

Abstract
Huntington's disease (HD) is an adult-onset neurodegenerative disorder involving motor dysfunction, cognitive deficits, and psychiatric disturbances that result from underlying striatal and cortical dysfunction and neuropathology. The YAC128 mouse model of HD reproduces both the motor deficits and selective degeneration observed in the human disease. However, the presence of cognitive impairment in this model has not been determined. Here, we report mild cognitive deficits in YAC128 mice that precede motor onset and progressively worsen with age. Rotarod testing revealed a motor learning deficit at 2 months of age that progresses such that by 12 months of age, untrained YAC128 mice are unable to learn the rotarod task. Additional support for cognitive dysfunction is evident in a simple swimming test in which YAC128 mice take longer to find the platform than wild-type (WT) controls beginning at 8 months of age. YAC128 mice also have deficits in open-field habituation and in a swimming T-maze test at this age. Strikingly, in the reversal phase of the swimming T-maze test, YAC128 mice take twice as long as WT mice to locate the platform, indicating a difficulty in changing strategy. At 12 months of age, YAC128 mice show decreased prepulse inhibition and habituation to acoustic startle. The clear pattern of cognitive dysfunction in YAC128 mice is similar to the symptoms and progression of cognitive deficits in human HD and provides both the opportunity to examine the relationship between cognitive dysfunction, motor impairment, and neuropathology in HD and to assess whether potential therapies for HD can restore cognitive function.
Keywords: Huntington's disease, cognition, behavior, mouse model, learning, memory
Introduction
Huntington's disease (HD) is the most common inherited neurodegenerative disorder affecting ∼1 in 10,000 individuals (Conneally, 1984). HD is caused by a CAG repeat expansion in the HD gene (Huntington's Disease Collaborative Research Group, 1993). Patients with HD have deficits in motor coordination, cognitive impairment, and psychiatric disturbances. In the brain, there is a characteristic degeneration of the caudate and putamen with a loss of GABAergic medium spiny projection neurons (Albin et al., 1992). The cortex is also severely affected, whereas the hippocampus and cerebellum are relatively spared until late in the disease (Cudkowicz and Kowall, 1990; Hedreen et al., 1991; Vonsattel and DiFiglia, 1998).
The clinical diagnosis of HD is dependent on characteristic choreiform movements that often herald the onset of the illness. This initial hyperkinesia is usually followed by bradykinesia, rigidity, dystonia, and eventually a complete inability to initiate voluntary movement. Even before clinical diagnosis, persons carrying the mutation demonstrate cognitive dysfunction. Specifically, presymptomatic HD carriers have deficits in shifting strategy, psychomotor speed, recognition memory, planning, and verbal fluency (Hahn-Barma et al., 1998; Lawrence et al., 1998a,b; Paulsen et al., 2001; Berrios et al., 2002; Snowden et al., 2002). At this point in time, the impairments are subtle, and many studies were unable to detect any difference in cognitive function before motor onset (Strauss and Brandt, 1990; Blackmore et al., 1995; de Boo et al., 1997, 1999).
As the disease progresses through motor onset, cognitive deficits worsen and become more widespread. Symptomatic HD patients show deficits in procedural learning (Heindel et al., 1988, 1989; Knopman and Nissen, 1991; Gabrieli et al., 1997; Schmidtke et al., 2002) and have been shown to have deficits in both long-term memory (Wilson et al., 1987; Rohrer et al., 1999) and working memory (Lawrence et al., 2000; Brown et al., 2001). One of the most common findings in HD patients is a difficulty in tasks requiring a shift in strategy (Lawrence et al., 1999; Ho et al., 2003). This type of response may stem from an inability to inhibit the previously learned response, a process thought to be mediated by the striatum (Mink, 1996). The loss of inhibition in HD patients is also demonstrated by impairments in prepulse inhibition (PPI) (Swerdlow et al., 1995).
The YAC128 mouse model of HD expresses full-length mutant huntingtin (htt) and recapitulates the motor dysfunction and neuropathology observed in human HD (Slow et al., 2003). Behaviorally, YAC128 mice show early deficits in motor coordination on the rotarod as well as biphasic hyperactive-hypoactive activity in an open field. Phenotypic onset of the disease occurs at 3 months of age with hyperkinesia. In the brain, striatal and cortical atrophy develop at ∼9 months of age and are followed by neuronal atrophy and neuronal loss.
Although the motor deficits and neuropathology in the YAC128 mice recapitulate symptoms of the human disease, cognitive function has not been assessed. As such, we designed these experiments to assess whether the cognitive deficits present in human HD are modeled in the YAC128 mice and to assess how they relate to motor dysfunction and neuropathology. Cognitive deficits have been demonstrated previously in the R6/2 mouse model of polyglutamine toxicity in which learning deficits were shown to precede motor dysfunction (Carter et al., 1999; Lione et al., 1999).
Overall, we show cognitive deficits early in the YAC128 mice that precede motor onset and progress to global cognitive impairment in symptomatic mice. The cognitive deficits we report are similar to those present in human HD patients and can be used both to dissect the relationship between cognitive impairment, motor dysfunction, and neuropathology in the natural history of YAC128 mice and to assess the efficacy of potential treatments for HD in preserving and or restoring cognitive function.
Materials and Methods
Animals. All of the experiments were performed on the YAC128 mouse model of HD maintained on the FVB/N strain background (Slow et al., 2003). Mice were group housed with littermates of mixed genotype. Mice were tested during the light phase of a normal light/dark cycle in which lights were turned off at 8:00 P.M. and on at 6:00 A.M. Experimenters were blind to the genotype of the mice. Unless stated otherwise, experiments were performed on 2-month-old presymptomatic mice and 8- to 9-month-old symptomatic mice. Some experiments were performed on 12-month-old mice. Experiments were performed with the approval of the animal care committee at the University of British Columbia.
Rotarod test of motor learning. Motor coordination and motor learning were assessed on an accelerating rotarod apparatus (UGO Basile, Comerio, Italy). In this test, mice must learn to run when placed on a rotating rod to prevent them from falling. Once the task is learned, the rotarod can be used to assess motor coordination and balance. Separate cohorts of mice were trained at 2, 7, or 12 months of age with three trials per day spaced 2 h apart for 3 d. Two-month-old mice were trained at a fixed speed of 24 rpm. Seven- and 12-month-old mice were trained with the rotarod accelerating from 5 to 40 rpm to facilitate learning in older animals. Mice were subsequently tested with three trials in 1 d spaced 2 h apart. Fixed-speed rotarod testing was completed at a speed of 24 rpm to a maximum time of 60 s. For the accelerating task, the rotarod was accelerated from 5 to 40 rpm over 5 min with a maximum score of 300 s. The latency to fall was recorded for each of the three trials and averaged to generate the overall time for each mouse. Mice trained at 2 and 7 months of age were tested bimonthly and monthly, respectively, until 12 months of age. Motor coordination was assessed bimonthly from 2 to 12 months of age at a fixed speed of 24 rpm or accelerating as described above.
Open-field habituation test of learning and memory. Open-field habituation was measured using an automated open-field system (San Diego Instruments, San Diego, CA). Because mice demonstrate less exploratory activity in a familiar environment, this simple test assesses the ability of the mouse to learn and remember the open-field chamber. Intrasession habituation is measured by the relative decrease in activity over time in a single open-field trial and is calculated as the difference in activity from the first interval divided by the activity of the first interval. Intersession habituation is the relative decrease in activity between repeated open-field trials and is calculated as the decrease in activity from the first trial divided by the level of activity in the first trial. The activity of mice in the open field was assessed in the dark during the light cycle and was measured automatically as the number of photobeam breaks during each trial. Mice that were naive to the open-field chamber were given five trials of 30 min duration. The first three trials were given at 9:00 A.M. on 3 consecutive days with the remaining trials given at 11:00 A.M. and 1:00 P.M. on the third day. On the first trial, activity was recorded for the duration of the 30 min period to assess intrasession habituation in 5 min intervals. The activity during the first 10 min of each of the five open-field trials was used to assess intersession habituation. For each trial, mice were allowed to remain in the testing chamber with the lights out for a total of 30 min to facilitate habituation.
Simple swimming test of procedural learning. A simple swimming test was designed to test procedural learning. In this test, mice were placed in the middle of a linear swimming chamber (76 × 13 cm; water depth, 9 cm; platform, 6 × 13 cm) facing away from an escape platform. Mice were trained to reach the platform in the shortest amount of time to escape from the water. Completion of this task involves learning and remembering the location of the platform or the route followed to reach the platform. On subsequent trials, mice must plan to turn around immediately for the shortest route to the platform. The amount of time required for the mouse to reach the platform and the initial swimming direction were recorded for each trial. Swimming toward the platform was arbitrarily given a score of 0, whereas swimming away from the platform was initially given a score of 1. Mice were trained at 2 months of age with three pairs of two consecutive trials spaced 2 h apart for 2 d. Mice were tested bimonthly with three tests per day spaced 2 h apart until 12 months of age.
Normal phase swimming T-maze test of procedural learning and memory. We developed a swimming T-maze test to assess procedural learning. In this test, mice were placed in the base of a water-filled T-maze with an escape platform located in the right arm of the maze (T-maze dimensions: arms, 38 × 14 cm; water depth, 7 cm; platform, 10 × 14 cm). Mice must learn to turn right after reaching the top of the T to reach the platform directly. The time to reach the platform and the path taken to reach the platform were recorded. Swimming right was arbitrarily given a score of 0, whereas swimming left was given a score of 1. Mice received four trials per day spaced 45 min apart for 3 d. To successfully complete this task, mice must remember either the location of or the path to the escape platform. Because FVB/N mice have severe retinal degeneration at the age tested (Taketo et al., 1991; Huerta et al., 1999), learning to swim to the correct arm of the maze likely relies on internal rather than external cues.
Reversal phase swimming T-maze test of strategy shifting. After 1 d of rest, we included a reversal phase to the swimming T-maze test to assess the ability of the mice to replace a previously learned strategy. For this test, the platform was switched to the left arm of the T-maze, and again the amount of time and the path to reach the platform were recorded. Swimming toward the platform was arbitrarily given a score of 0, whereas initially swimming away from the platform was given a score of 1. In addition, the total number of arm entries was noted for each mouse. Mice received four trials per day spaced 45 min apart for 3 d. After 3 d of reversal testing, the swimming speed of the mice was measured by blocking the stem of the T-maze and measuring the amount of time the mice take to swim the length of the top of the T to reach the platform. Mice were given five trials spaced 45 min apart. The last four trials were used to calculate swimming speed.
