

Abstract
Parkinson's disease (PD) is a neurodegenerative disorder characterized by selective loss of dopaminergic neurons and the presence of Lewy bodies. Previous reports have shown that α-synuclein deposited in brain tissue from individuals with synucleinopathy is extensively phosphorylated at Ser-129. Here, we investigate the role of phosphorylation of α-synuclein in the formation of inclusions involving synphilin-1 and parkin using site-directed mutagenesis to change Ser-129 of α-synuclein to alanine (S129A) to abolish phosphorylation at this site. Coexpression of wild-type α-synuclein and synphilin-1 in human neuroblastoma SH-SY5Y cells yielded cytoplasmic eosinophilic inclusions with some features resembling Lewy bodies, whereas coexpression of S129A α-synuclein and synphlin-1 formed few or no inclusions. Moreover, coexpression of parkin with α-synuclein and synphilin-1 formed more ubiquitinated inclusions, but these inclusions decreased with expression of S129A α-synuclein instead of wild-type α-synuclein. Coimmunoprecipitation assays revealed a decreased interaction of S129A α-synuclein with synphilin-1 compared with wild-type α-synuclein. Expression of S129A α-synuclein instead of wild-type α-synuclein also decreased the association of synphilin-1 and parkin and subsequently reduced the parkin-mediated ubiquitination of synphilin-1 and the formation of ubiquitinated inclusions. Treatment of SH-SY5Y cells with H2O2 increased α-synuclein phosphorylation and enhanced the formation of inclusions formed by coexpression of α-synuclein, synphilin-1, and parkin, whereas treatment with the casein kinase 2 inhibitor 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole had the opposite affect. These results indicate that phosphorylation of α-synuclein at S129 may be important for the formation of inclusions in PD and related α synucleinopathies.
Keywords: α-synuclein, Parkinson's disease, synphilin-1, parkin, ubiquitin, eosinophilic inclusion, Lewy body
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative disorder characterized by tremor, rigidity, and bradykinesia. The two pathological hallmarks of PD are selective loss of dopaminergic neurons in the substantia nigra and the presence of cytoplasmic eosinophilic inclusions termed Lewy bodies (Pollanen et al., 1993; Galvin et al., 1999; Chung et al., 2001b; Taylor et al., 2002; Braak et al., 2003; Dawson and Dawson, 2003). α-Synuclein is a major protein component in Lewy bodies and Lewy neurites of sporadic PD (Spillantini et al., 1997, 1998; Spillantini and Goedert, 2000; Kahle et al., 2001; Kotzbauer et al., 2001; Forman et al., 2004). Three different mutations in the α-synuclein gene (A53T, A30P, and E46K) cause autosomal-dominant hereditary PD (Polymeropoulos et al., 1997; Farrer et al., 1998; Kruger et al., 1998; Zarranz et al., 2004; Greenbaum et al., 2005). Recent reports have shown that α-synuclein in Lewy bodies is hyperphosphorylated at S129, the major phosphorylation site of α-synuclein (Fujiwara et al., 2002; Hasegawa et al., 2002). Hyperphosphorylated α-synuclein at S129 is also present in synucleinopathy lesions in α-synuclein transgenic mice and flies (Kahle et al., 2002; Neumann et al., 2002; Takahashi et al., 2003). These reports suggest a role for phosphorylation of α-synuclein in PD pathogenesis.
Lewy bodies are a permanent pathologic hallmark of PD, but the underlying molecular mechanisms leading to the formation of Lewy bodies are poorly understood (Galvin et al., 1999). We reported previously that synphilin-1, a protein that interacts with α-synuclein (Engelender et al., 1999), is highly enriched in Lewy bodies (Wakabayashi et al., 2000). Cotransfection of α-synuclein and synphilin-1 in human embryonic kidney 293 (HEK 293) cells yields cytoplasmic eosinophilic inclusions resembling Lewy bodies (Engelender et al., 1999; Lee et al., 2004). We also identified synphilin-1 as a substrate of parkin (a ubiquitin ligase, also present in Lewy bodies) (Chung et al., 2001a; Shimura et al., 2001; Schlossmacher et al., 2002). Recently, we demonstrated that parkin mediates the ubiquitination of proteins within the inclusion predominantly via lysine 63-linked polyubiquitin chain formation and showed that lysine 63-linked ubiquitination of synphilin-1 did not lead to synphilin-1 degradation but instead enhanced the formation of ubiquitinated inclusions (Lim et al., 2005). However, whether the phosphorylation of α-synuclein influences the formation of these cytoplasmic inclusions is unclear.
In this study, we investigated the role of α-synuclein phosphorylation in the formation of eosinophilic cytoplasmic inclusions using cultured cells. Expression of S129A α-synuclein (a mutant that cannot be phosphorylated at S129) decreased the formation of eosinophilic inclusions in cytosol arising from coexpression of synphlilin-1, relative to what was seen when expressing wild-type α-synuclein. Furthermore, expression of S129A α-synuclein instead of wild-type α-synuclein reduced the ubiquitinated inclusions formed by coexpression parkin and synphilin-1. Treatment with H2O2 increased α-synuclein phosphorylation and enhanced the formation of cytoplasmic inclusions, whereas treatment with the casein kinase 2 (CK2) inhibitor 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB) decreased these effects. These results strongly support the notion that phosphorylation of α-synuclein at S129 may be important for inclusion formation in PD pathogenesis.
Materials and Methods
Reagents. Media for cell culture and LipofectAMINE Plus reagent were from Invitrogen (Carlsbad, CA). Anti-α-synuclein monoclonal antibody (directed against amino acids 15-123 of rat α-synuclein but exhibiting good reaction with human α-synuclein) was from BD Biosciences (Palo Alto, CA). Anti-ubiquitin polyclonal antibody was from Dako (High Wycombe, UK); anti-myc polyclonal antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA); anti-phospho-S129 α-synuclein antibody was developed as described previously (Fujiwara et al., 2002). Cyanine 3 (Cy3)-conjugated goat anti-mouse IgG and fluorescein (FITC)-conjugated goat anti-rabbit IgG were from Jackson ImmunoResearch (West Grove, PA). H2O2 was from Sigma (St. Louis, MO); DRB was from Biomol (Plymouth Meeting, PA); 1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylmercapto) butadiene (U0126) was from Promega (Madison, WI); and wortmannin, 9,12-epoxy-1H-diindolo(1,2,3-fg:3′,2′,1′-kl)pyrrolo (3,4-i)(1,6)benzodiazocine-10-carboxylic acid,2, 3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-, hexyl ester, (9α,10β,12α) (KT5720), and 3-[1-[3-(amidinothio)propyl]-1H-indol-3-yl]-3-(1-methyl-1H-indol-3-yl)maleimide bisindolylmaleimide IX, methanesulfonate (Ro-318220) were from Calbiochem (La Jolla, CA).
