

Abstract
Cul3 belongs to the family of cullin proteins, which function as scaffold proteins of E3 ubiquitin ligase complexes. Here we show cell-autonomous involvement of Cul3 in axonal arborization and dendritic elaboration of Drosophila mushroom body neurons. Cul3 mutant neurons are defective in terminal morphogenesis of neurites. Interestingly, mutant axons often terminate around branching points. In addition, dendritic elaboration is severely affected in Cul3 mutant neurons. However, loss of Cul3 function does not affect extension of the axons that rarely arborize. Function of cullin-type proteins has been shown to require covalent attachment of Nedd8 (neural precursor cell-expressed developmentally downregulated), a ubiquitin-like protein. Consistent with this notion, Cul3 is inactivated by a mutation in its conserved neddylation site, and Nedd8 mutant neurons exhibit similar neuronal morphogenetic defects. Together, Cul3 plays an essential role in both axonal arborization and proper elaboration of dendrites and may require neddylation for its proper function.
Keywords: Drosophila, Cul3, Nedd8, neurite, arborization, mushroom body
Introduction
Most neurons have multiple synaptic partners. Branching/arborization of neurites facilitates such multiparty communications. In addition, arborization/elaboration of neurites determines the ultimate shapes of the neurons, changes of which can drastically alter the computational characteristics of the neurons (Koch and Segev, 2000). Branching and terminal arborization of neurites can be regulated by both extrinsic factors and intrinsic determinants (for review, see Caroni, 1997; Acebes and Ferrus, 2000; Keith and Wilson, 2001). Diverse target-derived diffusible factors have been shown to regulate terminal arborization and branch formation (Cohen-Cory and Fraser, 1995; Wang et al., 1999; Krylova et al., 2002; Dent et al., 2004). Elaboration of neurites is also an intrinsic property of developing neurons, as suggested by the phenomenon that cultured crayfish motor neurons exhibit their characteristic neuronal arborization patterns in the absence of target muscles (Arcaro and Lnenicka, 1995). However, the molecular mechanisms that govern terminal morphogenesis of neurons have just begun to be elucidated.
Mosaic screening is a powerful tool for identifying novel genes required for various aspects of neuronal morphogenesis (Newsome et al., 2000; Reuter et al., 2003; Zugates and Lee, 2004). It basically involves making homozygous mutant neurons within genetically heterozygous brains. MARCM (mosaic analysis with a repressible cell marker) permits visualization of homozygous mutant cells, making morphological analysis of mutant neurons possible in mosaic fly brains (Lee and Luo, 1999). MARCM labels cells based on lineage and birth timing; thus, it can also be used to characterize normal development of complex tissues (Lee and Luo, 1999; Kiger et al., 2000; Jefferis et al., 2001). MARCM analysis of the Drosophila mushroom body (MB), the olfactory learning and memory center (for review, see Davis, 2001; Heisenberg, 2003), has led to identification of various subtypes of MB neurons that are sequentially derived from common precursors (Lee et al., 1999; Zhu et al., 2003). By isolating mutations that disrupt MB development in mosaic organisms, we and others have identified genes involved in various poorly understood neuronal morphogenetic processes, such as remodeling of neuronal projections (Lee et al., 2000; Watts et al., 2003; Zheng et al., 2003) and divergent segregation of axonal branches (Wang et al., 2002).
Here we report recovery of a novel lethal mutation in the Drosophila Cullin3 [Cul3, also named guftagu (gft)] gene based on morphogenetic defects in MB axonal termini. Mutant MB clones show extension problems selectively in the axonal processes that normally exhibit multiple arbors. Interestingly, mutant processes often stall around the places at which arborization occurs. We also observe defective reelaboration of MB dendrites during metamorphosis. Compared with their wild-type counterparts, most remodeled Cul3 mutant dendrites are much shorter and fail to form typical claw-like structure at their termini. Cul3 belongs to the family of cullin proteins that form E3 ubiquitin ligase complexes together with other components (Ou et al., 2002). We further demonstrate that conserved Nedd8 (neural precursor cell-expressed developmentally downregulated) modification site is essential for Cul3 activity, and Nedd8 mutant MB neurons have similar phenotypes as Cul3 mutant neurons. Together, we suggest that Cul3-based ubiquitin ligase is required for neurite arborization, and its proper function may require neddylation.
Materials and Methods
Fly strains. GAL80 fly stocks used for generation of MARCM clones include the following: (1) hs-FLP; tubP-GAL80,FRT40A/CyO,y and (2) UAS-mCD8-GFP,hs-FLP;tubP-GAL80,FRT40A/CyO;GAL4-OK107; (3) UAS-mCD8-GFP,l(2L)MB397,FRT40A/CyO, (4) UAS-mCD8-GFP,l(2L)MB397,FRT40A,GAL4-201Y/CyO,y, and (5) Cul32,FRT40A/CyO,y were used for creation of Cul3 mutant MARCM clones; (6) Nedd8AN015,FRT40A/CyO,Kr-GFP was used for creation of Nedd8 mutant clones; (7) UAS-mCD8-GFP,FRT40A was used for generating wild-type clones; ectopic expression of transgenes in MARCM clones involves (8) UAS-GFP-Cul3 and (9) UAS-GFP-Cul3K717R.
Other fly stocks collected for this study include Df(2L)TE35BC-24 (BL-3588), Df(2L)b87e25 (BL-3138), Df(2L)r10 (BL-1491), Df(2L)rd9 (BL-3592), stc05441/CyO (BL-11408), and Cul306430/CyO (BL-12175).
MARCM-based genetic screen. UAS-mCD8-GFP,FRT40A male flies were subjected to chemical mutagenesis according standard procedures (Lewis and Bacher, 1968) with ethyl methanesulfonate (EMS) at 40 mm. Individual male progenies derived from mutagenized flies were then crossed with hs-FLP;tubP-GAL80,FRT40A/CyO;GAL4-OK107 for MARCM analysis of MB neurons homozygous for the mutations induced on the UAS-mCD8-GFP,FRT40A chromosome arms in the newly hatched larvae (NHL), as shown in the following genetic flow.

Mapping by complementation tests. After learning that the l(2L)MB397 line was homozygous lethal, we tried to map the lethal mutation(s) using the 2L deficiency kit from the Bloomington Stock Center (Indiana University, Bloomington, IN). l(2L)MB397 only failed to complement with Df(2L)TE35BC-24, localizing one lethal mutation to the region 35B04-35F07. Additional complementation tests using Df(2L) b87e25, Df(2L)r10, and Df(2L)rd9 indicated that the lethal mutation is located in either the 35B10-35C2 region or the 35C5-35D1 region. By crossing with existing lethal mutation within these regions, we finally found that l(2L)MB397 failed to complement with two independent Cul3 mutations, indicating that l(2L)MB397 carries a new recessive lethal allele of Cul3 (Cul38).
Molecular characterization of l(2L)MB397. To determine the nature of mutation of Cul38, genomic DNAs were isolated from both wild-type and l(2L)MB397 homozygous mutant second-instar larvae using standard procedures. Genomic DNAs for individual exons and their flanking intron splicing sites of Cul3 were then amplified by PCR and sequenced by the ABI sequencing system (Applied Biosystems, Foster City, CA).