PPI and habituation to acoustic startle. Acoustic startle PPI and habituation to acoustic startle were assessed using two SR-Lab Systems (San Diego Instruments). PPI is a test of sensorimotor gating and does not involve learning. PPI is measured as the percentage of decrease in startle intensity from a pulse-alone startle when a prepulse is given before the pulse. Habituation to acoustic startle is a test of learning of memory. If mice remember that there is no biological consequence associated with a repeated loud sound, they will show decreased startle in response. Before testing, the sensitivity of the two startle chambers was calibrated using a vibrating standardization unit at 700 V (San Diego Instruments). Mice were then placed into each startle chamber and given a 5 min acclimatization period with background noise alone (65 dB). Each mouse was presented with six trials (block 1) of a 40 ms, 120 dB noise burst (pulse alone). Subsequently, the mice experienced eight blocks of 6 trials (48 trials total), each block consisting of the following trial types: (1) no stimulus (background noise only); (2) a 40 ms, 120 dB noise burst alone; or (3-6) a 40 ms, 120 dB noise burst preceded 100 ms by a 20 ms prepulse (2, 4, 8, and 16 dB above background noise). The order of trials within each block of six trials was pseudorandomized, and four of the eight blocks contained one extra pulse-alone trial. The mice then received another six trials (block 10) of 40 ms, 120 dB noise burst (pulse alone). The intertrial interval was randomized throughout the entire session and ranged between 8 and 23 s. Each animal enclosure was wiped clean with ethanol between test subjects. Habituation analysis was measured using the average of four blocks of pulse-alone scores (block 1, pulse-alone startles from blocks 2-5, pulse-alone startles from blocks 6-9, block 10) and was calculated as the difference in startle between the first block and subsequent blocks divided by the startle in the first block. PPI was calculated from the average of six trials per prepulse as follows: PPI = [(pulse-alone startle) - (prepulse + pulse startle)]/pulse-alone startle.
Statistical analysis. Data are given as the mean ± SEM. Behavioral measures were analyzed by either repeated-measures ANOVA or two-factor ANOVA using software from SPSS (Chicago, IL). Within-subjects effects of age per trial per day and the interaction of genotype and age per trial per day as well as between-subjects effect of genotype were assessed. In case of significant differences between genotypes, post hoc comparisons between genotypes at individual points were assessed with the Student's t test. For these comparisons, the significance level was adjusted to account for errors of multiple measurements [significance level = 0.05/(number of measurements); e.g., for six trials, the significance level was 0.05/6 = 0.008]. Simple comparisons of one variable between two genotypes were assessed by the Student's t test with a significance level of 0.05. Comparisons of categorical data were performed with χ2 tests.
Results
YAC128 mice show impaired motor learning on the rotarod
Motor learning was assessed using the rotarod test of motor coordination. Three separate cohorts of mice were trained on the rotarod at 2, 7, or 12 months of age. The scores for the second and third days of training were recorded as well as the score on a subsequent test day. At 2 months of age, mice were trained and tested on the rotarod at a fixed speed of 24 rpm. In this test, the maximum score was 60 s, and normal mice were able to stay on the rotarod for the full duration of the test on most trials. Although both wild-type (WT) and YAC128 mice learned the rotarod task, WT mice learned the task more rapidly than YAC128 mice (Fig. 1A) (genotype: F(1,55) = 4.4; p = 0.041). Despite receiving equal amounts of training, WT mice were able to stay on the rotarod 10 s longer than YAC128 mice on each of the training days (Fig. 1A) (training day 2: WT, 43.7 ± 3.0 s; YAC128, 31.5 ± 2.8 s; p = 0.005). However, on the test day, YAC128 mice performed equally to WT mice on the rotarod (Fig. 1A) (p = 0.54), indicating that once they were trained, they were physically able to stay on the rod as long as WT mice. Thus, at 2 months of age, the motor coordination of YAC128 mice was normal, but there was a mild motor learning deficit whereby YAC128 mice required more training to reach the same level of performance as WT mice.
Figure 1.

YAC128 mice show deficits in motor learning and motor coordination on the rotarod. Three cohorts of mice were trained on the rotarod at 2, 7, or 12 months of age. Training consisted of three trials per day for 3 d. The scores for the second and third days were recorded. The mice were subsequently given three trials on each test day, and the scores were averaged. A, At 2 months of age, both WT and YAC128 mice learned the rotarod task, but WT mice learned the rotarod task more rapidly than YAC128 mice (day: F(2, 110) = 43.3, p < 0.001; genotype by day: F(2, 110) = 7.8, p = 0.001; genotype: F(1,55) = 4.4, p = 0.041; n = 27 WT, 30 YAC128). On the test day, YAC128 mice performed as well as WT mice on the rotarod, indicating that their motor coordination was equal to wild type but their motor learning was impaired (WT, 53.7 ± 2.2 s; YAC128, 55.6 ± 2.1 s; p = 0.54). B, At 7 months of age, YAC128 mice learned the rotarod task but did not perform as well as WT mice during training or on the test day (day: F(2,58) = 10.6, p < 0.001; genotype by day: F(2,58) = 0.9, p = 0.4; genotype: F(1,29) = 85.1, p < 0.001; p < 0.001; n = 16 YAC128, 15 WT). C, At 12 months of age, previously untrained YAC128 mice failed to learn the rotarod task, whereas WT mice were still able to learn the task (day: F(2,40) = 7.7, p = 0.001; genotype by day: F(2,40) = 7.7, p = 0.001; genotype: F(1,20) = 32.8, p < 0.001; n = 8 YAC128, 14 WT). D, YAC128 mice that had been trained at an earlier time point ran on the rotarod, indicating that 12-month-old YAC128 mice failed to run on the rotarod because of a learning deficit (mice trained at 2 months of age: WT, 272 ± 20 s; YAC128, 139 ± 14 s; p < 0.001; mice trained at 7 months of age: WT, 180 ± 15 s; YAC128, 88 ± 14 s; p < 0.001; mice trained at 12 months of age: WT, 160 ± 18 s; YAC128, 7 ± 24 s; p < 0.001). E, F, The onset of motor dysfunction was monitored bimonthly using a fixed-speed rotarod test at 24 rpm and an accelerating rotarod test. In both tests, YAC128 mice showed significant motor deficits compared with WT mice (24 rpm, genotype: F(1,15) = 17.2, p < 0.001; accelerating, genotype: F(1,15) = 16.1, p < 0.001; n = 8 WT, 9 YAC128). There was no difference in motor performance at 2 months of age between WT and YAC128 mice. Subsequently, motor coordination in YAC128 mice declined steadily from 4 to 12 months, whereas WT mice showed little change (24 rpm, age by genotype: F(5,75) = 7.1, p < 0.001; accelerating, age by genotype: F(5,75) = 10.2, p < 0.001). There was a trend toward decreased performance on both tests at 4 months of age, which became significant at 6 months of age (24 rpm: WT, 57 ± 6 s; YAC128, 27 ± 4 s; p = 0.001; accelerating: WT, 259 ± 16 s; YAC128, 164 ± 15 s; p = 0.001). sec, Seconds. Error bars represent SEM. **p < 0.01; ***p < 0.001.
At 7 and 12 months of age, mice were trained and tested in an accelerating rotarod test to a maximum score of 300 s. Seven-month-old mice from both genotypes learned the rotarod task, but YAC128 mice performed significantly worse than WT mice during training and on the test day (Fig. 1B) (day: F(2,58) = 10.6, p < 0.001; genotype: F(1,29) = 85.1, p < 0.001). Thus, even after 3 d of training, YAC128 mice were unable to maintain their balance on the rotarod as well as WT mice. Similarly, YAC128 mice trained at 12 months of age were significantly worse than WT mice during training and testing (Fig. 1C) (genotype: F(1,20) = 32.8; p < 0.001). At this age, the differences in rotarod performance were dramatic, because the YAC128 mice failed to demonstrate any improvement in performance with training and thereby showed a complete inability to stay on the rotarod even after 3 d of training (day: F(2,12) = 0.5; p = 0.6).
To determine whether the severe rotarod impairment in YAC128 mice at 12 months of age stemmed from a motor deficit or an inability to learn, we compared the performance of the cohorts of mice trained at 2, 7, and 12 months of age at the 12 month time point. There was a clear effect of age on rotarod performance indicating that either earlier training or more training results in improved performance (age: F(2,116) = 15.5; p < 0.001). Nonetheless, all three cohorts at 12 months of age demonstrated a clear rotarod deficit in the YAC128 mice compared with the WT mice (Fig. 1D) (genotype: F(1,58) = 70.5; p < 0.001). Examining the performance of mice trained before 12 months of age revealed that YAC128 mice trained at earlier time points were able to appropriately perform the rotarod task at 12 months of age (YAC128 mice trained at 2 months of age, 139 ± 14 s; YAC128 mice trained at 7 months of age, 88 ± 14 s; YAC128 mice trained at 12 months of age, 7 ± 24 s). This indicates that YAC128 mice were physically capable of staying on the rotarod at this late age. Thus, the complete failure of previously untrained YAC128 mice to maintain their balance on the rotarod at 12 months of age even after 3 d of training cannot be solely accounted for by deficiencies in motor coordination. In addition to motor impairment, YAC128 mice also had a substantial learning deficit at 12 months of age.
To compare the onset of the motor learning deficit with the development of motor dysfunction, YAC128 mice were followed bimonthly from 2 to 12 months of age on the rotarod. Mice were tested at a fixed speed of 24 rpm and in an accelerating rotarod test (Fig. 1E,F, respectively). In both tests, YAC128 mice performed significantly worse than WT mice (24 rpm, genotype: F(1,15) = 17.2, p < 0.001; accelerating, genotype: F(1,15) = 16.1, p < 0.001). YAC128 mice performed equally to WT mice at 2 months of age, when the learning deficit was first observed. At 4 months of age, there was a trend toward decreased rotarod performance in the YAC128 mice compared with wild type that reached significance at 6 months of age (p = 0.001). With increased age, the motor coordination of YAC128 mice on the rotarod worsened, whereas WT mice maintained a relatively constant level of performance (24 rpm, age by genotype: F(5,75) = 7.1, p < 0.001; accelerating, age by genotype: F(5,75) = 10.2, p < 0.001). Thus, the deficit in motor coordination on the rotarod develops at 4 months of age and progressively worsens over time. This deficit is preceded by mild impairments in motor learning that occur at 2 months of age. The motor learning deficit also worsens with time, leading to a complete inability to learn the task at 12 months of age.
YAC128 mice show impaired memory in a test of open-field habituation
Habituation is the decrease in response after repeated exposure to the same stimulus. Open-field habituation is measured by a decrease in exploratory activity with increased exposure to the open-field chamber within one trial or over multiple trials. Two-month-old presymptomatic and 8-month-old symptomatic YAC128 mice were given a total of five open-field trials in which their exploratory activity was measured over the first 10 min of the trial before an additional 20 min of time was provided to facilitate habituation.