Plasmids, site mutation, and transfection. Plasmids expressing α-synuclein, myc-synphilin-1, Flag-parkin, and mutant Flag-parkin (T240R, P471L, and Q311 stop) were described previously (Engelender et al., 1999; Chung et al., 2001a). The QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) was used to make point mutations of α-synuclein S129A and S129E, according to the manufacturer's protocols. All of the resulting constructs with the S129 site alteration were confirmed by sequencing. Transient transfections were performed with LipofectAMINE Plus (Invitrogen) according to the manufacturer's protocol.
Cell cultures, immunoprecipitation, and Western blot analysis. Undifferentiated human neuroblastoma SH-SY5Y cells were grown in DMEM containing 10% fetal bovine serum, 1× minimal essential medium non-essential amino acids, and 1× antibiotic-antimycotic (100 U/ml penicillin, 100 μg/ml streptomycin, and 2.5 μg/ml Fungizone) at 37°C under 5% CO2/95% air. Transfected cells were harvested in lysis buffer A [20 mm HEPES, pH 7.4, 2 mm EGTA, 50 mm β-glycerol phosphate, 1% Triton X-100, 10% glycerol, 1 mm dithiothreitol (DTT), 1 mm phenyl-methylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mm Na3VO4, and 5 mm NaF] as Triton-soluble fractions. Triton-insoluble fractions, which mainly contained the aggregated proteins, were dissolved in buffer A containing 2% SDS. Immunoprecipitations (IPs) from the transfected cell lysates were performed with anti-myc or anti-hemagglutinin (anti-HA) antibody and protein G PLUS/protein A-agarose (Amersham Biosciences, Arlington Heights, IL), then washed six times in lysis buffer A. The resulting immunoprecipitates and cell lysates were resolved on 4 -12% NuPAGE Bis-Tris gels or 10% SDS Tris-glycine gels and transferred onto polyvinylidene difluoride membranes (Invitrogen). The membranes were blocked in TBST (10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.1% Tween 20) containing 5% nonfat milk and then probed with different antibodies. Proteins were detected by using enhanced chemiluminescence reagents (NEN, Boston, MA).
Hematoxylin and eosin staining, immunohistochemistry, and thioflavin S staining. We performed hematoxylin and eosin (H & E) staining according to the manufacturer's instructions (Sigma). For immunohistochemistry, cells were fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.2% Triton X-100 for 15 min at room temperature and processed as described previously (Tanaka et al., 2001). Cell preparations were incubated with primary antibodies anti-α-synuclein, anti-myc, anti-phospho-S129 α-synuclein, or anti-ubiquitin, then with secondary antibodies Cy3-conjugated anti-mouse or FITC-conjugated anti-rabbit, nuclei were stained with 4′,6-diamidino-2-phenylindole, and all signals were analyzed by fluorescence microscopy (Axiovert 100; Zeiss, Oberkochen, Germany). For thioflavin S staining, transfected cells were washed with PBS and incubated with 0.01% thioflavin S (Sigma) for 8 min; cells were then fixed with 4% paraformaldehyde for 30 min for subsequent immunohistochemistry. The number of cells with eosinophilic inclusions or inclusions positive for both anti-ubiquitin and anti-phospho-S129 α-synuclein were counted in six different fields with ∼1000 cells in each experimental condition. Counts were done by an investigator blind to the experimental condition.
Immunoelectron microscopy. Seventy-two hours after transfection, cells were fixed in 4% paraformaldehyde plus 0.05% glutaraldeyde plus 0.1 m phosphate buffer, pH 7.2-7.2, overnight at 4°C. Cells were gently scraped and pelleted after a 30 min rinse in wash buffer (0.1 m phosphate plus 3% sucrose), then incubated for 30 min with 0.12% tannic acid (Mallinkrodt, Paris, KY). Cells were stained en bloc with 2% uranyl acetate in maleate for 30 min. After dehydration in ethanol, cells were embedded in LR Gold. Polymerized blocks were trimmed and cut into 70-80 nm sections collected on formvar-coated 200 mesh nickel grids. For immunolabeling, grids were treated with 50 mm NH4Cl in TBS buffer (50 mm Trizma base plus 150 mm NaCl, pH 7.4), blocked in 1% BSA/0.5% fish gelatin/TBST (10 mm Tris plus 500 mm NaCl plus 0.05% Tween 20, pH 7.2), and incubated with primary anti-phospho-α-synuclein antibody and with 12-nm gold-conjugated secondary antibody. Imaging was performed on a Phillips (Eindhoven, The Netherlands) CM 120 Transmission electron microscope operating at 80 kV.
Data analysis. Quantitative data are expressed as the arithmetic means ± SE based on at least three separate experiments performed in duplicate. The difference between two groups was statistically analyzed by Student's t test or an ANOVA (one way). A p value <0.05 was considered significant.
Results
Alteration of S129 site in α-synuclein
To study the role of phosphorylation in α-synuclein-mediated inclusion formation, the S129 residue in α-synuclein was mutagenized to alanine to abolish the phosphorylation site. As a comparison, we also mutagenized this site to glutamate, containing a negative charge, which mimics some of the effects of phosphorylation. We generated constructs with either the S129A or the S129E alteration using vector pRK5 containing wild-type α-synuclein or the PD-related mutations A53T or A30P of α-synuclein (all possible combinations resulting in nine different constructs, which were confirmed by sequencing). The constructs were transiently transfected into SH-SY5Y cells for 48 h, followed by 24 h of serum withdraw. Cells were then harvested, and cell lysates were subjected to duplicate immunoblotting using anti-phospho-S129 α-synuclein and anti-α-synuclein antibodies to detect phosphorylated and total α-synuclein, respectively. As depicted in Figure 1B, the anti-phospho-S129 α-synuclein antibody detected phosphorylated α-synuclein but not S129A α-synuclein or S129E α-synuclein. All of the constructs expressed similar levels of α-synuclein.
Figure 1.

Alteration of S129 site in α-synuclein. A, Schematic diagram of the α-synuclein construct. CMV, Cytomegalovirus. B, Cells were transfected with wild-type α-synuclein, S129A α-synuclein, or S129E α-synuclein, with or without A30P and A53T mutations, for 48 h. Cell lysates were subjected to immunoblotting using anti-α-synuclein or anti-phospho-S129 α-synuclein antibodies. WB, Western blot.