Construction of UAS-GFP-Cul3 and UAS-GFP-Cul3K717R. For pUAST-GFP-Cul3, Cul3 cDNAs amplified by reverse transcription-PCR using a pair of primers flanking the entire open reading frame were first cloned into the EcRV site of pBSK vector. After confirming the sequence, Cul3 cDNAs were connected to the 3′-end of GFP cDNA through a BglII site and were subsequently cloned into EcoRI-XhoI of pUAST. For pUAST-GFP-Cul3K717R, the above pBSK-Cul3 construct was used for PCR-based site-directed mutagenesis to generate a Lys-to-Arg substitution at Lys-717. Then the Cul3K717R cDNA was ligated to the GFP and cloned into pUAST using the same restriction sites as the wild-type Cul3 cDNAs.
Phenotypic analysis and rescue of MARCM clones. MARCM clones of MB neurons were induced and examined at specific developmental stages as described previously (Lee and Luo, 1999; Lee et al., 1999). Rescuing the phenotypes of the MARCM clones involves coexpression of UAS-GFP-Cul3 or UAS-GFP-Cul3K717R with UAS-mCD8-GFP in GAL80-minus neurons. MARCM clones were detected by rat anti-mCD8 monoclonal antibody (1:100; Caltag, Burlingame, CA). Fluorescent images were collected using a Zeiss (Oberkochen, Germany) LSM510 confocal microscope and processed with Adobe Photoshop (Adobe Systems, San Jose, CA). Midlines were labeled according to the positions of esophagi that were visualized by enhancing the intensity of images.
Results
l(2L)MB397 homozygous mutant MB neurons show axon terminal morphogenetic defects
Using MARCM technology (Fig. 1A), we have been screening for novel mutations that cause specific developmental defects in the MB neurons by generating homozygous mutant clones in otherwise phenotypically wild-type brains (Lee et al., 2000; Wang et al., 2002; Zheng et al., 2003). The MBs are paired neuropil structures in the Drosophila protocerebrum. One adult MB is derived from four indistinguishable neuroblasts (Nbs), each of which sequentially generates three major types of MB neurons that, respectively, project their axonal processes into three distinct sets of MB lobes (Fig. 1B) (Ito et al., 1997; Lee et al., 1999). γ neurons, born before the mid-third instar stage, extend axons medially toward the midline of protocerebrum into the γ lobes, whereas all later-derived MB neurons, including α′/β′ and α/β neurons, have bifurcated primary axons that project into specific pairs of perpendicularly oriented lobes (Lee et al., 1999). Using GAL4-OK107 (Connolly et al., 1996) in MARCM, one can selectively label all GAL80-minus MB neurons in mosaic brains. Our primary screening involved the induction of mitotic recombination in NHL, followed by analysis of MB clones at the adult stage. We observe three distinct sizes of clones: large multi-cellular Nb clones innervating all five axonal lobes (Fig. 1B, D) and single-cell/two-cell clones consisting purely of γ neurons (Fig. 1C,E). We screened 1200 mutagenized 2L chromosome arms and recovered one line, l(2L)MB397, that shows truncated γ and β′ axonal processes in homozygous mutant MB clones (Fig. 1D, E).
Figure 1.
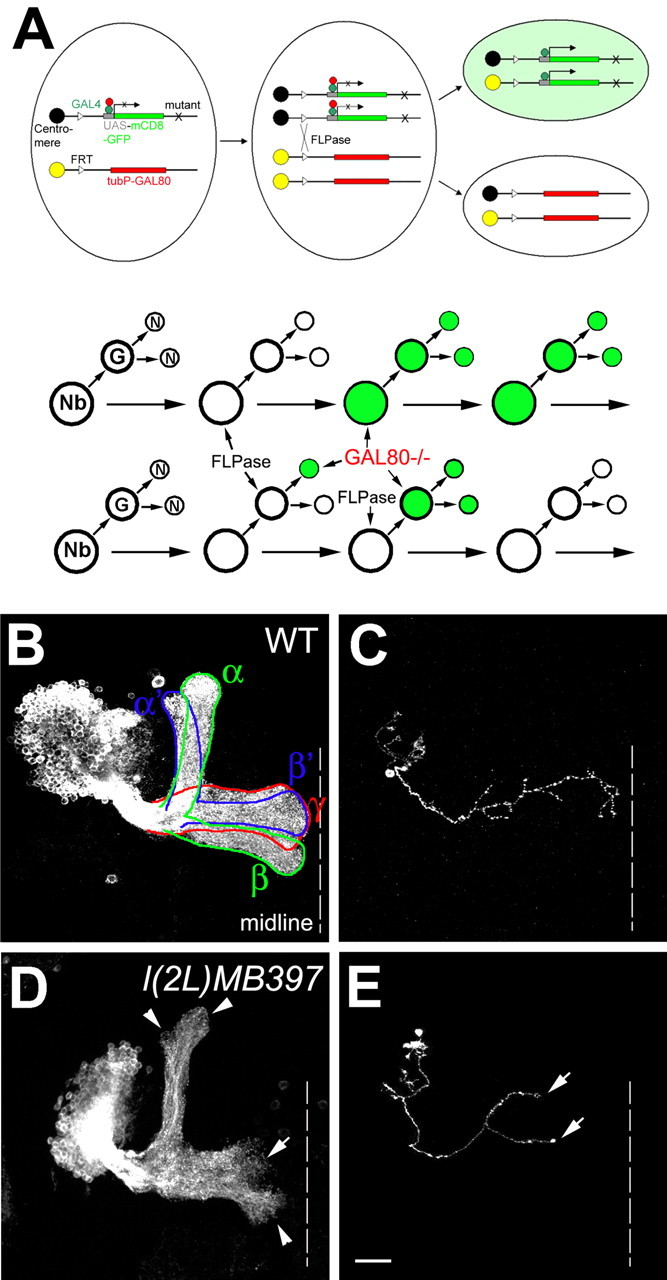
l(2L)MB397 exhibits axon terminal morphogenetic defects in MB neurons. A, Schematic diagram of MARCM. Top, Heat shock-induced FLP recombinase (FLPase) mediates the FRT site-specific mitotic recombination, which results in the expression of mCD8-GFP driven by GAL4 in one of the daughter cells attributable to loss of the repressor transgene GAL80. If the mother cell is heterozygous for a mutant gene (X), the daughter cell homozygous for the mutant gene will be positively labeled. Bottom, An Nb or single-cell/two-cell MARCM clone can be generated in Drosophila CNS, depending on whether GAL80 is lost in the newly generated Nb, in the postmitotic neurons (N), or in the ganglion mother cell (GMC). B, C, Wild-type Nb (WT; B) and single-cell (C) clones of MB neurons, generated in NHL, were examined in adults. Note that one Nb clone contains all five MB lobes (outlined by solid lines in B) and that all of the medial lobes (γ, β, and β′) can reach the midline (dashed lines in this and all of the following figures). D, E, l(2L)MB397 mutant Nb (D) and single-cell (E) clones, generated in NHL, were examined in adults. Note that the γ and β′ lobes are significantly shortened (arrow in D), but other lobes have only subtle morphological defects at the termini (arrowheads in D). Truncated γ axons are also observed in single-cell clones (arrows in E). Scale bar (in this and all of the following figures), 20 μm. All unilateral MB clones are oriented such that their medial processes project from left to right toward the midline. All images are processed from composite confocal images. Genotype: B, C, hs-FLP/X;UAS-mCD8-GFP, FRT40A/tubP-GAL80, FRT40A;GAL4-OK107/+; D, E, hs-FLP/X;l(2L)MB397,UAS-mCD8-GFP,FRT40A/tubP-GAL80,FRT40A;GAL4-OK107/+.