At 2 months of age, the activity of YAC128 mice did not differ from WT mice (WT, 163 ± 4 beam breaks; YAC128, 170 ± 4 beam breaks; p = 0.18). Within a 30 min open-field trial, both genotypes showed a decrease in activity between intervals, but the decreases in activity were equal (interval: F(5, 110) = 48.3, p < 0.001; genotype by interval: F(5, 110) = 1.0, p = 0.4; genotype: F(1,22) = 1.3, p = 0.3). Similarly, both groups showed equivalent decreases in activity between repeated open-field trials (trial: F(4,88) = 39.2, p < 0.001; genotype by trial: F(4,88) = 1.9, p = 0.1; genotype: F(1,22) = 1.5, p = 0.2). As a result, the extent of intrasession and intersession habituation was not different between YAC128 and WT mice (intrasession habituation, genotype: F(1,22) = 1.8, p = 0.2; intersession habituation, genotype: F(1,22) = 0.3, p = 0.57). Thus, presymptomatic YAC128 mice did not exhibit a learning and memory deficit in open-field habituation at 2 months of age.
Next, we examined symptomatic YAC128 mice at 8 months of age. At this time, YAC128 mice showed significant hypoactivity compared with WT mice (Fig. 2A) (interval 1: WT, 190 ± 11 beam breaks; YAC128, 145 ± 7.4 beam breaks; p = 0.008). Activity of both the WT and YAC128 mice declined in subsequent intervals in the 30 min open-field trial, with the activity of WT mice declining more rapidly than YAC128 mice (Fig. 2A,B) (interval: F(5,115) = 36.4, p < 0.001; genotype by interval: F(5,115) = 4.5, p < 0.001; genotype: F(1,23) = 8.1, p = 0.009). The initial difference in activity was eliminated, and for the last four intervals, the activity of WT and YAC128 mice did not differ (Fig. 2A). To account for the initial differences in activity, we calculated habituation as the difference in activity from the first trial divided by the activity of the first trial. This calculation revealed decreased intrasession habituation in the YAC128 mice compared with the WT mice (Fig. 2C) (interval: F(4,92) = 8.6, p < 0.001; genotype by interval: F(4,92) = 0.9, p = 0.4; genotype: F(1,23) = 9.7, p = 0.005).
Figure 2.
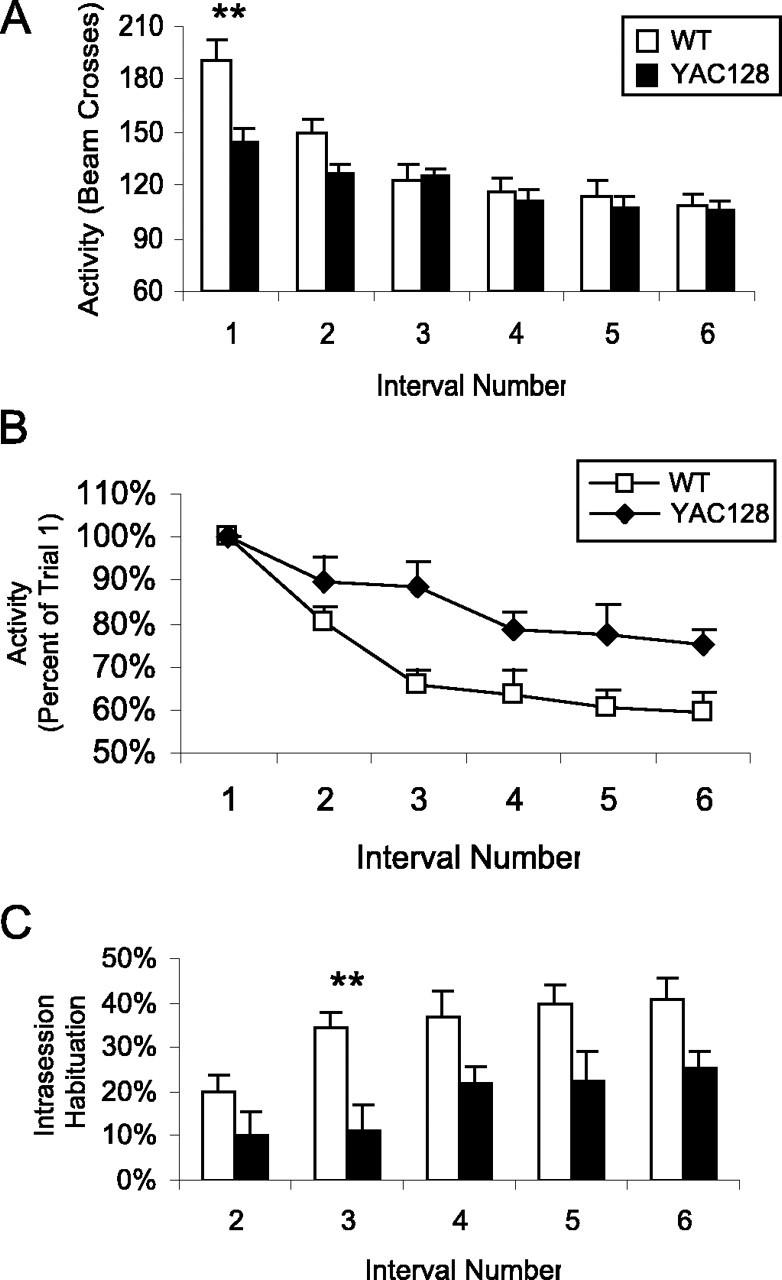
YAC128 mice show decreased intrasession open-field habituation. The activity of 8-month-old mice in an open field was tested for 30 min divided into six 5 min intervals. As mice became familiar with the open field, they showed less exploratory activity. A, Although the activity of both WT mice and YAC128 mice decreased during the 30 min trial, WT activity declined at a more rapid rate (interval: F(5,115) = 36.4, p < 0.001; genotype by interval: F(5,115) = 4.5, p < 0.001; genotype: F(1,23) = 8.1, p = 0.009). Initially, YAC128 mice showed significant hypoactivity compared with WT mice (interval 1: WT, 190 ± 12 beam breaks; YAC128, 145 ± 7 beam breaks; p = 0.008), but this difference was eliminated as the WT mice habituated to the open-field chamber (e.g., interval 3: WT, 123 ± 8 beam breaks; YAC128, 125 ± 5 beam breaks; p = 0.84). B, To control for differences in baseline activity levels, the activity for each interval was divided by the level of activity in the first interval. When the initial level of activity was controlled for, WT mice showed a greater decrease in activity from the first interval starting at interval 3 (percentage of original activity in interval 3: WT, 66%; YAC128, 89%; p = 0.004). C, As a result, YAC128 mice showed decreased intersession habituation compared with WT mice, in which habituation was measured as the difference in activity from the first interval divided by the activity in the first interval (interval: F(4,92) = 8.6, p < 0.001; genotype by interval: F(4,92) = 0.9, p = 0.4; genotype: F(1,23) = 9.7, p = 0.005; n = 12 WT, 13 YAC128). Error bars represent SEM. **p < 0.01.
In subsequent trials on the next 2 d, both YAC128 and WT mice demonstrated intersession habituation (Fig. 3A) (trial: F(4,92) = 60.8, p < 0.001; genotype by trial: F(4,92) = 2.5, p = 0.04; genotype: F(1,23) = 4.8, p = 0.04). Again, in the intersession comparison, YAC128 mice were hypoactive in the first trial, with the difference in activity between the WT and YAC128 mice declining with repeated trials (Fig. 3B). This resulted in a trend toward decreased intersession habituation in YAC128 mice compared with WT mice (trial: F(3,69) = 39.9, p < 0.001; genotype by trial: F(3,69) = 0.1, p = 1.0; genotype: F(1,23) = 3.2, p = 0.08). Accordingly, the rate of decline in activity per trial was significantly less in YAC128 mice compared with WT mice, indicating a deficit in intersession habituation (WT, 38.8 ± 5.0 beam breaks/trial; YAC128, 26.2 ± 3.2 beam breaks/trial; p = 0.05). Overall, symptomatic YAC128 mice showed decreased intrasession and intersession habituation compared with WT mice, suggesting a deficit in memory.
Figure 3.

YAC128 mice show decreased intersession open-field habituation. The activity of 8-month-old mice in an open field was tested once per day for 2 d, followed by three trials on the third day. Mice that recognized the open field is not a novel environment showed less activity with each trial. A, The activity of WT mice declined more rapidly than the activity of YAC128 mice, but both groups showed decreased activity over repeated trials (trial: F(4,92) = 60.8, p < 0.001; genotype by trial: F(4,92) = 2.5, p = 0.04; genotype: F(1,23) = 4.8, p = 0.04). Error bars represent SEM. **p < 0.01. B, YAC128 mice are hypoactive at 8 months of age as indicated by an initial 18% difference in activity between YAC128 and WT mice. With repeated open-field trials, the difference in activity between WT and YAC128 mice progressively decreased to 5% by the last trial. Examining the rate of decrease in activity per trial demonstrates that WT mice showed a more rapid habituation than YAC128 mice (WT, 38.8 ± 5.0 beam breaks/trial; YAC128, 26.2 ± 3.2 beam breaks/trial; p = 0.05; n = 12 WT, 13 YAC128).
YAC128 mice show cognitive deficits in a simple swimming test
A simple swimming test was designed to assess the ability of mice to learn and remember how to reach an escape platform. In this test, mice were placed in a linear swimming chamber facing away from the escape platform and learn to turn around and swim to the platform (Fig. 4A). Both WT and YAC128 mice trained at 2 months of age rapidly learned to turn immediately to reach the platform (Fig. 4B). Testing the same cohort of mice at 4 and 6 months of age revealed no differences in the time or path taken to reach the platform between YAC128 and WT mice. Here, it appears that the motor deficits detected on the rotarod starting at 4 months of age did not impair the swimming ability of the YAC128 mice even at 6 months of age.
Figure 4.