Expression of S129A α-synuclein decreases the formation of cytoplasmic eosinophilic inclusions
Because coexpression of α-synuclein and synphilin-1 yields cytoplasmic eosinophilic inclusions (Engelender et al., 1999), we studied the effect of α-synuclein phosphorylation on the formation of inclusions. When constructs encoding wild-type α-synuclein and synphilin-1 were cotransfected into SH-SY5Y cells for 72 h, we observed the formation of cytosolic phase-dense eosinophilic inclusions (Fig. 2A, middle), as described previously (Engelender et al., 1999). However, cells cotransfected with constructs encoding S129A α-synuclein and synphilin-1 had few or no inclusions (Fig. 2A, right), compared with the ∼6% of cells that had cytosolic eosinophilic inclusions when cotransfected with constructs encoding α-synuclein and synphilin-1 (Fig. 2B). Because the transfection efficiency was 30-50% (data not shown), the percentage of cells expressing α-synuclein and synphilin-1 that develop inclusions was probably 12-20%. In contrast, only ∼0.5% of cells had eosinophilic inclusions in the transfection group receiving the constructs encoding S129A α synuclein and synphilin-1 and in the group transfected with vector alone (pRK5) (Fig. 2B). Furthermore, we observed that coexpression of S129E α-synuclein and synphilin-1 resulted in a similar percentage of cells with inclusions as did coexpression of wild-type α-synuclein and synphilin-1. S129E α-synuclein may mimic some of the effects of phosphorylation as a result of the presence of the negatively charged residue. These results indicated that phosphorylation of α-synuclein at S129 enhanced eosinophilic inclusion formation.
Figure 2.
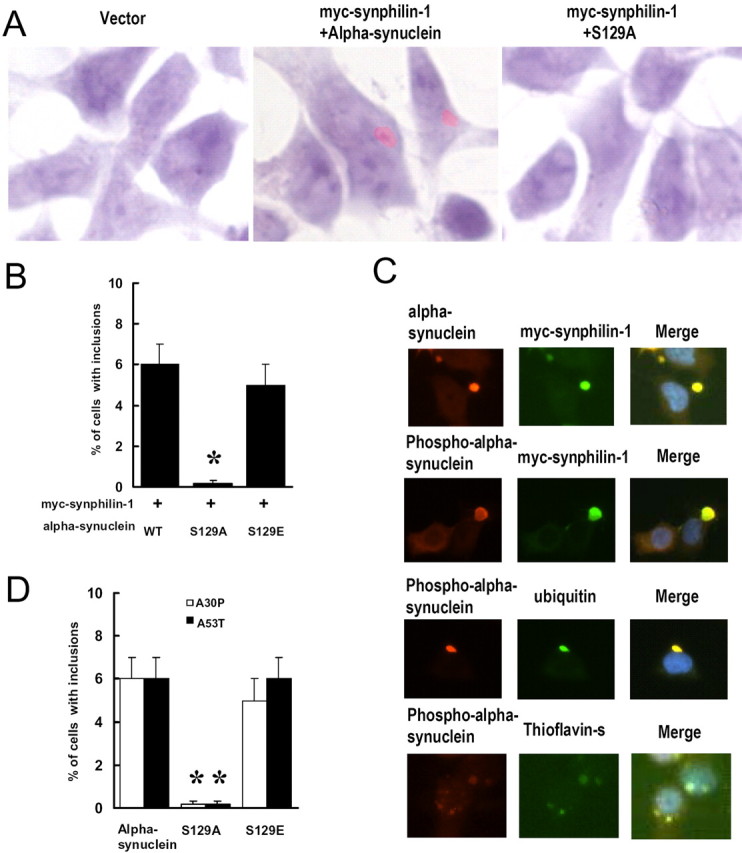
Expression of S129A α-synuclein decreases cytoplasmic eosinophilic inclusion formation. Cells were cotransfected with myc-synphilin-1 along with α-synuclein, S129A α-synuclein, or S129E α-synuclein. Seventy-two hours later, cells were subjected to H & E staining or immunocytochemical analysis. A, Representative cells stained with H & E show cytoplasmic eosinophilic inclusions. B, Cells with cytoplasmic eosinophilic inclusions were counted. Data are shown as the means ± SE from three separate experiments performed in duplicate. *p < 0.05 versus cells cotransfected with wild-type (WT) α-synuclein and myc-synphilin-1. C, Representative images of immunocytochemical analysis and thioflavin S staining in cells coexpressing wild-type α-synuclein and myc-synphilin-1. D, Cells were cotransfected with myc-synphilin-1 and A30P or A53T α-synuclein with or without the S129 site alteration for 72 h. Cells with inclusions were counted and shown as the means ± SE for three separate experiments performed in duplicate. *p < 0.05 versus cells cotransfected with myc-synphilin-1 and A30P or A53T α-synuclein.
To further study these cytoplasmic eosinophilic inclusions formed by coexpression of wild-type α-synuclein and synphilin-1, we performed double immunostaining for ubiquitin and α-synuclein or S129 phosphorylated α-synuclein costaining with thioflavin S immunohistochemistry analysis. As depicted in Figure 2C, these inclusions stained positive when using anti-α-synuclein, anti-synphilin-1,anti-phospho-S129 α-synuclein, and anti-ubiquitin antibodies similar to Lewy bodies. We found that ∼90% of α-synuclein/synphilin-1 inclusions also exhibited phospho-S129α-synuclein immunoreactivity. Previous reports showed that thioflavin S-positive staining in inclusions of rat cortical neurons with fibrillar structure (Rideout et al., 2004), a feature that is also present in Lewy bodies (Neumann et al., 2002). We found that ∼95% of phospho-S129 α-synuclein inclusions also stained positive for thioflavin S in our cell model (Fig. 2C, bottom). Furthermore, by immunoelectron microscopy, in cells coexpressing wild-type α-synuclein and synphilin-1, some fibrillar material in the inclusions stained positive when using an anti-phospho-S129 α-synuclein antibody (Fig. 3). Electron-dense cytoplasmic inclusions (1-5 μm in diameter) were identified that contained 7-12 nm filaments. Some filaments stained positive by anti-phospho-S129 α-synuclein antibody, whereas others did not. The filaments that did not label by anti-phosphorylated α-synuclein antibodies may contain other neurofilament proteins or perhaps contain phosphorylated α-synuclein that failed to bind the antibody as a result of limited epitope availability. Moreover, we also observed some phospho-S129 α-synuclein-positive granular and amorphous components. In contrast, when constructs encoding S129A α synuclein and synphilin-1 were cotransfected, or when the vector alone (pRK5) was transfected into cells, virtually none of these cells contained thioflavin S-positive inclusions or fibrillar inclusions staining with anti-phospho-S129 α-synuclein antibody, as determined by immunoelectron microscopy (data not shown).
Figure 3.
