

Abstract
Increased levels of mitochondrial-free calcium have been associated with several cell-death paradigms, such as excitotoxicity and ceramide-mediated neuronal death. In the latter, calcium is transferred from the endoplasmic reticulum to mitochondria by a mechanism that is only partly understood. We show here that CDK5 (cyclin-dependent kinase 5) plays a role. Free calcium levels in the endoplasmic reticulum and mitochondria were measured with fluorescent markers in C2-ceramide-treated primary cultures of mesencephalic neurons and differentiated pheochromocytoma PC12 cells. Calcium levels decreased in the endoplasmic reticulum as they increased in mitochondria. Both changes were blocked by the pharmacological and molecular CDK5 inhibitors roscovitine and a dominant-negative form of CDK5. Although the kinase did not mediate the transfer of calcium per se, which required the proapoptotic Bcl-2 family protein t-Bid (the truncated form of Bid), it facilitated the transfer by inducing the clustering of endoplasmic reticulum and mitochondria around the centrosome where they formed close contacts, as shown by immunocytochemistry and electron microscopy. Organelle clustering resulted from CDK5-dependent phosphorylation of the microtubule-associated protein tau on threonine 231. This caused its release from microtubules into the soluble fraction of cellular proteins, which appears to favor retrograde transport of the organelles. Mutation of threonine 231 to alanine, so that tau could not be phosphorylated at this site, prevented the ceramide-induced release of tau from microtubules, organelle clustering, the increase in mitochondrial-free calcium levels, and neuronal death, demonstrating the importance of the CDK5-dependent signaling cascade in this calcium-dependent cell-death mechanism.
Keywords: tau, apoptosis, phosphorylation, centrosome, microtubule, clustering
Introduction
Changes in free calcium levels in the endoplasmic reticulum (ER) and mitochondria have been shown to play important roles in neuronal death mechanisms (Duchen, 2000; Paschen, 2003; Verkhratsky and Toescu, 2003). Mitochondrial-free calcium levels increase during excitotoxic cell death, after entry of calcium into the cell through glutamate receptors (Stout et al., 1998). They also increase in ceramide-induced (Muriel et al., 2000; Pinton et al., 2001; Darios et al., 2003) or staurosporine-induced (Nutt et al., 2002b) neuronal death, but in this case, calcium is transferred to mitochondria from the ER (Szalai et al., 1999; Darios et al., 2003). The transfer takes place at contact sites between the organelles in which calcium concentrations reach high levels (Rizzuto et al., 1993). The exact mechanism of calcium transfer from ER to mitochondria during neuronal death has not been completely elucidated but appears to require the intervention of proapoptotic proteins such as Bax, Bak, t-Bid (the truncated form of Bid), and/or the voltage-dependent anion channel (Csordas et al., 2002; Nutt et al., 2002a; Rapizzi et al., 2002; Darios et al., 2003). Mitochondrial calcium uptake from the ER also requires close apposition between the organelles (Rizzuto et al., 1998; Csordas et al., 1999). During cell death induced by both exogenous or endogenous ceramide, it has been shown that the endoplasmic reticulum and mitochondria become closely associated early in the process, when mitochondrial-free calcium levels are increasing (Muriel et al., 2000). Regulation of the physical interaction between ER and mitochondria may therefore play an important role in ER to mitochondria calcium transfer.
ER and mitochondria are mobile in cells (Hajnoczky et al., 2000; Rintoul et al., 2003). Little is known about how they interact, although actin and microtubule networks appear to play a role (Reynolds and Rintoul, 2004). Among the proteins involved in microtubule-based cellular transport, the microtubule-associated protein tau is of particular interest. It has been implicated in both organelle trafficking (Sato-Harada et al., 1996; Ebneth et al., 1998; Mandelkow et al., 2004) and the susceptibility of neurons to cell death (Delobel et al., 2003). Tau binding to microtubules is reported to inhibit organelle trafficking (Sato-Harada et al., 1996; Ebneth et al., 1998) and is regulated by phosphorylation of specific amino acids (Illenberger et al., 1998; Sengupta et al., 1998; Wada et al., 1998; Biernat et al., 2002; Takahashi et al., 2003). Many protein kinases phosphorylate tau (Buee et al., 2000), including CDK5 (cyclin-dependent kinase 5), a serine/threonine kinase associated with postmitotic neurons (Nikolic et al., 1996) reported to play a role in neuronal death (Patrick et al., 1999; Ahlijanian et al., 2000; Zheng et al., 2005) upstream of mitochondrial dysfunction (Weishaupt et al., 2003).
These functions of CDK5 prompted us to investigate whether it plays a role in the rearrangement of mitochondria and ER observed during ceramide-mediated neuronal death and in the transfer of calcium from the ER to mitochondria that occurs during this process.
Materials and Methods
Constructs. Vectors for expression of p25 (pcDNA3-p25), CDK5 (pcDNA3-CDK5), and dominant-negative CDK5 (pcDNA3-DNK5) were kindly provided by Dr. L. H. Tsai (Harvard University School of Medicine, Cambridge, MA) (Tsai et al., 1994). The p25 and DNK5 (dominant-negative form of CDK5) cDNAs were subcloned in a pEGFP (enhanced green fluorescent protein) vector (BD Biosciences Clontech, Le Pont de Claix, France) to permit identification of transfected cells. The pcDNA-3-t-Bid expression vector was described previously (Darios et al., 2003). The full-length cDNA of human tau containing exons 2, 3, and 10 (htau40) subcloned in the mammalian expression vector pcDNA3 was kindly provided by Dr. G. V. Johnson (University of Alabama, Birmingham, AL) (Cho and Johnson, 2004), as was the construct in which threonine 231 was replaced by alanine, referred to as T231A tau.
Cell culture, treatment, and transfection. Animals were treated in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council 1996), the European Communities Council Directive 86/609/EEC, and the guidelines of the institutional ethics committee. All efforts were made to minimize the number of animals used and their suffering. Primary cultures of mesencephalon were prepared at embryonic day 15.5 from pregnant Wistar rats (Elevage Janvier, Le Genest St. Isle, France) that had been anesthetized and then decapitated. The ventral midbrain was dissected as described previously (Brugg et al., 1996), mechanically dissociated, and plated on polyethylenimine (1 mg/ml)-coated culture wells, in N5 medium, supplemented with 5% horse serum (HS) and 2.5% fetal calf serum (FCS), at a density of 5 × 104 cells/cm2. After 2 d in culture, FCS content was reduced to 0.5% to prevent astrocyte proliferation. All experiments were initiated at day 6, when cultures contain >98% of postmitotic neurons. To initiate the cell-death program, C2-ceramide (25 μm; Tebu-Bio, Le Perray en Yvelines, France) was added to N5 medium containing 1% horse serum. To inhibit CDK5 activity, roscovitine (10 μm; Sigma-Aldrich, Saint-Quentin Fallavier, France) was added 30 min before C2-ceramide.
For experiments requiring transfection of expression vectors, pheochromocytoma PC12 cells were used instead of primary cultures. The cells, plated at a density of 2 × 104 cells/cm2, were cultured in L15 medium supplemented with 2% horse serum and 100 ng/ml NGF (Alomone Labs, Tel Aviv, Israel) during 6 d to obtain differentiation, as described previously (Muriel et al., 2000; Darios et al., 2003). Experiments were performed in L15 medium containing 1% HS. PC12 cells were transfected with the liposome DMRIE-C (Invitrogen, Cergy Pontoise, France), according to the instructions of the manufacturer. Transfections were performed after 5 d of differentiation, and experiments in these cells were performed 24 h later. Control cells were transfected with the corresponding empty pEGFP or pcDNA3 vectors.
