

Abstract
The importance of the cAMP signaling pathway in the modulation of ethanol sensitivity has been suggested by studies in organisms from Drosophila melanogaster to man. However, the involvement of specific isoforms of adenylyl cyclase (AC), the molecule that converts ATP to cAMP, has not been systemically determined in vivo. Because AC1 and AC8 are the only AC isoforms stimulated by calcium, and ethanol modulates calcium flux by the NMDA receptor, we hypothesized that these ACs would be important in the neural response to ethanol. AC1 knock-out (KO) mice and double knock-out (DKO) mice with genetic deletion of both AC1 and AC8 display substantially increased sensitivity to ethanol-induced sedation compared with wild-type (WT) mice, whereas AC8 KO mice are only minimally more sensitive. In contrast, AC8 KO and DKO mice, but not AC1 KO mice, demonstrate decreased voluntary ethanol consumption compared with WT mice. DKO mice do not display increased sleep time compared with WT mice after administration of ketamine or pentobarbital, indicating that the mechanism of enhanced ethanol sensitivity in these mice is likely distinct from the antagonism of ethanol of the NMDA receptor and potentiation of the GABAA receptor. Ethanol does not enhance calcium-stimulated AC activity, but the ethanol-induced phosphorylation of a discrete subset of protein kinase A (PKA) substrates is compromised in the brains of DKO mice. These results indicate that the unique activation of PKA signaling mediated by the calcium-stimulated ACs is an important component of the neuronal response to ethanol.
Keywords: adenylyl cyclase, calcium, cAMP, ethanol, knock-out mouse, pharmacology, protein kinase A
Introduction
Ethanol is one of the most widely used neuroactive agents, but only within the last decade has significant progress been made toward understanding the mechanisms by which it affects neuronal function. Several studies have demonstrated that ethanol acts as an antagonist of the NMDA receptor and potentiates the effects of the GABAA receptor (Mehta and Ticku, 1988; Dildy and Leslie, 1989; Lovinger et al., 1989, 1990; Harris et al., 1995; Wallner et al., 2003).
More recent evidence has identified the cAMP signaling pathway as an important modulator of ethanol sensitivity. Genetic manipulations in Drosophila melanogaster and mice that decrease the activity of this pathway result in increased sensitivity to the sedative effects of ethanol (Moore et al., 1998; Thiele et al., 2000; Wand et al., 2001). To date, however, the specific role of any of the mammalian isoforms of adenylyl cyclase (AC) in modulating adult ethanol sensitivity in vivo has not been defined.
Adenylyl cyclase, the molecule that converts ATP to cAMP, is an essential component of the cAMP pathway and is normally activated by interaction with the stimulatory G-protein subunit, Gαs. However, this is not the case for AC1 and AC8, two of the 10 AC isoforms, which are the only AC isoforms primarily stimulated by calcium through activation of calmodulin (although several isoforms are calcium sensitive) (Tang et al., 1991; Cali et al., 1994; Wong et al., 1999; Chern, 2000; Wang and Storm, 2003). In situ hybridization studies have revealed that the mRNAs for AC1 and AC8 are expressed at high levels in distinct, but overlapping, regions in the brain, including cortex, hippocampus, cerebellum, thalamus, and hypothalamus (Xia et al., 1991; Muglia et al., 1999).
Mice with genetic deletions of AC1, AC8, or both AC1 and AC8 reveal the important role that these proteins play in neuronal function. AC8 knock-out (KO) mice and AC1 KO mice demonstrate a variety of deficits in long-term depression and long-term potentiation (LTP) (Villacres et al., 1998; Schaefer et al., 2000; Wang et al., 2003). Double knock-out (DKO) mice missing both AC1 and AC8 show an even more severe phenotype, displaying memory deficits and loss of the late phase of LTP in area CA1 of the hippocampus. Neither of these deficits is seen in AC1 KO or AC8 KO mice (Wong et al., 1999), indicating that AC1 and AC8 are each able to compensate for loss of the other calcium-stimulated AC isoform in certain neuronal functions.
Based on the deficits in neuronal function in AC KO mice and the importance of the cAMP pathway in the effects of ethanol, we hypothesized that mice deficient in AC1 and/or AC8 would have increased sensitivity to ethanol. Here, we demonstrate that these AC KO mice have increased sensitivity to the sedative effects of ethanol and consume less ethanol than wild-type (WT) mice without enhanced sensitivity to other pharmacologic agents that mimic components of the effects of ethanol on neurons. Finally, we demonstrate that ethanol-induced phosphorylation of a subset of protein kinase A (PKA) targets is compromised in the brains of DKO mice and suggest that the calcium-stimulated ACs may contribute to the homeostatic response to the inhibition by ethanol of neuronal function.
Materials and Methods
Animal husbandry
All mice were backcrossed at least nine generations to C57BL/6 mice from The Jackson Laboratory (Bar Harbor, ME). To generate mice for these experiments, we used progeny of homozygous mutants and C57BL/6 mice from The Jackson Laboratory bred in our colony. Analysis for chromosomal markers specific for C57BL/6 and 129/Sv strains indicated that 129/Sv genomic DNA was limited to a 16 centiMorgan (cM) region (1.1-16 cM) surrounding the AC1 locus on chromosome 11 and a 37 cM region (17.8-54.5 cM) surrounding the AC8 locus on chromosome 15. To assess the possibility that the 129/Sv genomic DNA surrounding the AC8 locus was contributing to the altered ethanol consumption in AC8 KO mice, we generated mice that were heterozygotes and homozygotes for this 129/Sv locus for analysis as described by Wolfer et al. (2002). We mated AC8 KO mice with 129/Sv mice and C57BL/6 mice with 129/Sv mice and used the F1 progeny of each cross for experiments. Mice were maintained on a 12 h light/dark schedule with ad libitum access to food and water except as indicated. All experiments were performed using male mice between 2 and 4 months of age. All mouse protocols were in accordance with the National Institutes of Health guidelines and were approved by the Animal Care and Use Committee of Washington University School of Medicine.
Loss of righting reflex assay
Mice were given an intraperitoneal injection of ethanol [20% (w/v) solution], pentobarbital (2 mg/ml; Abbott Labs, Chicago, IL), ketamine (10 mg/ml; Fort Dodge, Kansas City, MO), 4,5,6,7-tetrahydroisoxazolo(5,4-c)pyridin-3-ol (THIP) (3 mg/ml; Sigma, St. Louis, MO), or pentobarbital plus (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801) (5-30 μg/ml; Sigma) and placed on their back in a U-shaped trough after they lost the ability to right themselves. Sleep time is defined as the time until the mice regained the ability to right themselves onto three of four paws three times within 30 s. All drugs were dissolved in 0.9% saline.
Two-bottle ethanol and taste-preference tests
Mice were singly housed in the testing apparatus with access to two water bottles for 1 week before testing to habituate them to the environment. At the start of the test, one bottle was filled with water, whereas the second was filled with a 3% (v/v) ethanol solution. The ethanol concentration was increased every 6 d throughout the test (to 6, 10, and finally 20%). Each day, the weights of the food, liquid, and mice were recorded, and the positions of the bottles were switched to prevent side preference from confounding data interpretation. The taste-preference tests were performed in a similar manner, except that two concentrations of quinine and saccharin were presented versus water to each mouse for 4 d in a counterbalanced design.
Ataxia testing
Grid test. Mice were given an intraperitoneal injection of ethanol or saline and returned to their home cage. After 30 min, they were placed in the grid test apparatus for 10 min. Missteps (steps through the grid that contacted the floor below) were counted and normalized to total activity, as determined by beam breaks.