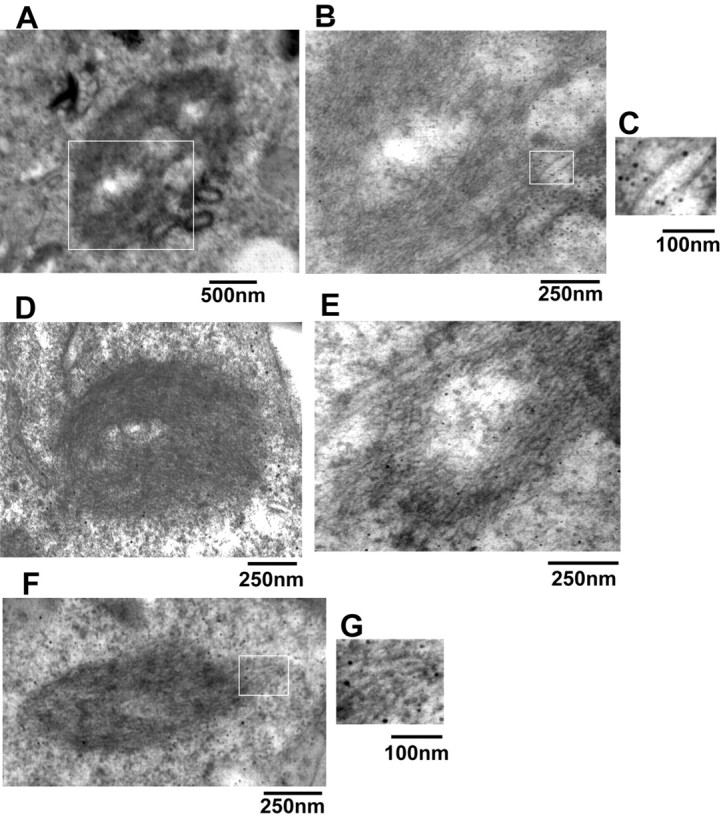
Immunoelectron microscopy analysis of inclusions in transfected cells. Cells were cotransfected with myc-synphilin-1 and α-synuclein, S129A α-synuclein, or S129E α-synuclein, and 72 h later, cells were subjected to immunoelectron microscopy. A and D-F are representative images of different shape cytoplasmic inclusions in cells coexpressing myc-synphilin-1 and wild-type α-synuclein. B represents a higher magnification of the rectangle in A. C and G represent the higher magnification of rectangles in B and F, which shows the gold-labeled filaments by using phospho-α-synuclein antibody.
A30P and A53T mutations in α-synuclein cause autosomal-dominant familial PD (Polymeropoulos et al., 1997; Kruger et al., 1998). To investigate the effect of PD-associated mutations of α-synuclein in the formation of these inclusions, cells were cotransfected for 72 h with myc-synphilin-1 and A30P or A53T α-synuclein with or without the S129 alteration. As shown in Figure 2D, the percentage of cells with inclusions was not altered by the PD-associated α-synuclein mutations. These results indicate that PD-related mutations of α-synuclein do not alter the effect of phosphorylation on inclusion formation.
Expression of S129A α-synuclein decreases the formation of ubiquitinated inclusions
Previously, we reported that coexpression of parkin, synuclein, and synphilin-1 resulted in the formation of ubiquitin-positive cytosolic inclusions in HEK 293 cells (Chung et al., 2001a). Here, cotransfected Flag-parkin, α-synuclein, and myc-synphilin-1 into SH-SY5Y cells for 72 h resulted in a dramatic increase in the formation of ubiquitinated inclusions with fibrillar structure by immunoelectron microscopy (data not shown) and immunohistochemistry assay, compared with what was seen in cells cotransfected only with α-synuclein and myc-synphilin-1 (Fig. 4A). The number of cells containing multiple inclusions also significantly increased with parkin cotransfection (Fig. 4B). However, when familial-linked parkin mutant constructs (T240R, P437L, and Q311 stop) were cotransfected with α-synuclein and myc-synphilin-1, cells with ubiquitinated inclusions decreased to the level of cells cotransfected only with α-synuclein and myc-synphilin-1 (Fig. 4C,D). These results indicated that expression of wild-type, but not mutant, parkin promoted the formation of ubiquitinated inclusions.
Figure 4.

Expression of S129A α-synuclein decreases the formation of ubiquitinated inclusions. A, B, Cells were transfected with various constructs for 72 h as indicated and subjected to immunocytochemical assay with anti-ubiquitin and anti-phospho-α-synuclein antibodies. Cells with ubiquitin-positive and phospho-α-synuclein-positive inclusions were counted. Data are shown as the means ± SE for three separate experiments performed in duplicate. *p < 0.05 versus cells cotransfected with α-synuclein and myc-synphilin-1. C, D, Cells cotransfected with α-synuclein, myc-synphilin-1 and Flag-parkin, or mutant parkin constructs were subjected to immunocytochemical assay 72 h later as described in A. Data are shown as the means ± SE for three separate experiments performed in duplicate. *p < 0.05 versus cells cotransfected with α-synuclein, myc-synphilin-1, and Flag-parkin. E, Cells were cotransfected with α-synuclein, S129A α-synuclein, or S129E α-synuclein plus myc-synphilin-1 and Flag-parkin and subjected to immunocytochemical assay 72 h later as described in A. Data are shown as the means ± SE for three separate experiments performed in duplicate. *p < 0.05 versus cells cotransfected with wild-type (WT) α-synuclein, myc-synphilin-1, and Flag-parkin.
To study the effect of alteration of S129 site in α-synuclein on the formation of these ubiquitinated inclusions, SH-SY5Y cells were cotransfected for 72 h with myc-synphilin-1, Flag-parkin, and α-synuclein, S129A α-synuclein, or S129E α-synuclein. As shown in Figure 4E, there was a fourfold decrease in cells with ubiquitinated inclusions when expressing of S129A α-synuclein compared with what was seen when expressing wild-type α-synuclein or S129E α-synuclein. These results indicate that phosphorylation of α-synuclein at S129 enhanced the formation of ubiquitinated inclusions.
We have shown previously that α-synuclein interacts with synphlin-1, and parkin interacts with and ubiquitinates synphilin-1 (Engelender et al., 1999; Chung et al., 2001a). Recently, we found that parkin ubiquitination of synphilin-1 within ubiquitinated inclusions formed by coexpression of α-synuclein, synphlin-1, and parkin occurs predominantly via K63-linkaged ubiquitination (Lim et al., 2005). To further study whether phosphorylation of α-synuclein alters the interaction among α-synuclein, synphlin-1, and parkin and influences the formation of inclusions, we conducted cotransfection experiments with myc-tagged synphilin-1 and HA-tagged α-synuclein with or without the S129 alteration and with or without Flag-tagged parkin followed by Coimmunoprecipitation assays. Compared with wild-type α-synuclein, S129A α-synuclein showed a decreased interaction with synphilin-1, regardless of parkin expression levels (Fig. 5). Moreover, expression of S129A α-synuclein decreased the association of parkin and synphilin-1 (Fig. 5B, anti-parkin panel) and further decreased parkin mediated-ubiquitination of synphilin-1 (Fig. 5A, B, anti-ubiquitin panel) compared with expression of wild-type α-synuclein. We did not detect any interaction between parkin and α-synuclein (data not shown) (Chung et al., 2001a) or any ubiquitination of α-synuclein (all forms) by parkin in our system (Fig. 5, anti-ubiquitin panel). Together, these results support the finding that expression S129A α-synuclein decreased the formation of these ubiquitinated inclusions.
