

Abstract
The present study investigated whether thrombin, a potent microglial activator, can induce reactive oxygen species (ROS) generation through activation of microglial NADPH oxidase and if this may contribute to oxidative damage and consequent neurodegeneration. Seven days after intrahippocampal injection of thrombin, Nissl staining and immunohistochemistry using the neuronal-specific nuclear protein NeuN revealed a significant loss in hippocampal CA1 neurons. In parallel, thrombin-activated microglia, assessed by OX-42 and OX-6 immunohistochemistry, and ROS production, assessed by hydroethidine histochemistry, were observed in the hippocampal CA1 area in which degeneration of hippocampal neurons occurred. Reverse transcription-PCR at various time points after thrombin administration demonstrated an early and transient expression of inducible nitric oxide synthase (iNOS) and several proinflammatory cytokines. Western blot analysis and double-label immunohistochemistry showed an increase in the expression of and the localization of iNOS within microglia. Additional studies demonstrated that thrombin induced the upregulation of membrane (gp91phox) and cytosolic (p47phox and p67phox) components, translocation of cytosolic proteins (p47phox, p67phox, and Rac1) to the membrane, and p67phox expression of the NADPH oxidase in microglia in the hippocampus in vivo, indicating the activation of NADPH oxidase. The thrombin-induced oxidation of proteins and loss of hippocampal CA1 neurons were partially inhibited by an NADPH oxidase inhibitor and by an antioxidant. To our knowledge, the present study is the first to demonstrate that thrombin-induced neurotoxicity in the hippocampus in vivo is caused by microglial NADPH oxidase-mediated oxidative stress. This suggests that thrombin inhibition or enhancing antioxidants may be beneficial for the treatment of neurodegenerative diseases, such as Alzheimer's disease, that are associated with microglial-derived oxidative damage.
Keywords: thrombin, hippocampus, microglia, NADPH oxidase, oxidative stress, Alzheimer's disease
Introduction
Growing evidence suggests that microvascular damage is involved in neuropathogenesis of Alzheimer's disease (AD) (Grammas, 2000; Borroni et al., 2002). AD patients may have increased blood-brain barrier (BBB) permeability, resulting in increased serum protein accumulation within the brain extracellular space, suggesting that certain blood-derived factors are capable of inducing various pathophysiological responses (Wardlaw et al., 2003).
Among blood-derived factors, thrombin is elevated, whereas the activity of thrombin inhibitor, protease nexin I, is sharply reduced in AD brains (Choi et al., 1995; Berzin et al., 2000). A correlation between the incidence of head trauma and the occurrence of AD also has indicated that physical injury to the BBB may exacerbate the onset of the disease through thrombin activation and accumulation in the brain (Uryu et al., 2002). In addition, thrombin induces cell death in hippocampal neurons (Donovan et al., 1997; Striggow et al., 2000). Although plasma prothrombin is the primary source of increased thrombin levels in the brain, prothrombin mRNA transcripts and local production of thrombin and its receptors in response to injury have been reported in rat, mouse, and human brain, including the hippocampus (Weinstein et al., 1995; Beilin et al., 2001). Collectively, these observations indicate that thrombin may act as an endogenous neurotoxin, leading to the hippocampal neurodegeneration that occurs in neurodegenerative diseases such as AD. This hypothesis is supported by recent results showing that thrombin-induced neurotoxicity in rat hippocampus is associated with cognitive deficits (Mhatre et al., 2004).
Thrombin-induced neurotoxicity is mediated by microglial activation and consequent production of toxic and inflammatory mediators (Carreño-Müller et al., 2003; Choi et al., 2003a). A number of studies have demonstrated that microglial activation is found in the brains of AD patients (Benveniste et al., 2001) and in the hippocampus of animal models of AD caused by administration of β-amyloid (Stepanichev et al., 2003).
It has been demonstrated that activated microglia produce reactive oxygen species (ROS) such as superoxide (O2-) and O2--derived oxidants, which may induce or exacerbate neurotoxicity by causing oxidative stress to neurons (Parvathenani et al., 2003; Wu et al., 2003). These studies also showed that ROS production was attributable to activation of microglial NADPH oxidase. Regarding this, several studies have demonstrated that brains of AD patients show evidence of oxidative stress, including oxidative modifications to proteins (Hensley et al., 1995), lipids (Palmer and Burns, 1994), and DNA (Mecocci et al., 1993). Collectively, these observations suggest that NADPH oxidase-mediated oxidative stress, possibly originating from activated microglia, contributes to the neurodegeneration that occurs in AD (Zekry et al., 2003). This hypothesis is strongly supported by the finding that, in the brain of AD patients, microglial NADPH oxidase is activated, resulting in the formation of ROS (Shimohama et al., 2000). Also, β-amyloid, the major component of the senile plaques in AD, activates NADPH oxidase in cultured rat microglia, leading to production of ROS (Qin et al., 2002).
NADPH oxidase is composed of cytosolic subunits (gp40phox, p47phox, p67phox, and GTP-binding protein P21-Rac1) and membrane subunits (gp91phox and gp22phox) (Cross and Segal, 2004). When microglia are activated, the entire cytosolic complex is translocated to the membrane, in which it assembles with the membrane-associated proteins and becomes activated. This activated NADPH oxidase is capable of producing O2-, which is rapidly converted to O2--derived oxidants, including hydrogen peroxide (H2O2), hydroxyl radicals, and peroxynitrite (Patel et al., 2005), which eventually lead to neurodegeneration.
The present study examines whether microglial NADPH oxidase could be activated by thrombin in the rat hippocampus in vivo and whether NADPH oxidase-derived ROS participate in thrombin-induced degeneration of hippocampal neurons in vivo.
Materials and Methods
Stereotaxic surgery and drug injection. All experiments were performed in accordance with approved animal protocols and guidelines established by Ajou University. Female Sprague Dawley (SD) rats (260-280 g) were anesthetized by injection of chloral hydrate (360 mg/kg, i.p.) and positioned in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA). A midline sagittal incision was made in the scalp, and holes were drilled in the skull over the lateral ventricles and dorsal hippocampus using the following coordinates: 0.8 mm posterior to bregma and 1.5 mm lateral to the midline for intracerebroventricular injections; and 3.8 mm posterior to bregma and 2.0 mm lateral to the midline for intrahippocampal injections according to the atlas of Paxinos and Watson (1998). The hole of the tip was directed vertically down to 3.6 mm beneath the surface of the brain for the ventricles and to 2.5 mm for the hippocampus. All injections were made using a Hamilton syringe equipped with a 26S gauge beveled needle and attached to a syringe pump (KDScientific, New Hope, PA). Infusions were made at a rate of 0.2 μl/min for thrombin (20 U in 4 μl of sterile PBS; Sigma, St. Louis, MO) and 0.5 μl/min for diphenylene iodonium (DPI) (100 μm in 20 μl of sterile saline; Calbiochem, La Jolla, CA) and for vehicle [PBS or bovine serum albumin (BSA) (200 μg in 4 μl of sterile saline) as controls]. Because PBS had the same effects as BSA, the data of PBS were used as controls in the current study. Thrombin was essentially free from other known activated factors such as lipopolysaccharide (LPS), determined by LPS blocker plymyxin B (Lee et al., 2005), as well as from plasminogen and plasmin (information from the manufacturer).