Ataxia battery. Mice were subjected to an additional battery of tests to assess ethanol-induced ataxia. The tests included in this battery are described below.
Balance beam. Experiments were performed as described by Crabbe et al. (2003a). Briefly, mice were given four training trials on a balance beam (width, 19 mm; length, 90 cm). The following day, mice were given one trial on the balance beam, and basal missteps were counted. Ten minutes after an intraperitoneal injection of 1.25 mg/kg ethanol, mice were given one trial on the balance beam, and missteps were counted.
Dowel and vertical screen tests. Experiments were performed as described by Crabbe et al. (2003b). Briefly, mice were given six trials on a 9.6 mm dowel to reach a criterion of 2 min without falling. Mice were then given an intraperitoneal injection of 1.75 mg/kg ethanol and immediately placed on a 15.8 mm dowel, and the latency to fall was recorded. Thirty minutes after injection, mice were tested for latency to fall from a vertical screen and then for latency to fall from a 9.6 mm dowel.
Observer-rated ataxia. Assessment of wobble and splay (measures of observer-rated ataxia) was performed as described previously (Metten et al., 2004). Briefly, mice were given an intraperitoneal injection of 3 g/kg ethanol. During the period of 10-11 min after injection, mice were placed on a flat surface and given a score between 0-5 for wobble and splay, based on defined criteria, by an observer blinded to genotype.
Blood ethanol concentration determination
Mice were given an intraperitoneal injection of ethanol to achieve a dose of 3.6 g/kg. Blood was collected by retro-orbital phlebotomy. The ethanol concentration of each sample was determined in duplicate using the Sigma Ethanol diagnostic kit (Sigma).
Adenylyl cyclase activity assays
Adenylyl cyclase activity assays were performed as described previously (Wong et al., 1999) using hippocampal, cerebellar, and cortical membrane protein homogenates.
Western blot analysis
Mice were killed by CO2 inhalation, brains were rapidly removed, and subregions dissected, if necessary, and frozen in liquid nitrogen. Brains were homogenized in a buffer containing 4% (w/v) 3-[(3-cholamido-propyl)dimethylammonio]-1-propanesulfonate, 2 m thiourea, 7 m urea, and 30 mm Tris, pH 8.5. One tablet of Complete protease inhibitor mixture (Roche Products, Indianapolis, IN) was added to 50 ml of lysis buffer. After homogenization, samples were centrifuged at 8000 × g for 10 min and the supernatants collected. Protein concentrations were determined using the 2D-Quant kit (Amersham Biosciences, Piscataway, NJ). Membrane fractions (for GluR1 Western blots) were isolated as described previously (Wong et al., 1999). Equal amounts of protein (50 μg) were submitted to SDS-PAGE and transferred to a nitrocellulose membrane. Each lane contained the brain lysate from a distinct animal. Membranes were probed with primary antibody at the following concentrations: anti-phosphorylated PKA substrate motif, 1:1000 (Cell Signaling Technology, Beverly, MA); anti-phospho GluR1, Serine 845, 1:1000 (Upstate Biotechnology, Lake Placid, NY); anti-phospho GluR1, Serine 831, 1:1000 (Upstate Biotechnology); and anti-GluR1, 1:1000 (Upstate Biotechnology). Antibodies were detected using HRP-conjugated goat anti-rabbit secondary antibody, and signals were visualized using chemiluminescence (SuperSignal West Dura kit; Pierce, Rockford, IL). Densitometric analysis was performed using Un-Scan-It (Silk Scientific, Orem, UT).
Statistical analysis
One-way and two-way ANOVA tests, followed by Bonferonni's post hoc tests, where appropriate, were done to evaluate statistical significance. F statistics and p values are indicated in the figures. Results are displayed as mean ± SEM.
Results
Enhanced sensitivity of adenylyl cyclase knock-out mice to the sedative effects of ethanol
To test the hypothesis that AC1 KO, AC8 KO, and DKO mice would demonstrate enhanced sensitivity to ethanol, we measured the sleep time of mice of each genotype after ethanol administration in the loss of righting reflex (LORR) test. At each of the three doses of ethanol tested, AC1 KO and DKO mice both demonstrated a significant increase in sleep time compared with WT mice, sleeping 1.5-fold and twofold longer, respectively (Fig. 1A). In addition, DKO mice slept significantly longer than AC1 KO mice at all three doses. In contrast, AC8 KO mice differed only slightly from WT mice in sleep time after ethanol administration, demonstrating a small, but statistically significant, increase in sleep time at the highest dose of ethanol tested.
Figure 1.
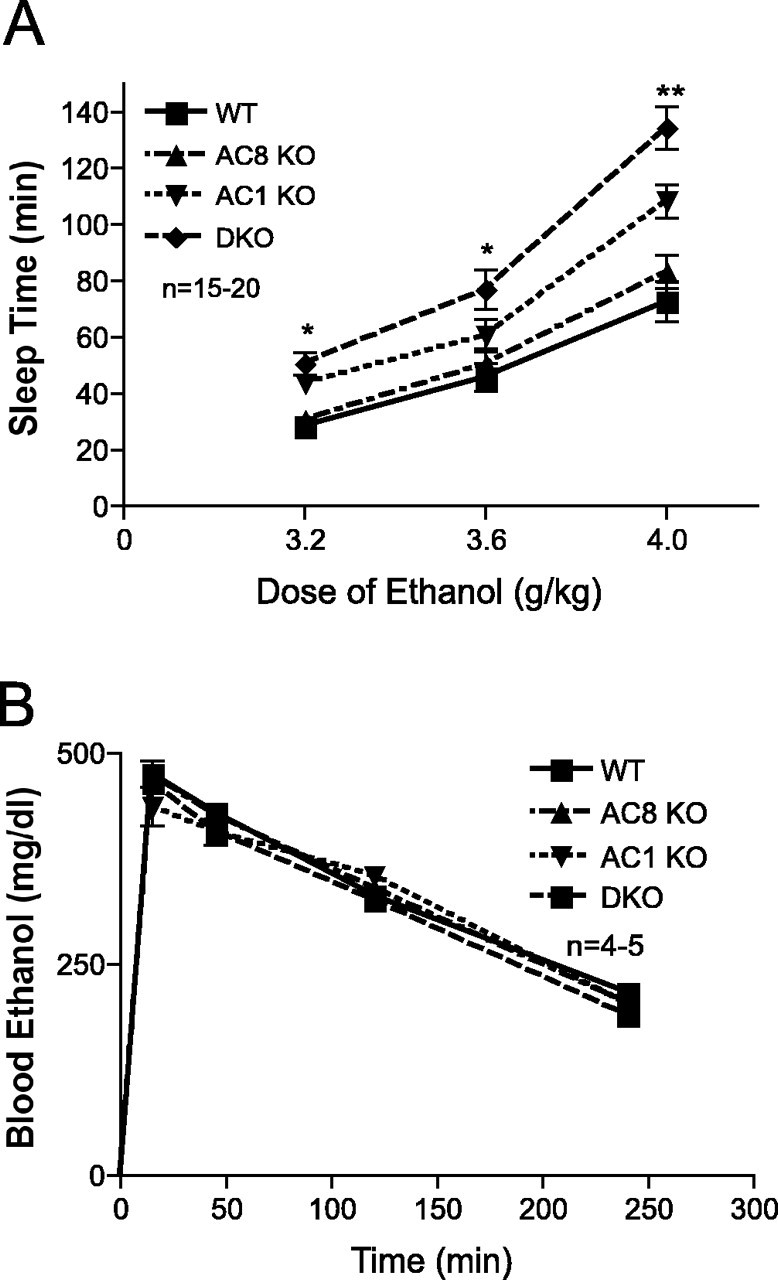
Adenylyl cyclase mutant mice demonstrate enhanced sensitivity to the sedative effects of ethanol without altered ethanol metabolism. A, Sleep-time measurement in the LORR assay for WT, AC8 KO, AC1 KO, and DKO mice after intraperitoneal injection of ethanol. There was a significant main effect of genotype (overall ANOVA, F(3,177) = 474.3; p < 0.0001; post hoc comparisons, *p < 0.001 for both DKO vs WT and AC1 KO vs WT; **p < 0.001 for DKO, AC1 KO, and AC8 KO vs WT), dose (overall ANOVA, F(2,177) = 2080; p < 0.0001), and dose times genotype (overall ANOVA, F(6,177) = 44.78; p < 0.0001; n = 15-20 mice/genotype). B, Blood ethanol concentration after intraperitoneal injection of 3.6 g/kg ethanol does not differ between WT, AC8 KO, AC1 KO, and DKO mice (n = 4-5 mice/genotype). Error bars represent SEM.