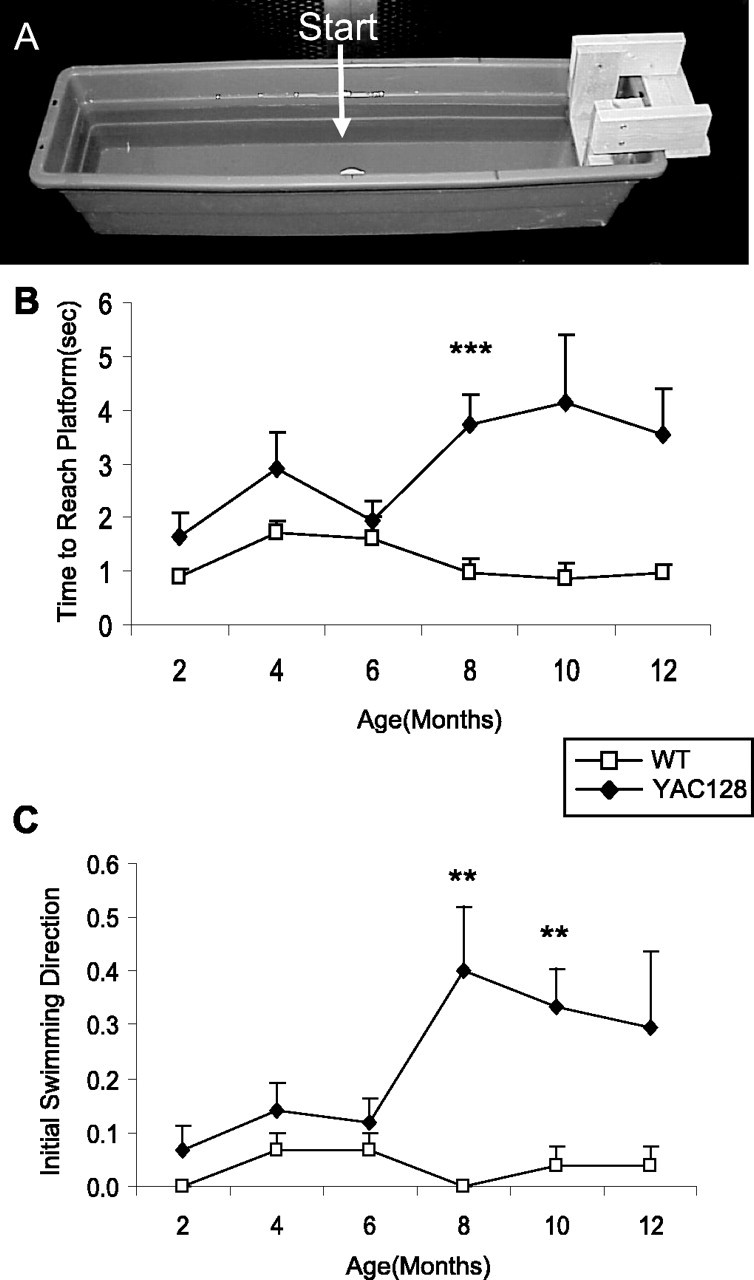
YAC128 mice show a cognitive deficit in a simple swimming test. Mice were placed at the start position facing away from the platform, and the time and path taken to reach the platform were recorded. The apparatus is shown in A. At 2 months of age, all mice learned to turn immediately and swim directly to the platform, and there was no significant difference in the amount of time required to reach the platform (WT, 0.9 ± 0.1 s; YAC128, 1.7 ± 0.4 s, p = 0.1). B, The amount of time needed to reach the platform remained equal between the YAC128 and WT mice until 8 months of age, when YAC128 mice took significantly longer to reach the platform (8 months of age: WT, 1.0 ± 0.3 s; YAC128, 3.7 ± 0.5 s; p < 0.001). Overall, YAC128 mice took significantly longer to reach the platform than WT mice (genotype: F(1,16) = 23.1, p < 0.001). sec, Seconds. C, The increased time to reach the platform resulted from the path taken to reach the platform. Mice swimming toward the platform initially were given a score of 0, whereas mice swimming away from the platform initially were given a score of 1. Starting at 8 months of age, YAC128 mice showed a number of errors in the path chosen to reach the platform, likely resulting from a memory deficit (errors in initial swimming direction: WT, 0 of 27 trials, 0 of 9 mice; YAC128, 12 of 30 trials, 7 of 10 mice; χ2 = 13.7; p < 0.001; n = 9 WT, 9 YAC128). Error bars represent SEM. **p < 0.01; ***p < 0.001.
Surprisingly, at 8 months of age, there was a dramatic increase in the amount of time YAC128 mice took to reach the platform compared with WT animals (Fig. 4B) (WT, 1.0 ± 0.3 s; YAC128, 3.7 ± 0.5 s; p < 0.001). To ascertain whether the increased latency to reach the platform stemmed from a cognitive deficit, we examined the path taken to the platform. In contrast to WT mice, YAC128 mice often failed to swim directly to the platform. To quantify this observation, we arbitrarily gave mice swimming toward the platform a score of 0 and mice swimming away from the platform a score of 1. Using this measurement, the YAC128 mice showed a significant deficit in initiating the task in the correct direction, first detected at 8 months of age and persisting to 12 months of age (Fig. 4C) (errors in initial swimming direction: WT, 0 of 27 trials, 0 of 9 mice; YAC128, 12 of 30 trials, 7 of 10 mice; χ2 = 13.7; p < 0.001). Overall, YAC128 mice took longer to reach the platform starting at 8 months of age, primarily as a result of choosing the incorrect direction to swim at the outset (time to reach platform, genotype: F(1,16) = 23.1; p < 0.001). A motor deficit that decreases swimming speed may also contribute to the time difference, although the swimming speed was unaffected at 6 months of age.
YAC128 mice show multiple cognitive deficits in swimming T-maze test
A swimming T-maze test was used to test procedural and spatial learning in symptomatic YAC128 mice at 8.5 months of age. We developed this test as a simple two-choice test of learning and memory to facilitate rapid training and testing in visually impaired mice. Initially, the escape platform was placed in the right arm of the T-maze. After two trials, both WT and YAC128 mice showed a large decrease in the amount of time required to reach the escape platform (Fig. 5A). By the third day, both genotypes had achieved a constant level of performance, with YAC128 mice taking significantly longer than WT mice to reach the platform (Fig. 5B) (genotype: F(1,23) = 11.5, p = 0.003; WT, 1.9 ± 0.1 s; YAC128, 3.0 ± 0.3 s).
Figure 5.

YAC128 mice show cognitive and motor deficits in swimming T-maze. Mice (8.5 months of age) were trained to swim to a platform located in the right arm of a swimming T-maze. Mice received 3 d of testing with four trials per day. The time and path to reach the platform were recorded. For the path to the platform, right was given a score of 0 and left was given a score of 1. A, After two trials, both YAC128 and WT mice showed a marked decrease in the time required to reach the platform, with minor improvements in time thereafter. B, During the first 2 d, there was no difference in the time required to reach the platform between YAC128 and WT mice. On the third day, YAC128 mice took significantly longer to reach the platform (trial: F(3,69) = 0.6, p = 0.6; genotype by trial: F(3,69) = 0.8, p = 0.5; genotype: F(1,23) = 11.5, p = 0.003; WT, 1.9 ± 0.1 s; YAC128, 3.0 ± 0.3 s). **p < 0.01. C, Closer inspection of the latency to reach the platform on day 3 reveals that WT mice showed very little variation, whereas the YAC128 group have several errant trials (errant trials >4 s: WT, 0 of 44 trials, 0 of 12 mice; YAC128, 10 of 52 trials, 6 of 13 mice; χ2 = 9.4; p = 0.002). D, Some of these errant trials are caused by choosing an indirect path to the platform. Initially, both groups demonstrate a chance level of selecting the correct arm to swim down to reach the platform. By the third day, WT mice always swam down the correct arm, whereas YAC128 mice had some trials in which the wrong arm was chosen (trials swimming down incorrect arm: WT, 0 of 44 trials, 0 of 12 mice; YAC128, 5 of 52 trials, 3 of 13 mice; χ2 = 4.5; p = 0.04; n = 12 WT, 13 YAC128). Error bars represent SEM. sec, Seconds.
To determine whether the increased latency to reach the platform resulted from a motor or cognitive deficit, we examined the path taken to reach the platform and individual swim times for the third day. This analysis revealed that YAC128 mice had several “errant” trials that were much slower than the average, whereas the times for WT mice were tightly bunched around the average (Fig. 5C) (errant trials >4 s: WT, 0 of 44 trials, 0 of 12 mice; YAC128, 10 of 52 trials, 6 of 13 mice; χ2 = 9.4; p = 0.002). To gain insight into the difference, we monitored the direction that each mouse turned after arrival at the stem of the T. Initially, approximately one-half of the mice, regardless of genotype, turned left (given a score of 1), and the other half turned right (given a score of 0). After training, mice learned to turn right to reach the platform, and by the third day, all of the WT mice turned right in all of their trials (Fig. 5D). In contrast, YAC128 mice still turned left in a small number of trials on the third day (WT, 0 of 44 incorrect path trials, 0 of 12 mice; YAC128, 5 of 52 incorrect path trials, 3 of 13 mice; χ2 = 4.5; p = 0.04). This accounts for only one-half of the errant trials observed. Observations suggest that the remainder of the difference in average time to reach the platform was caused by increased pausing and decreased swimming speed (assessed after the reversal phase of the T-maze). Nonetheless, the decision to take inappropriate paths to the platform indicates that a cognitive deficit contributed to the increased latency to reach the platform observed in YAC128 mice.
To assess the ability of YAC128 mice to change strategy, we incorporated a reversal phase into the swimming T-maze test by switching the platform from the right arm to the left arm of the maze. Mice that were trained to swim directly to the right arm would now have to change their strategy to find the shortest path to the platform. In the first trial after switching the platform to the left arm, there was a remarkably clear difference between the YAC128 and WT mice. All of the WT mice swam down the right arm initially, and after discovering that the platform was no longer present, immediately swam down the unvisited left arm of the maze and found the platform. Similar to the WT mice, all of the YAC128 mice entered the right arm of the T-maze initially. However, after discovering that the platform was not present in the right arm of the maze, the majority of the YAC128 mice swam back to the start of the T-maze where they had already been. At this point, some of the YAC128 mice returned to the right arm of the T-maze, whereas some swam directly to the platform in the left arm.
We measured this abnormality by quantifying the number of arms of the maze entered en route to the platform. Although all of the WT mice showed two arm entries, YAC128 mice showed anywhere from two to five arm entries to reach the platform (Fig. 6A). This pattern of response resulted in the YAC128 mice having more arm entries than WT mice (WT, two arm entries; YAC128, 2.9 ± 0.3 arm entries; χ2 = 9.0; p = 0.03) and taking twice as long to reach the platform as WT mice (Fig. 6B) (WT, 7.5 ± 0.4 s; YAC128, 15.6 ± 1.9 s; p < 0.001). With additional trials, both the WT and YAC128 mice learned to swim directly to the platform in the left arm, and by the last trial, swim times were similar to the last trial for the normal phase of the T-maze (Fig. 6B). Again, on day 3 of the reversal phase of the T-maze testing, YAC128 mice took longer than WT mice to reach the platform, but this difference did not reach significance, possibly because a constant level of performance had not been reached (genotype: F(1,23) = 3.9, p = 0.06; WT, 2.0 ± 0.2 s; YAC128, 3.4 ± 0.5 s). The difference in time to reach the platform on the third day resulted from choosing the incorrect arm more frequently (Fig. 6C) (not significant), an increased number of errant trials (data not shown), increased pausing, and decreased swimming speed (see below). It was not caused by an increased number of arm entries, which was equal after day 1 (Fig. 6D). Here, a clear deficit in changing strategy was apparent in the path and time that YAC128 mice took to reach the platform in the reversal of the T-maze test, which training reduces. The mild cognitive deficit observed in the normal phase of the T-maze test was also observed here.