Tissue preparation and immunohistochemistry. Animals were anesthetized with chloral hydrate (360 mg/kg, i.p.) at the indicated time points after injection and transcardially perfused with saline solution containing 0.5% sodium nitrate and heparin (10 U/ml), followed by fixation with 4% paraformaldehyde dissolved in 0.1 m phosphate buffer (PB). Brains were removed from the skull, postfixed overnight at 4°C in buffered 4% paraformaldehyde, and stored at 4°C in 30% sucrose solution until they sank. Brains were frozen sectioned using a sliding microtome into 40 μm coronal sections and collected in six separate series. Immunohistochemistry was performed using the avidin-biotin staining technique as described previously (Choi et al., 2003a,b). Briefly, free-floating serial sections were rinsed three times for 10 min in PBS and then pretreated for 5 min at room temperature (RT) in PBS containing 1% H2O2. Sections were then rinsed in PBS containing 0.3% Triton X-100 and 0.5% BSA and then preincubated for 1 h at RT in PBS containing 0.5% BSA. Next, the sections were incubated overnight with gentle shaking at RT with PBS containing 0.5% BSA and the following monoclonal primary antibodies: OX-42 (1:200; Serotec, Oxford, UK), which recognizes complement receptor 3; OX-6 (1:200; BD Biosciences, San Diego, CA), which recognizes major histocompatibility complex class II antigens and stains microglia; and the neuronal-specific nuclear protein NeuN (1:200; Serotec) as a general stain for neurons. Sections were then rinsed in PBS and incubated for 1 h at RT in 1:200 biotin-conjugated anti-mouse antibody in PBS containing 0.5% BSA. Sections were rinsed again and incubated for 1 h at RT in avidin-biotin complex solution (Vector Laboratories, Burlingame, CA). After rinsing three times in PBS, the signal was detected by incubating sections in 0.5 mg/ml 3,3′ diaminobenzidine in PB containing 0.003% H2O2. Sections were then rinsed in PBS, mounted on gelatin-coated slides, and viewed under a bright-field microscope (Olympus Optical, Tokyo, Japan). For Nissl staining, some of the hippocampus tissue was mounted on gelatin-coated slides, dried for 1 h at RT, stained with 0.5% cresyl violet (Sigma), dehydrated, coverslipped, and then analyzed under a bright-field microscope (Olympus Optical).
Double-immunofluorescence staining. For immunofluorescence staining, sections were processed as described previously (Choi et al., 2003a). Briefly, sections were mounted on gelatin-coated slides, dried for 1 h at RT, and washed twice in PBS. Slides were incubated for 30 min in PBS containing 0.2% Triton X-100. After blocking with 0.5% BSA, slides were incubated overnight at 4°C with antibodies selective for inducible nitric oxide (iNOS) (1:200; Upstate Biotechnology, Lake Placid, NY) and OX-42 (1:100; Serotec). After thorough rinsing in PBS, slides were covered for 1 h at RT with a mixture of FITC-conjugated anti-mouse IgG (1:100; Vector Laboratories) and Texas Red-conjugated anti-rabbit IgG (1:100; Vector Laboratories). Slides were also incubated overnight at 4°C with anti-p67phox antibody (1:200; BD Biosciences) and then exposed for 1 h at RT to fluorescein-conjugated lycopersicon esculentum (tomato) lectin (1:200; Vector Laboratories) and Texas Red-conjugated goat anti-mouse IgG (1:100; Vector Laboratories). Slides were then rinsed three times with PBS, coverslipped with Vectashield medium (Vector Laboratories), and analyzed using a confocal microscope (Olympus Optical).
Reverse transcription-PCR. Brain tissues from the ipsilateral hippocampus were dissected at the indicated time points after thrombin or PBS injection, and total RNA was extracted in a single step using RNAzol B (Tel-Test, Friendswood, TX) following the instructions of the manufacturer. Total RNA was reverse transcribed into cDNA using Superscript II reverse transcriptase (Invitrogen, Rockville, MD) and random primers (Promega, Madison, WI). The primer sequences used in this study were as follows: 5′-TGA TGT TCC CAT TAG ACA GC-3′ (forward) and 5′-GAG GTG CTG ATG TAC CAG TT-3′ (reverse) for interleukin-1β (IL-1β); 5′-AAA ATC TGC TCT GGT CTT CTG G-3′ (forward) and 5′-GGT TTG CCG AGT AGA CCT CA-3′ (reverse) for interleukin-6 (IL-6); 5′-GTA GCC CAC GTC GTA GCA AA-3′ (forward) and 5′-CCC TTC TCC AGC TGG GAG AC-3′ (reverse) for tumor necrosis factor-α (TNF-α); 5′-GCA GAA TGT GAC CAT CAT GG-3′ (forward) and 5′-ACA ACC TTG GTG TTG AAG GC-3′ (reverse) for iNOS; and 5′-TCCCTC AAG ATT GTC AGC AA-3′ (forward) and 5′-AGA TCC ACA ACG GAT ACA TT-3′ (reverse) for glyceraldehyde phosphate dehydrogenase. The PCR amplification consisted of 30 cycles of denaturation at 94°C for 30 s, annealing at 56°C for 30 s (for IL-1β, TNF-α, and iNOS) or 60°C for 30 s (for IL-6), and extension at 72°C for 90 s. PCR products were separated by electrophoresis on 1.5% agarose gels, after which the gels were stained with ethidium bromide and photographed. For semiquantitative analyses, the photographs were scanned using the Computer Imaging Device and accompanying software (Fujifilm, Tokyo, Japan).
Western blot analysis. Brain tissues from the ipsilateral hippocampus were dissected and homogenized in ice-cold lysis buffer containing the following (in mm): 20 Tris-HCl, pH 7.5, 1 EDTA, 5 MgCl2, 1 dithiothreitol, 0.1 phenylmethylsulfonyl fluoride, and protease inhibitor mixture (Sigma). The tissue homogenates was centrifuged at 4°C for 20 min at 14,000 × g, and the supernatant was transferred to a fresh tube. The extracts were frozen and kept at -80°C. For subcellular fractionation, protein extracts of both the cytosolic and membrane factions were prepared from the ipsilateral hippocampus at the indicated time points after thrombin injection. Tissues were gently homogenized using a glass homogenizer in ice-cold buffer consisting of the following (in mm): 20 HEPES, 250 sucrose, 10 KCl, 1.5 MgCl2, 2 EDTA, and protease inhibitor mixture (Sigma). Homogenates were centrifuged for 5 min at 500 × g at 4°C, and supernatants were collected and centrifuged for 20 min at 13,000 × g at 4°C. The pellets were further centrifuged for 1 h at 100,000 × g at 4°C, and the resulting supernatants and pellets were designated as the cytosolic and membrane fractions, respectively. Equal amounts of protein (50 μg) were mixed with loading buffer (0.125 m Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, 10% mercaptoethanol, and 0.002% bromophenol blue), boiled for 5 min, and separated by SDS-PAGE. After electrophoresis, proteins were transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA) using an electrophoretic transfer system (Bio-Rad, Hercules, CA). The membranes were washed with Tris-buffered saline solution containing 2.5 mm EDTA (TNE) and then blocked for 1 h in TNE containing 5% skim milk. The membranes were then incubated overnight at 4°C with one of the following the specific primary antibodies: rabbit anti-iNOS (1:1000; Upstate Biotechnology), mouse anti-Rac1 (1:5000; Upstate Biotechnology), mouse anti-p67phox (1:500; BD Biosciences), rabbit anti-p47phox (1:200; Santa Cruz Biotechnology, Santa Cruz, CA), and mouse anti-gp91phox (1:500; BD Biosciences). After washing, the membranes were incubated for 1 h at RT with secondary antibodies (1:2000; Amersham Biosciences, Arlington Heights, IL) and washed again. Finally, the blots were developed with enhanced chemiluminescence detection reagents (Amersham Biosciences). The blots were reprobed with antibodies against actin (1:2000; Santa Cruz Biotechnology). To determine the relative degree of membrane purification, the membrane fraction was subjected to immunoblotting for calnexin, a membrane marker, using a rabbit polyclonal antibody against calnexin (1:1000; Stressgen, Victoria, British Columbia, Canada). For semiquantitative analyses, the densities of bands on immunoblots were measured with the Computer Imaging Device and accompanying software (Fujifilm).
In situ detection of O2- and O2--derived oxidants. Hydroethidine histochemistry was performed for in situ visualization of O2- and O2- derived oxidants (Wu et al., 2003). Twenty-four hours after thrombin injection, hydroethidine (1 mg/ml in PBS containing 1% dimethylsulfoxide; Molecular Probes, Eugene, OR) was administered intraperitoneally. After 15 min, the animals were transcardially perfused with a saline solution containing 0.5% sodium nitrate and heparin (10 U/ml) and then fixed with 4% paraformaldehyde in 0.1 m PB. After fixation, the brains were cut into 40 μm slices using a sliding microtome. Sections were mounted on gelatin-coated slides, and the oxidized hydroethidine product, ethidium, was examined by confocal microscopy (Olympus Optical).