Adenylyl cyclase knock-out mice do not have altered ethanol metabolism
It is possible that the increased sensitivity of AC1 KO, AC8 KO, and DKO mice to the sedative effects of ethanol might be a result of altered metabolism of ethanol in these mice and not alterations in neuronal function. To test this possibility, we administered a single dose of 3.6 g/kg ethanol to mice and measured blood ethanol concentration at time points ranging from 15 min to 4 h after ethanol injection. There were no differences between genotypes in blood ethanol concentration at any time after administration (Fig. 1B).
Decreased voluntary ethanol consumption in adenylyl cyclase knock-out mice
We next assessed voluntary ethanol consumption in a two-bottle choice test, in which singly housed mice were given a choice between two bottles, one containing water and one containing ethanol at various concentrations. As shown in Figure 2A, AC8 KO and DKO mice consumed significantly less ethanol than WT mice at higher ethanol concentrations. In contrast, AC1 KO mice did not differ from WT mice in ethanol consumption at any concentration, despite the fact that they demonstrated increased sleep time after ethanol administration. AC8 KO and DKO mice, but not AC1 KO mice, also displayed a significantly lower preference for ethanol (Fig. 2B). It should be noted that there were no differences between DKO and AC8 KO mice with respect to ethanol consumption or preference. Furthermore, there were no differences between genotypes in total fluid consumption during the test (Fig. 2C). Although AC1 KO and DKO mice did not differ from WT mice in total food consumption, AC8 KO mice did consume significantly more food than WT mice when corrected for body weight (Fig. 2D). It is possible that the altered ethanol consumption in AC8 KO and DKO mice may reflect alterations in caloric intake and/or requirements in these mice. However, if the decrease in ethanol consumption in AC8 KO mice was a result of decreased caloric requirements, one would expect these mice to consume less, not more, food than WT mice. Furthermore, DKO mice consumed less ethanol than WT mice but did not differ from WT mice in food consumption. In conjunction, these results indicate that the altered ethanol consumption in AC8 KO and DKO mice is not attributable to alterations in caloric intake in these mice. Finally, it is possible that the diminished ethanol consumption in AC8 KO mice is a result of the presence of flanking 129/Sv genomic DNA instead of the deletion of AC8. To address this possibility, we generated mice that were either homozygous or heterozygous for 129/Sv genomic DNA at this locus, using a method described by Wolfer et al. (2002), and demonstrated that there was no difference in ethanol consumption between mice heterozygous or homozygous for 129/Sv genomic DNA at this locus (see supplementary data, available at www.jneurosci.org as supplemental material).
Figure 2.
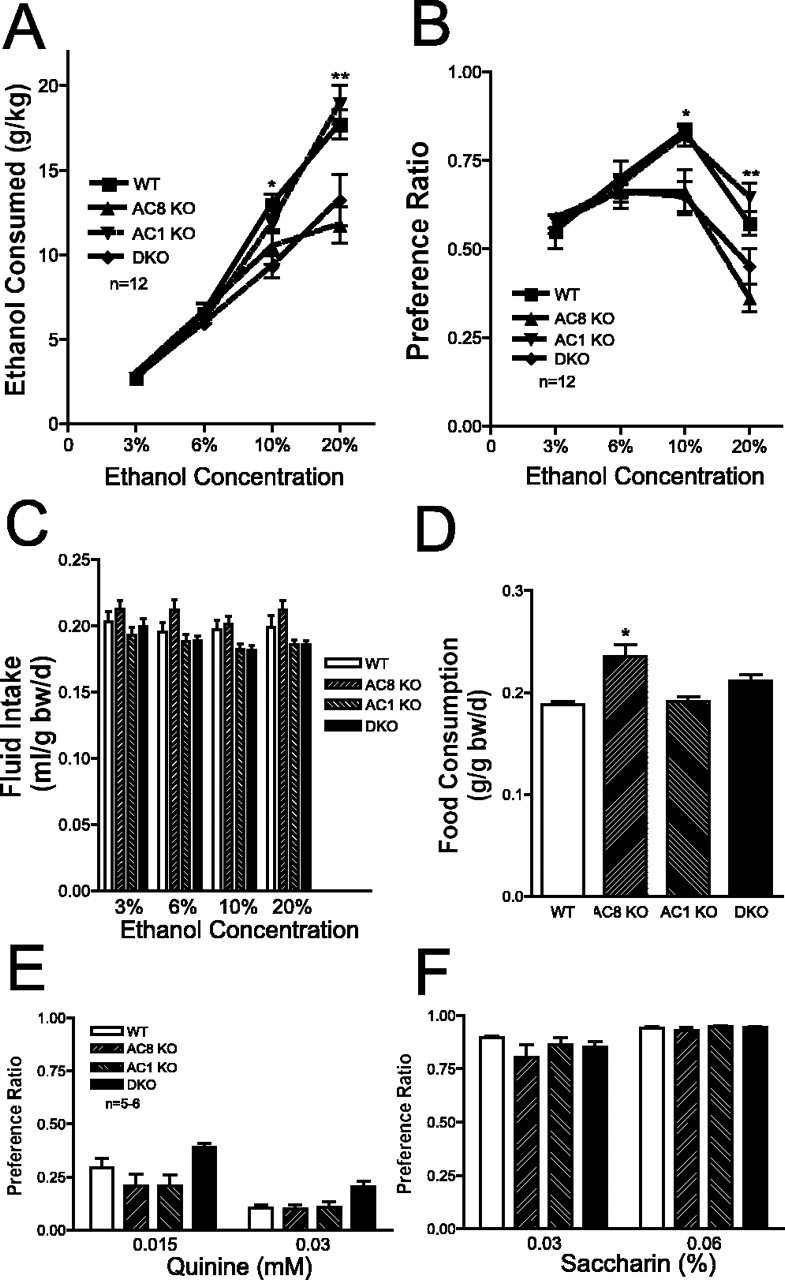
Adenylyl cyclase mutant mice demonstrate reduced voluntary ethanol consumption without altered taste preference. A, Consumption, normalized to bodyweight, of 3, 6, 10, and 20% ethanol solutions by WT, AC8 KO, AC1 KO, and DKO mice. There were significant differences between genotypes (overall ANOVA, F(3,176) = 66.40; p < 0.0001; post hoc comparisons, *p < 0.001 for DKO vs WT; **p < 0.001 for DKO vs WT and AC8 KO vs WT; n = 12 mice/genotype). B, Ethanol preference (volume of ethanol consumed/total volume of fluid consumed) for 3, 6, 10, and 20% ethanol solutions by WT, AC8 KO, AC1 KO, and DKO mice. There were significant differences between genotypes (overall ANOVA, F(3,176) = 8.840; p < 0.0001; post hoc comparisons, *p < 0.01 DKO vs WT and AC8 KO vs WT; **p < 0.001 AC8 KO vs WT; n = 12 mice/genotype). C, Total daily fluid intake, normalized to bodyweight, by WT, AC8 KO, AC1 KO, and DKO mice (n = 12 mice/genotype). D, Daily food consumption, normalized to body weight, by WT, AC8 KO, AC1 KO, and DKO mice (n = 12 mice/genotype; overall ANOVA, F(3,44) = 9.849; p < 0.0001; post hoc comparisons, *p < 0.001 vs WT). E, Quinine preference (volume of quinine solution consumed/total volume of fluid consumed) for 0.15 and 0.03 mm quinine solutions by WT, AC8 KO, AC1 KO, and DKO mice (n = 5-6). F, Saccharin preference (volume of saccharin solution consumed/total volume of fluid consumed) for 0.03 and 0.06% saccharin solutions by WT, AC8 KO, AC1 KO, and DKO mice (n = 5-6). Error bars represent SEM.