The axon terminal defects of l(2L)MB397 are observed in mutant Nb clones, as well as in single-cell/two-cell clones of mutant γ neurons. First, mutant Nb clones acquired shorter γ and β′ lobes than wild-type clones (Fig. 1D). It appears that most γ processes in the mutant Nb clones are only approximately two-thirds the length of wild-type γ processes (Figs. 1D, arrow, 2I). Second, in contrast to the truncated γ and β′ lobes, only subtle morphological defects are observed in the α, α′, and β lobes. These three lobes extend as far as the wild-type counterparts, but their tips are rough and less dense (Fig. 1D, arrowheads). Third, ∼76% of single-cell/two-cell clones of mutant γ neurons, unlike their wild-type controls, fail to project axons to the tips of the γ lobes (Fig. 1, compare E, arrows, with C). Together, l(2L)MB397 selectively affects terminal morphogenesis of MB axons, especially the γ and β′ processes.
Figure 2.
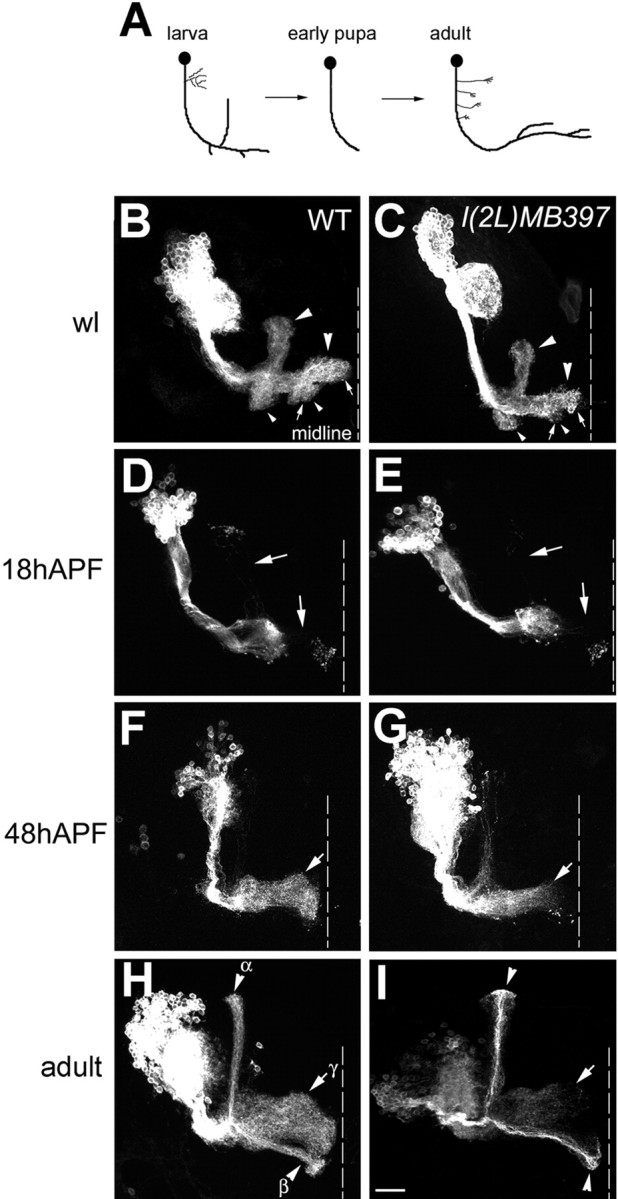
l(2L)MB397 impedes reextension of γ axons during metamorphosis of the MBs. A, Schematic drawing of γ neuron remodeling. B-I, GAL4-201Y-labeled wild-type (WT; B, D, F, H) and l(2L)MB397 mutant (C, E, G, I) Nb clones were generated in NHL and examined at the wandering larval stage (B, C), 18 h APF (D, E), 48 h APF (F, G), and in adults (H, I). Larval MB neurons have two perpendicular oriented lobes (large arrowheads in B and C) and two short bundles of terminal branches (small arrowheads in B and C). l(2L)MB397 mutant clones exhibit subtle deformation of lobe termini at the wandering larval (wl) stage (arrows in C compared with arrows in B), normal pruning of larval lobes by 18 h APF (arrows in E compared with arrows in D) but incomplete axon regeneration, even after eclosion (arrows in G and I compared with arrows in F and H). Note that late-pupal-born α/β axons have no obvious terminal morphogenetic defects (arrowheads in I compared with arrowheads in H). Genotype: B, D, F, H, FRTG13,UAS-mCD8-GFP,GAL4-201Y/FRTG13,hs-FLP,tubP-GAL80; C, E, G, I, hs-FLP/X;l(2L)MB397,UAS-mCD8-GFP,FRT40A,GAL4-201Y/tubP-GAL80,FRT40A.
Short axons of l(2L)MB397 mutant γ neurons result from extension defect
Formation of the γ lobe involves pruning of larval-specific axonal branches, followed by reextension of the pruned γ axons (Fig. 2A) (Lee et al., 1999). Thus, it is possible that mutant γ axons stop short because of reextension problems. Alternatively, short γ processes might be derived from partially pruned larval-specific axonal branches or result from degeneration of fully reextended axons. To distinguish these possibilities, we examined the morphologies of mutant γ neurons at different developmental stages.
GAL4-201Y-marked Nb clones were generated in NHL and analyzed at different developmental stages. GAL4-201Y selectively labels γ neurons and late-pupal-born α/β neurons (Yang et al., 1995), permitting detailed examination of γ neurons through metamorphosis. In wandering larvae, l(2L)MB397 mutant clones acquire larval MB-specific morphological features, including two perpendicularly oriented lobes (Fig. 2 B, C, large arrowheads), as well as two short bundles of terminal branches (Fig. 2 B, C, small arrowheads). No dramatic morphogenetic defects are observed in l(2L)MB397 mutant clones at this stage, except subtle deformation of the termini of the medial axon bundles (Fig. 2, compare arrows in B, C; similar to the terminal morphogenetic defects of adult MBs). By 18 h after puparium formation (APF), most larval-specific axonal branches are pruned in mutant clones and look morphologically indistinguishable from their wild-type controls (Fig. 2 D, E, arrows). In contrast to normal pruning, we observe severe regeneration problems in l(2L)MB397 mutant clones. For instance, by 48 h APF, wild-type clones have reextended axons to the midline (Fig. 2 F, arrow), but regrowth of the majority of mutant axons is incomplete and appears unsynchronized (Fig. 2G, arrow). In addition, no age-dependent morphological changes could be detected in l(2L)MB397 mutant clones after eclosion (Fig. 2, compare arrows in H, I; and data not shown). These results suggest that l(2L)MB397 mutant γ appear truncated because of reextension problems rather than partial pruning or progressive degeneration.