Detection of protein oxidation. The extent of protein oxidation was assessed by measuring protein carbonyl levels with an OxyBlot protein oxidation detection kit (Chemicon, Temecula, CA) according to the protocol of the manufacturer with some modifications (Singhal et al., 2002). Protein samples were prepared from rat brains harvested 48 h after injection with thrombin in the absence or presence of DPI (100 μm, i.c.v.) or trolox (50 mg/kg, i.p.). Subsequently, protein samples (15 μg) were mixed in a microcentrifuge tube with 5 μl of 12% SDS and 10 μl of 1× 2,4-dinitrophenylhydrazine (DNPH) solution. Ten microliters of 1× neutralization solution (a kit component) was added instead of the DNPH solution as the negative control. Tubes were incubated at RT for 15 min and then mixed with 7.5 μl of neutralization solution. Next, the samples were mixed in equal volumes of SDS sample buffer and separated by SDS-PAGE. After electrophoresis, proteins were transferred to polyvinylidene difluoride membranes (Millipore). The membranes were then blocked for 1 h at RT in Tris-buffered saline containing 0.1% Tween 20 and 1% BSA. Membranes were incubated overnight at RT with the anti-DNPH antibody (1:150) and then incubated at RT for 1 h with secondary antibodies (1:300). Blots were developed using enhanced chemiluminescence reagents (Amersham Biosciences). Proteins that underwent oxidative modification (i.e., carbonyl group formation) were identified as a band in the samples derivatized with DNPH. The optical density of the bands was measured using the Computer Imaging Device and accompanying software (Fujifilm). Levels of protein carbonyls were quantified and expressed as the fold increase versus untreated controls.
Counting of hippocampal CA1 neurons. The number of CA1 neurons was assessed at three levels of the dorsal hippocampus. Specifically, alternate sections were obtained at 3.3, 3.6, 4.16, and 4.3 mm posterior to the bregma, and two regions from each level (n = 8 regions for each animal) were used to count cells in the CA1 region. The number of neurons within the CA1 layer was counted using a light microscope (Olympus Optical) at a magnification of 400× and expressed as the number of CA1 neurons per millimeter of linear length as described previously with some modifications (Candelario-Jalil et al., 2003). To maintain consistency across animals, a rectangular box (0.5 × 0.05 mm) was centered over the CA1 cell layer beginning 1.5 mm lateral to the midline. Only neurons with normal visible nuclei were counted. The mean number of CA1 neurons per millimeter for both hemispheres was calculated for each treatment group.
Glial cultures. Microglia were cultured from the cerebral cortices as described previously (Shin et al., 2004). Briefly, cerebral cortices were isolated from postnatal day 1 SD rats, and meninges and blood vessels were removed. Cortices were minced, and cells were dissociated for 30 min at 25°C in PBS containing 0.25% trypsin and 1 mm EDTA. Digestion was terminated by adding an equal volume of MEM containing 10% fetal bovine serum (FBS) (HyClone, Logan, UT), and cells were triturated to obtain a single cell suspension. Cells were grown for 2 weeks in 75 cm2 T-flasks (0.5 hemisphere per flask). The microglia were then detached from the flasks by mild shaking and filtered through a nylon mesh to remove astrocytes and clumped cells. Cells were plated into 24-well plates (1 × 105 cells per well) or 35 mm culture dishes (5 × 105 cells per well). After 1 h, the culture medium was changed to MEM containing 5% FBS. Primary astrocytes were cultured from the cerebral cortices of postnatal day 1 SD rats as described previously (Shin et al., 2004). Briefly, the cortices were triturated into single cells in MEM containing 10% FBS and grown for 2 weeks in 75 cm2 T-flasks (0.5 hemisphere per flask). To prepare pure astrocytes, microglia were detached by mild shaking, and the cells remaining in the flask were harvested with 0.1% trypsin. Astrocytes were plated in a 24-well plate and cultured in MEM supplemented with 5% FBS.
Determination of O2- production. Intracellular production of O2- was assayed by the reduction of nitroblue tetrazolium (Calbiochem) as described previously with some modifications (McDonald et al., 1997). Microglia-enriched cultures in 24-well plates were treated with thrombin (40 U/ml). After 12 h, nitroblue tetrazolium (1 mg/ml) was added, and the cells were incubated for 60 min. After treatment, microscopic examination verified the generation of insoluble formazan as dark purple granules. The medium was removed, and the formazan was dissolved in dimethylsulfoxide. The lysate was transferred to a 96-well plate, and the absorbance at 570 nm was measured with a spectrophotometer. To determine the effect of NADPH oxidase inhibitor on O2- production, cultures were preincubated with vehicle or DPI (0.1-5 μm) for 1 h before the addition of thrombin.
Statistical analysis. All values are expressed as mean ± SEM. Statistical significance (p < 0.05 for all analyses) was assessed by ANOVA using Instat 3.05 (GraphPad Software, San Diego, CA), followed by Student-Newman-Keuls analyses.
Results
Thrombin induces neurodegeneration and microglial activation in the hippocampus
Thrombin (20 U) or vehicle (PBS) as a control was unilaterally injected into the CA1 layer of the rat hippocampus. Seven days later, brains were removed and coronal sections were processed for immunohistochemistry. Neurons were labeled with an antibody against a nuclear protein (NeuN) that is expressed in the nuclei of neurons. Compared with PBS-treated CA1 layer of the hippocampus (Fig. 1A), there was a significant loss of NeuN-immunopositive neurons in the thrombin-treated CA1 layer of hippocampus (Fig. 1B). Thrombin-induced neurodegeneration was further confirmed by staining for Nissl substance using sections adjacent to those used for NeuN immunostaining. Consistent with the results of NeuN immunostaining, there was a dramatic reduction of Nissl-stained cells in the CA1 area of the hippocampus in thrombin-treated animals (Fig. 1D) compared with PBS-treated animals (Fig. 1C).
Figure 1.

Thrombin-induced neurotoxicity in the CA1 layer of rat hippocampus in vivo. PBS (A, C) or thrombin (B, D; 20 U/4 μl) was unilaterally injected into the CA1 layer of the hippocampus. Animals were killed 7 d after injection, brains were removed, and coronal sections (40 μm) were cut using a sliding microtome. Every sixth serial section was selected and processed for NeuN immunostaining or Nissl staining. A, B, NeuN immunostaining in the CA1 layer of hippocampus. Note that there is a significant reduction of NeuN-immunopositive cells in thrombin-injected hippocampus. C, D, CA1 layer of the hippocampus stained for Nissl substance (cresyl violet). The results are representative of six to eight animals per group. Dotted lines indicate the CA1 layer of the hippocampus. Scale bar, 200 μm.
Recent findings, including ours, have indicated that thrombin activates rat microglia in culture (Möller et al., 2000; Ryu et al., 2000; Suo et al., 2002) and in the substantia nigra in vivo (Carreño-Müller et al., 2003; Choi et al., 2003a). This leads to an increased production of proinflammatory cytokines, iNOS, and NO, which have been proposed to play a pathological role in several neurodegenerative disorders, including AD (Akiyama et al., 2000; Benveniste et al., 2001). In the current study, we examined the activation of microglia in the CA1 area of the hippocampus after thrombin injection in vivo. Rat brain sections were obtained 24 h after thrombin injection and immunostained with OX-42 and OX-6 antibodies, which recognize complement receptor 3 and major histocompatibility complex II, respectively. In PBS-injected CA1 layer of the hippocampus, OX-42-immunopositive microglia exhibited the typical ramified morphology of resting microglia (Fig. 2A, a). In contrast, in the thrombin-injected hippocampus, the majority of OX-42-immunopositive microglia displayed an activated morphology, including larger cell bodies with short, thick, or no processes (Fig. 2B, b). Similar patterns were observed for OX-6 immunohistochemical staining, which was performed on sections adjacent to those used for OX-42 immunostaining. These results show that thrombin treatment caused the appearance of many OX-6-immunopositive cells in the CA1 layer of the hippocampus (Fig. 2D, d), in which hippocampal neurons were degenerated (Fig. 1). However, OX-6-immunopositive cells were not observed in PBS-treated controls (Fig. 2C,c).
Figure 2.