Taste preference is not altered in adenylyl cyclase knock-out mice
It is possible that the altered ethanol consumption and preference in DKO and AC8 KO mice is attributable to global changes in their ability to detect differences in taste. To test this possibility, we repeated the two-bottle preference test, but instead of ethanol, we presented the mice with a choice of quinine, a bitter tastant, or saccharin, a sweet tastant, versus water. AC8 KO, AC1 KO, and DKO mice did not differ from WT mice in their preference for quinine (Fig. 2E) or saccharin (Fig. 2F), indicating that alterations in global taste sensation are not responsible for the reduced ethanol consumption in these mice.
Adenylyl cyclase knock-out mice do not demonstrate enhanced ataxia after ethanol administration
We assessed the sensitivity of DKO and WT mice to the ataxic effects of ethanol by using several different tests, because other studies have shown that ethanol differentially alters motor function across an array of tests for ataxia (Crabbe et al., 2003a,b; Metten et al., 2004). We first assessed ethanol-induced ataxia by measuring missteps through a cage grid. Both WT and DKO mice showed a significant increase in the ataxia ratio after administration of 2 g/kg ethanol compared with saline administration (Fig. 3A). However, there was no difference between genotypes in this test, indicating that DKO mice do not display increased sensitivity to the ataxic effects of ethanol. We next assessed sensitivity to ethanol-induced ataxia by measuring hindfoot missteps on a 19-mm-wide balance beam before and after administration of 1.25 g/kg ethanol (Fig. 3B). Ethanol administration resulted in a significant increase in missteps, but this was equivalent in WT and DKO mice. In addition, we assessed the latency to fall from a vertical screen 30 min after administration of 1.75 g/kg ethanol (Fig. 3C) and observer-rated ataxia 10 min after administration of 3 g/kg ethanol (Fig. 3D). In both cases, ethanol-induced ataxia was equivalent in WT and DKO mice. In addition, we assessed the latency of mice to fall from 15.8 and 9.6 mm dowels immediately after and 30 min after administration of 1.75 g/kg ethanol, respectively. In both cases, DKO mice did not demonstrate enhanced ethanol-induced ataxia (data not shown). In summary, ethanol-induced ataxia, as measured by an array of tests of motor function, was equivalent in WT and DKO mice. Error bars represent SEM.
Figure 3.

Adenylyl cyclase mutant mice do not demonstrate enhanced ethanol-induced ataxia. A, Ataxia ratio for WT and DKO mice 30 min after intraperitoneal injection of ethanol (2 g/kg) or saline (0 g/kg). Ataxia ratio is defined as grid step-throughs/locomotor activity counts (n = 8-9 mice/genotype). There was a significant effect of ethanol treatment but not of genotype (overall ANOVA, F(1,29) = 15.79; p < 0.001). B, Hindfoot missteps on 19 mm balance beam before and 10 min after intraperitoneal ethanol injection (n = 11 mice/genotype). There was a significant effect of ethanol treatment but not of genotype (overall ANOVA, F(1,40) = 12.69; p < 0.001). C, Latency to fall from vertical screen 30 min after intraperitoneal injection of ethanol (n = 11 mice/genotype). D, Observer rated ataxia for “splay” 10-11 min after intraperitoneal injection of ethanol (n = 11 mice/genotype). Error bars represent SEM.
Adenylyl cyclase knock-out mice do not demonstrate enhanced sensitivity to pharmacologic agents that mimic the actions of ethanol
As described above, two of the well characterized effects of ethanol on neurons include antagonism of the excitatory NMDA receptor and potentiation of the inhibitory GABAA receptor. We hypothesized that DKO mice, which display enhanced sensitivity to the sedative effects of ethanol, might also have increased sensitivity to the sedative effects of an NMDA receptor antagonist or a GABAA receptor modulator. To test the sensitivity of mice to an NMDA receptor antagonist, we measured the sleep time of WT and DKO mice in the LORR assay after administration of ketamine. As shown in Figure 4A, DKO mice did not differ from WT mice in sleep time after two different doses of ketamine. We then examined the sensitivity of WT and DKO mice to pentobarbital, a GABAA receptor modulator. DKO mice did not differ from WT mice in sleep time after administration of two different doses of pentobarbital (Fig. 4B). Some studies have shown differences in the sedative effects of ethanol and pentobarbital in mice with deletions of particular GABAA receptor subunits (Blednov et al., 2003; Kralic et al., 2003). To address this possibility and confirm that DKO mice did not show increased sleep time after activation of GABAA receptors, we measured sleep time after administration of THIP, a direct agonist of the GABAA receptor. DKO mice did not differ from WT mice in sleep time after administration of 60 mg/kg THIP (Fig. 4C), confirming the results seen with administration of pentobarbital.
Figure 4.

Adenylyl cyclase mutant mice do not demonstrate enhanced sensitivity to NMDA receptor antagonists or GABAA receptor modulators. A, Sleep time in WT and DKO mice after intraperitoneal injection of ketamine (n = 9-10 mice/genotype). B, Sleep time in WT and DKO mice after intraperitoneal injection of pentobarbital (n = 9-10 mice/genotype). C, Sleep time in WT and DKO mice after intraperitoneal injection of THIP (n = 7-8 mice/genotype). D, Sleep time in WT and DKO mice after intraperitoneal injection of 30 mg/kg pentobarbital plus the indicated dose of MK-801 (n = 7-15 mice/genotype). There was a significant effect of MK-801 dose but not of genotype (overall ANOVA, F(3,80) = 33.48; p < 0.0001). Error bars represent SEM.
It is possible that the enhanced sensitivity of DKO mice to the sedative effects of ethanol is a result of synergism between the antagonism by ethanol of the NMDA receptor and potentiation of the GABAA receptor. To test this possibility, we assessed the ability of increasing concentrations of MK-801, an NMDA antagonist, to enhance pentobarbital-induced loss of righting (Fig. 4D). Although MK-801 administration alone is unable to induce loss of righting reflex (data not shown), increasing concentrations of MK-801 resulted in equivalent increases in pentobarbital-induced sedation in WT and DKO mice.