l(2L)MB397 mutant axons often stall around the places at which arborization normally occurs
Because l(2L)MB397 mutation selectively affects γ and β′ lobes, it is unlikely that l(2L)MB397 generally blocks axon extension. Furthermore, it is also unlikely that l(2L)MB397 specifically impedes reextension of axons in that β′ lobes do not undergo pruning and reextension as γ lobes. Given that most γ and β′ processes undergo repeated arborization and few arbors exist in the other three lobes, one possibility is that failure in axon arborization underlies l(2L)MB397 terminal morphogenetic defects of mutant axons. To examine this possibility, we collected isolated single-cell clones of γ neurons, generated in NHL, for detailed phenotypic analysis. We observe that mutant γ axons have few arbors. For quantitative analysis, we arbitrarily defined major arbors as branches longer than 15 μm (Fig. 3F-H, arrows). Because fewer axon arbors in the mutant γ neurons could result from extension defects instead of arborization problems, our initial analysis was focused on the mutant γ neurons that had extended axons to the lobe termini. We found that 57% (12 of 21) of the fully extended mutant γ neurons have no major axon arbor (Fig. 3A). In contrast, only 15% (15 of 100) of wild-type γ neurons have axons without major arbors (Fig. 3E). These phenomena suggest that arborization is suppressed in mutant γ axons. Interestingly, when we analyzed all single-cell clones (including those whose axons did not reach lobe termini), similar percentages of γ neurons (14% of mutant clones versus 15% of wild-type clones) acquired fully extended “arbor-free” axons, regardless of their genotypes. Thus, l(2L)MB397 mutation might cause axons to stall during the process of branching but have no effect on the axons that do not form arbors. In support of this notion, mutant GAL4-201Y-positive late-pupal-born α/β axons, which do not arborize (S. Zhu and T. Lee, unpublished data), have no obvious terminal morphogenetic defects (Fig. 2, compare arrowheads in H, I).
Figure 3.

l(2L)MB397 mutant γ axons have few arbors and stall randomly within γ lobes. Single-cell clones of l(2L)MB397 mutant (A-D) and wild-type (WT; E-H) γ neurons, generated in NHL, were examined in adults. Mutant γ axons can extend to the tip of γ lobe (A) or stall at any position with in the γ lobes (arrows in B-D). Few arbors can be found even in fully extended mutant γ processes (A). In contrast, only a small part of wild-type γ axons have no major arbors (E). The majority of wild-type γ axons often have multiple major arbors that are randomly distributed along the γ lobes (arrowheads in F-H). Genotype: A-D, hs-FLP/X;l(2L)MB397,UAS-mCD8-GFP,FRT40A/tubP-GAL80,FRT40A;GAL4-OK107/+; E-H, hs-FLP/X;UAS-mCD8-GFP,FRT40A/tubP-GAL80,FRT40A;GAL4-OK107/+.
To examine further whether mutant axons stall during the process of branching, we tried to determine where axon arborization normally occurs and where mutant axons have stalled in single-cell clones of early-larval-born γ neurons, as well as α′/β′ neurons. We found that mutant γ processes could terminate within any part of the γ lobes (Fig. 3B-D) (see Fig. 5). Analogously, wild-type γ axons form arbors at stochastic positions (Fig. 3F-H) (see Fig. 5). However, such complex and highly variable axon arborization patterns make it difficult to ascribe l(2L)MB397 the terminal morphogenetic defects of axons to extension or arborization problems. Unlike γ neurons, most α′/β′ neurons exhibit invariant patterns of axon arborization. Typical β′ processes acquire two widely separate clusters of arbors, one within the proximal one-third segment of the β′ lobe and another within the distal one-third segment (Figs. 4A, 5), whereas their sister neurites, the α′ processes, rarely branch (Fig. 4A). To determine whether axons tend to stall at the positions at which arborization normally occurs in mutant α′/β′ neurons, we collected 100 isolated single-cell clones of α′/β′ neurons after brief induction of mitotic recombination in l(2L)MB397 heterozygous mid-third-instar larvae and analyzed their phenotypes. First, only two α′ processes stop short, consistent with the subtle phenotypes in the α′ lobes of mutant Nb clones (data not shown). Second, truncation occurs in one-quarter of mutant β′ processes. Low penetrance of phenotypes in single-cell clones of mutant α′/β′ neurons, which undergo morphogenesis shortly after mitotic recombination, is probably attributable to perdurance of preexisting wild-type molecules. However, in the mutant γ neurons, wild-type molecules may not exist any more when reextension of their pruned axons occurs several days after loss of heterozygosity. Third, in great contrast with mutant γ axons that often stall randomly within γ lobes, all truncated β′ axons stall within either the proximal one-third segments (Figs. 4B, 5) or the distal one-third segments (Figs. 4C, 5) of the β′ lobes, close to the positions at which two clusters of arbors form in wild-type β′ processes. Together, we suggest that l(2L)MB397 mutant axons often stall around branching points, and it is possible that mutant neurites become truncated because of defects in arborization rather than extension.
Figure 5.

Comparison of ending points of l(2L)MB397 mutant axons and arborization points of wild-type axons. The distance from the ending points of l(2L)MB397 mutant axons or the branch points of wild-type axons to the midline were measured and compared. In the wild-type γ axons, each dot represents the distance from the first branch point of a single neuron to the midline of the brain. In the l(2L)MB397 mutant γ axons, each dot represents the distance from the ending points of axons to the midline. Only truncated l(2L)MB397 mutant γ axons without major axon arbors were examined. In the case of wild-type β′ axons, the distance from the proximal arborization point (the right cluster) or the first arborization point of the distal cluster of arbors (the left cluster) to the midline were measured separately. Note that ending points of l(2L)MB397 mutant β′ axons fall into two clusters, close to the arborization point of wild-type β′ axons. Numbers in parentheses represent the total number of neurons analyzed.
Figure 4.

l(2L)MB397 mutant β′ axons often terminate around the places at which arborization normally occurs. Single-cell clones of wild-type (WT; A) and l(2L)MB397 mutant (B, C) α′/β′ neurons, generated at the mid-third-instar larval stage, were examined in adults. Wild-type β′ axons form arbors within the proximal one-third segment (arrowhead in A) and the distal one-third segment (arrow in A). Mutant β′ axons stall either near the proximal position of the arborization (arrowhead in B) or close to the distal locus of the arborization (arrow in C). Genotype: A, hs-FLP/X;UAS-mCD8-GFP,FRT40A/tubP-GAL80,FRT40A;GAL4-OK107/+; B, C, hs-FLP/X;l(2L)MB397,UAS-mCD8-GFP,FRT40A/tubP-GAL80,FRT40A;GAL4-OK107/+.