CDK5 activity. Cell lysates were prepared in 100 μl of 20 mm 3-(N-morpholino)propanesulfonic acid (MOPS), pH 7.2, containing 0.3 m NaCl, 1 mm MgCl2, 1 mm EGTA, 0.1 mm EDTA, 0.5% Nonidet P-40, 1 mm dithiothreitol, and 2% Complete Mini inhibitor cocktail (Roche, Mannheim, Germany). Lysates were cleared by centrifugation at 4°C (18,000 × g, 30 min). An anti-CDK5 antibody (C8, 2 μl; Santa Cruz Biotechnology, Santa Cruz, CA) was added to the extracts, and, after 1 h of incubation at 4°C, 10 μl of protein A-Sepharose (Amersham Biosciences, Saclay, France) was added to the mixture. After additional incubation for 10 h at 4°C, the beads were removed from the extracts, washed twice with 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, and 0.05% Tween-20, and washed three times with 20 mm MOPS, pH 7.2, 1 mm MgCl2, 1 mm EGTA, and 0.1 mm EDTA. Bead-bound CDK5 was then used to phosphorylate the CDK5 substrate histone H1 by incubation for 30 min at 30°C with 20 mm MOPS, pH 7.2, 1 mm MgCl2, 0.1 mm [γ-32P]ATP (10 μCi of [γ-32P]dATP), and 0.4 mg/ml histone H1. The reaction was terminated by the addition of SDS-PAGE sample buffer and then boiled for 5 min. The proteins were fractionated by SDS-PAGE. Phosphorylated histone H1 was visualized with a phospho-imager (FLA2000; Fuji, Tokyo, Japan) and quantified with the dedicated software.
Immunocytochemistry. Cells were fixed in 4% paraformaldehyde for 25 min and analyzed by standard immunocytochemical procedures. Primary antibodies were monoclonal anti-cytochrome c clone 6H2.B4 (1:100; PharMingen, Hamburg, Germany), polyclonal anti-glucose-regulated protein 78 (GRP78) (1:400; Stressgen, Victoria, British Columbia, Canada) and polyclonal anti-γ-tubulin (1:500; kindly provided by Dr. M. Bornens, Curie Institute, Paris, France). Secondary antibodies were Alexa Fluor 488-conjugated goat anti-mouse IgG (1: 600; Interchim, Montluçon, France) and cyanine 3-conjugated goat anti-rabbit IgG (1:800; Jackson ImmunoResearch, Sohum, UK).
Western blot. Total cell extracts were prepared by lysis in 150 mm NaCl, 50 mm Tris-HCl, 0.5% Triton X-100, 0.1% SDS, 200 nm okadaic acid, 50 mg/ml sodium fluoride, 2 mm orthovanadate, 25 mm β-glycerophophate (all from Sigma), and 2% Complete Mini inhibitor cocktail. Alternatively, microtubule-free extracts were prepared by lysis at 4°C in 20 mm HEPES, pH 7.5, 250 mm sucrose, 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 1 mm DTT, 200 nm okadaic acid, 50 mg/ml sodium fluoride, 2 mm orthovanadate, 25 mm β-glycerophophate, and 2% Complete Mini inhibitor mixture. The supernatants were recovered after centrifugation at 100,000 × g for 30 min at 4°C.
The proteins were separated by SDS-PAGE, blotted onto nitrocellulose membranes, and incubated with the primary antibody. We used the following antibodies: Tau-5, directed against total tau protein, regardless of its phosphorylation status (1:500: Chemicon, Euromedex, Souffelweyersheim, France), AT8 (tau phosphorylated on S202/T205, 1:500; Innogenetics, Gent, Belgium), AT180 (tau phosphorylated on T231, 1:200; Innogenetics), anti-α-tubulin clone DM1A (1:1000; Sigma), or anti-p35 C19 (1:1000; Santa Cruz Biotechnology). After incubation with appropriate secondary anti bodies coupled to horseradish peroxidase (1: 150,000; Jackson ImmunoResearch), immunoreactivity was revealed with the Super Signal chemiluminescence detection kit (Pierce, Rockford, IL).
Calcium imaging. Mitochondrial-free calcium levels ([Ca2+]mit) were quantified with rhod-2 AM (5 μm, 550 nm excitation, 630 nm emission; Molecular Probes, Eugene, OR), a positively charged rhodamine derivative that accumulates in mitochondria. The method of quantification was described previously (Muriel et al., 2000; Darios et al., 2003). Briefly, the cells were viewed with an inverted fluorescence microscope equipped with a video camera and an image analysis system (Compix Hamamatsu, Paris, France). Images were acquired with a 60× objective. Fluorescence was quantified over the whole surface of each cell body in six to eight randomly chosen fields per culture well (three wells per condition). The mean intensity of fluorescence per cell was calculated and expressed as the percentage of the corresponding control.
Endoplasmic reticulum-free calcium levels ([Ca2+]er) were quantified in a buffer containing 125 mm NaCl, 5 mm KCl, 1 mm MgSO4, 1 mm Na2HPO4, 5.5 mm glucose, 20 mm NaHCO3, 2 mm l-glutamine, and 20 mm HEPES, pH 7.4, with mag-fura-2 AM (4 μm, 340/380 nm excitation, 510 nm emission; Molecular Probes), a low-affinity ratiometric probe that is suitable for detecting high intraluminal [Ca2+]er levels (Solovyova et al., 2002). The intensity of mag-fura-2 fluorescence, excited at 340 and 380 nm, was determined on paired images using the Compix Hamamatsu software as above. The 340/380 ratios were calculated offline. The mean ratio per cell was then calculated and expressed as the percentage of the corresponding control.
Quantification of organelle clustering. The distribution of ER (labeled with an anti-GRP78 antibody) and mitochondria (labeled with an anti-cytochrome c antibody or with the calcium probe rhod-2) was quantified by image analysis. Unsaturated images were acquired with a 60× objective on an inverted fluorescence microscope equipped with a video camera and an image analysis system (Compix Hamamatsu). Mitochondria or ER were considered to be clustered when a fluorescent patch larger than 10 μm2 had a relative intensity of fluorescence at least 30% higher than the mean fluorescence in the rest of the cytoplasm. All of the cells in six to eight randomly chosen fields per well (∼200 cells per well) were analyzed in each condition (three wells per condition). The results are the mean of three independent experiments.
Electron microscopy. Cells were fixed with 2% paraformaldehyde and 1% glutaraldehyde for 20 min and postfixed with 2.5% glutaraldehyde for 30 min, then with 1% osmium tetroxide, and embedded in Epon. Ultrathin sections (50 nm) were cut and stained with uranyl acetate for examination under a Jeol (Peabody, MA) 1200 EX electron microscope at 80 kV. Ultrathin sections were analyzed for each experimental condition at the same cellular level. The entire cross-section of randomly chosen cells was analyzed at 15,000× magnification (approximately six to eight micrographs per cell). All micrographs corresponding to each cell were analyzed, and all mitochondria or ER that appeared on more than one micrograph were marked to avoid analyzing the same organelle more than once. To estimate the mean distance between ER and mitochondria, the minimum distance between the mitochondrial outer membrane and the nearest ER membrane was measured for all mitochondria, as described previously (Pacher et al., 2000), with the analysis software (Soft Imaging System, Münster, Germany). At least 15 independent cells were analyzed for each condition, and ∼50 mitochondria were analyzed on each section.
Statistics. Data are expressed as mean ± SEM. Statistical analysis was performed by ANOVA. Differences between groups were analyzed with the Student-Newman-Keuls test. Results were considered to be statistically significant at p < 0.05. Experiments were performed in triplicate and repeated at least three times.