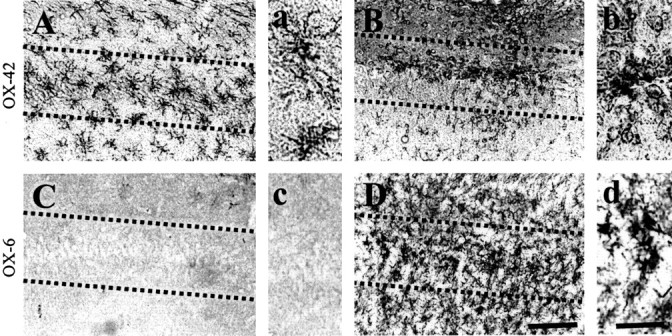
Thrombin-induced microglial activation in the CA1 area of the hippocampus. PBS (A, C) or thrombin (B, D; 20 U/4 μl) was unilaterally injected into the CA1 layer of the hippocampus. Animals were killed 24 h after injection, brains were removed, and coronal sections were cut using a sliding microtome. Brain sections were immunostained with OX-42 (A, B) or OX-6 (C, D) antibodies to identify microglia. a-d show higher magnifications of A-D, respectively. Note the significant microglial activation in the thrombin-treated CA1 area of the hippocampus compared with the PBS-treated control (A, C). These results are representative of six to eight animals per group. Dotted lines indicate the CA1 cell layer of the hippocampus. Scale bars: A-D, 200 μm; a-d, 50 μm.
Because these results indicate that thrombin induces neurotoxicity and microglial activation, we further investigated whether intrahippocampal injection of thrombin produced microglia-derived iNOS and proinflammatory cytokines (IL-1β, IL-6, and TNF-α). Reverse transcription-PCR analysis showed a significant induction in the expression of mRNAs for IL-1β, IL-6, and TNF-α (Fig. 3A) as well as iNOS (Fig. 3B) as early as 4 h after thrombin injection. Western blot analysis also showed that thrombin enhanced the expression of iNOS protein, with maximal levels attained 12 h after injection, followed by a return to normal levels 48 h after injection (Fig. 3C). For the identification of cell types expressing iNOS protein, we performed double-immunofluorescent staining with a combination of antibodies against iNOS and OX-42 in hippocampal sections obtained 12 h after thrombin injection. Simultaneous imaging of immunofluorescence on the same tissue sections revealed that iNOS immunoreactivity was localized within OX-42-immunopositive microglia (Fig. 3D).
Figure 3.
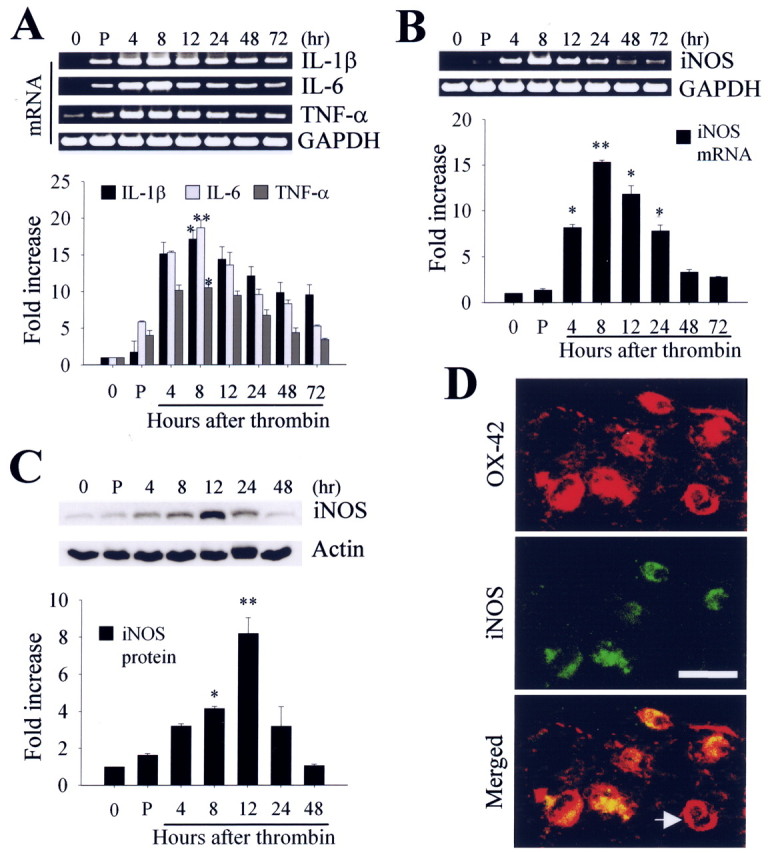
A, B, Reverse transcription-PCR analysis of thrombin-induced mRNA expression of proinflammatory cytokines and iNOS in the hippocampus. Animals were decapitated after intrahippocampal injection of thrombin (20 U/4 μl), and total RNA was isolated in the ipsilateral hippocampus at the indicated time points. C, Western blot analysis of iNOS expression in hippocampus at indicated time points after intrahippocampal thrombin injection. In A-C, untreated (0 h) or PBS-treated (4 h) hippocampus was used as a control. Error bars represent the mean ± SEM for four to five samples per time point. *p < 0.05, **p < 0.01 compared with control according to ANOVA and Student-Newman-Keuls analyses. P, PBS. D, Colocalization of iNOS immunoreactivity within the activated microglia in the CA1 area of hippocampus. The sections of rat hippocampus were prepared 12 h after intrahippocampal injection of thrombin (20 U/4 μl) and then immunostained simultaneously with iNOS and OX-42 as a marker of microglia. Images were captured from the same area and merged. Note the absence of iNOS expression in some microglia (indicated by an arrow). Scale bar, 25 μm.
Thrombin induces the NADPH oxidase-mediated production of ROS by activated microglia in the hippocampus
Recent studies have suggested that activated microglia produce O2- and O2--derived oxidants (Bal-Price et al., 2002; Parvathenani et al., 2003). These ROS can lead to in vivo and in vitro degeneration of dopaminergic neurons (Gao et al., 2003a,b; Wu et al., 2003; Zhang et al., 2004) and developing Purkinje cells (Marín-Teva et al., 2004). Thus, we investigated whether thrombin-activated microglia produced ROS in the hippocampus in vivo. To examine this possibility, we performed in situ analysis of ROS production by hydroethidine histochemistry (Wu et al., 2003). The fluorescent products of oxidized hydroethidine (ethidium; red fluorescence) were increased significantly 48 h after thrombin injection (Fig. 4A, bottom) compared with PBS-injected controls (Fig. 4A, top). Importantly, thrombin-induced ethidium accumulation was predominantly present in the CA1 layer of the hippocampus, in which neurodegeneration (Fig. 1) and microglial activation (Fig. 2) were observed.
Figure 4.