Ethanol does not alter calcium-stimulated adenylyl cyclase activity
We hypothesized that ethanol might have a direct effect on the activity of calcium-stimulated adenylyl cyclases. To test this possibility, we isolated membranes from the cortices of WT and DKO mice and assayed adenylyl cyclase activity in response to increasing concentrations of calcium (Fig. 5A). Increasing concentrations of calcium resulted in increased adenylyl cyclase activity in membranes from WT cortices. As demonstrated previously (Wong et al., 1999), membranes prepared from DKO brain did not demonstrate significant calcium-stimulated AC activity (data not shown). When the assay was repeated in the presence of 50 mm ethanol, calcium-stimulated adenylyl cyclase activity was un-altered. Similar results were obtained using membranes prepared from the cerebellum (Fig. 5B) and the hippocampus (Fig. 5C).
Figure 5.

Ethanol does not enhance calcium-stimulated adenylyl cyclase activity. A, Calcium-stimulated adenylyl cyclase activity in membrane preparations from WT cortices was assayed in the presence and absence of 50 mm ethanol (n = 5 samples/condition). B, Calcium-stimulated adenylyl cyclase activity in membrane preparations from WT cerebelli was assayed in the presence and absence of 50 mm ethanol (n = 5 samples/condition). C, Calcium-stimulated adenylyl cyclase activity in membrane preparations from WT hippocampi was assayed in the presence and absence of 50 mm ethanol (n = 5 samples/condition). Error bars represent SEM.
Ethanol-induced phosphorylation of multiple PKA substrates is compromised in the DKO brain
Because we found no difference between WT and DKO mice in sensitivity to NMDA receptor antagonists or GABAA receptor agonists, and ethanol does not directly alter calcium-stimulated AC activity, we hypothesized that the enhanced sensitivity to ethanol-induced sedation displayed by DKO mice results from altered signal transduction in the brains of these mice after ethanol administration. To address this possibility, we assessed the extent of phosphorylation of PKA substrates in the whole brain by Western blot analysis using an antibody directed against the phosphorylated form of a PKA consensus phosphorylation motif. We harvested brains from WT mice when they regained consciousness (as assessed by righting reflex) after injection of 4 g/kg ethanol, between 55 and 74 min after injection. We then harvested brains from DKO mice at matching times after ethanol injection. Importantly, the DKO mice were still unable to right themselves at these time points. We also harvested brains from WT and DKO mice at matching times after saline injection. Western blot analysis of whole-brain lysates demonstrated that ethanol treatment induced the phosphorylation of multiple proteins, as indicated by the increased intensity of immunoreactive bands (Fig. 6A). Interestingly, the ethanol-induced phosphorylation of a subset of proteins at PKA consensus sites was significantly greater in WT brain lysates than in DKO brain lysates (Fig. 6A). Several other proteins showed a significant increase in ethanol-induced phosphorylation, and a trend (0.05 < p < 0.1) toward increased phosphorylation in WT brain compared with DKO brain. Conversely, there was a separate subgroup of proteins that demonstrated ethanol-induced phosphorylation that was equivalent in the WT and DKO brain (Fig. 6A). In addition, there was no difference in phosphorylation levels of PKA substrates between WT and DKO brain lysates from mice injected with saline. These results indicate the existence of a subset of proteins for which the PKA-dependent phosphorylation after ethanol treatment is regulated specifically by the calcium-stimulated ACs.
Figure 6.

Ethanol-induced phosphorylation of a subset of PKA substrates is compromised in DKO brain. A, Western blot analysis of lysates of whole brain from WT and DKO mice treated with ethanol or saline display increased intensity of several bands detected by an antibody to the phosphorylated PKA consensus motif after ethanol treatment. Each lane contains 50 μg of protein from the brain lysate of a unique mouse. The asterisk indicates bands that were increased in intensity after ethanol treatment to a greater extent in WT brain than in DKO brain (treatment, p < 0.05; genotype, p < 0.05). The numbers in parentheses indicate the fold difference in mean intensity of indicated bands in WT ethanol group versus DKO ethanol group. The # symbol indicates bands that were increased equivalently in WT and DKO brain after ethanol treatment (treatment, p < 0.05; genotype, p > 0.05). B, Levels of GluR1 phosphorylated at serine 845, a site phosphorylated by PKA, in brains from WT and DKO mice treated with saline or ethanol. C, Normalized intensity of GluR1 phosphorylated at serine 845 in brains from WT and DKO mice treated with saline or ethanol (n = 3 samples/genotype). There was a significant effect of ethanol treatment but not of genotype (overall ANOVA, F(1,8) = 39.31; p < 0.0005). D, Levels of GluR1 phosphorylated at serine 831, a site phosphorylated by PKC and CaMKII, in brains from WT and DKO mice treated with saline or ethanol. E, Normalized intensity of GluR1 phosphorylated at serine 831 in brains from WT and DKO mice treated with saline or ethanol (n = 3 samples/genotype). MW, Molecular weight. Error bars represent SEM.
The function of the AMPA receptor is regulated by PKA-dependent phosphorylation of its GluR1 subunit on serine 845 and might be a potentially important target of the calcium-stimulated ACs after ethanol administration (Roche et al., 1996; Banke et al., 2000; Ehlers, 2000). We analyzed the ability of ethanol to regulate phosphorylation of this residue in whole-brain lysates from WT and DKO mice (Fig. 6B). Ethanol treatment induced a significant increase in the phosphorylation of this residue in both WT and DKO brain lysates (Fig. 6C). In contrast, phosphorylation of GluR1 at serine 831, which is regulated by protein kinase C and calmodulin-dependent kinase II (Roche et al., 1996), was unaffected by ethanol treatment in both WT and DKO brain lysates (Fig. 6D, E). These studies provide the first evidence that the PKA-dependent phosphorylation of the AMPA receptor can be modulated in vivo by ethanol treatment.
Ethanol-induced phosphorylation of a subset of PKA substrates is regulated by AC1 and AC8 in a brain region-specific manner
Because the calcium-stimulated ACs and other proteins that regulate the activity of the cAMP signal transduction system are expressed differentially throughout the brain, it is possible that the ethanol-induced increase in PKA substrate phosphorylation is modulated differently in various brain regions. To examine this possibility, we harvested the cortex and cerebellum from WT and DKO mice 45 min after administration of 4 g/kg ethanol and determined the extent of phosphorylation of PKA substrates by Western blot analysis. It should be noted that in this experiment, neither WT nor DKO mice treated with ethanol had regained consciousness when they were killed, eliminating the possibility that differences in protein phosphorylation levels between WT and DKO lysates reflect the different state of consciousness at the time they were killed, as might be possible for the previous experiment. In the cortex, there were several proteins that were phosphorylated after ethanol administration to a greater extent in WT mice than in DKO mice (Fig. 7A). In addition, there were proteins that demonstrated an equivalent increase in phosphorylation in WT and DKO cortex after ethanol administration (Fig. 7A). In contrast, ethanol-induced phosphorylation of PKA substrates in the cerebellum was equivalent between WT and DKO mice for all proteins examined (Fig. 7B). These results demonstrate that the calcium-stimulated ACs modulate the ethanol-induced phosphorylation of a subset of PKA substrates in the cortex but not in the cerebellum.
Figure 7.