Results
Activation of CDK5 is necessary for ceramide-mediated cell death
To study the implication of CDK5 in calcium-dependent neuronal death, we treated primary cultures of mesencephalon (>98% postmitotic neurons) with C2-ceramide (25 μm). An increase in CDK5 activity was observed within the first 5 min of C2-ceramide treatment (Fig. 1A) and was prevented by roscovitine, a CDK inhibitor that is specific for CDK5 in postmitotic neurons (Niethammer et al., 2000). The increase in CDK5 activity was not attributable to increased expression of CDK5 (Fig. 1B) but to cleavage of the CDK5 activator p35 into the more potent p25 (Patrick et al., 1999) that occurred in parallel with the increase in CDK5 activity (Fig. 1B). Inhibition of CDK5 by roscovitine reduced the level of neuronal death observed in C2-ceramide-treated cell cultures, indicating that CDK5 played a role in the apoptotic process (Fig. 1C).
Figure 1.
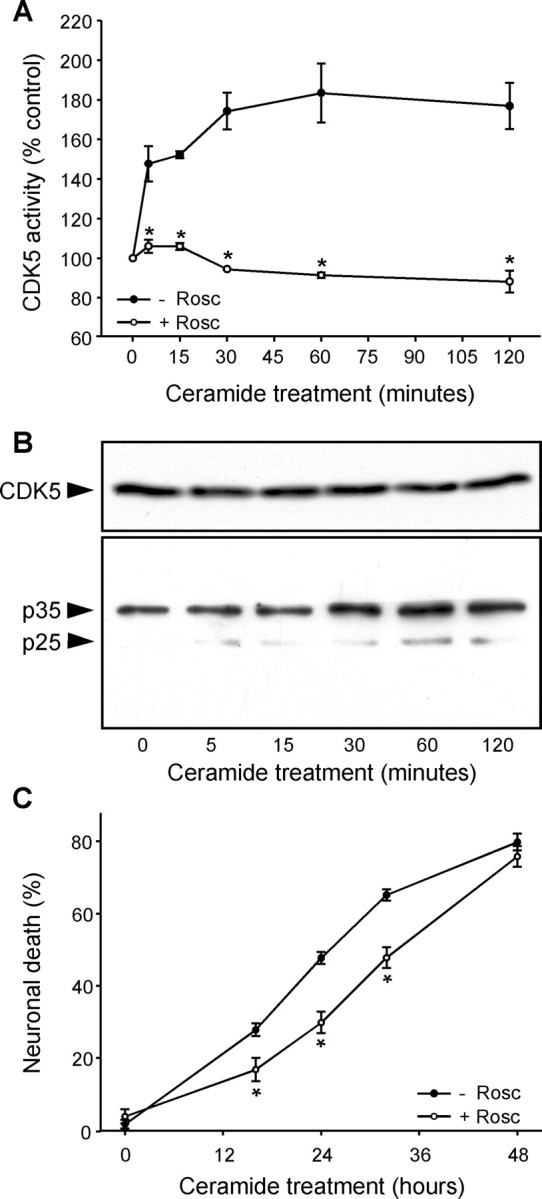
CDK5 activation is an early step in C2-ceramide-mediated neuronal death. A, An increase in CDK5 activity was observed in primary cultures of mesencephalon during the first minutes of C2-ceramide treatment and was blocked by the CDK inhibitor roscovitine (10 μm; Rosc). B, CDK5 protein levels were unchanged by C2-ceramide treatment, but its activator p25 was produced by proteolytic cleavage of p35 with the same time course as the increase in CDK5 activity. C, Inhibition of CDK5 activity by roscovitine reduced C2-ceramide-induced neuronal death. *p < 0.05, significantly different than without roscovitine.
Activation of CDK5 contributes to the transfer of calcium from the ER to mitochondria
To determine whether CDK5 activation was implicated in the transfer of calcium from the ER to mitochondria, we first analyzed free calcium levels in mitochondria ([Ca2+]mit) with the fluorescent cation rhod-2 that accumulates in the matrix of the organelle. In ceramide-treated primary cultures of mesencephalon, [Ca2+]mit increased (Fig. 2A,B). The increase was not attributable to an increase in the uptake of cation rhod-2 by mitochondria, because the membrane potential of mitochondria decreases as [Ca2+]mit increases (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). To demonstrate that the increase in [Ca2+]mit was attributable to a transfer of calcium from the ER, [Ca2+]er was measured with the fluorescent marker mag-fura-2, which is specific for this organelle (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). [Ca2+]er decreased as [Ca2+]mit increased (Fig. 2A,B), strongly suggesting that calcium was transferred from the ER to mitochondria in ceramide-treated cells. Both the decrease in [Ca2+]er and the increase in [Ca2+]mit were significantly reduced by the CDK5 inhibitor roscovitine in primary cultures of mesencephalon (Fig. 2A) and DNK5 (Tsai et al., 1994) in PC12 cells (Fig. 2B). This shows that CDK5 is implicated in the transfer of calcium from the ER to mitochondria during ceramide-induced cell death.
Figure 2.

CDK5 activation is followed by a transfer of calcium from ER to mitochondria. A, B, C2-ceramide induced an increase in [Ca2+]mit (measured with rhod-2), whereas [Ca2+]er (quantified with mag-fura-2) decreased. These effects were reduced when CDK5 was inhibited by roscovitine (Rosc) in mesencephalic neurons (A) or by expression of DNK5 in neuronally differentiated PC12 cells (B). *p < 0.05, significantly different than without roscovitine or DNK5.
The intensity of rhod-2 fluorescence was low in control cells and appeared as small spots that were distributed throughout the cells. In ceramide-treated cells, however, rhod-2 fluorescence was more intense in larger patches localized near the nucleus than in the rest of the cell (Fig. 3). Similar images were obtained when mitochondria were labeled with an antibody against cytochrome c (Fig. 4), suggesting that the pattern is intrinsic to the intracellular distribution of mitochondria, not to the selective uptake of calcium by a subpopulation of the organelle. The regional distribution of rhod-2 fluorescence attributable to ceramide treatment was CDK5 dependent, because it was inhibited by both roscovitine (Fig. 3A) and by DNK5 (Fig. 3B). Thus, CDK5 played a role in both the increase in [Ca2+]mit and its localization. This raises the question as to whether the role of CDK5 in the transfer of calcium from ER to mitochondria is direct or is secondary to the redistribution of ER and mitochondria in the cells.
Figure 3.

The C2-ceramide-induced increase in [Ca2+]mit is regionally localized. rhod-2 fluorescence ([Ca2+]mit) becomes concentrated in patches (white arrowheads) after C2-ceramide treatment in mesencephalic neurons (A) and PC12 cells (B). The patches were not observed in the presence of the CDK5 inhibitors roscovitine (Rosc) and DNK5 (A, B). rhod-2 accumulation in the nucleolus (#) was unrelated to treatments. Scale bars, 10 μm. *p < 0.05, significantly different than control.
Figure 4.

CDK5 activation causes clustering of ER and mitochondria. A, Expression of CDK5 and its activator p25 increased CDK5 activity in PC12 cells. B, C, Expression of CDK5 and its activator p25 in PC12 cells was sufficient to induce the clustering (arrowheads) of ER, labeled with anti-GRP78 antibody, and mitochondria (Mito), labeled with anti-cytochrome c antibody. D-F, The clustering of ER and mitochondria attributable to C2-ceramide treatment was inhibited by roscovitine (Rosc) in mesencephalic neurons (D, E) and by DNK5 expression in PC12 cells (F). Scale bars, 10 μm. *p < 0.05, significantly different than in corresponding controls.
