptic and postsynaptic components in isolated hippocampal neurons. J Neurosci 20: 8344-8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter H (1995) Measurements of exocytosis from single presynaptic nerve terminals reveal heterogeneous inhibition by Ca2+-channel blockers. Neuron 14: 773-779. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM, Stewart MG (1999) Hippocampal synapses: do they talk to their neighbours? Trends Neurosci 22: 382-388. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Smith SJ (1995) Vesicle pool mobilization during action potential firing at hippocampal synapses. Neuron 14: 983-989. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Reuter H, Wendland B, Schweizer FE, Tsien RW, Smith SJ (1993) The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron 11: 713-724. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Smith SJ, Reuter H (1996) The timing of synaptic vesicle endocytosis. Proc Natl Acad Sci USA 93: 5567-5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA (2001) Calcium accelerates endocytosis of vSNAREs at hippocampal synapses. Nat Neurosci 4: 129-136. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, De Angelis D, Rothman JE, Ryan TA (2000) The use of pHluorins for optical measurements of presynaptic activity. Biophys J 79: 2199-2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Tsujimoto T (1995) Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proc Natl Acad Sci USA 92: 846-849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Wesseling JF (1999) Identification of a novel process limiting the rate of synaptic vesicle cycling at hippocampal synapses. Neuron 24: 1017-1028. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Williams JH (2000) “Kiss and run” exocytosis at hippocampal synapses. Proc Natl Acad Sci USA 97: 12828-12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Rhee JS, Rosenmund C, Jahn R (2000) Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 407: 189-194. [DOI] [PubMed] [Google Scholar]
- Walker MC, Ruiz A, Kullmann DM (2001) Monosynaptic GABAergic signaling from dentate to CA3 with a pharmacological and physiological profile typical of mossy fiber synapses. Neuron 29: 703-715. [DOI] [PubMed] [Google Scholar]
- Wu L-G (2004) Kinetic regulation of vesicle endocytosis at synapses. Trends Neurosci 27: 548-554. [DOI] [PubMed] [Google Scholar]
- Wu L-G, Betz WJ (1996) Nerve activity but not intracellular calcium determines the time course of endocytosis at the frog neuromuscular junction. Neuron 17: 769-779. [DOI] [PubMed] [Google Scholar]
- Wu L-G, Saggau P (1997) Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci 20: 204-212. [DOI] [PubMed] [Google Scholar]
- Yoon K-W, Rothman SM (1991) Adenosine inhibits excitatory but not inhibitory synaptic transmission in the hippocampus. J Neurosci 11: 1375-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Petersen CCH, Nicoll RA (2000) Effects of reduced vesicular filling on synaptic transmission in rat hippocampal neurones. J Physiol (Lond) 525: 195-206. [DOI] [PMC free article] [PubMed] [Google Scholar]