Figure 5.

Expression of S129A α-synuclein decreases its interaction with synphilin-1. A, B, Lysates prepared from cells transfected with various constructs as indicated were subjected to IP with anti-HA or anti-myc followed by anti-myc, anti-α-synuclein, anti-parkin, and anti-ubiquitin immunoblotting. In the anti-ubiquitin panel, there was clear ubiquitin immunoreactivity above 97 kDa (migration position of synphilin-1) in lane 4 compared with the other lanes, whereas in all lanes, there was an absence of ubiquitin immunoreactivity around 15 kDa (migration position of α-synuclein). The experiment was repeated three times with similar results. WT, Wild type.
Oxidative stress induces α-synuclein phosphorylation at S129 and increases the inclusion formation
Oxidative stress is hypothesized to play a role in PD and other neurodegenerative diseases, potentially through the activation of kinases and other signaling cascades (Hashimoto et al., 1999; Finkel and Holbrook, 2000; Giasson et al., 2000; Kanda et al., 2000; Zhang et al., 2000; Lee et al., 2002). To examine the role of oxidative stress on α-synuclein phosphorylation, SH-SY5Y cells were cotransfected with α-synuclein, synphilin-1, and parkin for 48 h, followed by treatment with either 100 or 150 μm H2O2. Twenty-four hours later, we observed that the expression levels of α-synuclein, synphilin-1, and parkin was the same in all experimental conditions (data not shown), but treatment with H2O2 induced a twofold to threefold increase of α-synuclein phosphorylation (Fig. 6A) and a twofold increase in cells with ubiquitinated inclusions compared with untreated control cells (Fig. 6B). Virtually 95% of the ubiquitinated inclusions stained positive with anti-phospho-S129 α-synuclein antibodies (Fig. 6C); in contrast, H2O2 treatment of cells expressing S129A α-synuclein had no effect on the formation of ubiquitinated inclusions (Fig. 6A, B). These results suggest that oxidative stress can induce α-synuclein phosphorylation and the inclusion formation.
Figure 6.
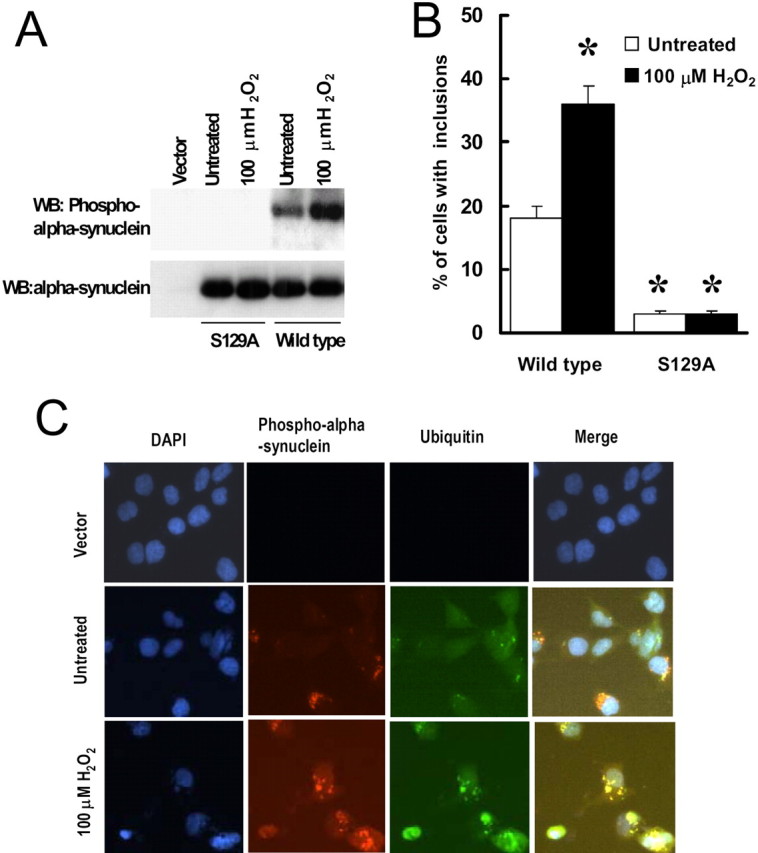
Oxidative stress increases α-synuclein phosphorylation at S129 and increases the formation of cytoplasmic inclusions. A, B, Cells were cotransfected with myc-synphilin-1, Flag-parkin, and α-synuclein with or without S129 alteration for 48 h and then treated with 100 μm H2O2. Twenty-four hours later, cell lysates were prepared and subjected to anti-α-synuclein and anti-phospho-S129 α-synuclein immunoblotting (A). WB, Western blot. Cells were subjected to immunocytochemical assay with anti-phospho-S129 and anti-ubiquitin antibodies (B). Cells with ubiquitin-positive and phospho-α-synuclein-positive inclusions were counted. Data are shown as the means ± SE for three separate experiments performed in duplicate. *p < 0.05 versus untreated cells transfected with wild-type α-synuclein, myc-synphilin-1, and Flag-parkin. C, Representative images of immunocytochemical assay in cells that were cotransfected with wild-type α-synuclein, myc-synphilin-1, Flag-parkin, or transfected with the vector alone for 48 h and were then either left untreated or were treated with H2O2 for 24 h. DAPI, 4′,6-Diamidino-2-phenylindole.
CK2 inhibitor DRB blocks α-synuclein phosphorylation at S129 and decreases the inclusion formation
Previous reports showed that residue S129 of α-synuclein can be phosphorylated by CK1 and CK2 (Okochi et al., 2000). To further explore the role of S129 phosphorylation on the inclusion formation, SH-SY5Y cells were transfected with α-synuclein, synphilin-1, and parkin for 48 h, whereupon cells were treated with 100 μm H2O2 in the presence or absence of 50 μm DRB, a CK2 inhibitor. As observed 24 h later, DRB blocked H2O2-induced α-synuclein phosphorylation in both Triton X-100-soluble and -insoluble fractions (Fig. 7A). Triton X-100-insoluble fractions were dissolved in buffer A containing 2% SDS and represent the aggregated portion of phosphorylated α-synuclein. Furthermore, treatment with DRB significantly decreased the number of cells with H2O2-induced ubiquitinated inclusions (Fig. 7B). On the other hand, treatment with DRB had no effect in cells coexpressing S129A α-synuclein, synphilin-1, and parkin. We also observed that inhibitors of protein kinase C (PKC; Ro-318220), protein kinase A (PKA; KT5720), mitogen-activated protein kinase kinase 1 (MEK1; U0126), or phosphoinositide 3-kinase (PI3K; wortmannin) had no effect on α-synuclein phosphorylation (Fig. 7C) or the inclusion formation (data not shown). These results suggest that CK2 is activated by H2O2 leading to S129 α-synuclein phosphorylation and enhances the inclusion formation.