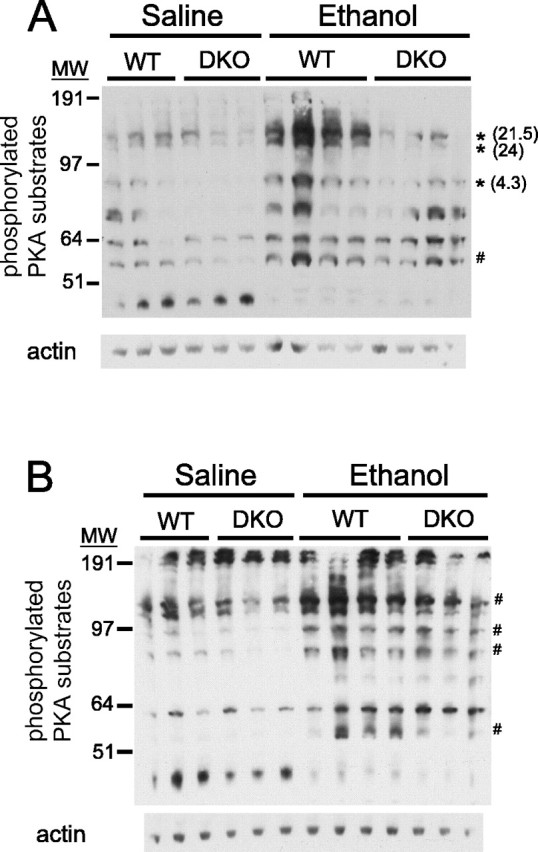
Ethanol-induced phosphorylation of a subset of PKA substrates is compromised in the cortex, but not the cerebellum, of DKO mice. Western blots of lysates of cortex (A) or cerebellum (B) isolated from WT and DKO mice treated with ethanol or saline are shown, indicating increased intensity of several bands detected by an antibody to the phosphorylated PKA consensus motif after ethanol treatment. The asterisk indicates bands that were increased after ethanol treatment to a greater extent in the WT brain than in the DKO brain (treatment, p < 0.05; genotype, p < 0.05). The number in parentheses indicates the fold difference in mean intensity of indicated bands in WT ethanol group versus DKO ethanol group. The # symbol indicates bands that were increased equivalently in WT and DKO brain after ethanol treatment (treatment, p < 0.05; genotype, p > 0.05). There were no bands in cerebellar samples that were increased after ethanol treatment to a greater extent in WT mice than in DKO mice. MW, Molecular weight.
Discussion
Here, we demonstrate that AC1 KO and DKO mice have increased sensitivity to the sedative effects of ethanol compared with WT mice, whereas AC8 KO mice show only a slight increase in sensitivity. In addition, AC8 KO and DKO mice, but not AC1 KO mice, have lower voluntary ethanol consumption than WT mice. Furthermore, DKO mice do not have enhanced sensitivity to ketamine and pentobarbital, pharmacologic agents that mimic components of the effects of ethanol on neurons. Finally, we demonstrate that whereas ethanol is without effect on calcium-stimulated AC activity, the ethanol-induced phosphorylation of a subset of PKA substrates is compromised in the brains of DKO mice.
The deficits in learning and memory and LTP that are found in DKO mice, but not in AC1 KO or AC8 KO mice, indicate that loss of one calcium-stimulated AC isoform can be compensated for by the remaining calcium-stimulated AC isoform (Wong et al., 1999). Consistent with this, we find that AC1 KO mice display sleep times after ethanol administration that are intermediate between those of WT and DKO mice. If AC1 was the only calcium-stimulated AC important in modulating the response to the sedative effects of ethanol, one would expect that AC1 KO and DKO mice would display identical sleep times after ethanol administration. Because this is not the case, we conclude that both AC1 and AC8 are contributors to this response to ethanol.
Surprisingly, there was a dissociation between alterations in ethanol-induced sedation and voluntary ethanol consumption among genotypes. In general, knock-out mice with altered ethanol sensitivity demonstrate an inverse relationship between sensitivity to ethanol-induced sedation and voluntary ethanol consumption (Thiele et al., 1998, 2000; Hodge et al., 1999; Bowers and Wehner, 2001; Wand et al., 2001; Naassila et al., 2002, 2004; Spanagel et al., 2002) (but see Yaka et al., 2003). Our results show that whereas both AC1 and AC8 modulate sensitivity to ethanol-induced sedation, voluntary ethanol consumption is modulated solely by AC8. One possible explanation lies in the differential expression of AC1 and AC8 within the brain: AC8 is highly expressed in the thalamus, hypothalamus, and brainstem, whereas AC1 is not (Xia et al., 1991; Muglia et al., 1999). Calcium-stimulated AC activity in these regions may be important for determining the magnitude of rewarding or aversive effects of ethanol or transducing them into a behavioral response.
Several groups have provided evidence that alterations in the cAMP signaling system modulate sensitivity to ethanol, consistent with our results. Drosophila with mutations that decrease the activity of the cAMP signaling pathway, including rutabaga, the Drosophila homolog of calcium-stimulated AC, demonstrates increased sensitivity to ethanol (Moore et al., 1998). Mice with decreases in Gαs or PKA activity display increased sensitivity to ethanol, whereas mice with increased PKA activity demonstrate decreased sensitivity (Thiele et al., 2000; Wand et al., 2001). The results described here provide additional support for the importance of the cAMP signaling cascade in the regulation of ethanol sensitivity in adult mice and identify specific mammalian AC isoforms important for this regulation.
Because our strategy of backcrossing the AC1 and AC8 KO mice to C57BL/6 permits mutation-flanking 129/Sv DNA regions to be carried along with the targeted loci (Wu et al., 1995; Schaefer et al., 2000), it is possible that the effects we describe here could be attributable to the presence of genes linked to the loci for AC1 and/or AC8. Analysis of quantitative trait loci (QTL) has identified several chromosomal regions that modulate ethanol-induced sedation and consumption (Crabbe, 2002). Among these are regions on chromosomes 11 and 15, where the loci for AC1 and AC8 are located, respectively. The QTL for ethanol-induced sedation on chromosome 11 has been efficiently recombined away from the locus for AC1 during generation of lines (see Materials and Methods for details) and cannot contribute to the effects we described here (Bennett et al., 2002). In contrast, a QTL for ethanol consumption on chromosome 15 is linked to our 129/Sv-derived locus for AC8, and it is possible that the altered ethanol consumption in AC8 KO mice may be associated with the presence of a different gene associated with this QTL (Vadasz et al., 2000). This alternative is unlikely, because we demonstrated no difference in ethanol consumption between mice heterozygous at this locus compared with mice homozygous at this locus for 129/Sv sequences. The most parsimonious explanation is that the QTL on chromosome 15 may be caused by differences in the activity of AC8 between inbred strains of mice.
The deficits displayed by DKO mice in NMDA-dependent phenomena have led to the suggestion that activation of the NMDA receptor couples to activation of the calcium-stimulated ACs (Chetkovich and Sweatt, 1993; Wong et al., 1999). If true, then the antagonism by ethanol of the NMDA receptor should result in decreased activation of AC1 and AC8 and might explain the enhanced sensitivity of DKO mice to ethanol. However, DKO mice did not demonstrate increased sleep time compared with WT mice after administration of NMDA receptor antagonists or GABAergic modulators, including ketamine, pentobarbital, THIP, or a mixture of pentobarbital and MK-801. We conclude that the enhanced sensitivity of DKO mice to ethanol-induced sedation is independent of the antagonism by ethanol of the NMDA receptor and potentiation of the GABAA receptor. Furthermore, these results suggest that loss of AC1 and AC8 does not increase sensitivity to all sedative agents but is specific for ethanol-induced sedation.