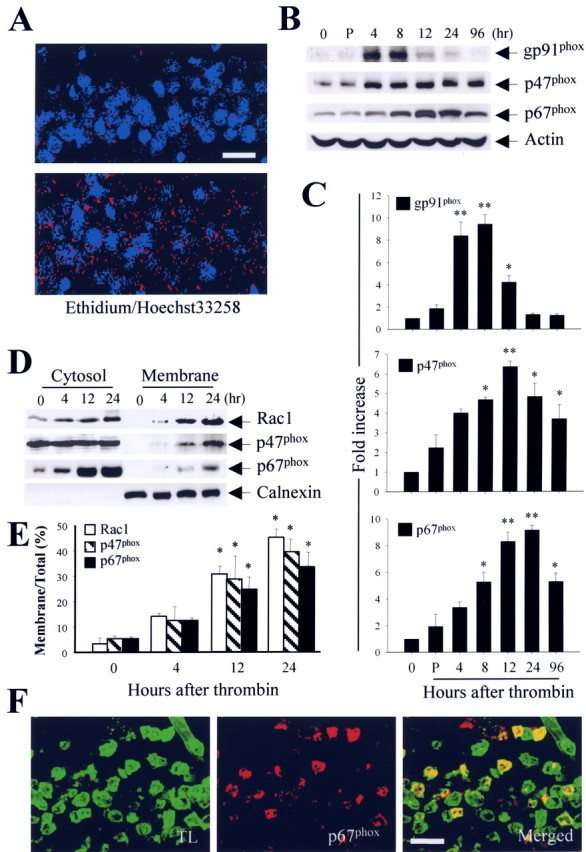
A, In situ visualization of thrombin-induced O2- and O2--derived oxidant production in the CA1 layer of the hippocampus. Animals were injected with hydroethidine (1 μg/μl, i.p.) 48 h after intrahippocampal injection of thrombin (20 U/4 μl). After 15 min, the animals were killed, and sections of hippocampus were prepared for hydroethidine histochemistry to detect the extracellular superoxide. Confocal micrographs show ethidium fluorescence (red) in PBS-injected (top) or thrombin-injected (bottom) CA1 region of the hippocampus. Nuclei were counterstained with Hoechst 33258 (blue). B, Western blot analysis showing upregulation of membrane-bound subunit gp91phox and cytosolic subunits p47phox and p67phox in thrombin-treated hippocampus. Animals were decapitated after intrahippocampal injection of thrombin (20 U) at the indicated time points, and the hippocampi were isolated immediately from the ipsilateral hemisphere. Untreated (0 h) or PBS-treated (4 h) hippocampus was used as a control. Tissue lysates were analyzed by Western blot analysis with gp91phox, p47phox, and p67phox antibodies. C, Error bars represent the mean ± SEM from four to five samples per time point. *p < 0.05, **p < 0.01 compared with control according to ANOVA and Student-Newman-Keuls analyses. D, Translocation of cytosolic subunits (Rac1, p47phox, and p67phox) from the cytosol to the plasma membrane after intrahippocampal injection of thrombin, indicating activation of NADPH oxidase in the hippocampus. Animals were decapitated after intrahippocampal injection of thrombin (20 U) at the indicated time points, and hippocampi were isolated immediately from the ipsilateral hemisphere. Tissue lysates were fractionated and analyzed by immunoblot analysis with p67phox antibody. The membrane protein calnexin was used to normalize the data. E, Error bars represent the mean ± SEM for four to five samples per time point. The levels of NADPH oxidase subunits in the cytosol and membrane were determined by densitometric scanning of Western blots, and the levels of those subunits in the membrane were expressed as a percentage of the total level of each subunit. *p < 0.05, **p < 0.01 compared with control according to ANOVA and Student-Newman-Keuls analyses. F, Localization of p67phox immunoreactivity in activated microglia within the CA1 region of the hippocampus treated with thrombin. The sections of hippocampus were prepared 12 h after thrombin injection (20 U) and then simultaneously stained with an antibody against p67phox and a maker for microglia (tomato lectin). Confocal images were captured from the same area and merged. TL, Tomato lectin. Scale bars: A, 100 μm; F, 50 μm.
Recent studies have demonstrated that NADPH oxidase is a significant source of ROS during inflammation (Bal-Price et al., 2002; Gao et al., 2003a,b; Parvathenani et al., 2003; Wu et al., 2003; Zhang et al., 2004). We therefore investigated whether thrombin-induced ROS production was mediated by activation of the microglial NADPH oxidase in the hippocampus in vivo. Western blotting showed that, compared with nontreated (0 h) or PBS-treated (4 h after injection) controls, thrombin caused a significant and time-dependent upregulation of the total levels of gp91phox, p47phox, and p67phox proteins (Fig. 4B, C). Although expression of p47phox and p67phox proteins was maintained up to 96 h after thrombin injection, gp91phox protein expression returned to normal levels 24 h after thrombin injection.
The translocation of NADPH oxidase subunits from the cytosol to the plasma membrane indicates activation of NADPH oxidase, which leads to ROS production (Babior, 1999; Cross and Segal, 2004). For this reason, tissue samples isolated at different times after intrahippocampal thrombin injection were separated into membrane and cytosolic components and examined by Western blotting. After thrombin treatment, levels of cytosolic NADPH oxidase subunits (p47phox, p67phox, and Rac1) were significantly increased in membrane components (Fig. 4D, E), indicating translocation and activation of the NADPH oxidase complex.
To further determine the identity of the cell type expressing p67phox protein in the CA1 layer of the hippocampus, brain sections from animals treated for 12 h with thrombin were simultaneously immunostained for p67phox and stained for microglia (tomato lectin). Confocal microscopy showed that p67phox immunoreactivity was localized within activated microglia (Fig. 4F).
Oxidative damage to proteins is significantly increased in the hippocampus of AD patients (Hensley et al., 1995; Lyras et al., 1997; Markesbery and Carney, 1999; Castegna et al., 2003). To examine the extent of thrombin-induced oxidative damage, we analyzed protein carbonyls levels in the hippocampus (Singhal et al., 2002). The carbonyl levels were assessed by Western blotting, and the band intensities were compared. The level of protein carbonyls was significantly increased in the hippocampus 48 h after intrahippocampal injection of thrombin compared with PBS-injected controls (Fig. 5A, B). Next, we assessed NADPH oxidase-related oxidative damage. Pretreatment with an NADPH oxidase inhibitor, DPI (100 μm, i.c.v.), significantly reduced the levels of thrombin-induced protein carbonyls in the hippocampus (Fig. 5A, B), despite the lack of specificity (Irani et al., 1997; Li and Trush, 1998) and neurotoxicity (Gao et al., 2002) of this inhibitor. In addition, trolox (50 mg/kg, i.p.), an antioxidant, also reduced the levels of thrombin-induced protein carbonyls in the hippocampus (Fig. 5A, B), although trolox is well characterized as a lipid peroxidation breaker (Mak and Weglicki, 2004).
Figure 5.

A, Thrombin induces protein oxidation in the hippocampus. Animals were decapitated 48 h after intrahippocampal injection of thrombin (20 U) in the absence or presence of the NADPH oxidase inhibitor DPI (100 μm, i.c.v.) or the antioxidant trolox (50 mg/kg, i.p.). The hippocampi were isolated immediately from the ipsilateral hemisphere. Samples were analyzed by Western blotting for protein carbonyls as markers of oxidatively modified proteins. B, Bars represent the means ± SEM of four to five samples. *p < 0.05, **p < 0.01 compared with control according to ANOVA and Student-Newman-Keuls analyses. C, Western blot analysis showing expression of p67phox protein in rat cortical cultures of microglia or astrocytes treated for 6 h in the absence or presence of thrombin (40 U/ml). D, Effect of DPI on thrombin-induced production of O2- in cultured microglia. Cultures of microglia were pretreated for 1 h with DPI (0.1-5 μm), after which they were treated for 12 h with thrombin (40 U/ml). Next, the cells were treated for 1 h with nitroblue tetrazolium (NBT; 1 mg/ml), the medium was removed, and the resulting formazan was dissolved in dimethylsulfoxide. The lysates were transferred to a 96-well plate, and the absorbance was measured at 570 nm in a spectrophotometer. Error bars represent the mean ± SEM of triplicate cultures in the three separate platings. **p < 0.01 compared with untreated control cultures; ##p < 0.01 compared with cultures treated with thrombin only (ANOVA and Student-Newman-Keuls analyses).
In the separate experiments, basal levels and thrombin-enhanced (40 U/ml for 6 h) expression of p67phox protein were detected in primary cultures of microglia but not astrocytes (Fig. 5C). Consistent with the in vivo data, pretreatment of cultured microglia with DPI (0.5-5 μm) caused a dose-dependent decrease in ROS production as revealed by a reduction of nitroblue tetrazolium (Fig. 5D). In contrast, DPI (5 μm) alone had no effect. These results further confirm that thrombin-induced activation of microglia oxidase could contribute to the production of ROS.
NADPH oxidase contributes to thrombin-induced neurodegeneration in the hippocampus in vivo
We hypothesized that ROS generated by thrombin-induced activation of NADPH oxidase contributed to neuronal cell death in the hippocampus in vivo. To test this hypothesis, we investigated whether DPI or trolox altered the effects of thrombin on hippocampal neurons in vivo. NeuN immunohistochemical analysis showed that pretreatment with DPI (100 μm, i.c.v.) (Fig. 6C) or trolox (50 mg/kg, i.p.) (Fig. 6D) partially protected neurons in the CA1 layer of the hippocampus against thrombin-induced neurotoxicity (Fig. 6B) (see also Fig. 1). Based on the percentage of NeuN-immunopositive cells on the ipsilateral versus the contralateral side, DPI and trolox were found to increase the number of NeuN-immunopositive neurons in the CA1 layer of the hippocampus by 28% (p < 0.05) and 31% (p < 0.05), respectively (Fig. 6E). Treatment with DPI or trolox alone, however, did not influence neuronal survival, although DPI was found to be neurotoxic (Gao et al., 2002).
Figure 6.