The localized transfer of calcium from ER to mitochondria during cell death implicates CDK5-dependent organelle clustering
To determine whether CDK5 participates directly in the C2-ceramide-induced transfer of calcium from the ER to mitochondria, we coexpressed CDK5 with its activator p25 in PC12 cells. A high level of CDK5 activity was detected 24 h after transfection (Fig. 4A), but no increase in [Ca2+]mit was observed (data not shown). This suggests that CDK5 activation, necessary for the ceramide-induced increase in [Ca2+]mit as seen above (Fig. 2), was not sufficient in itself to produce this effect, which requires concurrent ceramide-dependent activation of caspase-8 that cleaves Bid to t-Bid (Darios et al., 2003) (see below).
We therefore assessed whether CDK5 activation was responsible for the redistribution of ER and mitochondria. The organelles were labeled with anti-GRP78 and anti-cytochrome c antibodies, respectively, and the distribution was quantified as described in Materials and Methods. Expression of CDK5 and its activator p25 was sufficient to induce the clustering of both ER and mitochondria, suggesting that CDK5 was responsible for the regional localization of the increase in [Ca2+]mit (Fig. 4B,C), although not the increase itself. Conversely, in C2-ceramide-treated cells, inhibition of CDK5 by roscovitine (Fig. 4D,E) or expression of DNK5 (Fig. 4F) prevented relocalization of the organelles.
To determine whether the CDK5-dependent clustering of ER and mitochondria contributed to the formation of close contacts between the organelles observed previously on electron micrographs of C2-ceramide-treated cells (Muriel et al., 2000), we measured the distance between mitochondria and the closest segment of ER in cells treated with C2-ceramide in the presence and in the absence of the molecular inhibitor DNK5. As illustrated in Figure 5, ER and mitochondria were closer together in C2-ceramide-treated cells than in control cells (Fig. 5A,B). This effect of C2-ceramide was reduced by expression of the dominant-negative form of CDK5, indicating that it resulted from activation of the kinase. Consistently, coexpression of CDK5 with its activator p25 was sufficient in itself to bring ER and mitochondria closer together (Fig. 5C).
Figure 5.
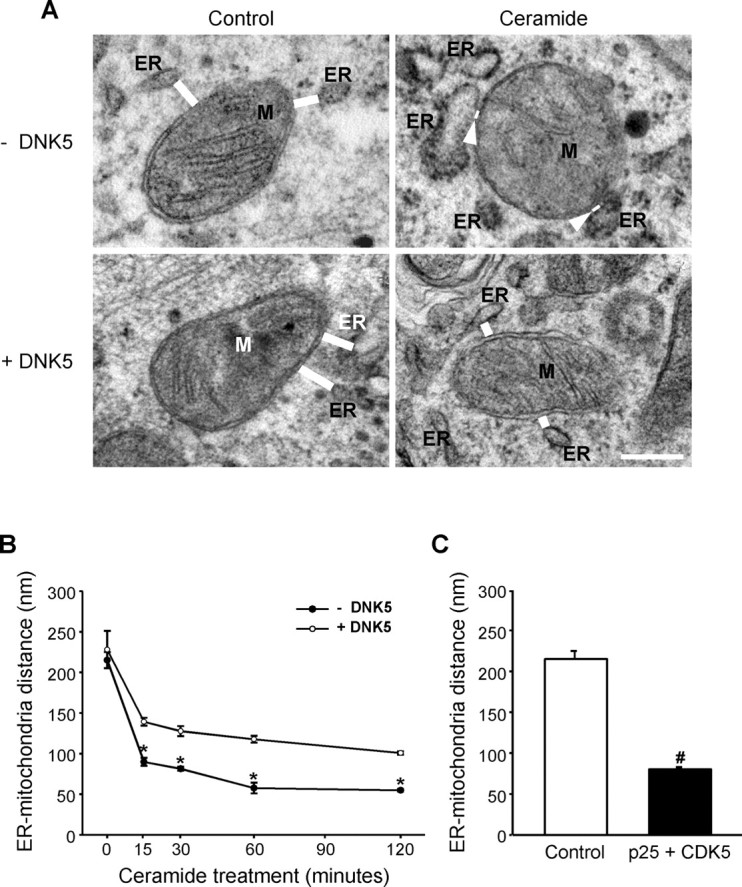
CDK5 activation by C2-ceramide brings ER and mitochondria closer together. A, Electron micrographs of PC12 cells treated with C2-ceramide in the absence or presence of the CDK5 inhibitor DNK5. C2-ceramide decreased the distance between ER and mitochondria (M; distances shown by white bars and arrowheads). DNK5 expression prevented the decrease in distance between organelles. Scale bar, 500 nm. Note the swelling of ER that occurs early after ceramide treatment. B, Time course of the decrease in the distance between ER and mitochondria in C2-ceramide-treated cells measured on electron micrographs. *p < 0.05, significantly lower than in DNK5-expressing cells. C, Expression of CDK5 and its activator p25 decreased the distance between ER and mitochondria. #p < 0.05, significantly lower than in control cells.
Because CDK5 activation induced clustering of ER and mitochondria but was unable to mediate the transfer of calcium between those organelles, we analyzed its relationship to t-Bid, a proapoptotic member of the Bcl-2 family that is known to be involved in the transfer of calcium in ceramide-mediated cell death (Csordas et al., 2002; Darios et al., 2003). As shown in Figure 6A, the expression of t-Bid induced an increase in [Ca2+]mit. However, coexpression of t-Bid with CDK5 and p25 induced a greater increase in [Ca2+]mit (Fig. 6A), suggesting that both molecular pathways contributed to the increase in [Ca2+]mit during ceramide-mediated cell death. Furthermore, whereas t-Bid induced an increase in [Ca2+]mit that was homogeneous throughout the cells (Fig. 6B), coexpression of t-Bid with CDK5/p25 reproduced the localized increase in [Ca2+]mit observed in C2-ceramide-treated cells (Fig. 6B,C).
Figure 6.

CDK5-mediated clustering of organelles enhances the t-Bid-induced increase in [Ca2+]mit. A, Expression of CDK5 and its activator p25 potentiated the t-Bid-induced increase in [Ca2+]mit. B, C, Expression of t-Bid alone caused a homogeneous increase in [Ca2+]mit, whereas coexpression t-Bid with p25/CDK5 causes a localized increase (white arrowhead). rhod-2 accumulation in the nucleolus (#) is unrelated to treatments. *p < 0.05, significantly different than in control; #p < 0.05, significantly different than in t-Bid-expressing cells.
Together, these results showed that CDK5 was responsible for the redistribution of mitochondria and ER observed during ceramide-mediated neuronal death. This results in the formation of privileged contacts between the organelles and potentiates the transfer of calcium mediated by t-Bid.
ER and mitochondria are translocated to the centrosome during neuronal death
Both the actin or microtubule networks are known to play a role in organelle trafficking (Reynolds and Rintoul, 2004). To determine whether they contribute to the CDK5-dependent clustering of ER and mitochondria during ceramide-mediated neuronal death, we examined the effects of depolymerization on the process. Nocodazole that depolymerizes microtubules prevented the C2-ceramide-induced clustering of ER and mitochondria (Fig. 7A), which is the CDK5-dependent component of the ceramide-induced increase in [Ca2+]mit but not the t-Bid-dependent component (Fig. 7B). Cytochalasin D that depolymerizes actin filaments had no effect on either parameter (Fig. 7A,B). This shows that the CDK5-dependent potentiation of the ceramide-induced increase in [Ca2+]mit was attributable to movement of ER and mitochondria that was independent of the actin cytoskeleton but required an intact microtubule system.
Figure 7.