We show that the ability of ethanol to induce the phosphorylation of a subset of PKA substrates was compromised in the brains of DKO mice. Previous work has demonstrated that ethanol treatment is able to induce the nuclear translocation of PKA, cAMP response element (CRE)-binding protein phosphorylation, and CRE-mediated transcription (Yang et al., 1996; Constantinescu et al., 1999, 2002). These results are the first to demonstrate that the ability of ethanol to induce PKA activation and phosphorylation of PKA substrates is mediated, in part, by the calcium-stimulated ACs. Furthermore, they provide the first evidence linking the calcium-stimulated cyclases to phosphorylation of specific PKA targets. In addition, these results provide additional evidence that there are discrete spatiotemporal pools of cAMP within the cell, with consequences on specific down-stream targets, paralleling work by others in cardiac myocytes and neurons (Davare et al., 2001; Zaccolo and Pozzan, 2002). At present, the exact mechanism leading to the activation of AC1 and AC8 after ethanol exposure remains to be elucidated. It is possible that ethanol administration leads to elevation of intracellular calcium, resulting in the activation of AC1 and AC8. Conversely, ethanol exposure may result in increased Gαs activity through indirect stimulation of adenosine receptors, among others (Choi et al., 2004). Examining the sensitivity of mice with genetic deletions of the other members of the AC family will provide additional information about this process.
Interestingly, the ethanol-induced phosphorylation of a subset of PKA substrates is compromised in the cortex, but not the cerebellum, of DKO mice. These results correlate well with the behavioral response of the mice to ethanol, because DKO mice display enhanced sensitivity to the sedative, but not the ataxic, effects of ethanol. We hypothesize that the sedative effects of ethanol are mediated through inhibition of cortical function, although its ataxic effects are mediated through inhibition of cerebellar activity, explaining the dissociation in the behavioral expression of enhanced ethanol sensitivity in the DKO mice.
Interventions that perturb the levels of neuronal activity result in compensatory, homeostatic alterations that counteract the effects of such perturbations (Davis and Bezprozvanny, 2001). We propose that the ethanol-mediated induction of phosphorylation of PKA substrates is part of a homeostatic mechanism, initiated at least in part by AC1 and AC8. This activation counteracts the inhibitory effects of ethanol on neuronal activity, such that knock-out mice missing AC1 and/or AC8 have defects in this homeostatic process. However, it remains possible that the phosphorylation may be part of a response to ethanol without contributing to a homeostatic restoration of activity. It is likely that AMPA receptors are one of the targets of this homeostatic process, because treatment with ethanol leads to phosphorylation of the GluR1 subunit of the AMPA receptor at a residue regulated by PKA but not at a residue regulated by PKC and calmodulin-dependent kinase II. Phosphorylation of serine 845 of the GluR1 receptor has been demonstrated to have multiple effects on AMPA receptor activity, including potentiation of the peak current and open probability of the receptor, and increasing the rate of reinsertion of the receptor in the plasma membrane (Roche et al., 1996; Banke et al., 2000; Ehlers, 2000). Thus, the increased phosphorylation of GluR1 at serine 845 induced by ethanol treatment would result in increased activity of the AMPA receptor, counteracting the inhibitory effect of ethanol on neuronal activity. Although our results suggest that this phosphorylation is not regulated by the calcium-stimulated ACs, another study (Lu et al., 2003) has shown that barrelless mice, which have a spontaneous mutation of AC1, have decreased phosphorylation of this residue in synaptosomes isolated from somatosensory cortex of neonatal mice. It is likely that the AC1-mediated regulation of phosphorylation of this residue is lost during development, explaining the difference in our results. Although our results suggest that phosphorylation of GluR1 at serine 845 is more likely regulated by Gαs-stimulated ACs, it nonetheless provides support for the idea that regulation of PKA activity by multiple sources is a critical component of the homeostatic response to ethanol-mediated inhibition of neuronal activity. The future characterization of targets uniquely regulated by the calcium-stimulated ACs will provide additional insight into the mechanism of the neuronal response to the inhibitory effects of ethanol.
Footnotes
This work was supported by National Institutes of Health Grants AA12957 and AG18876. J.W.M. thanks the Washington University Medical Scientist Training Program for financial support.
Correspondence should be addressed to Dr. Louis J. Muglia, Department of Pediatrics and Molecular Biology and Pharmacology, Washington University School of Medicine, 660 South Euclid Avenue, Campus Box 8208, St. Louis, MO 63110. E-mail: Muglia_L@kids.wustl.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254118-09$15.00/0
References
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF (2000) Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci 20: 89-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett B, Beeson M, Gordon L, Carosone-Link P, Johnson TE (2002) Genetic dissection of quantitative trait loci specifying sedative/hypnotic sensitivity to ethanol: mapping with interval-specific congenic recombinant lines. Alcohol Clin Exp Res 26: 1615-1624. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Jung S, Alva H, Wallace D, Rosahl T, Whiting PJ, Harris RA (2003) Deletion of the alpha1 or beta2 subunit of GABAA receptors reduces actions of alcohol and other drugs. J Pharmacol Exp Ther 304: 30-36. [DOI] [PubMed] [Google Scholar]
- Bowers BJ, Wehner JM (2001) Ethanol consumption and behavioral impulsivity are increased in protein kinase Cγ null mutant mice. J Neurosci 21: RC180(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cali JJ, Zwaagstra JC, Mons N, Cooper DM, Krupinski J (1994) Type VIII adenylyl cyclase. A Ca2+/calmodulin-stimulated enzyme expressed in discrete regions of rat brain. J Biol Chem 269: 12190-12195. [PubMed] [Google Scholar]
- Chern Y (2000) Regulation of adenylyl cyclase in the central nervous system. Cell Signal 12: 195-204. [DOI] [PubMed] [Google Scholar]
- Chetkovich DM, Sweatt JD (1993) NMDA receptor activation increases cyclic AMP in area CA1 of the hippocampus via calcium/calmodulin stimulation of adenylyl cyclase. J Neurochem 61: 1933-1942. [DOI] [PubMed] [Google Scholar]
- Choi DS, Cascini MG, Mailliard W, Young H, Paredes P, McMahon T, Diamond I, Bonci A, Messing RO (2004) The type 1 equilibrative nucleoside transporter regulates ethanol intoxication and preference. Nat Neurosci 7: 855-861. [DOI] [PubMed] [Google Scholar]
- Constantinescu A, Diamond I, Gordon AS (1999) Ethanol-induced translocation of cAMP-dependent protein kinase to the nucleus. Mechanism and functional consequences. J Biol Chem 274: 26985-26991. [DOI] [PubMed] [Google Scholar]
- Constantinescu A, Gordon AS, Diamond I (2002) cAMP-dependent protein kinase types I and II differentially regulate cAMP response element-mediated gene expression: implications for neuronal responses to ethanol. J Biol Chem 277: 18810-18816. [DOI] [PubMed] [Google Scholar]
- Crabbe JC (2002) Alcohol and genetics: new models. Am J Med Genet 114: 969-974. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Metten P, Yu CH, Schlumbohm JP, Cameron AJ, Wahlsten D (2003a) Genotypic differences in ethanol sensitivity in two tests of motor incoordination. J Appl Physiol 95: 1338-1351. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Cotnam CJ, Cameron AJ, Schlumbohm JP, Rhodes JS, Metten P, Wahlsten D (2003b) Strain differences in three measures of ethanol intoxication in mice: the screen, dowel and grip strength tests. Genes Brain Behav 2: 201-213. [DOI] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW (2001) A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science 293: 98-101. [DOI] [PubMed] [Google Scholar]