Figure 6.

YAC128 mice show cognitive deficits in the reversal phase of the swimming T-maze test. Mice (8.5 months of age) were trained to swim to a platform located in the right arm of a swimming T-maze. After 3 d of four trials each day, the platform was switched to the left arm of the maze. The time to reach the platform, the path to the platform, and the total number of arm entries were recorded. A, On the first trial of the reversal phase, all WT mice swam right initially then swam straight to the platform in the left arm for a total of two arm entries. In contrast, most YAC128 mice swam back to the start of the maze before either retracing their steps or exploring the left arm of the maze leading to anywhere from two to five arm entries before reaching the platform. This resulted in YAC128 mice showing a greater number of arm entries compared with WT mice (WT, 2 arm entries; YAC128, 2.9 ± 0.3 arm entries; χ2 = 9.0; p = 0.03). B, YAC128 mice also showed a greater latency to reach the platform on the first trial of the reversal phase, despite all mice choosing the same initial swimming direction (WT, 7.5 ± 0.4 s; YAC128, 15.6 ± 1.9 s; p < 0.001). In subsequent trials, both groups improved their latency to reach the platform as they learn to swim directly to the platform in the left arm of the T-maze. On the third day, there was a trend toward an increased latency to reach the platform in YAC128 mice compared with WT mice (trial: F(3,69) = 5.9, p = 0.001; genotype by trial: F(3,69) = 1.7, p = 0.2; genotype: F(1,23) = 3.9, p = 0.06; WT, 2.0 ± 0.2 s; YAC128, 3.4 ± 0.5 s). sec, Seconds. C, This is partially because YAC128 mice continued to choose indirect paths to the platform in the last two trials, whereas all WT mice swam directly to the platform (trials swimming down the incorrect arm: WT, 0 of 22 trials, 0 of 12 mice; YAC128, 5 of 26 trials, 3 of 13 mice; χ2 = 4.7; p = 0.03). D, The difference in latency to reach the platform on the third day did not result from an increased number of arm entries that were equal after day 1. n = 12 WT, 13 YAC128. Error bars represent SEM. **p < 0.01, ***p < 0.001.
Deficits of YAC128 mice in swimming tests are primarily cognitive
To determine the relative contributions of cognitive and motor dysfunction to the increased latency to reach the platform in our swimming tests, we measured swimming speed in mice at 8.5 months of age. The stem of the T-maze was blocked off, and the time for mice to swim from the right arm of the maze to the platform in the left arm of the maze was measured (Fig. 7A). Assuming that mice, on average, swim to the middle of the T before turning, the distance is identical to that traveled in the T-maze tests, but differences in the amount of time required for mice to turn around a corner were not determined. In this assessment, the average latency of YAC128 mice to reach the platform was significantly longer than that of WT mice (WT, 1.6 ± 0.1 s; YAC128, 1.9 ± 0.1 s; p = 0.03), indicating that YAC128 mice had a slower swimming speed than WT mice, likely because of a deficit in motor coordination (WT, 48.2 ± 2.7 cm/s; YAC128, 39.1 ± 2.0 cm/s; p = 0.01). The difference in straight swimming time allows estimation of the contribution of motor deficits to the overall increased latency to reach the platform in YAC128 mice. As predicted, the difference in a straight swimming time of 0.3 s only accounts for a small part of the difference between YAC128 and WT mice on the final days of either normal phase or reversal phase T-maze testing, in which the differences were 1.1 and 1.4 s, respectively. Although part of this difference may stem from YAC128 mice taking longer to turn the corner (not determined), the remainder of the difference is most likely accounted for by cognitive dysfunction.
Figure 7.
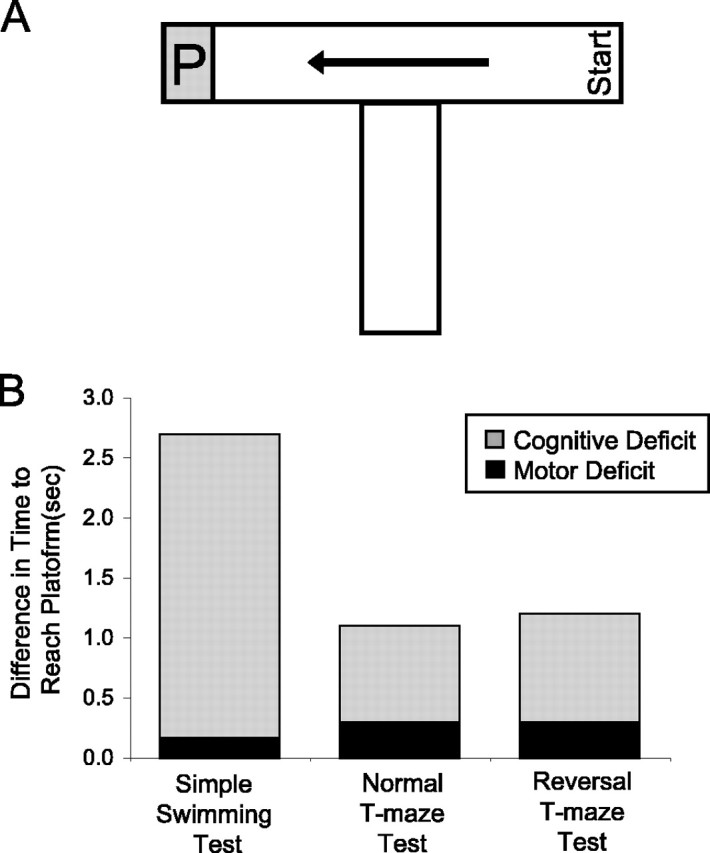
Cognitive deficits are primarily responsible for increased latencies to reach the platform in swimming tests. A, The swimming speed of the mice was assessed by measuring how long the mice took to swim linearly to the platform (P) along the top of the T in the swimming T-maze. YAC128 mice swam slower than WT mice, indicating a deficit in motor coordination at 8.5 months of age (WT, 48.2 ± 2.7 cm/s; YAC128, 39.1 ± 2.0 cm/s; p = 0.01). B, Using swimming speed to calculate the difference in latency to reach the platform predicted by motor deficit alone and comparing this to the actual difference observed reveals that motor dysfunction only accounts for a small percentage of the difference. Most of the difference in the latency to reach the platform results from cognitive deficits. n = 12 WT, 13 YAC128. sec, Seconds.
Similarly, we used the swimming speed calculated in the T-maze apparatus, to estimate the relative contributions of motor and cognitive deficits to the differences observed in the simple swimming test. We calculated the predicted difference in latency to reach the platform in the simple swimming test, based on motor dysfunction alone, as 0.2 s. This accounts for only 5% of the actual difference observed in the time to reach the platform at 8 months of age. Comparing the difference in latency to reach the platform between YAC128 and WT mice in the three swimming tests revealed that cognitive factors account for the majority of the difference observed (73-95%) (Fig. 7B). Thus, by measuring swimming speed in a non-cognitive test, we showed that motor dysfunction is present at this age but only accounts for a small proportion of the overall performance deficit in the simple swimming test and swimming T-maze tests. The remainder of the difference is caused by cognitive dysfunction, which is most apparent in the reversal phase of the swimming T-maze test in which a change in strategy is required.
Presymptomatic YAC128 mice show difficulty in shifting strategy
Because presymptomatic HD patients show mild cognitive symptoms that worsen as the disease progresses, we tested a cohort of 2-month-old presymptomatic YAC128 mice in the swimming T-maze to determine whether the cognitive deficits in symptomatic YAC128 mice were present at this young age. In the normal phase of the test, both YAC128 and WT mice rapidly learned to swim to the platform, and there was no difference in the time taken to reach the platform in any of the 12 trials (Fig. 8A). However, switching the location of the platform to the opposite arm of the maze revealed a difference in changing strategy between the YAC128 and WT mice even at this age. Although the difference was not as dramatic as we observed in symptomatic YAC128 mice, 2-month-old YAC128 mice took significantly longer than WT mice to reach the platform during the reversal phase of the swimming T-maze test (WT, 4.9 ± 0.4 s; YAC128, 9.5 ± 1.2 s; p = 0.003). Again, none of the 12 WT mice reentered the stem of the T-maze, whereas 3 of the 12 YAC128 mice retraced their path before reaching the platform (Fig. 8B). With additional trials, YAC128 mice were able to reach the platform as fast as WT mice (data not shown). At this age, there was no significant difference in swimming speed between the YAC128 and WT mice (WT, 45 ± 6 cm/s; YAC128, 45 ± 6 cm/s; p = 0.99). Overall, this demonstrated that presymptomatic YAC128 mice had difficulty in changing strategy but did not show any cognitive deficit in the normal phase of the swimming T-maze test. As with the rotarod test at this age, no deficit in motor coordination was detected in measuring swimming speed.
Figure 8.