Figure 7.

CK2 inhibitor DRB blocks α-synuclein phosphorylation at S129 and decreases the formation of cytoplasmic inclusions. A, Cells were cotransfected with α-synuclein, myc-synphilin-1, and Flag-parkin for 48 h and then treated with 100 μm H2O2 in either the presence or absence of 50 μm DRB. Twenty-four hours later, Triton X-100-soluble fractions, Triton X-100-insoluble but SDS-soluble fractions, and SDS-soluble fractions (including Triton X-100-soluble and Triton X-100-insoluble fractions) of cell lysates were subjected to anti-α-synuclein and anti-phospho-S129 α-synuclein immunoblotting. The experiment was repeated three times with similar results. B, Cells were cotransfected with myc-synphilin-1, Flag-parkin, and α-synuclein, with or without S129 alterations, for 48 h, followed by 100 μm H2O2 treatment in presence or absence of 50 μm DRB. Cells were collected 24 h later for immunocytochemical assay with anti-phospho-S129 and anti-ubiquitin antibodies. Cells with ubiquitin-positive and phospho-α-synuclein-positive inclusions were counted. Data are shown as the means ± SE for three separate experiments performed in duplicate. *p < 0.05 versus untreated cells transfected with wild-type α-synuclein, myc-synphilin-1, and Flag-parkin. #p < 0.05 versus 100 μm H2O2-treated cells transfected with wild-type α-synuclein, myc-synphilin-1, and Flag-parkin. C, Cells were cotransfected with α-synuclein, myc-synphilin-1, and Flag-parkin for 48 h and treated with 100 μm H2O2 in presence or absence of 2 μm KT5720, 5 μm U0126, 200 nm wortmannin, or 1 μm Ro-318220. Cell lysates were prepared 24 h later and were subjected to anti-α-synuclein and anti-phospho-S129 α-synuclein immunoblotting. The experiment was repeated three times with similar results.
Discussion
We studied the role of phosphorylation of α-synuclein in the formation of eosinophilic inclusions in cytosol arising by coexpression of α-synuclein, synphilin-1, and parkin. We found that expressing S129A α-synuclein (and thus blocking phosphorylation at S129) decreased the formation of these inclusions. Moreover, expression of S129A α-synuclein instead of wild-type α-synuclein impairs the formation of ubiquitinated inclusions formed by coexpression parkin and synphilin-1. In this regard, PD-associated mutations in parkin were also found to disrupt the formation of ubiquitinated inclusions. By coimmunoprecipitation assays, we found that S129A α-synuclein interacted less strongly with synphilin-1 than did wild-type α-synuclein. Expression of S129A α-synuclein also decreased the association of synphilin-1 and parkin and led to a decrease in the parkin-mediated ubiquitination of synphilin-1 compared with the effects of expressing wild-type α-synuclein. In addition, oxidative stress enhanced α-synuclein phosphorylation at S129 and increased the abundance of ubiquitinated inclusions. Finally, treatment with the CK2 inhibitor DRB blocked α-synuclein phosphorylation and decreased the formation of inclusions. These results provide strong evidence that phosphorylation of α-synuclein at S129 regulates the inclusion formation.
Human Lewy bodies are spherical eosinophilic inclusion bodies present in cytosol, which contain abnormal neurofilament proteins including α-synuclein. They can be found in specific regions of the nervous system, where their frequencies, size, and structures differ (Gibb et al., 1991). The well defined human Lewy body has a central core of circular structures and a halo of radiating filaments (7-20 nm in diameter) at the periphery (Gibb et al., 1991; Giasson et al., 2000). The cytoplasmic inclusions in the cell model described here mostly resembled Lewy bodies in PD but differed in some aspects. Like Lewy bodies, these inclusions were eosinophilic, and most of them had a round shape. They were immunoreactive to anti-phosphorylated α-synuclein, anti-synphilin-1, and anti-ubiquitin antibodies. Moreover, these inclusions contained thioflavin S-positive material. Using immunoelectron microscopy, most inclusions were found to contain fibrillar structures that stained positive with anti-phosphorylated α-synuclein antibodies. Similar to Lewy bodies, these inclusions had filaments as well as amorphous and granular aggregates. Unlike Lewy bodies, the filaments in these inclusions appeared less prominent, and not all the inclusions were round.
Our findings further demonstrate that phosphorylation of α-synuclein at S129 enhanced the formation of eosinophilic inclusions, in keeping with recent reports that α-synuclein phosphorylation at S129 promotes fibril formation in vitro (Fujiwara et al., 2002). Although there are four serine residues in human α-synuclein (at positions 9, 42, 87, and 129) (Ueda et al., 1993), only serine 129 of α-synuclein is a major site of phosphorylation in synucleinopathy lesions in human brain (Okochi et al., 2000; Fujiwara et al., 2002). By coexpression of synphlin-1 and S129A α-synuclein (wherein the S129 phosphorylation site is abolished) with or without parkin, we found that S129A α-synuclein decreased its interaction with synphilin-1 compared with wild-type α-synuclein. Expression of S129A α-synuclein instead of wild-type α-synuclein also decreased the association of synphilin-1 and parkin and subsequently reduced the parkin-mediated ubiquitination of synphilin-1. These results support the finding that expression S129A α-synuclein decreased the formation of ubiquitinated inclusions. In addition, we also found that coexpression of parkin with α-synuclein and synphilin-1 increased the formation of ubiquitinated inclusions (Fig. 4A), and familial parkin mutations T240R, P437L, and Q311 stop impaired the formation of ubiquitinated inclusions (Fig. 4C). Together, these results suggest that phosphorylation of α-synuclein at S129 and parkin play an important role in the formation of ubiquitinated inclusions. Whether these inclusions protect against or contribute to cytotoxicity is the subject of intense current debate. However, a recent report shows that cells containing α-synuclein/synphilin-1 inclusions are mostly present in surviving cells and less so in apoptotic cells, suggesting that these inclusions may play a protective role in cell death by sequestering toxic molecular species (Tanaka et al., 2004). In our cell model, we do not detect any toxicity (data not shown).