- Davis GW, Bezprozvanny I (2001) Maintaining the stability of neural function: a homeostatic hypothesis. Annu Rev Physiol 63: 847-869. [DOI] [PubMed] [Google Scholar]
- Dildy JE, Leslie SW (1989) Ethanol inhibits NMDA-induced increases in free intracellular Ca2+ in dissociated brain cells. Brain Res 499: 383-387. [DOI] [PubMed] [Google Scholar]
- Ehlers MD (2000) Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28: 511-525. [DOI] [PubMed] [Google Scholar]
- Harris RA, Proctor WR, McQuilkin SJ, Klein RL, Mascia MP, Whatley V, Whiting PJ, Dunwiddie TV (1995) Ethanol increases GABAA responses in cells stably transfected with receptor subunits. Alcohol Clin Exp Res 19: 226-232. [DOI] [PubMed] [Google Scholar]
- Hodge CW, Mehmert KK, Kelley SP, McMahon T, Haywood A, Olive MF, Wang D, Sanchez-Perez AM, Messing RO (1999) Supersensitivity to allosteric GABAA receptor modulators and alcohol in mice lacking PKCepsilon. Nat Neurosci 2: 997-1002. [DOI] [PubMed] [Google Scholar]
- Kralic JE, Wheeler M, Renzi K, Ferguson C, O'Buckley TK, Grobin AC, Morrow AL, Homanics GE (2003) Deletion of GABAA receptor alpha 1 subunit-containing receptors alters responses to ethanol and other anesthetics. J Pharmacol Exp Ther 305: 600-607. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF (1989) Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 243: 1721-1724. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF (1990) NMDA receptor-mediated synaptic excitation selectively inhibited by ethanol in hippocampal slice from adult rat. J Neurosci 10: 1372-1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HC, She WC, Plas DT, Neumann PE, Janz R, Crair MC (2003) Adenylyl cyclase I regulates AMPA receptor trafficking during mouse cortical “barrel” map development. Nat Neurosci 6: 939-947. [DOI] [PubMed] [Google Scholar]
- Mehta AK, Ticku MK (1988) Ethanol potentiation of GABAergic transmission in cultured spinal cord neurons involves gamma-aminobutyric acidA-gated chloride channels. J Pharmacol Exp Ther 246: 558-564. [PubMed] [Google Scholar]
- Metten P, Best KL, Cameron AJ, Saultz AB, Zuraw JM, Yu CH, Wahlsten D, Crabbe JC (2004) Observer-rated ataxia: rating scales for assessment of genetic differences in ethanol-induced intoxication in mice. J Appl Physiol 97: 360-368. [DOI] [PubMed] [Google Scholar]
- Moore MS, DeZazzo J, Luk AY, Tully T, Singh CM, Heberlein U (1998) Ethanol intoxication in Drosophila: genetic and pharmacological evidence for regulation by the cAMP signaling pathway. Cell 93: 997-1007. [DOI] [PubMed] [Google Scholar]
- Muglia LM, Schaefer ML, Vogt SK, Gurtner G, Imamura A, Muglia LJ (1999) The 5′-flanking region of the mouse adenylyl cyclase type VIII gene imparts tissue-specific expression in transgenic mice. J Neurosci 19: 2051-2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naassila M, Ledent C, Daoust M (2002) Low ethanol sensitivity and increased ethanol consumption in mice lacking adenosine A2A receptors. J Neurosci 22: 10487-10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naassila M, Pierrefiche O, Ledent C, Daoust M (2004) Decreased alcohol self-administration and increased alcohol sensitivity and withdrawal in CB1 receptor knockout mice. Neuropharmacology 46: 243-253. [DOI] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL (1996) Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 16: 1179-1188. [DOI] [PubMed] [Google Scholar]
- Schaefer ML, Wong ST, Wozniak DF, Muglia LM, Liauw JA, Zhuo M, Nardi A, Hartman RE, Vogt SK, Luedke CE, Storm DR, Muglia LJ (2000) Altered stress-induced anxiety in adenylyl cyclase type VIII-deficient mice. J Neurosci 20: 4809-4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanagel R, Siegmund S, Cowen M, Schroff KC, Schumann G, Fiserova M, Sillaber I, Wellek S, Singer M, Putzke J (2002) The neuronal nitric oxide synthase gene is critically involved in neurobehavioral effects of alcohol. J Neurosci 22: 8676-8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WJ, Krupinski J, Gilman AG (1991) Expression and characterization of calmodulin-activated (type I) adenylylcyclase. J Biol Chem 266: 8595-8603. [PubMed] [Google Scholar]
- Thiele TE, Marsh DJ, Ste Marie L, Bernstein IL, Palmiter RD (1998) Ethanol consumption and resistance are inversely related to neuropeptide Y levels. Nature 396: 366-369. [DOI] [PubMed] [Google Scholar]
- Thiele TE, Willis B, Stadler J, Reynolds JG, Bernstein IL, McKnight GS (2000) High ethanol consumption and low sensitivity to ethanol-induced sedation in protein kinase A-mutant mice. J Neurosci 20: RC75(1-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadasz C, Saito M, Balla A, Kiraly I, Vadasz Jr C, Gyetvai B, Mikics E, Pierson D, Brown D, Nelson JC (2000) Mapping of quantitative trait loci for ethanol preference in quasi-congenic strains. Alcohol 20: 161-171. [DOI] [PubMed] [Google Scholar]
- Villacres EC, Wong ST, Chavkin C, Storm DR (1998) Type I adenylyl cyclase mutant mice have impaired mossy fiber long-term potentiation. J Neurosci 18: 3186-3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner M, Hanchar HJ, Olsen RW (2003) Ethanol enhances alpha 4 beta 3 delta and alpha 6 beta 3 delta gamma-aminobutyric acid type A receptors at low concentrations known to affect humans. Proc Natl Acad Sci USA 100: 15218-15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wand G, Levine M, Zweifel L, Schwindinger W, Abel T (2001) The cAMP-protein kinase A signal transduction pathway modulates ethanol consumption and sedative effects of ethanol. J Neurosci 21: 5297-5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Storm DR (2003) Calmodulin-regulated adenylyl cyclases: cross-talk and plasticity in the central nervous system. Mol Pharmacol 63: 463-468. [DOI] [PubMed] [Google Scholar]
- Wang H, Pineda VV, Chan GC, Wong ST, Muglia LJ, Storm DR (2003) Type 8 adenylyl cyclase is targeted to excitatory synapses and required for mossy fiber long-term potentiation. J Neurosci 23: 9710-9718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfer DP, Crusio WE, Lipp HP (2002) Knockout mice: simple solutions to the problems of genetic background and flanking genes. Trends Neurosci 25: 336-340. [DOI] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR (1999) Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron 23: 787-798. [DOI] [PubMed] [Google Scholar]
- Wu ZL, Thomas SA, Villacres EC, Xia Z, Simmons ML, Chavkin C, Palmiter RD, Storm DR (1995) Altered behavior and long-term potentiation in type I adenylyl cyclase mutant mice. Proc Natl Acad Sci USA 92: 220-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia ZG, Refsdal CD, Merchant KM, Dorsa DM, Storm DR (1991) Distribution of mRNA for the calmodulin-sensitive adenylate cyclase in rat brain: expression in areas associated with learning and memory. Neuron 6: 431-443. [DOI] [PubMed] [Google Scholar]
- Yaka R, Tang KC, Camarini R, Janak PH, Ron D (2003) Fyn kinase and NR2B-containing NMDA receptors regulate acute ethanol sensitivity but not ethanol intake or conditioned reward. Alcohol Clin Exp Res 27: 1736-1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Diehl AM, Wand GS (1996) Ethanol exposure alters the phosphorylation of cyclic AMP responsive element binding protein and cyclic AMP responsive element binding activity in rat cerebellum. J Pharmacol Exp Ther 278: 338-346. [PubMed] [Google Scholar]
- Zaccolo M, Pozzan T (2002) Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 295: 1711-1715. [DOI] [PubMed] [Google Scholar]