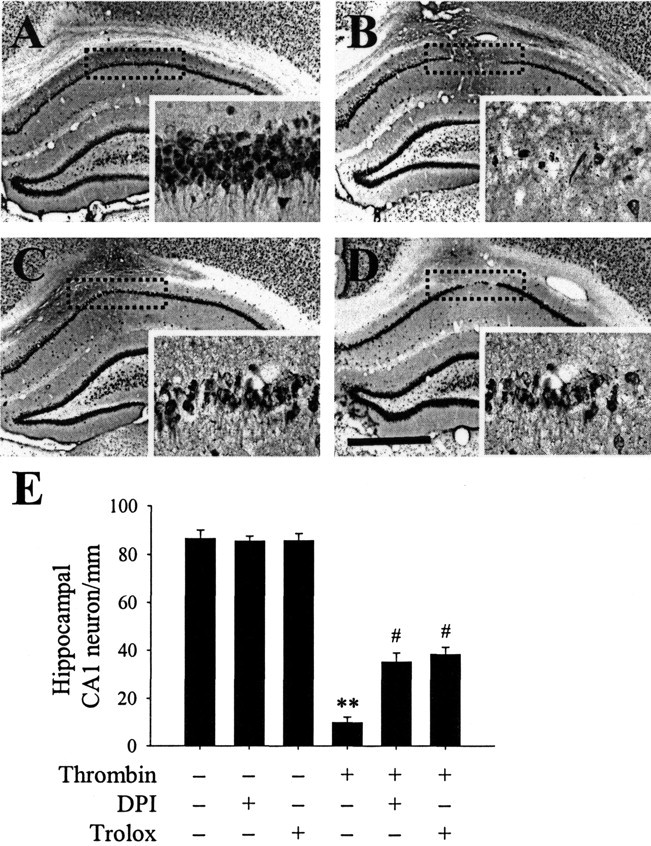
DPI or trolox prevents thrombin-induced neuronal death in the hippocampus. PBS or thrombin (20 U) was unilaterally injected into the hippocampus in either the absence or presence of the NADPH oxidase inhibitor DPI (100 μm, i.c.v.) or the antioxidant trolox (50 mg/kg, i.p.). Animals were killed 7 d after injection. The brains were removed, and coronal sections (40 μm) were cut using a sliding microtome. Every sixth serial section was selected and processed for NeuN immunostaining. Shown are images of a hippocampus treated with PBS alone (A), thrombin (B), DPI plus thrombin (C), or trolox plus thrombin (D). Insets show magnified photomicrographs of the area in the CA1 layer marked by dotted rectangles. The results are representative of six to eight animals per group. Scale bar, 1 mm. E, Number of NeuN-immunopositive neurons in the CA1 layer of hippocampi treated with thrombin in either the absence or presence of DPI or trolox. As controls, DPI or trolox alone was injected. Error bars represent the means ± SEM from six to eight samples per group. **p < 0.01 compared with untreated hippocampi; #p < 0.05 compared with hippocampi treated with thrombin only (ANOVA and Student-Newman-Keuls analyses).
Discussion
In the present study, we found that intrahippocampal injection of thrombin leads to the upregulation and activation of NADPH oxidase within microglia in vivo. This results in ROS production and subsequent oxidative modification of proteins in the hippocampus. This oxidative stress is prevented by DPI and trolox, leading to an increase in neuronal survival. Collectively, our data demonstrate that oxidative stress originating from NADPH oxidase within activated microglia is associated with thrombin-induced neurodegeneration in the hippocampus in vivo.
Several studies in cultured cells have revealed that thrombin induces the degeneration of hippocampal neurons (Striggow et al., 2000) and spinal motor neurons (Turgeon et al., 1998). We have shown recently that thrombin is neurotoxic to dopaminergic neurons in mesencephalic cultures (Choi et al., 2003b). These numerous in vitro findings of thrombin-induced neurotoxicity are further confirmed by the current results showing that intrahippocampal injection of thrombin produces a substantial loss of hippocampal CA1 neurons in vivo, as revealed by NeuN immunostaining and Nissl staining. This is quite similar to our recent finding that intranigral injection of thrombin causes a loss of dopaminergic neurons in the substantia nigra in vivo (Choi et al., 2003a,b). Interestingly, accumulating data in AD brains also show an increase in thrombin immunoreactivity in neuritic plaques (Akiyama et al., 1992) and a decrease in the level of major brain inhibitor protease nexin I (Vaughan et al., 1994; Choi et al., 1995). In addition, thrombin has been reported to modulate the production of amyloid protein precursor and its cleavage into fragments, which is found in the amyloid plaques of AD brains (Igarashi et al., 1992; Chong et al., 1994). Collectively, these results indicate that thrombin may contribute to the neuropathological process in AD brains, including the hippocampus. This hypothesis is fully supported by the recent findings that thrombin-induced neurotoxicity in the hippocampus is associated with cognitive impairments, including deficits in learning and memory (Mhatre et al., 2004).
The progression of AD is characterized by activation of microglia (Cagnin et al., 2001; Zekry et al., 2003). This leads to the increased production of ROS and resulting oxidative insult (Benveniste et al., 2001; Coraci et al., 2002), which plays a key role in neurodegeneration (Gao et al., 2003a,b; Wu et al., 2003; Zhang et al., 2004). NADPH oxidase is a multicomponent enzyme, and its activation is responsible for generating ROS in microglia (Babior, 1999; Cross and Segal, 2004; Patel et al., 2005). These ROS can cross the cell membranes and cause neurodegeneration via oxidative damage, such as protein oxidation (Lyras et al., 1997; Markesbery and Carney, 1999). The results of the present study showed that thrombin induced the upregulation of gp91phox, p47phox, and p67phox proteins and caused the translocation of cytosolic NADPH oxidase subunits (p47phox, p67phox, and Rac1) to the plasma membrane in microglia within the hippocampus in vivo. In thrombin-treated hippocampus, there was not only an activation of microglial NADPH oxidase but also a significant enhancement of ROS production and protein oxidation. Our results are quite comparable with recent reports that seizure-induced hippocampal damage may be attributable to activation of microglial NADPH oxidase and consequent production of ROS (Patel et al., 2005) and that microglial NADPH oxidase-deficient mice are resistant to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity because of the absence of ROS production and resulting oxidative damage (Wu et al., 2003). Also, Block et al. (2004) demonstrated recently that nanometer size diesel exhaust particles induce neurotoxicity through the activation of microglial NADPH oxidase and consequent oxidative stress. Together with the effect of thrombin on microglial activation (Möller et al., 2000; Ryu et al., 2000; Suo et al., 2002; Carreño-Müller et al., 2003; Choi et al., 2003a), these results collectively suggest that thrombin activates microglial NADPH oxidase, which stimulates ROS production and thereby results in neurodegeneration in the hippocampus in vivo. This interpretation is further complemented by our present findings that both DPI, an NADPH oxidase inhibitor, and trolox, an antioxidant, protect against thrombin-induced neurodegeneration in the hippocampus in vivo.
Although our results point to a likely role of microglial NADPH oxidase, recent findings demonstrate that the amyloid protein precursor-induced loss of cortical neurons in cultures is mediated by neuronal NADPH oxidase (Niikura et al., 2004) and that β-amyloid-induced neurotoxicity in cultured hippocampal neurons is mediated by astrocyte NADPH oxidase (Abramov et al., 2004). Therefore, it is likely that NADPH oxidase originating from neurons (Serrano et al., 2003) or astrocytes (Noh and Koh, 2000; Tammariello et al., 2000) may also participate in thrombin-induced neurotoxicity. However, we did not confirm direct evidence of whether NADPH oxidase is expressed in neurons or astrocytes in the hippocampus in vivo, although the present study shows an absence of NADPH oxidase expression in cultured cortical astrocytes.
Apart from the role of NADPH oxidase, other microglial-derived proinflammatory cytokines or cytotoxic factors may be involved in thrombin-induced neurotoxicity. It has been shown that the levels of IL-1β (Cacabelos et al., 1994), IL-6 (Hüll et al., 1996), and TNF-α (Perry et al., 2001) are upregulated in AD brains. Moreover, NO, catalyzed by iNOS, may also be involved in the pathological processes of AD (Lüth et al., 2002), although the presence of iNOS in human microglia is disputed (Colasanti et al., 1995; Zhao et al., 1998; Heneka et al., 2001). The neurotoxic effects of NO are, in general, attributed to its reaction with O2- to form peroxynitrite, which can cause oxidative damage to proteins and other macromolecules, all of which are seen in the brain of AD patients (Koppal et al., 1999; Torreilles et al., 1999). The involvement of such factors is supported by our present observations showing the expression of iNOS and proinflammatory cytokines, including IL-1β, IL-6, and TNF-α, in thrombin-treated hippocampus in vivo.