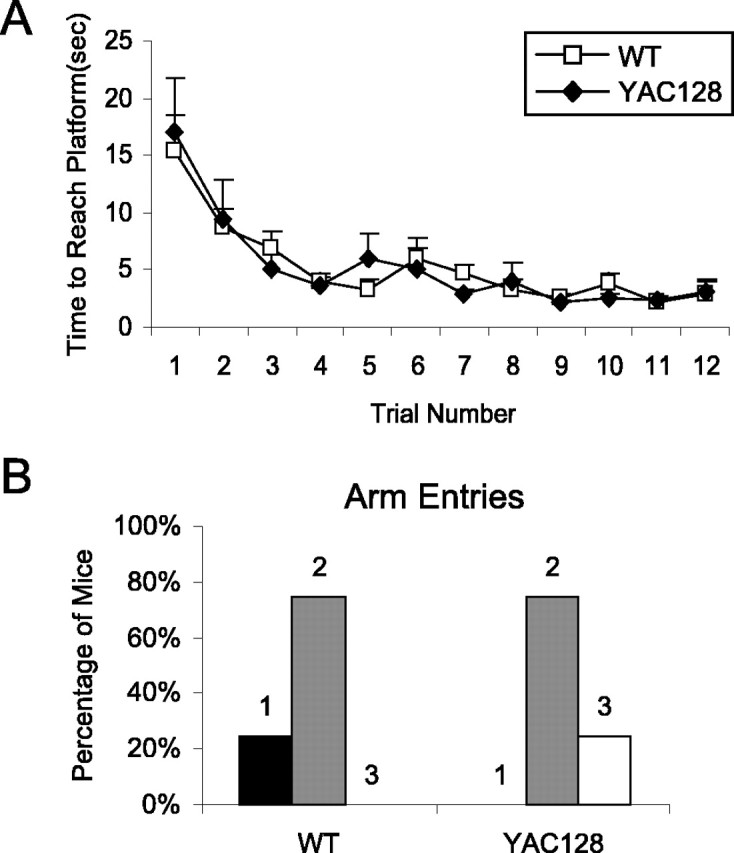
Presymptomatic YAC128 mice show cognitive deficit in strategy shifting. Two-month-old YAC128 mice were tested in the swimming T-maze test with reversal. A, During the normal phase of the test, both YAC128 and WT mice learned to swim to the platform, and no differences were observed (trial: F(11,242) = 11.0, p < 0.001; genotype by trial: F(11,242) = 0.3, p = 1.0; genotype: F(1,22) = 0.0, p = 0.9). During the reversal phase, when the location of the platform was switched, YAC128 mice required significantly more time to find the platform than WT mice (WT, 4.9 ± 0.4 s; YAC128, 9.5 ± 1.2 s; p = 0.003). Error bars represent SEM. sec, Seconds. B, After the platform was switched to the opposite arm of the T-maze, none of the WT mice swam down the stem of the T-maze. In contrast, 3 of the 12 YAC128 mice retraced their steps to the starting position before swimming to the platform. This resulted in the YAC128 mice exhibiting more arm entries en route to the platform (χ2 = 6.0; p = 0.05). At this age, the swimming speed of YAC128 mice is equal to that of WT mice, indicating that motor coordination is normal at this time point (WT, 45 ± 6 cm/s; YAC128, 45 ± 6 cm/s; p = 0.99). n = 12 WT, 12 YAC128.
YAC128 mice show decreased sensorimotor gating in the PPI test
Based on observations of decreased PPI in human HD patients (Swerdlow et al., 1995), we tested symptomatic YAC128 mice at 9 and 12 months of age to determine whether this difference was evident in the mouse model. Using the same apparatus, we also assessed habituation to acoustic startle. Acoustic startle is a fast response to a loud noise stimulus that can be measured by a movement-sensitive apparatus. Mice exposed to repeated, high-intensity sounds learn to startle less when they recognize the sound and remember that there is no harmful event associated with the sound. This habituation to acoustic startle can be used to assess learning and memory. When mice are exposed to a quieter sound before the loud stimulus, they will startle less than they would for the loud stimulus alone. This PPI is not learned but rather measures sensorimotor gating. At 9 months of age, there was no difference between WT and YAC128 mice in PPI or habituation to acoustic startle (data not shown).
YAC128 mice tested at 12 months of age showed significantly less PPI compared with WT mice, indicating a deficit in sensorimotor gating (Fig. 9A) (genotype: F(1,85) = 62.7, p < 0.001; prepulse: F(3,255) = 37.4, p < 0.001). At 12 months of age, YAC128 mice also showed decreased habituation to acoustic startle. With repeated pulses, both WT and YAC128 mice showed a decreased response, but the habituation was greater in WT mice than in YAC128 mice (Fig. 9B) (block: F(2,88) = 3.9, p = 0.025; genotype by block: F(2,88) = 0.3, p = 0.7; genotype: F(1,44) = 7.9, p = 0.008). The decreased habituation to acoustic startle reflects a deficit in learning and memory, whereas the decreased PPI suggests a difficulty in inhibiting the motor response to the sound. In both cases, the acoustic startle observed in YAC128 mice may have been influenced by emotional state, because this is known to impact startle response magnitude and emotional changes are common in patients with HD.
Figure 9.
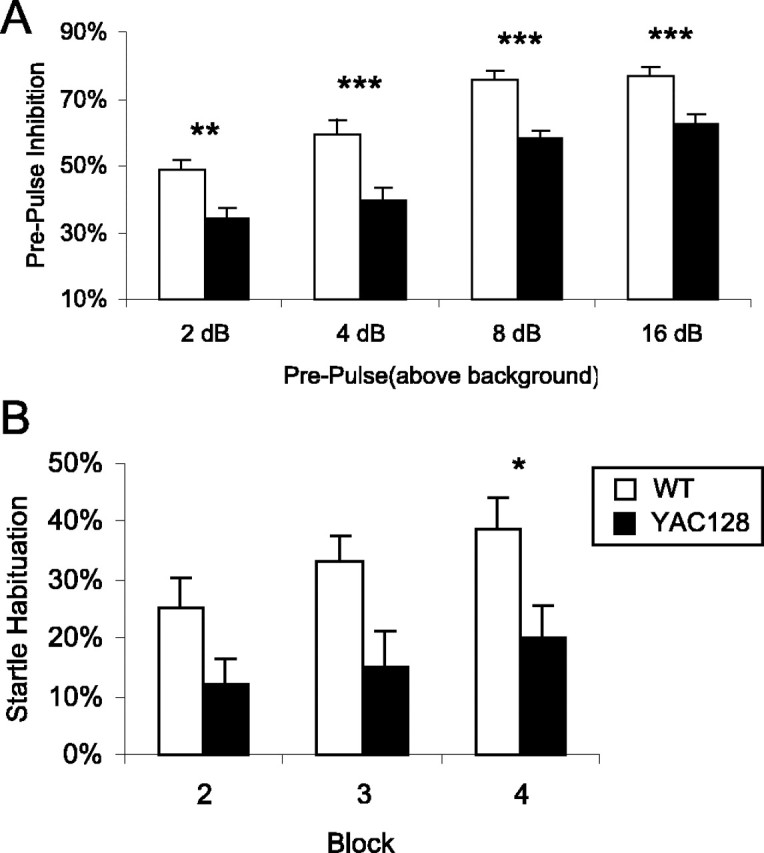
YAC128 mice show decreased PPI and decreased habituation to acoustic startle. A, PPI was measured as the percentage of decrease in the amount of startle with a prepulse compared with the amount of startle with no prepulse. At 9 months of age, there was not a significant difference between WT and YAC128 mice (n = 11 WT, 13 YAC128; data not shown). At 12 months of age, YAC128 mice showed a deficit in PPI at 2, 4, 8, and 16 dB above background (genotype: F(1,85) = 62.7, p < 0.001; prepulse: F(3,255) = 37.4, p < 0.001; n = 39 WT, 48 YAC128). B, At this age, YAC128 mice also showed a decreased habituation to acoustic startle compared with WT mice (block: F(2,88) = 3.9, p = 0.025; genotype by block: F(2,88) = 0.3, p = 0.7; genotype: F(1,44) = 7.9, p = 0.008; n = 21 WT, 25 YAC128). Error bars represent SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
In this study, we have demonstrated clear cognitive deficits in the YAC128 mouse model that precede the onset of motor deficits and progressively worsen with age. Based on cognitive impairments present in human HD, we adapted and validated behavioral tests to dissociate cognitive from motor dysfunction in YAC128 mice. We showed deficits in procedural learning, memory, sensorimotor gating, and strategy shifting, all of which are present in patients with HD (Heindel et al., 1988; Swerdlow et al., 1995; Lawrence et al., 2000; Watkins et al., 2000; Ho et al., 2003). Importantly, the pattern of symptom development is similar to human HD with limited cognitive deficits early that are followed by widespread impairment after motor onset.
Cognitive deficits in presymptomatic YAC128 mice
Presymptomatic HD patients show mild cognitive deficits in procedural learning, memory, planning, and shifting strategy (Hahn-Barma et al., 1998; Lawrence et al., 1998a,b; Snowden et al., 2002). These deficits worsen with the advancement of the disease and are also found in symptomatic HD patients (Bamford et al., 1995; Lawrence et al., 1999; Ho et al., 2003). In YAC128 mice, disease onset occurs at 3 months of age with hyperkinesia. Accordingly, we tested for presymptomatic cognitive deficits in 2-month-old YAC128 mice.
At 2 months of age, YAC128 mice showed a learning deficit on the rotarod test of motor coordination (Lalonde et al., 1995; Gerlai et al., 1996; Le Marec and Lalonde, 1997; Hyde et al., 2001; Dubois et al., 2002; McFadyen et al., 2003). At this age, YAC128 mice do not have a motor deficit, because trained YAC128 mice performed as well as WT mice. Because YAC128 mice require more training to reach the same level of performance, it is clear that they learn slower than WT mice. Presymptomatic YAC128 mice also showed a deficit in strategy shifting whereby they took longer to reach the platform in the reversal phase of the swimming T-maze test, despite swimming as fast as WT mice. At this time point, YAC128 mice showed normal cognition in the simple swimming test, the normal phase of the swimming T-maze test, and in tests of habituation.
Because the earliest behavioral abnormality reported previously in YAC128 mice was hyperactivity at 3 months of age, cognitive dysfunction is present before any other behavioral symptom. Similar findings have been reported in human HD and the R6/2 mouse model (Hahn-Barma et al., 1998; Lawrence et al., 1998a,b; Snowden et al., 2002). Because HD is characterized by motor dysfunction, it is not surprising that cognitive tasks involving motor function deteriorate first, especially when HD patients show deficits in several paradigms of skill learning (Heindel et al., 1988, 1989; Gabrieli et al., 1997; Schmidtke et al., 2002; Knopman and Nissen, 1991). Similarly, difficulty in changing strategy is one of the earliest reported cognitive symptoms in HD (Lawrence et al., 1998a).
Cognitive deficits in symptomatic YAC128 mice
Symptomatic HD patients show widespread cognitive dysfunction (Backman et al., 1997). In our assessment of symptomatic YAC128 mice, we examined abilities deficient in both presymptomatic and symptomatic HD patients, namely procedural learning, memory, and strategy shifting (Hahn-Barma et al., 1998; Lawrence et al., 1998a,b; Snowden et al., 2002). We also followed the progression of motor impairment for comparison with the development of cognitive dysfunction.
Motor abilities were assessed using the rotarod test. YAC128 mice showed normal performance at 2 months of age, but by 4 months of age, YAC128 mice were unable to stay on the rotarod as long as WT mice. Thereafter, YAC128 rotarod performance decreased steadily with age, whereas WT mice maintained a consistent level of performance. Decreased motor function on the rotarod is the most common symptom among animal models of HD (Carter et al., 1999; Schilling et al., 1999; Laforet et al., 2001; Lin et al., 2001; von Horsten et al., 2003).