The clustering of ER and mitochondria and the increase in [Ca2+]mit in C2-ceramide-treated cells requires an intact microtubule system and takes place around the centrosome. A, B, Depolymerization of microtubules with nocodazole (1 μm; Noco) prevented the clustering of ER and mitochondria (A) and the increase in [Ca2+]mit (B) in C2-ceramide-treated (Cer) mesencephalic neurons, whereas depolymerization of the actin cytoskeleton with cytochalasin D (2 μm; Cyto D) had no effect. *p < 0.05, significantly different than in control cells; #p < 0.05, significantly different than in C2-ceramide-treated cells. C, Coimmunolabeling of mitochondria and α-tubulin showed that the organelles cluster around the centrosome. Scale bar, 5 μm.
This observation suggests that the region in which the organelles are concentrated may correspond to microtubule organizing center, or centrosome. To test this hypothesis, we coimmunolabeled, in C2-ceramide-treated cells, mitochondria and the centrosome with anti-cytochrome c and anti-γ-tubulin antibodies, respectively. As shown in Figure 7C, mitochondria clustered around the centrosome, consistent with microtubule-dependent transport.
CDK5-dependent phosphorylation of tau on threonine 231 mediates the reorganization of the ER and mitochondria
The microtubule-associated protein tau is a substrate of CDK5 (Baumann et al., 1993) and has been implicated in organelle trafficking (Sato-Harada et al., 1996; Ebneth et al., 1998; Mandelkow et al., 2004). We therefore examined the phosphorylation of tau during ceramide-mediated cell death with a battery of antibodies against epitopes in the tau protein that might be phosphorylated by CDK5. Total tau levels in whole-cell lysates, detected with the tau-5 antibody, did not change during C2-ceramide treatment, indicating that its expression was not affected. Tau phosphorylated on threonine 231 (T231; numbered according to the human tau isoform containing 441 amino acids), labeled by the antibody AT180, appeared within the first 15 min of treatment (Fig. 8A). The phosphorylation of T231 was reduced by the CDK5 inhibitor roscovitine, confirming that it was phosphorylated by this enzyme under our conditions, in accordance with published data (Hamdane et al., 2003). No phosphorylation of tau was observed with the epitope-specific antibodies used: AT270 (T181), AT-8 (S202/T205), and AD2 (S396/S404) (data not shown).
Figure 8.
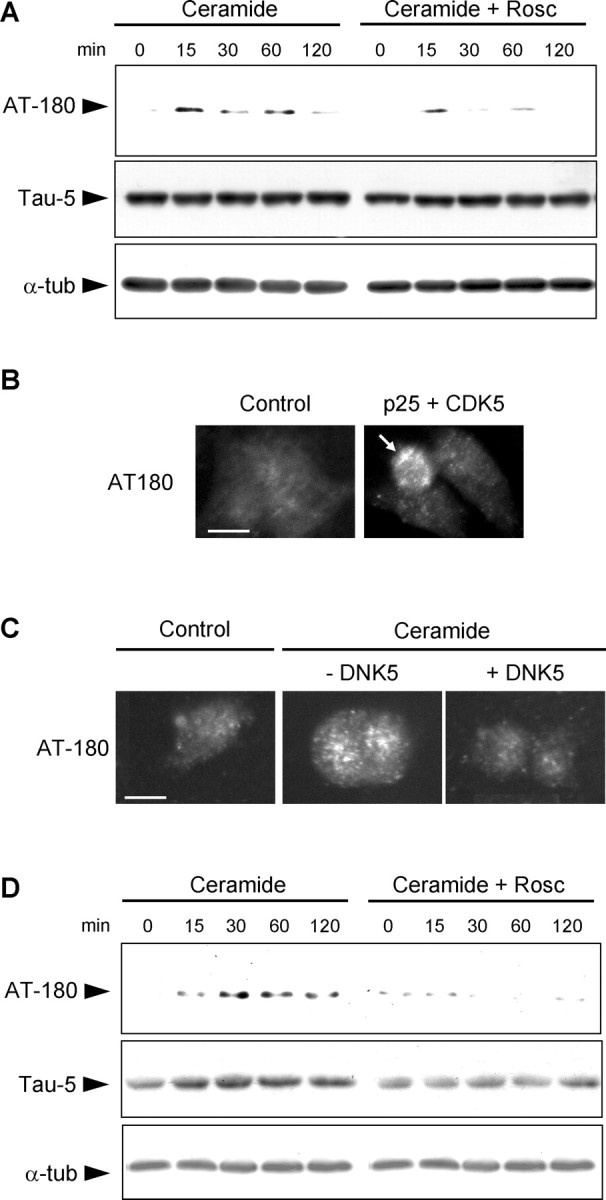
CDK5 phosphorylates tau on threonine 231. A, Western blot of whole-cell lysates shows that phosphorylation of tau on T231 (AT180 immunoreactivity) was detected within 15 min in C2-ceramide-treated mesencephalic neurons and was prevented by roscovitine (Rosc). Total tau levels (Tau-5 immunoreactivity) and total α-tubulin (α-tub) levels did not change. B, Expression of CDK5 and its activator p25 in PC12 cells resulted in the phosphorylation of T231 in transfected cells (arrow). C, Inhibition of CDK5 by expression of DNK5 in PC12 cells prevented the C2-ceramide-induced phosphorylation of T231. Scale bar, 10 μm. D, Tau phosphorylated on T231 (AT180 immunoreactivity) was recovered in the microtubule-free fraction of soluble proteins from mesencephalic neurons treated with C2-ceramide, indicating that it had been released from microtubules. Inhibition of CDK5 by roscovitine prevented the appearance of T231-phosphorylated tau in the microtubule-free fraction. Total tau levels (Tau-5 immunoreactivity) increased accordingly, indicating that tau was released from microtubule after C2-ceramide treatment. α-Tubulin levels remained constant in this microtubule-free fraction, indicating that microtubules were not depolymerized during the treatment.
To further confirm that T231 was phosphorylated by CDK5, we looked for AT180 immunoreactivity in cells expressing CDK5 and its activator p25. Activation of CDK5 in transfected cells resulted in the appearance of AT180 immunoreactivity (Fig. 8B), indicating that T231 was phosphorylated by the kinase. Conversely, expression of DNK5 in C2-ceramide-treated cells prevented the appearance of AT180 immunoreactivity (Fig. 8C). CDK5 was therefore responsible for T231 phosphorylation during ceramide-mediated neuronal death.
We then assessed the consequences of tau phosphorylation on T231, which has been shown to decrease the binding of tau to microtubules (Sengupta et al., 1998; Cho and Johnson, 2004). To determine whether tau was released from microtubules after C2-ceramide-treatment, we examined a microtubule-free fraction of cell lysates for the presence of unbound tau. Tau phosphorylated on T231 increased in this fraction during C2-ceramide treatment (Fig. 8D), as did the levels of total tau. In both cases, the increase was prevented by roscovitine. As in whole-cell lysates, the levels of free α-tubulin in microtubule-free fraction did not increase during C2-ceramide treatment, indicating that the microtubules were not depolymerized during the treatment (Fig. 8D). These results suggest that phosphorylation on T231 induces the displacement of tau from microtubules into the soluble fraction. However, phosphorylation and release of tau from microtubules was not associated with aggregation of the protein (data not shown).
To further demonstrate that phosphorylation on T231 was necessary for the release of tau from microtubules during ceramide-mediated cell death, we expressed wild-type tau or tau in which threonine 231 was mutated to alanine (T231A) and could no longer be phosphorylated at this site (Cho and Johnson, 2004). In cells expressing wild-type tau, C2-ceramide treatment increased the level of total tau in the microtubule-free fraction (Fig. 9A). This was not the case in cells expressing tau T231A (Fig. 9A), showing that phosphorylation of T231 is required for the release of tau from microtubules during C2-ceramide treatment.