To our knowledge, the present study is the first to demonstrate that thrombin causes damage to hippocampal neurons through the activation of microglial NADPH oxidase and consequent oxidative stress. Combined with extensive clinical findings and in vivo and in vitro experimental data, the results from this study strongly suggest that thrombin can act as an endogenous neurotoxin and that inhibitors of thrombin or antioxidants can be useful agents for treating oxidative stress-mediated neurodegenerative diseases such as AD.
Footnotes
This work was supported by funds from the Korea Science and Engineering Foundation (KOSEF) (Brain Disease Research Center), KOSEF Grant 1999-2-210-002-5, the Neurobiology Research Program of the Korean Ministry of Science and Technology, and a grant from the Brain Research Center of the 21st Century Frontier Research Program from the Korea Ministry of Science and Technology.
Correspondence should be addressed to Byung Kwan Jin, Brain Disease Research Center, Ajou University School of Medicine, Suwon 443-721, Korea. E-mail: bkjin@ajou.ac.kr.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254082-•$15.00/0
References
- Abramov AY, Canevari L, Duchen MR (2004) β-Amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci 24: 565-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Ikeda K, Kondo H, McGeer PL (1992) Thrombin accumulation in brains of patients with Alzheimer's disease. Neurosci Lett 146: 152-154. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21: 383-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babior BM (1999) NADPH oxidase: an update. Blood 93: 1464-1476. [PubMed] [Google Scholar]
- Bal-Price A, Matthias A, Browin GC (2002) Stimulation of NADPH oxidase in activated rat microglia removes nitric oxide but induces peroxynitrite production. J Neurochem 80: 73-80. [DOI] [PubMed] [Google Scholar]
- Beilin O, Gurwitz D, Korczyn AD, Chapman J (2001) Quantitative measurements of mouse brain thrombin-like and thrombin inhibition activities. NeuroReport 12: 2347-2351. [DOI] [PubMed] [Google Scholar]
- Benveniste EN, Nguyen VT, O'Keefe GM (2001) Immunological aspects of microglia: relevance to Alzheimer's disease. Neurochem Int 39: 381-391. [DOI] [PubMed] [Google Scholar]
- Berzin TM, Zipser BD, Rafii MS, Kuo-Leblane V, Yancopoulos GD, Glass DJ, Fallon JR, Stopa EG (2000) Agrin and microvascular damage in Alzheimer's disease. Neurobiol Aging 21: 349-355. [DOI] [PubMed] [Google Scholar]
- Block ML, Wu X, Pei Z, Li G, Wang T, Qin L, Wilson B, Yang J, Hong JS, Veronesi B (2004) Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: the role of microglia, phagocytosis, and NADPH oxidase. FASEB J 18: 1618-1620. [DOI] [PubMed] [Google Scholar]
- Borroni B, Akkawi N, Martini G, Colciaghi F, Prometti P, Rozzini L, Di Luca M, Lenzi GL, Romanelli G, Caimi L, Padovani A (2002) Microvascular damage and platelet abnormalities in early Alzheimer's disease. J Neurol Sci 203-204: 189-193. [DOI] [PubMed] [Google Scholar]
- Cacabelos R, Alvarez XA, Fernandez-Novoa L, Franco A, Mangues R, Pellicer A, Nishimura T (1994) Brain interleukin-1 beta in Alzheimer's disease and vascular dementia. Methods Find Exp Clin Pharmacol 16: 141-151. [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB (2001) In-vivo measurement of activated microglia in dementia. Lancet 358: 461-467. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, González-Falcón A, García-Cabrera M, Álvarez D, Al-Dalain S, Martínez G, León OS, Springer JE (2003) Assessment of the relative contribution of COX-1 and COX-2 isoforms to ischemia-induced oxidative damage and neurodegeneration following transient global cerebral ischemia. J Neurochem 86: 545-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreño-Müller E, Herrera AJ, de Pablos RM, Tomás-Camardiel M, Venero JL, Cano J, Machado A (2003) Thrombin induces in vivo degeneration of nigral dopaminergic neurons along with the activation of microglia. J Neurochem 84: 1201-1214. [DOI] [PubMed] [Google Scholar]
- Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA (2003) Proteomic identification of nitrated proteins in Alzheimer's disease brain. J Neurochem 85: 1394-1401. [DOI] [PubMed] [Google Scholar]
- Choi BH, Kim RC, Vaughan PJ, Lau A, Van Nostrand WE, Cotman CW, Cunningham DD (1995) Decrease in protease nexins in Alzheimer's disease. Neurobiol Aging 16: 557-562. [DOI] [PubMed] [Google Scholar]
- Choi SH, Joe EH, Kim SU, Jin BK (2003a) Thrombin-induced microglial activation produces degeneration of nigral dopaminergic neurons in vivo J Neurosci 23: 5877-5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Lee DY, Ryu JK, Kim J, Joe EH, Jin BK (2003b) Thrombin induces nigral dopaminergic neurodegeneration in vivo by altering expression of death-related proteins. Neurobiol Dis 14: 181-193. [DOI] [PubMed] [Google Scholar]
- Chong YH, Jung JM, Choi W, Park CW, Choi KS, Suh YH (1994) Bacterial expression, purification of full length and carboxyl terminal fragment of Alzheimer amyloid precursor protein and their proteolytic processing by thrombin. Life Sci 4: 1259-1268. [DOI] [PubMed] [Google Scholar]
- Colasanti M, Di Pucchio T, Persichini T, Sogos V, Presta M, Lauro GM (1995) Inhibition of inducible nitric oxide synthase mRNA expression by basic fibroblast growth factor in human microglial cells. Neurosci Lett 195: 45-48. [DOI] [PubMed] [Google Scholar]
- Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, Luster AD, Silverstein SC, El-Khoury JB (2002) CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer's disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol 160: 101-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross AR, Segal AW (2004) The NADPH oxidase of professional phagocytes—prototype of the NOX electron transport chain systems. Biochim Biophys Acta 1657: 1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan FM, Pike CJ, Cotman CW, Cunningham DD (1997) Thrombin induces apoptosis in cultured neurons and astrocytes via a pathway requiring tyrosine kinase and RhoA activities. J Neurosci 17: 5316-5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Hong JS, Zhang W, Liu B (2002) Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci 22: 782-790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Liu B, Hong JS (2003a) Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J Neurosci 23: 6181-6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Liu B, Zhang W, Hong JS (2003b) Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson's disease. FASEB J 17: 1954-1956. [DOI] [PubMed] [Google Scholar]
- Grammas P (2000) A damaged microcirculation contributes to neuronal cell death in Alzheimer's disease. Neurobiol Aging 21: 199-205. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Wiesinger H, Dumitrescu-Ozimek L, Riederer P, Feinstein DL, Klockgether T (2001) Neuronal and glial coexpression of argininosuccinate synthetase and inducible nitric oxide synthase in Alzheimer's disease. J Neuropathol Exp Neurol 60: 906-916. [DOI] [PubMed] [Google Scholar]
- Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, Aksenova M, Gabbita SP, Wu JF, Carney JM, Lovell M, Markesbery WR, Butterfield DA (1995) Brain regional correspondence between Alzheimer's disease histopathology and biomarkers of protein oxidation. J Neurochem 65: 2146-2156. [DOI] [PubMed] [Google Scholar]
- Hüll M, Fiebich BL, Lieb K, Strauss S, Berger SS, Volk B, Bauer J (1996) Interleukin-6-associated inflammatory processes in Alzheimer's disease: new therapeutic options. Neurobiol Aging 17: 795-800. [DOI] [PubMed] [Google Scholar]
- Igarashi K, Murai H, Asaka J (1992) Proteolytic processing of amyloid beta protein precursor (APP) by thrombin. Biochem Biophys Res Commun 185: 1000-1004. [DOI] [PubMed] [Google Scholar]
- Irani K, Xia Y, Zweier JL, Dollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, Goldschmidt-Clermont PJ (1997) Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 275: 1649-1652. [DOI] [PubMed] [Google Scholar]