At 7 months of age, YAC128 mice learned the rotarod task but were unable to reach the same level of performance as WT mice. By 12 months of age, the motor learning deficit originally detected at 2 months of age progressed into a severe learning deficit that completely prevented YAC128 mice from learning the rotarod task. At this age, previously untrained YAC128 mice could not perform the rotarod task, whereas mice trained earlier were physically able to run on the rotarod. Although environmental enrichment improves motor function (Hockly et al., 2002), the fact that learning on the rotarod is thought to result from changes in motor strategy and not increased motor function (Buitrago et al., 2004) suggests that the reason that YAC128 mice trained at 7 months of age performed better than YAC128 mice trained at 12 months of age is because of differences in cognitive rather than physical ability.
In the simple swimming test, YAC128 mice took longer to reach the platform than WT mice beginning at 8 months of age. Only 5% of this difference were accounted for by differences in swimming speed. The remainder resulted from cognitive deficits that predisposed the YAC128 mice to choose an indirect path to the platform. Because these mice were unable to rely on visual cues for navigation (Taketo et al., 1991; Huerta et al., 1999), this is likely a deficit in procedural rather than spatial memory. Memory deficits have also been reported in human HD (Wilson et al. 1987; Rohrer et al., 1999; Lawrence et al., 2000; Brown et al., 2001). Validation for this test comes from the ability of WT mice to learn to swim directly to the platform and remember this skill in subsequent trials.
Symptomatic YAC128 mice also showed deficits in open-field habituation, a simple test of learning and memory (Gerlai et al., 1996; Bolivar et al., 2000; Cook et al., 2002). At 8 months of age, YAC128 mice were hypoactive compared with WT mice. Although YAC128 mice demonstrated intrasession and intersession habituation, in both cases WT mice showed a more rapid decline in activity, because the difference in activity between WT and YAC128 mice diminished with repeated trials. Although we have attempted to control for the initial difference in activity in our calculation of habituation, it is possible that the initial hypoactivity in YAC128 mice contributed to their decreased habituation. However, the observation of memory deficits in the simple swimming test and in habituation to acoustic startle supports a memory deficit as the cause of decreased open-field habituation in symptomatic YAC128 mice. Validation for the use of open-field habituation for memory testing comes from experiments showing more rapid habituation in rodents treated with drugs prescribed for memory disorders (Platel and Porsolt, 1982).
We also demonstrated cognitive deficits in a swimming T-maze test at 8.5 months of age in which trained YAC128 mice took longer to reach the platform than WT mice. A motor deficit in swimming speed is present at this age but does not account for the difference observed. Although WT mice all swam directly to the platform on the third day, YAC128 mice showed multiple errant trials in which they swam down the wrong arm, possibly as a result of a memory deficit. The use of this test is validated by the ability of WT mice to improve their latency to reach the platform and choose the correct arm in every trial. Similarly, cognitive function has been assessed in a water plus-maze (Dobkin et al., 2000).
Symptomatic YAC128 mice showed a dramatic cognitive deficit in the reversal phase of the swimming T-maze test. When the location of the platform was changed, WT mice all rapidly adapted to the new task, reaching the platform without retracing their path. In contrast, most YAC128 mice returned to the start of the T-maze after discovering the platform was not present in the right arm. This difference in response strategy provides a clear manifestation of cognitive dysfunction in YAC128 mice. Remarkably, not a single WT mouse had difficulty in changing strategy, whereas difficulty was the norm in YAC128 mice. Interestingly, difficulties in changing strategy are present in HD patients (Lawrence et al., 1999; Ho et al., 2003).
At 12 months of age, YAC128 mice had decreased PPI compared with wild type. This represents a decreased ability to inhibit a motor response. Because motor inhibition is thought to be mediated by the striatum, this finding is in line with the striatal pathology reported in YAC128 mice (Mink, 1996; Slow et al., 2003). Decreased PPI has been reported in human HD patients and mouse models of polyglutamine toxicity (Swerdlow et al., 1995; Carter et al., 1999). We also demonstrated decreased habituation to acoustic startle in 12-month-old YAC128 mice, providing additional evidence for impaired memory. Because 9-month-old YAC128 mice had an equivalent motor deficit on the rotarod as 12-month-old YAC128 mice and did not show deficits in PPI or habituation to acoustic startle, the deficits in 12-month-old animals were likely cognitive and not related to the motor dysfunction at this age.
Cognitive deficits have been reported previously in animal models expressing N-terminal fragments of htt. The R6/2 mouse model shows deficits in learning and PPI but has normal open-field habituation (Lione et al., 1999; Murphy et al., 2000; Bolivar et al., 2003). Similarly, a rat transgenic model of HD exhibits a spatial memory deficit in the radial maze test (von Horsten et al., 2003). Our findings in YAC128 mice extend these results to demonstrate cognitive dysfunction in a mouse model expressing full-length mutant htt.
YAC128 mice show deficits similar to animals with striatal lesions
Animal research has demonstrated two distinct forms of learning that are mediated by different systems within the brain. Cognitive learning involves the acquisition of knowledge about the environment, such as the generation of a spatial map, which is then used to influence subsequent actions. Stimulus-response learning involves procedural learning in response to the stimulus. Lesions involving the lateral striatum eliminate stimulus-response learning, whereas those involving the medial striatum or hippocampus affect cognitive learning (Packard and McGaugh, 1996; Devan and White, 1999; DeCoteau and Kesner, 2000). Studies involving striatal damage caused by the mitochondrial toxin 3NP have shown perseverance in a lever-pressing task and a T-maze alteration task as well as a spatial learning deficit in lesioned animals (Shear et al., 1998; El Massioui et al., 2001). Thus, the demonstration of procedural learning deficits and perseverance in the YAC128 mice is in accordance with the observed striatal pathology in these mice, because similar deficits have been achieved in animals with striatal lesions.
Conclusion
We demonstrate a clear pattern of cognitive dysfunction in the YAC128 mouse model of HD that complements the motor dysfunction and selective neuropathology present in these mice (summarized in supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Cognitive deficits arise before motor onset and progress with age. These cognitive impairments are similar to deficits observed in human HD and can be used as outcome measures in therapeutic trials to assess the effect of potential treatments on cognition.
Footnotes
This work was supported by grants from the Canadian Institutes of Health Research (CIHR), the Huntington's Disease Society of America, and the High Q Foundation. J.M.V.R. is supported by the CIHR and the Michael Smith Foundation for Health Research. B.R.L. is supported by the CIHR, the Huntington Society of Canada, the Hereditary Disease Foundation, and the Canadian Genetic Diseases Network. M.R.H. is a Killam University Professor and holds a Canada Research Chair in Human Genetics.
Correspondence should be addressed to Dr. Michael R. Hayden, Centre for Molecular Medicine and Therapeutics, British Columbia Research Institute, 980 West 28th Avenue, Vancouver, British Columbia, V5Z 4H4 Canada. E-mail: mrh@cmmt.ubc.ca.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254169-12$15.00/0
References
- Albin RL, Reiner A, Anderson KD, Dure LS, Handelin B, Balfour R, Whetsell Jr WO, Penney JB, Young AB (1992) Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington's disease. Ann Neurol 31: 425-430. [DOI] [PubMed] [Google Scholar]
- Backman L, Robins-Wahlin TB, Lundin A, Ginovart N, Farde L (1997) Cognitive deficits in Huntington's disease are predicted by dopaminergic PET markers and brain volumes. Brain 120: 2207-2217. [DOI] [PubMed] [Google Scholar]
- Bamford KA, Caine ED, Kido DK, Cox C, Shoulson I (1995) A prospective evaluation of cognitive decline in early Huntington's disease: functional and radiographic correlates. Neurology 45: 1867-1873. [DOI] [PubMed] [Google Scholar]
- Berrios GE, Wagle AC, Markova IS, Wagle SA, Rosser A, Hodges JR (2002) Psychiatric symptoms in neurologically asymptomatic Huntington's disease gene carriers: a comparison with gene negative at risk subjects. Acta Psychiatr Scand 105: 224-230. [DOI] [PubMed] [Google Scholar]
- Blackmore L, Simpson SA, Crawford JR (1995) Cognitive performance in UK sample of presymptomatic people carrying the gene for Huntington's disease. J Med Genet 32: 358-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolivar VJ, Caldarone BJ, Reilly AA, Flaherty L (2000) Habituation of activity in an open field: a survey of inbred strains and F1 hybrids. Behav Genet 30: 285-293. [DOI] [PubMed] [Google Scholar]
- Bolivar VJ, Manley K, Messer A (2003) Exploratory activity and fear conditioning abnormalities develop early in R6/2 Huntington's disease transgenic mice. Behav Neurosci 117: 1233-1242. [DOI] [PubMed] [Google Scholar]
- Brown RG, Redondo-Verge L, Chacon JR, Lucas ML, Channon S (2001) Dissociation between intentional and incidental sequence learning in Huntington's disease. Brain 124: 2188-2202. [DOI] [PubMed] [Google Scholar]
- Buitrago MM, Schulz JB, Dichgans J, Luft AR (2004) Short and long-term motor skill learning in an accelerated rotarod training paradigm. Neurobiol Learn Mem 81: 211-216. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ (1999) Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci 19: 3248-3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conneally PM (1984) Huntington disease—genetics and epidemiology. Am J Hum Genet 36: 506-526. [PMC free article] [PubMed] [Google Scholar]
- Cook MN, Bolivar VJ, McFadyen MP, Flaherty L (2002) Behavioral differences among 129 substrains: implications for knockout and transgenic mice. Behav Neurosci 116: 600-611. [PubMed] [Google Scholar]