l(2L)MB397 MB phenotypes are caused by a splicing mutation in Drosophila Cul3
To determine whether a lethal mutation in 2L is responsible for l(2L)MB397 MB phenotypes, we first identified and mapped an l(2L)MB397 lethal mutation to the 35B10-35D01 cytogenomic region based on complementation experiments with the 2L deficiency kit (see Materials and Methods). Additional complementation tests using additional deficiency lines mapped the lethal mutation to either 35B10-35C02 or 35C05-35D01. By crossing with existing lethal mutations within these two regions, we found that l(2L)MB397 fails to complement with two previously identified Drosophila Cul3 [Cul3, also named guftagu (gft)] loss-of-function alleles (Cul32 and Cul306430) (Fig. 6A). Cul3 belongs to the family of cullin proteins, which function as E3 ubiquitin ligases by forming complexes with other proteins (Ou et al., 2002; Chiba and Tanaka, 2004). In the fly genome, there are six different cullin genes: Cul1 (also called lin19), Cul2, Cul3, Cul4, Cul5, and CG11261, which is close to Cul1 and Cul2 (Ou et al., 2002). The Cul3 gene consists of 11 exons and encodes a protein of 773 amino acids. Sequence analysis demonstrated that the l(2L)MB397 chromosome has a point mutation at the splice acceptor site of the intron between exon 6 and exon 7 of Cul3 (Fig. 6B). This splicing defect may lead to truncation and/or open reading frame shift of Cul3 after amino acid 516, which may eliminate several C-terminal conserved domains, including Roc1 binding site, Nedd8 modification site (also see below), and Cullin motif (Wada et al., 1999; Mistry et al., 2004). To confirm that all l(2L)MB397 MB phenotypes resulted from loss of Cul3 function, we created clones of MB neurons that are homozygous for Cul32 and observed similar axon terminal morphogenetic defects as in l(2L)MB397 mutant clones (Fig. 6C). Moreover, l(2L)MB397 MB phenotypes were fully rescued in the presence of an autonomously expressed wild-type Cul3 cDNA (Fig. 6D). Thus, l(2L)MB397 is a novel Cul3 mutation (Cul38).
Figure 6.
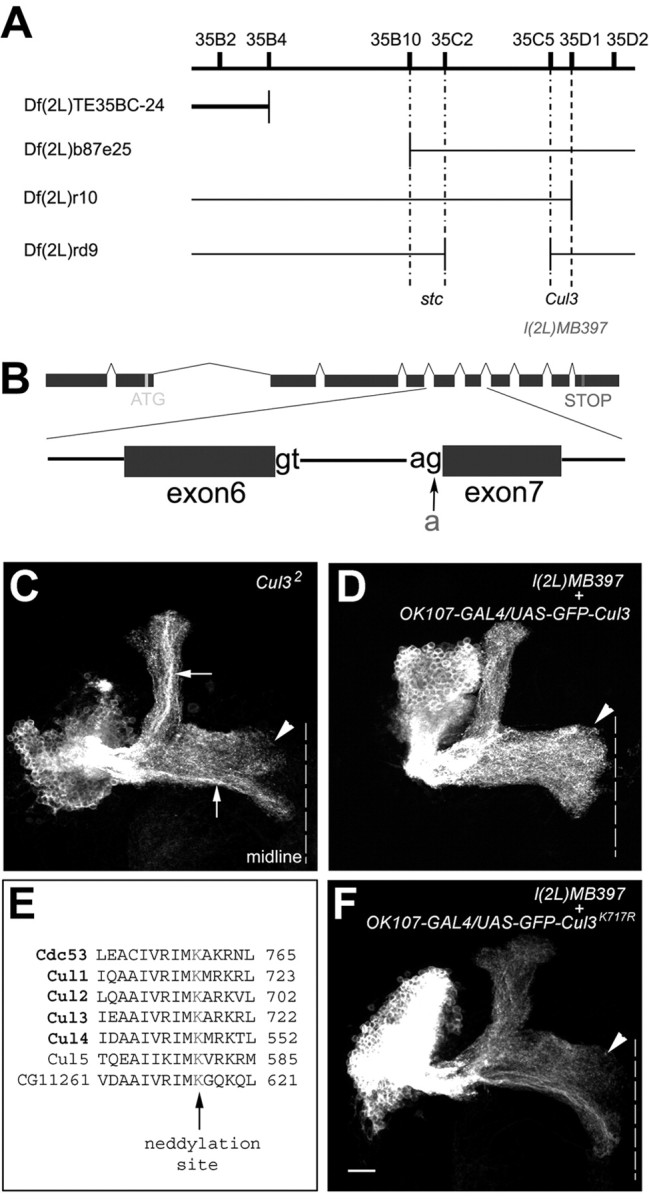
l(2L)MB397 carries a splicing mutation in Cul3, and the conserved neddylation site is essential for Cul3 activity. A, l(2L)MB397 fails to complement with the deficiency line whose deficiency region is indicated by the gap in the thick line but complemented with the deficiency lines whose deficiency regions are indicated by gaps in thin lines. Additional complementation tests assign l(2L)MB397 to the Cul3 complementation group. B, l(2L)MB397 carries a g-to-a mutation at the acceptor site of the intron between exon 6 and exon 7 of Cul3. C, The Cul32 mutant Nb clone shows terminal morphogenetic defects in the γ and β′ lobes (arrowhead). Also note the dense axon fascicles within the α and β lobes (arrows). D, All of the medial lobes are fully extended (arrowhead) in the l(2L)MB397 mutant Nb clone in which GFP-tagged wild-type Cul3 is autonomously expressed. E, Partial sequence alignment among yeast Cul1 (Cdc53) and six Drosophila cullin proteins. Consensus neddylation sites are highlighted in red. F, The terminal defects of the γ and β′ lobes (arrowhead) remain in the l(2L)MB397 mutant Nb clones that autonomously express GFP-tagged Cul3K717R. Genotype: C, hs-FLP,UAS-mCD8-GFP/X;Cul32,FRT40A/tubP-GAL80,FRT40A;GAL4-OK107/+; D, hs-FLP/UAS-GFP-Cul3;l(2L)MB397,UAS-mCD8-GFP,FRT40A/tubP-GAL80,FRT40A;GAL4-OK107/+; F, hs-FLP/X;l(2L)MB397,UAS-mCD8-GFP,FRT40A/tubP-GAL80,FRT40A;UAS-GFP-Cul3K717R/+;GAL4-OK107/+.
Neddylation may be required for Cul3 function
It has been shown that Cul1-based ubiquitin ligase activity requires the covalent attachment of Nedd8, a small ubiquitin-like protein, to a lysine residue in the C-terminal region of Cul1 (Amir et al., 2002) (for review, see Chiba and Tanaka, 2004). Biochemical studies have also demonstrated that all known members of human cullin family proteins can be conjugated to Nedd8 in vitro (Hori et al., 1999). Sequence alignment reveals that Cul3, like other cullin proteins, has a putative Nedd8 conjugation site (Lys-717) (Fig. 6E) (Wada et al., 1999). To examine possible involvement of Nedd8 covalent modification in regulating Cul3 function, we investigated whether mutation of Lys-717 to arginine in Cul3 eliminates its biological activity. Analogous mutations have inactivated other cullin proteins (Wada et al., 1999; Amir et al., 2002). We repeated earlier rescue experiments using a mutated Cul3 cDNA, encoding Cul3K717R. Multiple independently inserted transgenic lines were checked, and no mutant Cul3K717R transgenic line could rescue Cul38 MB phenotypes (Fig. 6F). These results indicate that normal Cul3 function requires an intact Nedd8 conjugation site. Elimination of neddylation site could at least partially explain the loss of function of Cul38 allele.