Oxidative stress is believed to be involved in the pathogenesis of PD and other neurodegenerative diseases (Hashimoto et al., 1999; Finkel and Holbrook, 2000; Giasson et al., 2000; Kanda et al., 2000; Zhang et al., 2000; Dev et al., 2003). In this study, we found that treatment with H2O2 in cells coexpressing α-synuclein, synphilin-1, and parkin enhanced phosphorylation of α-synuclein at S129 and significantly increased cytoplasmic inclusions, compared with untreated control cells (Fig. 4). These results suggest that oxidative stress can enhance eosinophilic inclusion formation via phosphorylation of α-synuclein at S129 and may contribute to Lewy body pathology.
Previous reports have shown three kinases can phosphorylate α-synuclein at S129, including CK1, CK2, and G-protein-coupled receptor kinases (Okochi et al., 2000; Pronin et al., 2000). In this study, we used DRB, a relatively specific CK2 inhibitor, to block H2O2-induced α-synuclein phosphorylation at S129 and found that DRB also decreased the formation of cytoplasmic inclusions induced by H2O2, whereas inhibitors of PKC, PKA, MEK1, and PI3K failed to do so (Fig. 7). These results are consistent with reports that DRB blocks the binding of α-synuclein and synphilin-1 and reduces the percentage of cells that contain cytoplasmic inclusions in 293T cells (Lee et al., 2004). CK2s are ubiquitously expressed, constitutively active kinases, which are localized in the nucleus and the cytosol (Tuazon and Traugh, 1991; Bennett et al., 1993; Tawfic and Ahmed, 1994; Gross et al., 1995). CK2 phosphorylation was reported to be activated in substantia nigra neurons in which dopaminergic neurons specifically degenerate during PD (Desdouits et al., 1995). These findings, together with our data presented here, suggest that CK2 may play a role in PD pathogenesis by phosphorylation of α-synuclein.
Protein phosphorylation may play an important role in the pathogenesis of neurodegenerative diseases. In Alzheimer's disease, brain dysfunction and degeneration are linked to the accumulation of neurofibrillary tangles containing hyperphosphorylated microtubule-associated protein tau (Lee et al., 2001). Enhanced phosphorylation of tau by glycogen synthase kinase 3β induces filamentous tau inclusions and accelerates tau-induced neurodegeneration in transgenic flies and mice (Lucas et al., 2001; Jackson et al., 2002). In spinocerebellar ataxia type 1, ataxin-1 is phosphorylated at serine 776. Substitution of this S776 residue with alanine (A776) greatly diminishes the ability of mutant ataxin-1 to aggregate in Chinese hamster ovary cells (Emamian et al., 2003). Ataxin-1[82Q]-A776 transgenic mice have a reduced rate of nuclear inclusion formation and a less pathogenic phenotype as measured by rotarod performance compared with ataxin-1[82Q]-S776 transgenic mice (Emamian et al., 2003). In a fly model, phosphorylation of S776 is required for ataxin-1 to interact with protein 14-3-3. This interaction stabilizes ataxin-1, stimulates ataxin-1 inclusion formation, and aggravates neurodegeneration in spinocerebellar ataxia type 1 flies (Chen et al., 2003). Together, our findings regarding the phosphorylation of α-synuclein at S129 may reflect a common mechanism underlying inclusion formation in other neurodegenerative diseases.
In summary, we show that phosphorylation of α-synuclein at S129 increased its interaction with synphilin-1, increased the interaction between synphilin-1 and parkin, and subsequently enhanced the parkin-mediated ubiquitination of synphilin-1 and the formation of eosinophilic cytoplasmic inclusions, which were similar, although not identical to Lewy bodies seen in neurons in PD. Our findings advance our understanding of the formation of inclusions in PD pathogenesis and in other α-synucleinopathy diseases.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS38377, the Udall Parkinson's Disease Research Center, and the National Institutes of Health. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. We thank Drs. Myriam Gorospe, Yusen Liu, and Michelle A. Poirier for helpful discussions. We thank Michael J. Delannoy and Leonard Monfredo for technical help.
Correspondence should be addressed to Dr. Christopher A. Ross, Division of Neurobiology, Department of Psychiatry, The Johns Hopkins University School of Medicine, Ross Research Building, Room 618, 720 Rutland Avenue, Baltimore, MD 21205. E-mail: caross@jhu.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255544-09$15.00/0
References
- Bennett MK, Miller KG, Scheller RH (1993) Casein kinase II phosphorylates the synaptic vesicle protein p65. J Neurosci 13: 1701-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del TK, Rub U, de Vos RA, Jansen SEN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24: 197-211. [DOI] [PubMed] [Google Scholar]
- Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, Aitken A, Skoulakis EM, Orr HT, Botas J, Zoghbi HY (2003) Interaction of Akt-phosphorylated ataxin-1 with 14 -3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell 113: 457-468. [DOI] [PubMed] [Google Scholar]
- Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM (2001a) Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med 7: 1144 -1150. [DOI] [PubMed] [Google Scholar]
- Chung KK, Dawson VL, Dawson TM (2001b) The role of the ubiquitin-proteasomal pathway in Parkinson's disease and other neurodegenerative disorders. Trends Neurosci 24: S7-14. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL (2003) Molecular pathways of neurodegeneration in Parkinson's disease. Science 302: 819 -822. [DOI] [PubMed] [Google Scholar]
- Desdouits F, Cohen D, Nairn AC, Greengard P, Girault JA (1995) Phosphorylation of DARPP-32, a dopamine- and cAMP-regulated phosphoprotein, by casein kinase I in vitro and in vivo. J Biol Chem 270: 8772-8778. [DOI] [PubMed] [Google Scholar]
- Dev KK, Hofele K, Barbieri S, Buchman VL, van der PH (2003) Part II: alpha-synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology 45: 14 -44. [DOI] [PubMed] [Google Scholar]
- Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, Zoghbi HY, Clark HB, Orr HT (2003) Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 38: 375-387. [DOI] [PubMed] [Google Scholar]
- Engelender S, Kaminsky Z, Guo X, Sharp AH, Amaravi RK, Kleiderlein JJ, Margolis RL, Troncoso JC, Lanahan AA, Worley PF, Dawson VL, Dawson TM, Ross CA (1999) Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat Genet 22: 110 -114. [DOI] [PubMed] [Google Scholar]
- Farrer M, Wavrant-De Vrieze F, Crook R, Boles L, Perez-Tur J, Hardy J, Johnson WG, Steele J, Maraganore D, Gwinn K, Lynch T (1998) Low frequency of alpha-synuclein mutations in familial Parkinson's disease. Ann Neurol 43: 394 -397. [DOI] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408: 239 -247. [DOI] [PubMed] [Google Scholar]
- Forman MS, Trojanowski JQ, Lee VM (2004) Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med 10: 1055-1063. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T (2002) Alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4: 160 -164. [DOI] [PubMed] [Google Scholar]