- Cudkowicz M, Kowall NW (1990) Degeneration of pyramidal projection neurons in Huntington's disease cortex. Ann Neurol 27: 200-204. [DOI] [PubMed] [Google Scholar]
- de Boo GM, Tibben A, Lanser JB, Jennekens-Schinkel A, Hermans J, Maat-Kievit A, Roos RA (1997) Early cognitive and motor symptoms in identified carriers of the gene for Huntington disease. Arch Neurol 54: 1353-1357. [DOI] [PubMed] [Google Scholar]
- de Boo GM, Tibben AA, Hermans JA, Jennekens-Schinkel A, Maat-Kievit A, Roos RA (1999) Memory and learning are not impaired in presymptomatic individuals with an increased risk of Huntington's disease. J Clin Exp Neuropsychol 21: 831-836. [DOI] [PubMed] [Google Scholar]
- DeCoteau WE, Kesner RP (2000) A double dissociation between the rat hippocampus and medial caudoputamen in processing two forms of knowledge. Behav Neurosci 114: 1096-1108. [DOI] [PubMed] [Google Scholar]
- Devan BD, White NM (1999) Parallel information processing in the dorsal striatum: relation to hippocampal function. J Neurosci 19: 2789-2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobkin C, Rabe A, Dumas R, El Idrissi A, Haubenstock H, Brown WT (2000) Fmr1 knockout mouse has a distinctive strain-specific learning impairment. Neuroscience 100: 423-429. [DOI] [PubMed] [Google Scholar]
- Dubois M, Strazielle C, Eyer J, Lalonde R (2002) Sensorimotor functions in transgenic mice expressing the neurofilament/heavy-LacZ fusion protein on two genetic backgrounds. Neuroscience 112: 447-454. [DOI] [PubMed] [Google Scholar]
- El Massioui N, Ouary S, Cheruel F, Hantraye P, Brouillet E (2001) Perseverative behavior underlying attentional set-shifting deficits in rats chronically treated with the neurotoxin 3-nitropropionic acid. Exp Neurol 172: 172-181. [DOI] [PubMed] [Google Scholar]
- Gabrieli JD, Stebbins GT, Singh J, Willingham DB, Goetz CG (1997) Intact mirror-tracing and impaired rotary-pursuit skill learning in patients with Huntington's disease: evidence for dissociable memory systems in skill learning. Neuropsychology 11: 272-281. [DOI] [PubMed] [Google Scholar]
- Gerlai R, Millen KJ, Herrup K, Fabien K, Joyner AL, Roder J (1996) Impaired motor learning performance in cerebellar En-2 mutant mice. Behav Neurosci 110: 126-133. [DOI] [PubMed] [Google Scholar]
- Hahn-Barma V, Deweer B, Durr A, Dode C, Feingold J, Pillon B, Agid Y, Brice A, Hahn-Barma V, Deweer B, Durr A, Dode C, Feingold J, Pillon B, Agid Y, Brice A, Dubois B (1998) Are cognitive changes the first symptoms of Huntington's disease? A study of gene carriers. J Neurol Neurosurg Psychiatry 64: 172-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedreen JC, Peyser CE, Folstein SE, Ross CA (1991) Neuronal loss in layers V and VI of cerebral cortex in Huntington's disease. Neurosci Lett 133: 257-261. [DOI] [PubMed] [Google Scholar]
- Heindel WC, Butters N, Salmon DP (1988) Impaired learning of a motor skill in patients with Huntington's disease. Behav Neurosci 102: 141-147. [DOI] [PubMed] [Google Scholar]
- Heindel WC, Salmon DP, Shults CW, Walicke PA, Butters N (1989) Neuropsychological evidence for multiple implicit memory systems: a comparison of Alzheimer's, Huntington's, and Parkinson's disease patients. J Neurosci 9: 582-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AK, Sahakian BJ, Brown RG, Barker RA, Hodges JR, Ane MN, Snowden J, Thompson J, Esmonde T, Gentry R, Moore JW, Bodner T (2003) Profile of cognitive progression in early Huntington's disease. Neurology 61: 1702-1706. [DOI] [PubMed] [Google Scholar]
- Hockly E, Cordery PM, Woodman B, Mahal A, van Dellen A, Blakemore C, Lewis CM, Hannan AJ, Bates GP (2002) Environmental enrichment slows disease progression in R6/2 Huntington's disease mice. Ann Neurol 51: 235-242. [DOI] [PubMed] [Google Scholar]
- Huerta JJ, Llamosas MM, Cernuda-Cernuda R, Garcia-Fernandez JM (1999) Spatio-temporal analysis of light-induced Fos expression in the retina of rd mutant mice. Brain Res 834: 122-127. [DOI] [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72: 971-983. [DOI] [PubMed] [Google Scholar]
- Hyde LA, Crnic LS, Pollock A, Bickford PC (2001) Motor learning in Ts65Dn mice, a model for Down syndrome. Dev Psychobiol 38: 33-45. [DOI] [PubMed] [Google Scholar]
- Knopman D, Nissen MJ (1991) Procedural learning is impaired in Huntington's disease: evidence from the serial reaction time task. Neuropsychologia 29: 245-254. [DOI] [PubMed] [Google Scholar]
- Laforet GA, Sapp E, Chase K, McIntyre C, Boyce FM, Campbell M, Cadigan BA, Warzecki L, Tagle DA, Reddy PH, Cepeda C, Calvert CR, Jokel ES, Klapstein GJ, Ariano MA, Levine MS, DiFiglia M, Aronin N (2001) Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington's disease. J Neurosci 21: 9112-9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde R, Bensoula AN, Filali M (1995) Rotorod sensorimotor learning in cerebellar mutant mice. Neurosci Res 22: 423-426. [DOI] [PubMed] [Google Scholar]
- Lawrence AD, Hodges JR, Rosser AE, Kershaw A, ffrench-Constant C, Rubinsztein DC, Robbins TW, Sahakian BJ (1998a) Evidence for specific cognitive deficits in preclinical Huntington's disease. Brain 121: 1329-1341. [DOI] [PubMed] [Google Scholar]
- Lawrence AD, Weeks RA, Brooks DJ, Andrews TC, Watkins LH, Harding AE, Robbins TW, Sahakian BJ (1998b) The relationship between striatal dopamine receptor binding and cognitive performance in Huntington's disease. Brain 121: 1343-1355. [DOI] [PubMed] [Google Scholar]
- Lawrence AD, Sahakian BJ, Rogers RD, Hodge JR, Robbins TW (1999) Discrimination, reversal, and shift learning in Huntington's disease: mechanisms of impaired response selection. Neuropsychologia 37: 1359-1374. [DOI] [PubMed] [Google Scholar]
- Lawrence AD, Watkins LH, Sahakian BJ, Hodges JR, Robbins TW (2000) Visual object and visuospatial cognition in Huntington's disease: implications for information processing in corticostriatal circuits. Brain 123: 1349-1364. [DOI] [PubMed] [Google Scholar]
- Le Marec N, Lalonde R (1997) Sensorimotor learning and retention during equilibrium tests in Purkinje cell degeneration mutant mice. Brain Res 768: 310-316. [DOI] [PubMed] [Google Scholar]
- Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ (2001) Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet 10: 137-144. [DOI] [PubMed] [Google Scholar]
- Lione LA, Carter RJ, Hunt MJ, Bates GP, Morton AJ, Dunnett SB (1999) Selective discrimination learning impairments in mice expressing the human Huntington's disease mutation. J Neurosci 19: 10428-10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadyen MP, Kusek G, Bolivar VJ, Flaherty L (2003) Differences among eight inbred strains of mice in motor ability and motor learning on a rotorod. Genes Brain Behav 2: 214-219. [DOI] [PubMed] [Google Scholar]
- Mink JW (1996) The basal ganglia: focused selection and inhibition of competing motor programs. Prog Neurobiol 50: 381-425. [DOI] [PubMed] [Google Scholar]
- Murphy KP, Carter RJ, Lione LA, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ (2000) Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human Huntington's disease mutation. J Neurosci 20: 5115-5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard MG, McGaugh JL (1996) Inactivation of hippocampus or caudate nucleus with lidocaine differentially affects expression of place and response learning. Neurobiol Learn Mem 65: 65-72. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Zhao H, Stout JC, Brinkman RR, Guttman M, Ross CA, Como P, Manning C, Hayden MR, Shoulson I (2001) Clinical markers of early disease in persons near onset of Huntington's disease. Neurology 57: 658-662. [DOI] [PubMed] [Google Scholar]
- Platel A, Porsolt RD (1982) Habituation of exploratory activity in mice: a screening test for memory enhancing drugs. Psychopharmacology (Berl) 78: 346-352. [DOI] [PubMed] [Google Scholar]
- Rohrer D, Salmon DP, Wixted JT, Paulsen JS (1999) The disparate effects of Alzheimer's disease and Huntington's disease on semantic memory. Neuropsychology 13: 381-388. [DOI] [PubMed] [Google Scholar]
- Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR (1999) Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet 8: 397-407. [DOI] [PubMed] [Google Scholar]
- Schmidtke K, Manner H, Kaufmann R, Schmolck H (2002) Cognitive procedural learning in patients with fronto-striatal lesions. Learn Mem 9: 419-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shear DA, Dong J, Gundy CD, Haik-Creguer KL, Dunbar GL (1998) Comparison of intrastriatal injections of quinolinic acid and 3-nitropropionic acid for use in animal models of Huntington's disease. Prog Neuropsychopharmacol Biol Psychiatry 22: 1217-1240. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR (2003) Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet 12: 1555-1567. [DOI] [PubMed] [Google Scholar]
- Snowden JS, Craufurd D, Thompson J, Neary D (2002) Psychomotor, executive, and memory function in preclinical Huntington's disease. J Clin Exp Neuropsychol 24: 133-145. [DOI] [PubMed] [Google Scholar]
- Strauss ME, Brandt J (1990) Are there neuropsychologic manifestations of the gene for Huntington's disease in asymptomatic, at-risk individuals? Arch Neurol 47: 905-908. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Paulsen J, Braff DL, Butters N, Geyer MA, Swenson MR (1995) Impaired prepulse inhibition of acoustic and tactile startle response in patients with Huntington's disease. J Neurol Neurosurg Psychiatry 58: 192-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taketo M, Schroeder AC, Mobraaten LE, Gunning KB, Hanten G, Fox RR, Roderick TH, Stewart CL, Lilly F, Hansen CT (1991) FVB/N: an inbred mouse strain preferable for transgenic analyses. Proc Natl Acad Sci USA 88: 2065-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Horsten S, Schmitt I, Nguyen HP, Holzmann C, Schmidt T, Walther T, Bader M, Pabst R, Kobbe P, Krotova J, Stiller D, Kask A, Vaarmann A, Rathke-Hartlieb S, Schulz JB, Grasshoff U, Bauer I, Vieira-Saecker AM, Paul M, Jones L, Lindenberg KS, et al. (2003) Transgenic rat model of Huntington's disease. Hum Mol Genet 12: 617-624. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M (1998) Huntington disease. J Neuropathol Exp Neurol 57: 369-384. [DOI] [PubMed] [Google Scholar]
- Watkins LH, Rogers RD, Lawrence AD, Sahakian BJ, Rosser AE, Robbins TW (2000) Impaired planning but intact decision making in early Huntington's disease: implications for specific fronto-striatal pathology. Neuropsychologia 38: 1112-1125. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Como PG, Garron DC, Klawans HL, Barr A, Klawans D (1987) Memory failure in Huntington's disease. J Clin Exp Neuropsychol 9: 147-154. [DOI] [PubMed] [Google Scholar]