To directly examine the requirement of Nedd8 for MB morphogenesis, we further investigated whether and how loss of Nedd8 affects MB development. MARCM clones of MB neurons that are homozygous for a Nedd8 loss-of-function allele (Nedd8AN015) (Ou et al., 2002) were generated in NHL and examined at the adult stage. Knocking out Nedd8 may disable various cullin proteins with distinct functions (Amir et al., 2002). Consistent with this notion, Nedd8 mutant Nb clones exhibit more complex phenotypes than Cul3 mutant clones. For instance, Nedd8 mutant MB Nb clones consist of only small numbers of γ-type neurons (Fig. 7C), indicating severe defects in MB neurogenesis. Such failure in MB neurogenesis might result from loss of Cul1 ubiquitin ligase activity that leads to cell cycle arrest at the G1/S transition attributable to abnormal accumulation of Cul1 ligase substrates, such as p27Kip1 (Tsvetkov et al., 1999). Nevertheless, axonal extension defects are observed in Nedd8 mutant clones as in Cul3 mutant clones (Fig. 7C). γ neurons homozygous for Nedd8AN015 fail to extend their axons to the midline in both Nb clones and single-cell/two-cell clones (Fig. 7, compare A, B with C, D). Therefore, with respect to γ neurons, Nedd8 mutant clones qualitatively phenocopy Cul3 mutant clones, further suggesting that proper function of Cul3 requires Nedd8 modification.
Figure 7.

Nedd8 mutant MB clones show axon terminal morphogenetic defects. Wild-type (WT; A, B) and Nedd8AN015 mutant (C, D) clones of MB neurons, generated in NHL, were examined in adults. Only a small number of γ neurons exist in the mutant Nb clone (C), and most mutant γ axons stall before reaching the midline (e.g., arrows in C and D). Genotype: A, B, hs-FLP/X;UAS-mCD8-GFP,FRT40A/tubP-GAL80,FRT40A;GAL4-OK107/+; C, D, hs-FLP,UAS-mCD8-GFP/X;Nedd8AN015,FRT40A/tubP-GAL80, FRT40A;GAL4-OK107/+.
Mutation of Cul3 blocks reelaboration of γ dendrites
Analogous mechanisms may underlie arborization of axons and elaboration of dendrites (for review, see Keith and Wilson, 2001; Jan and Jan, 2003). To determine the role(s) of Cul3 in MB dendritic morphogenesis, we examined and tried to quantify patterns of dendritic elaboration in isolated single-cell clones of Cul3 mutant γ neurons. Like γ axons, dendrites of γ neurons also undergo remodeling during metamorphosis (Lee et al., 1999, 2000; Zheng et al., 2003), and we recognized stage-specific dendritic elaboration patterns in larval versus adult γ neurons (Zhu et al., 2003). First, consistent with normal pruning of larval neurites in Cul3 mutant Nb clones, most adult mutant neurons possess three to five primary dendritic processes, comparable with what γ neurons normally have after remodeling (Fig. 8, compare A, B with C, D) (Zhu et al., 2003). Second, in contrast to no change in the numbers of primary dendrites, the averaged total dendritic length of mutant γ neurons is only 32.9 ± 15.9 μm (n = 25) (Fig. 8C,D), much shorter than 61.3 ± 24.9 μm (n = 25), the normal dendritic length per γ neuron (Fig. 8A, B). We noticed that quite a few (42.5%; n = 33) mutant dendrites are extremely short and hardly separable from their primary neurites (Fig. 8D). In addition, terminal elaboration looks abnormal in mutant dendrites. Unlike wild-type dendrites that mostly terminate with typical claw-like structures (Fig. 8A, B, arrows), Cul3 mutant γ dendrites often stall with dense irregular endings (Fig. 8D, arrows). These results demonstrate that elaboration of adult-specific dendritic branches is also impaired in Cul38 mutant γ neurons.
Figure 8.

Poor elaboration of dendrites in Cul3 mutant γ neurons. Close-up views of the dendritic regions of MARCM-labeled adult γ neurons. Wild-type γ neurons (WT; A, B) acquired longer dendritic branches than Cul3 mutant γ neurons (C, D). Arrows in A and B point to the typical claw-like structures at the ends of wild-type dendritic processes. Note that mutant dendrites often stall with dense irregular endings (arrows in D). Genotype: A, B, hs-FLP/X;UAS-mCD8-GFP,FRT40A/tubP-GAL80,FRT40A;GAL4-OK107/+; C, D, hs-FLP/X;Cul38,UAS-mCD8-GFP,FRT40A/tubP-GAL80, FRT40A;GAL4-OK107/+.
Discussion
Like phosphorylation, ubiquitination of target proteins is a potent posttranslational modification pathway that actively controls stability, trafficking, and activity of proteins involved in various cellular processes, such as cell cycle progression, apoptosis, and signal transduction (for review, see Pickart, 2001; Glickman and Ciechanover, 2002). More recently, the ubiquitin-proteasome system has been implicated in governing various aspects of neural development, including synaptogenesis, axon guidance, and axon pruning (Wan et al., 2000; DiAntonio et al., 2001; Keleman et al., 2002; Myat et al., 2002; Watts et al., 2003; Konishi et al., 2004; Liao et al., 2004). Here we demonstrate that Cul3 is a positive regulator for axon arborization and dendritic development in MB neurons. Distinct MB axons arborize differentially. Interestingly, mutation of Cul3 selectively blocked terminal morphogenesis of the axons that arborize extensively. Axons that rarely arborize were minimally affected, and mutant axons often stalled around the positions at which arborization normally occurs. Furthermore, Cul3 mutant β lobes, which only show subtle phenotypes at the tips, basically project along the right path and do not curl around and project away from the midline. These observations suggest involvement of Cul3 in proper arborization rather than general extension or midline attraction/repulsion of axons. Requirement of Cul3 for reelaboration of γ dendrites implies that common mechanisms may be used to govern branching of both axons and dendrites.
Cullin family proteins function as E3 ubiquitin ligases, which target specific substrates for degradation (Chiba and Tanaka, 2004). Cul3 promotes axon arborization likely via increasing turnover of its target protein(s). So far, there is one documented Drosophila Cul3 substrate, the full-length Cubitus interruptus (Ci155) (Ou et al., 2002; Mistry et al., 2004). Cul3-dependent degradation of Ci155 has been shown to modulate Hedgehog signaling in the posterior cells of Drosophila eye discs (Ou et al., 2002; Mistry et al., 2004). However, overexpression of Ci155 through development did not affect either wild-type or Cul3 mutant MB neurons (Zhu and Lee, unpublished results), suggesting that Ci155 is not involved in Cul3-governed neurite terminal morphogenesis. In Caenorhabditis elegans, Cul3 mediates meiosis-to-mitosis transition by targeting meiotic-1 (MEI-1)/Katanin for degradation (Pintard et al., 2003). Interestingly, Katanin60 (Kat60), the smaller subunit of the Katanin heterodimeric ATPase, possesses microtubule severing activities and exists in neuronal processes, as well as the centrosomes (Ahmad et al., 1999). Further inspired by the phenomena that levels of Kat60 are critical for axonal outgrowth (Karabay et al., 2004), we examined the candidacy of Kat60 for the physiological substrate of Cul3 in MB neurons. Again, we observe that GAL4-OK107-driven overexpression of Kat60 exert no detectable effect on either wild-type or Cul3 mutant MB morphogenesis (our unpublished results). In addition, Nicola et al. (2003) have reported that crossing a Kat60 UY element line with 7B P{GAL4} causes the dorsal lobes of Drosophila MBs to stop short (Nicolai et al., 2003), not mimicking Cul3 mutant phenotypes at all. Therefore, in the MBs, Kat60 is unlikely to be the substrate of Cul3, either.