- Koppal T, Drake J, Yatin S, Jordan B, Varadarajan S, Bettenhausen L, Butterfield DA (1999) Peroxynitrite-induced alterations in synaptosomal membrane proteins: insight into oxidative stress in Alzheimer's disease. J Neurochem 72: 310-317. [DOI] [PubMed] [Google Scholar]
- Lee DY, Oh YJ, Jin BK (2005) Thrombin-activated microglia contribute to death of dopaminergic neurons in rat mesencephalic cultures: dual roles of mitogen-activated protein kinase signaling pathways. Glia, in press. [DOI] [PubMed]
- Li Y, Trush MA (1998) Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun 253: 295-299. [DOI] [PubMed] [Google Scholar]
- Lüth HJ, Munch G, Arendt T (2002) Aberrant expression of NOS isoforms in Alzheimer's disease is structurally related to nitrotyrosine formation. Brain Res 953: 135-143. [DOI] [PubMed] [Google Scholar]
- Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell (1997) As assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer's diseases. J Neurochem 68: 2061-2069. [DOI] [PubMed] [Google Scholar]
- Mak T, Weglicki WB (2004) Potent antioxidant properties of 4-hydroxy-propranolol. J Pharmacol Exp Ther 308: 85-90. [DOI] [PubMed] [Google Scholar]
- Marín-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M (2004) Microglia promote the death of developing Purkinje cells. Neuron 41: 535-547. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, Carney JM (1999) Oxidative alterations in Alzheimer's disease. Brain Pathol 9: 133-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald DR, Brunden KR, Landreth GE (1997) Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci 17: 2284-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF (1993) Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann Neurol 34: 609-616. [DOI] [PubMed] [Google Scholar]
- Mhatre M, Nguyen A, Kashani S, Pham T, Adesina A, Grammas P (2004) Thrombin, a mediator of neurotoxicity and memory impairment. Neurobiol Aging 25: 783-793. [DOI] [PubMed] [Google Scholar]
- Möller T, Hanisch UK, Ransom BR (2000) Thrombin-induced activation of cultured rodent microglia. J Neurochem 75: 1539-1547. [DOI] [PubMed] [Google Scholar]
- Niikura T, Yamada M, Chiba T, Aiso S, Matsuoka M, Nishimoto I (2004) Characterization of V642I-AβPP-induced cytotoxicity in primary neurons. J Neurosci Res 77: 54-62. [DOI] [PubMed] [Google Scholar]
- Noh KM, Koh JY (2000) Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. J Neurosci 20: RC111(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AM, Burns MA (1994) Selective increase in lipid peroxidation in the inferior temporal cortex in Alzheimer's disease. Brain Res 645: 338-342. [DOI] [PubMed] [Google Scholar]
- Parvathenani LK, Tertyshnikova S, Greco CR, Roberts SB, Robertson B, Posmantur R (2003) P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer's disease. J Biol Chem 278: 13309-13317. [DOI] [PubMed] [Google Scholar]
- Patel M, Li QY, Chang LY, Crapo J, Liang LP (2005) Activation of NADPH oxidase and extracellular superoxide production in seizure-induced hippocampal damage. J Neurochem 92: 123-131. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998) The rat brain in stereotaxic coordinates. San Diego: Academic.
- Perry RT, Collins JS, Wiener H, Acton R, Go RC (2001) The role of TNF and its receptors in Alzheimer's disease. Neurobiol Aging 22: 873-883. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Cooper C, Liu B, Wilson B, Hong JS (2002) Microglia enhance beta-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J Neurochem 83: 973-983. [DOI] [PubMed] [Google Scholar]
- Ryu JY, Pyo HK, Jou IR, Joe EH (2000) Thrombin induces NO release from cultured rat microglia via protein kinase C, mitogen-activated protein kinase, and NF-κB. J Biol Chem 275: 29955-29959. [DOI] [PubMed] [Google Scholar]
- Serrano F, Kolluri NS, Wientjes FB, Card JP, Klann E (2003) NADPH oxidase immunoreactivity in the mouse brain. Brain Res 988: 193-198. [DOI] [PubMed] [Google Scholar]
- Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A, Chiba S, Perry G, Smith MA, Fujimoto S (2000) Activation of NADPH oxidase in Alzheimer's disease brains. Biochem Biophys Res Commun 273: 5-9. [DOI] [PubMed] [Google Scholar]
- Shin WH, Lee DY, Park KW, Kim SU, Yang MS, Joe EH, Jin BK (2004) Microglia expressing interleukin-13 undergo cell death and contribute to neuronal survival in vivo. Glia 46: 142-152. [DOI] [PubMed] [Google Scholar]
- Singhal AB, Wang X, Sumii T, Mori T, Lo EH (2002) Effects of normobaric hyperoxia in a rat model of focal cerebral ischemia-reperfusion. J Cereb Blood Flow Metab 22: 861-868. [DOI] [PubMed] [Google Scholar]
- Stepanichev MY, Zdobnova IM, Yakovlev AA, Onufriev MV, Lazareva NA, Zarubenko II, Gulyaeva NV (2003) Effects of tumor necrosis factor-alpha central administration on hippocampal damage in rat induced by amyloid beta-peptide (25-35). J Neurosci Res 71: 110-120. [DOI] [PubMed] [Google Scholar]
- Striggow F, Riek M, Breder J, Henrich-Noack P, Reymann KG, Reiser G (2000) The protease thrombin is an endogenous mediator of hippocampal neuroprotection against ischemia at low concentrations but cause degeneration at high concentrations. Proc Natl Acad Sci USA 97: 2264-2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suo Z, Wu M, Ameenuddin S, Anderson HE, Zoloty JE, Citron BA, Andrade-Gordon P, Festoff BW (2002) Participation of protease-activated receptor-1 in thrombin-induced microglia activation. J Neurochem 80: 655-666. [DOI] [PubMed] [Google Scholar]
- Tammariello SP, Quinn MT, Estus S (2000) NADPH oxidase contributes directly to oxidative stress and apoptosis in nerve growth factor-deprived sympathetic neurons. J Neurosci 20: RC53(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torreilles F, Salman-Tabcheh S, Guerin M, Torreilles J (1999) Neurodegenerative disorders: the role of peroxynitrite. Brain Res Brain Res Rev 30: 153-163. [DOI] [PubMed] [Google Scholar]
- Turgeon VL, Lloyd ED, Wang S, Festoff BW, Houenou LJ (1998) Thrombin perturbs neurite outgrowth and induces apoptotic cell death in enriched chick spinal motoneuron cultures through caspase activation. J Neurosci 18: 6882-6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Laurer H, Mclntosh T, Pratico D, Martinez D, Leight S, Lee VM, Trojanowski JQ (2002) Repetitive mild brain trauma accelerates Aβ deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J Neurosci 22: 446-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan PL, Su J, Cotman CW, Cunningham D (1994) Protease nexin-1, a potent thrombin inhibitor, is reduced around cerebral blood vessels in Alzheimer's disease. NeuroReport 5: 2529-2533. [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Sandercock PA, Dennis MS, Starr J (2003) Is breakdown of the blood-brain barrier responsible for lacunar stroke, leukoaraiosis, and dementia? Stroke 34: 806-812. [DOI] [PubMed] [Google Scholar]
- Weinstein JR, Gold SJ, Cunningham DD, Gall CM (1995) Cellular localization of thrombin receptor mRNA in rat brain: expression by mesencephalic dopaminergic neurons and codistribution with prothrombin mRNA. J Neurosci 15: 2906-2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S (2003) NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc Natl Acad Sci USA 100: 6145-6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zekry D, Epperson TK, Krause KH (2003) A role of NOX NADPH oxidase in Alzheimer's disease and other types of dementia? IUBMB Life 55: 307-313. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang T, Qin L, Gao HM, Wilson B, Ali SF, Zhang W, Hong JS, Liu B (2004) Neuroprotective effect of dextromethorphan in the MPTP Parkinson's disease model: role of NADPH oxidase. FASEB J 18: 589-591. [DOI] [PubMed] [Google Scholar]
- Zhao ML, Liu JS, He D, Dickson DW, Lee SC (1998) Inducible nitric oxide synthase expression is selectively induced in astrocytes isolated from adult human brain. Brain Res 813: 402-405. [DOI] [PubMed] [Google Scholar]