- Galvin JE, Lee VM, Schmidt ML, Tu PH, Iwatsubo T, Trojanowski JQ (1999) Pathobiology of the Lewy body. Adv Neurol 80: 313-324. [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM (2000) Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290: 985-989. [DOI] [PubMed] [Google Scholar]
- Gibb WR, Scott T, Lees AJ (1991) Neuronal inclusions of Parkinson's disease. Mov Disord 6: 2-11. [DOI] [PubMed] [Google Scholar]
- Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI (2005) The E46K mutation in alpha-synuclein increases amyloid fibril formation. J Biol Chem 280: 7800-7807. [DOI] [PubMed] [Google Scholar]
- Gross SD, Hoffman DP, Fisette PL, Baas P, Anderson RA (1995) A phosphatidylinositol 4,5-bisphosphate-sensitive casein kinase I alpha associates with synaptic vesicles and phosphorylates a subset of vesicle proteins. J Cell Biol 130: 711-724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Fujiwara H, Nonaka T, Wakabayashi K, Takahashi H, Lee VM, Trojanowski JQ, Mann D, Iwatsubo T (2002) Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J Biol Chem 277: 49071-49076. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Hsu LJ, Xia Y, Takeda A, Sisk A, Sundsmo M, Masliah E (1999) Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. NeuroReport 10: 717-721. [DOI] [PubMed] [Google Scholar]
- Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, Geschwind DH (2002) Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila Neuron 34: 509 -519. [DOI] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Odoy S, Okamoto N, Jacobsen H, Iwatsubo T, Trojanowski JQ, Takahashi H, Wakabayashi K, Bogdanovic N, Riederer P, Kretzschmar HA, Haass C (2001) Selective insolubility of alpha-synuclein in human Lewy body diseases is recapitulated in a transgenic mouse model. Am J Pathol 159: 2215-2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Spooren W, Fuss B, Mallon B, Macklin WB, Fujiwara H, Hasegawa M, Iwatsubo T, Kretzschmar HA, Haass C (2002) Hyperphosphorylation and insolubility of alpha-synuclein in transgenic mouse oligodendrocytes. EMBO Rep 3: 583-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda S, Bishop JF, Eglitis MA, Yang Y, Mouradian MM (2000) Enhanced vulnerability to oxidative stress by alpha-synuclein mutations and C-terminal truncation. Neuroscience 97: 279 -284. [DOI] [PubMed] [Google Scholar]
- Kotzbauer PT, Trojanowsk JQ, Lee VM (2001) Lewy body pathology in Alzheimer's disease. J Mol Neurosci 17: 225-232. [DOI] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet 18: 106 -108. [DOI] [PubMed] [Google Scholar]
- Lee G, Tanaka M, Park K, Lee SS, Kim YM, Junn E, Lee SH, Mouradian MM (2004) Casein kinase II-mediated phosphorylation regulates alpha-synuclein/synphilin-1 interaction and inclusion body formation. J Biol Chem 279: 6834 -6839. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Shin SY, Choi C, Lee YH, Lee SJ (2002) Formation and removal of alpha-synuclein aggregates in cells exposed to mitochondrial inhibitors. J Biol Chem 277: 5411-5417. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24: 1121-1159. [DOI] [PubMed] [Google Scholar]
- Lim KL, Chew KCM, Tan JMM, Wang C, Chung KK, Tanaka Y, Smith W, Engelender S, Ross CA, Dawson VL, Dawson TM (2005) Parkin mediates non-classic, proteasomal-independent, ubiquitination of spynphilin-1:implication for Lewy body formation. J Neurosci 25: 2002-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J (2001) Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J 20: 27-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W, Muller V, Odoy S, Fujiwara H, Hasegawa M, Iwatsubo T, Trojanowski JQ, Kretzschmar HA, Haass C (2002) Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J Clin Invest 110: 1429 -1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T, Meijer L, Kahle PJ, Haass C (2000) Constitutive phosphorylation of the Parkinson's disease associated alpha-synuclein. J Biol Chem 275: 390 -397. [DOI] [PubMed] [Google Scholar]
- Pollanen MS, Dickson DW, Bergeron C (1993) Pathology and biology of the Lewy body. J Neuropathol Exp Neurol 52: 183-191. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276: 2045-2047. [DOI] [PubMed] [Google Scholar]
- Pronin AN, Morris AJ, Surguchov A, Benovic JL (2000) Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J Biol Chem 275: 26515-26522. [DOI] [PubMed] [Google Scholar]
- Rideout HJ, Dietrich P, Wang Q, Dauer WT, Stefanis L (2004) alpha-Synuclein is required for the fibrillar nature of ubiquitinated inclusions induced by proteasomal inhibition in primary neurons. J Biol Chem 279: 46915-46920. [DOI] [PubMed] [Google Scholar]
- Schlossmacher MG, Frosch MP, Gai WP, Medina M, Sharma N, Forno L, Ochiishi T, Shimura H, Sharon R, Hattori N, Langston JW, Mizuno Y, Hyman BT, Selkoe DJ, Kosik KS (2002) Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am J Pathol 160: 1655-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ (2001) Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science 293: 263-269. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M (2000) The alpha-synucleinopathies: Parkinson's disease, dementia with Lewy bodies, and multiple system atrophy. Ann NY Acad Sci 920: 16 -27:16 -27. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388: 839 -840. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95: 6469 -6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Kanuka H, Fujiwara H, Koyama A, Hasegawa M, Miura M, Iwatsubo T (2003) Phosphorylation of alpha-synuclein characteristic of synucleinopathy lesions is recapitulated in alpha-synuclein transgenic Drosophila Neurosci Lett 336: 155-158. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo T, Mouradian MM (2004) Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J Biol Chem 279: 4625-4631. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa A, Dawson L, Dawson TM, Ross CA (2001) Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet 10: 919 -926. [DOI] [PubMed] [Google Scholar]
- Tawfic S, Ahmed K (1994) Association of casein kinase 2 with nuclear matrix. Possible role in nuclear matrix protein phosphorylation. J Biol Chem 269: 7489 -7493. [PubMed] [Google Scholar]
- Taylor JP, Hardy J, Fischbeck KH (2002) Toxic proteins in neurodegenerative disease. Science 296: 1991-1995. [DOI] [PubMed] [Google Scholar]
- Tuazon PT, Traugh JA (1991) Casein kinase I and II-multipotential serine protein kinases: structure, function, and regulation. Adv Second Messenger Phosphoprotein Res 23: 123-164. [PubMed] [Google Scholar]
- Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T (1993) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA 90: 11282-11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Engelender S, Yoshimoto M, Tsuji S, Ross CA, Takahashi H (2000) Synphilin-1 is present in Lewy bodies in Parkinson's disease. Ann Neurol 47: 521-523. [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez TE, del Ser T, Munoz DG, de Yebenes JG (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55: 164-173. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Dawson VL, Dawson TM (2000) Oxidative stress and genetics in the pathogenesis of Parkinson's disease. Neurobiol Dis 7: 240 -250. [DOI] [PubMed] [Google Scholar]