Figure 9.
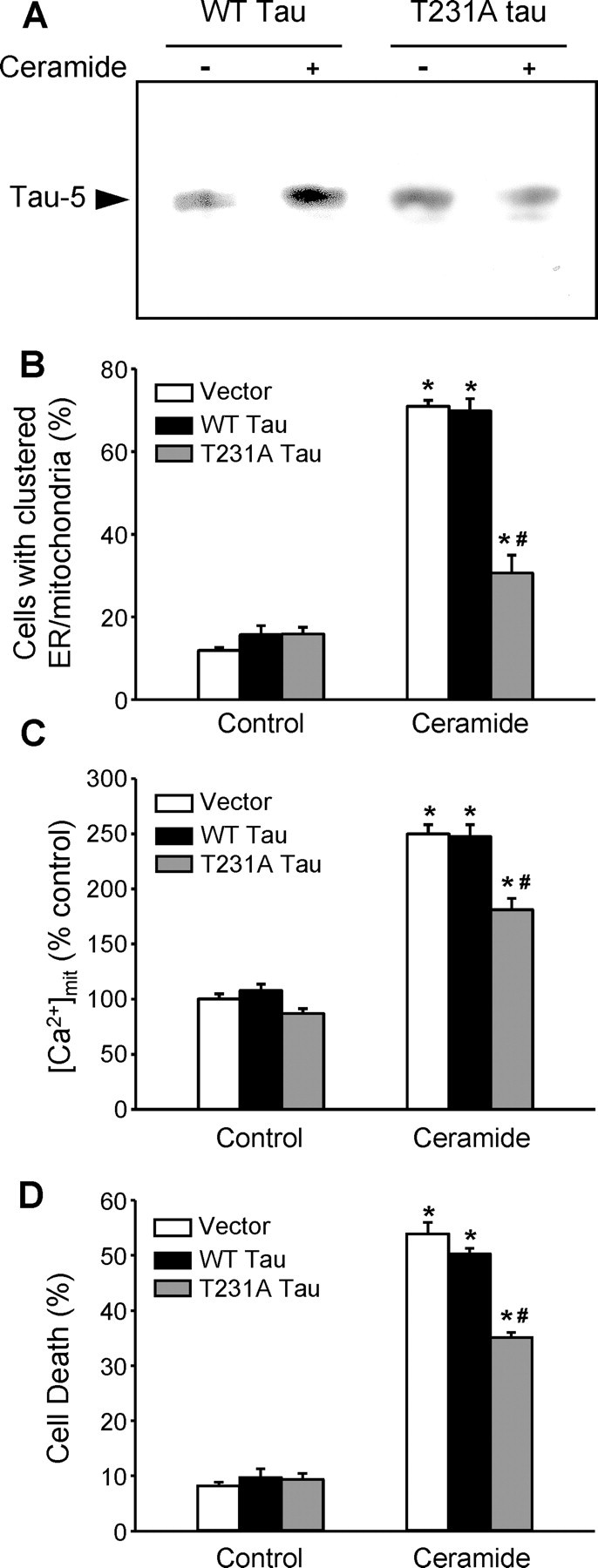
Phosphorylation of tau on T231 contributes to the clustering of mitochondria and ER, the increase in [Ca2+]mit, and cell death in C2-ceramide-treated PC12 cells. A, In PC12 cells expressing wild-type (WT) human tau or human tau with a T231A mutation that cannot be phosphorylated at this position, only wild-type tau was released in the microtubule-free soluble fraction of C2-ceramide-treated cells, indicating that release of tau from microtubules did not occur in the absence of T231 phosphorylation. B, The C2-ceramide-induced clustering of ER and mitochondria was reduced in cells expressing T231A tau. C, The C2-ceramide-induced increase in [Ca2+]mit was reduced in cells expressing T231A tau. D, Cell death was reduced in cells expressing T231A tau. *p < 0.05, significantly different than corresponding controls; #p < 0.05, significantly different than in C2-ceramide-treated cells transfected with empty vector or expressing wild-type tau.
Finally, to assess the relationship between tau phosphorylation, organelle clustering, and the increase in [Ca2+]mit, we expressed wild-type tau or tau T231A in PC12 cells. Tau T231A reduced the ceramide-induced clustering of ER and mitochondria around the centrosome compared with wild-type tau (Fig. 9B). It also prevented the CDK5-dependent component of the ceramide-induced increase in [Ca2+]mit (Fig. 9C) and the cell death that ensues (Fig. 9D). These results demonstrate that CDK5-dependent phosphorylation of tau on T231 was responsible for these events.
Discussion
In this study, we used pharmacological and molecular inhibitors of CDK5, roscovitine and DNK5, to show that CDK5 plays a role in the lethal transfer of calcium from ER to mitochondria that occurs during ceramide-mediated neuronal death. CDK5 did not itself mediate the transfer of calcium from ER to mitochondria. However, it enhanced the transfer mediated by proapoptotic proteins such as t-Bid by promoting the reorganization of the ER/mitochondria network. This involved the phosphorylation of tau on T231, which induced the release of tau from microtubules facilitating organelle movement.
Calcium transfer from ER to mitochondria
Calcium can be transferred from the ER to mitochondria at contact sites between the organelles through the formation of microdomains in which calcium reaches high concentrations (Rizzuto et al., 1993; Hajnoczky et al., 1995; Rizzuto et al., 1998). Ceramide is a well studied inducer of this form of calcium transfer (Szalai et al., 1999; Pinton et al., 2001; Rapizzi et al., 2002). In our previous study and in others, t-Bid was shown to play an essential role in the transfer of calcium from the ER to mitochondria (Csordas et al., 2002; Darios et al., 2003), although the mechanism has not been fully elucidated. Bak or Bax, which are also proapoptotic members of the Bcl-2 family, may also be involved (Nutt et al., 2002a), perhaps after t-Bid-dependent oligomerization and the formation of transmembrane channels (Eskes et al., 2000; Wei et al., 2000), although the voltage-dependent anion channel has been implicated as well (Rapizzi et al., 2002).
We now show that CDK5 activation is also implicated in the ceramide-mediated neuronal death pathway in which it enables the transfer of calcium from the ER to mitochondria to reach lethal levels. CDK5 was not directly responsible for calcium transfer, because CDK5 activation in the absence of overexpressed t-Bid or Bid cleavage by ceramide-dependent caspase-8 activation did not induce an increase in [Ca2+]mit, and CDK5 inhibition did not prevent the increase in [Ca2+]mit induced by t-Bid expression (data not shown). CDK5 did, however, enhance the formation of close contacts between the ER and mitochondria, which is the morphological basis for calcium transfer.
These contacts occur under physiological conditions (Rizzuto et al., 1998; Reynolds and Rintoul, 2004). However, they are reinforced during ceramide-mediated cell death (Muriel et al., 2000). We have now shown that the reinforcement of these contacts is CDK5 dependent, because the decrease in distance between the organelles was inhibited in the presence of a pharmacological inhibitor of the enzyme or a dominant-negative form of the kinase. Conversely, activation of CDK5 alone, by coexpression of the kinase and its activator p25, was sufficient to decrease the distance between the organelles, confirming that the process is mediated by CDK5. A similar association between ER and mitochondria has been reported to occur in the presence of micromolar cytosolic calcium (Wang et al., 2000; Yi et al., 2004), a condition in which the CDK5 activator p35 can be cleaved to p25 by the calcium-activated calpain (Patrick et al., 1999).