Levels of cell adhesion molecules (CAMs) may help regulate defasciculation as well as arborization of axons. As reported by Rutishauser's laboratory, blocking functions of L1 and neural CAM (N-CAM) leads to abnormal defasciculation and excessive arborization of neurites in developing chicken embryos (Landmesser et al., 1988). Conversely, aberrant accumulation of cell adhesion molecules, such as Fasciclin II, the Drosophila homolog of N-CAM, promotes fasciculation of axons and impedes neurite arborization (Lin and Goodman, 1994). Interestingly, Cul3 mutant Nb clones occasionally contain dense axon fascicles within normally homogeneous MB lobes (Fig. 6C, arrows), suggesting involvement of Cul3 in fasciculation/defasciculation of axons as well. It is possible that Cul3 mediates neurite arborization and prevents abnormal fasciculation of axons via a common mechanism, perhaps involving regulation of dynamic changes in the levels of cell adhesion molecules.
Given the involvement of cullins in more and more biological processes, it is no surprise that activities of cullin proteins need to be dynamically regulated. One common mechanism for regulation of cullin family proteins involves Nedd8 modification, a cascade of enzymatic reactions similar to ubiquitination (Chiba and Tanaka, 2004). Previous studies have suggested that Nedd8 regulates Cul1 ligase activity by facilitating the dissociation of p120 (CAND1) from Cul1 and enhancing recruitment of E2 ubiquitin to the Cul1-based ubiquitin ligase complex termed SCF (Skp1-Cul1/Cdc53-F-box) (Kawakami et al., 2001; Liu et al., 2002). In C. elegans, neddylation is required in Cul3 to target MEI-1 for degradation at the meiosis-to-mitosis transition (Pintard et al., 2003). However, in the developing Drosophila eye discs, Cul3-mediated degradation of Ci155 only partially requires Nedd8 modification (Ou et al., 2002), contrasting with the essential role of Nedd8 in Cul3-governed MB neurite terminal morphogenesis. Nedd8 was originally isolated from neural precursor cells, and its expression is developmentally downregulated (Kumar et al., 1993). Together with the requirement of Nedd8 for multiple aspects of MB development, neddylation of cullin family proteins is likely used repeatedly to govern diverse neuronal developmental processes. Studying how neddylation of cullins is regulated as well as identifying the physiological substrates of cullins promises to help further elucidate the molecular mechanisms underlying development of the brain.
Footnotes
This work was supported by National Institutes of Health Grant NS42049 (T.L.). T.L. is a Klingenstein fellow and a Sloan fellow. We thank C. T. Chien for Nedd8AN015 flies, the Bloomington Stock Center for other mutant flies, and S. Siechen and members of the Lee laboratory, especially C. T. Zugates, for comments on this manuscript.
Correspondence should be addressed to Tzumin Lee, Department of Cell and Structural Biology, University of Illinois, Urbana, IL 61801. E-mail: tzumin@life.uiuc.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254189-09$15.00/0
References
- Acebes A, Ferrus A (2000) Cellular and molecular features of axon collaterals and dendrites. Trends Neurosci 23: 557-565. [DOI] [PubMed] [Google Scholar]
- Ahmad FJ, Yu W, McNally FJ, Baas PW (1999) An essential role for katanin in severing microtubules in the neuron. J Cell Biol 145: 305-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Iwai K, Ciechanover A (2002) The NEDD8 pathway is essential for SCF(beta-TrCP)-mediated ubiquitination and processing of the NF-kappa B precursor p105. J Biol Chem 277: 23253-23259. [DOI] [PubMed] [Google Scholar]
- Arcaro KF, Lnenicka GA (1995) Intrinsic differences in axonal growth from crayfish fast and slow motoneurons. Dev Biol 168: 272-283. [DOI] [PubMed] [Google Scholar]
- Caroni P (1997) Intrinsic neuronal determinants that promote axonal sprouting and elongation. BioEssays 19: 767-775. [DOI] [PubMed] [Google Scholar]
- Chiba T, Tanaka K (2004) Cullin-based ubiquitin ligase and its control by NEDD8-conjugating system. Curr Protein Pept Sci 5: 177-184. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S, Fraser SE (1995) Effects of brain-derived neurotrophic factor on optic axon branching and remodelling in vivo. Nature 378: 192-196. [DOI] [PubMed] [Google Scholar]
- Connolly JB, Roberts IJ, Armstrong JD, Kaiser K, Forte M, Tully T, O'Kane CJ (1996) Associative learning disrupted by impaired Gs signaling in Drosophila mushroom bodies. Science 274: 2104-2107. [DOI] [PubMed] [Google Scholar]
- Davis RL (2001) Mushroom bodies, Ca2+ oscillations, and the memory gene amnesiac. Neuron 30: 653-656. [DOI] [PubMed] [Google Scholar]
- Dent EW, Barnes AM, Tang F, Kalil K (2004) Netrin-1 and semaphorin 3A promote or inhibit cortical axon branching, respectively, by reorganization of the cytoskeleton. J Neurosci 24: 3002-3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAntonio A, Haghighi AP, Portman SL, Lee JD, Amaranto AM, Goodman CS (2001) Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature 412: 449-452. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A (2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82: 373-428. [DOI] [PubMed] [Google Scholar]
- Heisenberg M (2003) Mushroom body memoir: from maps to models. Nat Rev Neurosci 4: 266-275. [DOI] [PubMed] [Google Scholar]
- Hori T, Osaka F, Chiba T, Miyamoto C, Okabayashi K, Shimbara N, Kato S, Tanaka K (1999) Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene 18: 6829-6834. [DOI] [PubMed] [Google Scholar]
- Ito K, Awano W, Suzuki K, Hiromi Y, Yamamoto D (1997) The Drosophila mushroom body is a quadruple structure of clonal units each of which contains a virtually identical set of neurones and glial cells. Development 124: 761-771. [DOI] [PubMed] [Google Scholar]
- Jan YN, Jan LY (2003) The control of dendrite development. Neuron 40: 229-242. [DOI] [PubMed] [Google Scholar]
- Jefferis GS, Marin EC, Stocker RF, Luo L (2001) Target neuron prespecification in the olfactory map of Drosophila Nature 414: 204-208. [DOI] [PubMed] [Google Scholar]
- Karabay A, Yu W, Solowska JM, Baird DH, Baas PW (2004) Axonal growth is sensitive to the levels of katanin, a protein that severs microtubules. J Neurosci 24: 5778-5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami T, Chiba T, Suzuki T, Iwai K, Yamanaka K, Minato N, Suzuki H, Shimbara N, Hidaka Y, Osaka F, Omata M, Tanaka K (2001) NEDD8 recruits E2-ubiquitin to SCF E3 ligase. EMBO J 20: 4003-4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith CH, Wilson MT (2001) Factors controlling axonal and dendritic arbors. Int Rev Cytol 205: 77-147. [DOI] [PubMed] [Google Scholar]