The CDK5-dependent decrease in the distance between ER and mitochondria during ceramide-mediated cell death was correlated with the CDK5-dependent clustering of the organelles around the centrosome. When this was blocked by nocodazole treatment, inhibition of CDK5, or expression of tau T231A that cannot be phosphorylated by CDK5, calcium transfer was reduced. It was not completely eliminated, however, because t-Bid was still present. It remains to be determined whether the greater density of organelles around the centrosome after CDK5 activation increases the probability that ER and mitochondria would form close contacts, or whether the formation of specialized contacts between the organelles is itself a regulated process. In the first case, cell death would occur by chance because mitochondria and ER are crowded together. Potentiation of the transfer of calcium from the ER to mitochondria and the ensuing cell death would simply be the unfortunate consequences of their increased proximity. In the second case, the movement of ER and mitochondria toward the centrosome would be part of a coordinated process, the very aim of which would be to increase mitochondrial-free calcium levels to the point at which they induce cell death. To answer this question, it will be necessary to determine what lies upstream activation of both CDK5 and t-Bid in this cell-death model and how mitochondria and ER recognize each other to form the privileged contacts required for this process. In this respect, a recent study has shown that the ER protein PACS-2 (phosphofurin acidic cluster sorting protein-2) plays a role in the association between ER and mitochondria, leading to Bid cleavage (Simmen et al., 2005).
Tau phosphorylation and reorganization of the ER/mitochondria network
We have shown here that the translocation of ER and mitochondria toward the centrosome is the consequence of CDK5 activation and phosphorylation of tau on T231. The specificity of this phosphorylation shows that organelle movement is part of a regulated process, not the result of generalized dysfunction in dying neurons. Indeed, in C2-ceramide-treated cells, neuronal death is not observed until much later (∼12 h after the beginning of treatment), whereas CDK5 activation, tau phosphorylation, and the increase in [Ca2+]mit all occur within the first 30 min.
T231-phosphorylated tau was found in the microtubule-free soluble protein fraction of cell extracts, in accordance with previous results showing that phosphorylation of this epitope decreases the binding affinity of tau for microtubules (Sengupta et al., 1998; Cho and Johnson, 2004). However, phosphorylation of tau on other sites (S202/T205, S214, and S262) has also been shown to decrease the binding affinity of tau for microtubules (Illenberger et al., 1998; Wada et al., 1998; Biernat et al., 2002; Takahashi et al., 2003). Kinases other than CDK5 may also play a role in tau phosphorylation (Sergeant et al., 2005) and may explain the partial effect of CDK5 inhibition. One of the most studied kinase, GSK3β (glycogen synthase kinase-3), phosphorylates tau on several epitopes (Sperber et al., 1995) and has been reported to play a role in the regulation of tau binding to microtubules (Cho and Johnson, 2004) and neuronal death (Hetman et al., 2000). GSK3β inhibition, however, had no effect in our cell culture model (data not shown). Perhaps the phosphorylation of S235, which has been suggested to be a prerequisite for T231 phosphorylation by GSK3β (Cho and Johnson, 2004), does not occur in our model.
Because tau has been proposed to consolidate microtubules (Drechsel et al., 1992), tau phosphorylation on T231 may decrease microtubule stability. However, depolymerization of microtubules does not seem to occur after C2-ceramide treatment (data not shown). On the contrary, treatment with nocodazole to depolymerize microtubules blocked both clustering of ER and mitochondria as well as mitochondrial calcium uptake, indicating that the microtubule network must be intact for the cell-death process to proceed.
Tau binding to microtubules has also been proposed to inhibit dynein-dependent and more importantly kinesin-dependent (Ebneth et al., 1998; Trinczek et al., 1999; Stamer et al., 2002) transport, resulting in a net clustering of mitochondria toward the centrosome. In our study, to the contrary, clustering was correlated with a decrease in tau binding to microtubules after CDK5 activation and phosphorylation of tau on T231. Expression of T231A tau that prevented its release from microtubules partially inhibited C2-ceramide-induced clustering of ER and mitochondria, suggesting that a decrease in tau binding to microtubules was necessary for organelle movement. This is consistent with previous studies showing that phosphorylation of tau by cAMP-dependent protein kinase or MARK (MT-affinity-regulating kinase)/Par1 (protease-activated receptor 1) kinase decreases tau binding to microtubules and facilitates movement along this network (Sato-Harada et al., 1996; Mandelkow et al., 2004).
It should be noted, however, that the effect of T231A tau expression on organelle clustering was only partial. Other CDK5 substrates might, therefore, also be involved; for example, Nudel, a protein that associates with cytoplasmic dynein (Niethammer et al., 2000; Sasaki et al., 2000). Phosphorylation of Nudel by CDK5 has been proposed to activate dynein-dependent transport (Niethammer et al., 2000), which could also explain the clustering of transported organelles toward the centrosome after CDK5 activation. In addition, multiple kinases [GSK3beta, MARK, PKA (protein kinase A), tau-tubulin kinase, SAPK (stress-activated protein kinase), CaMPK-II (calmodulin-dependent protein kinase II), Casein kinases I and II,...] and protein phosphatases (1, 2A, 2B, 2C,...) regulate tau phosphorylation and have been associated with neurodegeneration or cell stress (Sergeant et al., 2005). The contributions of these enzymes to the mitochondrial and ER clustering in the ceramide-dependent neuronal death pathway now need to be elucidated.
Mislocalization of mitochondria, observed in glutamate-treated primary cultures of forebrain neurons, has been hypothesized to contribute to neuronal death because of a defect in ATP distribution (Rintoul et al., 2003). We described here an alternative mechanism in which CDK5-dependent phosphorylation of tau, and possibly other substrates, contributes to an abnormal clustering of ER and mitochondria around the centrosome. Although this could alter the ATP supply to some parts of the neurons, it also potentiates the transfer of calcium from ER to mitochondria to levels that lead to mitochondrial dysfunction, cytochrome c release, caspase activation, and apoptosis (Duchen, 2000; Darios et al., 2003).
Footnotes
F.D. was supported by a Verum Foundation fellowship. We thank Dr. L. H. Tsai for the p25, CDK5, and DNK5 constructs, Dr. G. V. Johnson for the WT tau and T231A constructs, and Dr. M. Bornens for the polyclonal anti-γ-tubulin antibody.
Correspondence should be addressed to Dr. Merle Ruberg, Institut National de la Santé et de la Recherche Médicale U679 (formerly U289), Hôpital de la Salpêtrière, 47 Boulevard de l'Hôpital, 75013 Paris, France. E-mail: ruberg@ccr.jussieu.fr.
F. Darios's present address: Medical Research Council Laboratory of Molecular Biology, Hills Road, Cambridge CB2 2QH, UK. E-mail: darios@mrc-lmb.cam.ac.uk.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254159-10$15.00/0
References
- Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD (2000) Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci USA 97: 2910-2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann K, Mandelkow EM, Biernat J, Piwnica-Worms H, Mandelkow E (1993) Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett 336: 417-424. [DOI] [PubMed] [Google Scholar]
- Biernat J, Wu YZ, Timm T, Zheng-Fischhofer Q, Mandelkow E, Meijer L, Mandelkow EM (2002) Protein kinase MARK/PAR-1 is required for neurite outgrowth and establishment of neuronal polarity. Mol Biol Cell 13: 4013-4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugg B, Michel PP, Agid Y, Ruberg M (1996) Ceramide induces apoptosis in cultured mesencephalic neurons. J Neurochem 66: 733-739. [DOI] [PubMed] [Google Scholar]
- Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 33: 95-130. [DOI] [PubMed] [Google Scholar]
- Cho JH, Johnson GV (2004) Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3beta (GSK3beta) plays a critical role in regulating tau's ability to bind and stabilize microtubules. J Neurochem 88: 349-358. [DOI] [PubMed] [Google Scholar]
- Csordas G, Thomas AP, Hajnoczky G (1999) Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J 18: 96-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Madesh M, Antonsson B, Hajnoczky G (2002) tcBid promotes Ca2+ signal propagation to the mitochondria: control of Ca2+ permeation through the outer mitochondrial membrane. EMBO J 21: 2198-2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darios F, Lambeng N, Troadec JD, Michel PP, Ruberg M (2003) Ceramide increases mitochondrial free calcium levels via caspase 8 and Bid: role in initiation of cell death. J Neurochem 84: 643-654. [DOI] [PubMed] [Google Scholar]
- Delobel P, Mailliot C, Hamdane M, Sambo AV, Begard S, Violleau A, Delacourte A, Buee L (2003) Stable-tau overexpression in human neuroblastoma cells: an open door for explaining neuronal death in tauopathies. Ann NY Acad Sci 1010: 623-634. [DOI] [PubMed] [Google Scholar]
- Drechsel DN, Hyman AA, Cobb MH, Kirschner MW (1992) Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell 3: 1141-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR (2000) Mitochondria and calcium: from cell signalling to cell death. J Physiol (Lond) 529: 57-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E (1998) Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol 143: 777-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskes R, Desagher S, Antonsson B, Martinou JC (2000) Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol 20: 929-935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP (1995) Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82: 415-424. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Csordas G, Madesh M, Pacher P (2000) The machinery of local Ca2+ signalling between sarco-endoplasmic reticulum and mitochondria. J Physiol (Lond) 529: 69-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdane M, Sambo AV, Delobel P, Begard S, Violleau A, Delacourte A, Bertrand P, Benavides J, Buee L (2003) Mitotic-like tau phosphorylation by p25-Cdk5 kinase complex. J Biol Chem 278: 34026-34034. [DOI] [PubMed] [Google Scholar]
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z (2000) Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J Neurosci 20: 2567-2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illenberger S, Zheng-Fischhofer Q, Preuss U, Stamer K, Baumann K, Trinczek B, Biernat J, Godemann R, Mandelkow EM, Mandelkow E (1998) The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: implications for Alzheimer's disease. Mol Biol Cell 9: 1495-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow EM, Thies E, Trinczek B, Biernat J, Mandelkow E (2004) MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J Cell Biol 167: 99-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muriel MP, Lambeng N, Darios F, Michel PP, Hirsch EC, Agid Y, Ruberg M (2000) Mitochondrial free calcium levels (Rhod-2 fluorescence) and ultrastructural alterations in neuronally differentiated PC12 cells during ceramide-dependent cell death. J Comp Neurol 426: 297-315. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee MS, Morabito M, Tsai LH (2000) NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron 28: 697-711. [DOI] [PubMed] [Google Scholar]
- Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH (1996) The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev 10: 816-825. [DOI] [PubMed] [Google Scholar]
- Nutt LK, Pataer A, Pahler J, Fang B, Roth J, McConkey DJ, Swisher SG (2002a) Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca2+ stores. J Biol Chem 277: 9219-9225. [DOI] [PubMed] [Google Scholar]
- Nutt LK, Chandra J, Pataer A, Fang B, Roth JA, Swisher SG, O'Neil RG, McConkey DJ (2002b) Bax-mediated Ca2+ mobilization promotes cytochrome c release during apoptosis. J Biol Chem 277: 20301-20308. [DOI] [PubMed] [Google Scholar]
- Pacher P, Csordas P, Schneider T, Hajnoczky G (2000) Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J Physiol (Lond) 529: 553-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschen W (2003) Mechanisms of neuronal cell death: diverse roles of calcium in the various subcellular compartments. Cell Calcium 34: 305-310. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402: 615-622. [DOI] [PubMed] [Google Scholar]
- Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R (2001) The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J 20: 2690-2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, Tuft RA, Fogarty KE, Rizzuto R (2002) Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol 159: 613-624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds IJ, Rintoul GL (2004) Mitochondrial stop and go: signals that regulate organelle movement. Sci STKE 2004: PE46. [DOI] [PubMed] [Google Scholar]
- Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ (2003) Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci 23: 7881-7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Murgia M, Pozzan T (1993) Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262: 744-747. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280: 1763-1766. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Shionoya A, Ishida M, Gambello MJ, Yingling J, Wynshaw-Boris A, Hirotsune S (2000) A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron 28: 681-696. [DOI] [PubMed] [Google Scholar]
- Sato-Harada R, Okabe S, Umeyama T, Kanai Y, Hirokawa N (1996) Microtubule-associated proteins regulate microtubule function as the track for intracellular membrane organelle transports. Cell Struct Funct 21: 283-295. [DOI] [PubMed] [Google Scholar]
- Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K (1998) Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys 357: 299-309. [DOI] [PubMed] [Google Scholar]
- Sergeant N, Delacourte A, Buee L (2005) Tau protein as a differential biomarker of tauopathies. Biochim Biophys Acta 1739: 179-197. [DOI] [PubMed] [Google Scholar]
- Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, Thomas G (2005) PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J 24: 717-729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyova N, Veselovsky N, Toescu EC, Verkhratsky A (2002) Ca2+ dynamics in the lumen of the endoplasmic reticulum in sensory neurons: direct visualization of Ca2+-induced Ca2+ release triggered by physiological Ca2+ entry. EMBO J 21: 622-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperber BR, Leight S, Goedert M, Lee VM (1995) Glycogen synthase kinase-3 beta phosphorylates tau protein at multiple sites in intact cells. Neurosci Lett 197: 149-153. [DOI] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM (2002) Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol 156: 1051-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ (1998) Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci 1: 366-373. [DOI] [PubMed] [Google Scholar]
- Szalai G, Krishnamurthy R, Hajnoczky G (1999) Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J 18: 6349-6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Saito T, Hisanaga S, Pant HC, Kulkarni AB (2003) Tau phosphorylation by cyclin-dependent kinase 5/p39 during brain development reduces its affinity for microtubules. J Biol Chem 278: 10506-10515. [DOI] [PubMed] [Google Scholar]
- Trinczek B, Ebneth A, Mandelkow EM, Mandelkow E (1999) Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J Cell Sci 112: 2355-2367. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Delalle I, Caviness Jr VS, Chae T, Harlow E (1994) p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 371: 419-423. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Toescu EC (2003) Endoplasmic reticulum Ca2+ homeostasis and neuronal death. J Cell Mol Med 7: 351-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y, Ishiguro K, Itoh TJ, Uchida T, Hotani H, Saito T, Kishimoto T, Hisanaga S (1998) Microtubule-stimulated phosphorylation of tau at Ser202 and Thr205 by cdk5 decreases its microtubule nucleation activity. J Biochem (Tokyo) 124: 738-746. [DOI] [PubMed] [Google Scholar]
- Wang HJ, Guay G, Pogan L, Sauve R, Nabi IR (2000) Calcium regulates the association between mitochondria and a smooth subdomain of the endoplasmic reticulum. J Cell Biol 150: 1489-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, Korsmeyer SJ (2000) tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev 14: 2060-2071. [PMC free article] [PubMed] [Google Scholar]
- Weishaupt JH, Kussmaul L, Grotsch P, Heckel A, Rohde G, Romig H, Bahr M, Gillardon F (2003) Inhibition of CDK5 is protective in necrotic and apoptotic paradigms of neuronal cell death and prevents mitochondrial dysfunction. Mol Cell Neurosci 24: 489-502. [DOI] [PubMed] [Google Scholar]
- Yi M, Weaver D, Hajnoczky G (2004) Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol 167: 661-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YL, Kesavapany S, Gravell M, Hamilton RS, Schubert M, Amin N, Albers W, Grant P, Pant HC (2005) A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J 24: 209-220. [DOI] [PMC free article] [PubMed] [Google Scholar]