- Keleman K, Rajagopalan S, Cleppien D, Teis D, Paiha K, Huber LA, Technau GM, Dickson BJ (2002) Comm sorts robo to control axon guidance at the Drosophila midline. Cell 110: 415-427. [DOI] [PubMed] [Google Scholar]
- Kiger AA, White-Cooper H, Fuller MT (2000) Somatic support cells restrict germline stem cell self-renewal and promote differentiation. Nature 407: 750-754. [DOI] [PubMed] [Google Scholar]
- Koch C, Segev I (2000) The role of single neurons in information processing. Nat Neurosci [Suppl] 3: 1171-1177. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Stegmuller J, Matsuda T, Bonni S, Bonni A (2004) Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science 303: 1026-1030. [DOI] [PubMed] [Google Scholar]
- Krylova O, Herreros J, Cleverley KE, Ehler E, Henriquez JP, Hughes SM, Salinas PC (2002) WNT-3, expressed by motoneurons, regulates terminal arborization of neurotrophin-3-responsive spinal sensory neurons. Neuron 35: 1043-1056. [DOI] [PubMed] [Google Scholar]
- Kumar S, Yoshida Y, Noda M (1993) Cloning of a cDNA which encodes a novel ubiquitin-like protein. Biochem Biophys Res Commun 195: 393-399. [DOI] [PubMed] [Google Scholar]
- Landmesser L, Dahm L, Schultz K, Rutishauser U (1988) Distinct roles for adhesion molecules during innervation of embryonic chick muscle. Dev Biol 130: 645-670. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L (1999) Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22: 451-461. [DOI] [PubMed] [Google Scholar]
- Lee T, Lee A, Luo L (1999) Development of the Drosophila mushroom bodies: sequential generation of three distinct types of neurons from a neuroblast. Development 126: 4065-4076. [DOI] [PubMed] [Google Scholar]
- Lee T, Marticke S, Sung C, Robinow S, Luo L (2000) Cell-autonomous requirement of the USP/EcR-B ecdysone receptor for mushroom body neuronal remodeling in Drosophila Neuron 28: 807-818. [DOI] [PubMed] [Google Scholar]
- Lewis EB, Bacher F (1968) Method of feeding ethylmethane sulfonate (EMS) to Drosophila males. Dros Info Ser 43: 193. [Google Scholar]
- Liao EH, Hung W, Abrams B, Zhen M (2004) An SCF-like ubiquitin ligase complex that controls presynaptic differentiation. Nature 430: 345-350. [DOI] [PubMed] [Google Scholar]
- Lin DM, Goodman CS (1994) Ectopic and increased expression of Fasciclin II alters motoneuron growth cone guidance. Neuron 13: 507-523. [DOI] [PubMed] [Google Scholar]
- Liu J, Furukawa M, Matsumoto T, Xiong Y (2002) NEDD8 modification of CUL1 dissociates p120(CAND1), an inhibitor of CUL1-SKP1 binding and SCF ligases. Mol Cell 10: 1511-1518. [DOI] [PubMed] [Google Scholar]
- Mistry H, Wilson BA, Roberts IJ, O'Kane CJ, Skeath JB (2004) Cullin-3 regulates pattern formation, external sensory organ development and cell survival during Drosophila development. Mech Dev 121: 1495-1507. [DOI] [PubMed] [Google Scholar]
- Myat A, Henry P, McCabe V, Flintoft L, Rotin D, Tear G (2002) Drosophila Nedd4, a ubiquitin ligase, is recruited by Commissureless to control cell surface levels of the roundabout receptor. Neuron 35: 447-459. [DOI] [PubMed] [Google Scholar]
- Newsome TP, Asling B, Dickson BJ (2000) Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development 127: 851-860. [DOI] [PubMed] [Google Scholar]
- Nicolai M, Lasbleiz C, Dura JM (2003) Gain-of-function screen identifies a role of the Src64 oncogene in Drosophila mushroom body development. J Neurobiol 57: 291-302. [DOI] [PubMed] [Google Scholar]
- Ou CY, Lin YF, Chen YJ, Chien CT (2002) Distinct protein degradation mechanisms mediated by Cul1 and Cul3 controlling Ci stability in Drosophila eye development. Genes Dev 16: 2403-2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM (2001) Mechanisms underlying ubiquitination. Annu Rev Biochem 70: 503-533. [DOI] [PubMed] [Google Scholar]
- Pintard L, Kurz T, Glaser S, Willis JH, Peter M, Bowerman B (2003) Neddylation and deneddylation of CUL-3 is required to target MEI-1/Katanin for degradation at the meiosis-to-mitosis transition in C. elegans Curr Biol 13: 911-921. [DOI] [PubMed] [Google Scholar]
- Reuter JE, Nardine TM, Penton A, Billuart P, Scott EK, Usui T, Uemura T, Luo L (2003) A mosaic genetic screen for genes necessary for Drosophila mushroom body neuronal morphogenesis. Development 130: 1203-1213. [DOI] [PubMed] [Google Scholar]
- Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H (1999) p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol 9: 661-664. [DOI] [PubMed] [Google Scholar]
- Wada H, Yeh ET, Kamitani T (1999) Identification of NEDD8-conjugation site in human cullin-2. Biochem Biophys Res Commun 257: 100-105. [DOI] [PubMed] [Google Scholar]
- Wan HI, DiAntonio A, Fetter RD, Bergstrom K, Strauss R, Goodman CS (2000) Highwire regulates synaptic growth in Drosophila Neuron 26: 313-329. [DOI] [PubMed] [Google Scholar]
- Wang J, Zugates CT, Liang IH, Lee C-HJ, Lee T (2002) Drosophila Dscam is required for divergent segregation of sister branches and suppresses ectopic bifurcation of axons. Neuron 33: 559-571. [DOI] [PubMed] [Google Scholar]
- Wang KH, Brose K, Arnott D, Kidd T, Goodman CS, Henzel W, Tessier-Lavigne M (1999) Biochemical purification of a mammalian slit protein as a positive regulator of sensory axon elongation and branching. Cell 96: 771-784. [DOI] [PubMed] [Google Scholar]
- Watts RJ, Hoopfer ED, Luo L (2003) Axon pruning during Drosophila metamorphosis: evidence for local degeneration and requirement of the ubiquitin-proteasome system. Neuron 38: 871-885. [DOI] [PubMed] [Google Scholar]
- Yang MY, Armstrong JD, Vilinsky I, Strausfeld NJ, Kaiser K (1995) Subdivision of the Drosophila mushroom bodies by enhancer-trap expression patterns. Neuron 15: 45-54. [DOI] [PubMed] [Google Scholar]
- Zheng X, Wang J, Haerry TE, Wu AY, Martin J, O'Connor MB, Lee CH, Lee T (2003) TGF-beta signaling activates steroid hormone receptor expression during neuronal remodeling in the Drosophila brain. Cell 112: 303-315. [DOI] [PubMed] [Google Scholar]
- Zhu S, Chiang AS, Lee T (2003) Development of the Drosophila mushroom bodies: elaboration, remodeling and spatial organization of dendrites in the calyx. Development 130: 2603-2610. [DOI] [PubMed] [Google Scholar]
- Zugates CT, Lee T (2004) Genetic mosaic analysis in the nervous system. Curr Opin Neurobiol 14: 647-653. [DOI] [PubMed] [Google Scholar]