

Abstract
Mutations in the microtubule-associated protein tau gene have been linked to neurofibrillary tangle (NFT) formation in several neurodegenerative diseases known as tauopathies; however, no tau mutations occur in Alzheimer's disease, although this disease is also characterized by NFT formation and cell death. Importantly, the mechanism of tau-mediated neuronal death remains elusive. Aged mice expressing nonmutant human tau in the absence of mouse tau (htau mice) developed NFTs and extensive cell death. The mechanism of neuron death was investigated in htau mice, and surprisingly, the presence of tau filaments did not correlate directly with death within individual cells, suggesting that cell death can occur independently of NFT formation. Our observations show that the mechanism of neurodegeneration involved reexpression of cell-cycle proteins and DNA synthesis, indicating that nonmutant tau pathology and neurodegeneration may be linked via abnormal, incomplete cell-cycle reentry.
Keywords: cell cycle, neurofibrillary tangle, cell death, transgenic, nonmutant tau, neurodegeneration
Introduction
Neurofibrillary tangles (NFTs) are composed of hyperphosphorylated aggregates of the microtubule-associated protein tau and are coincident with cell death in several neurodegenerative diseases known as tauopathies, of which Alzheimer's disease (AD) is one of the most notable. In AD, the neuronal death and NFT formation are accompanied by another pathological hallmark, β-amyloid deposits, derived from processing of the amyloid precursor protein.
Mutations in the tau gene have been linked to NFT formation in a number of tauopathies in which no β-amyloid deposits develop (Hutton et al., 1998; Spillantini et al., 1998; Poorkaj et al., 2001). However, no mutations have been found in the tau gene in AD, and the relationship between tau pathology, amyloid deposition, and neuronal degeneration is difficult to determine (Hardy and Higgins, 1992; Taylor et al., 2002). Cell death generally occurs via perpetuating signals that evolve into one of several distinct pathways, typically categorized as either necrosis or apoptosis. These pathways are classically grouped based on stereotyped changes in nuclear morphology (Kerr, 1972; Leist and Jaattela, 2001), principally with respect to chromatin condensation, and on the presence or absence of the classic cell executioners: the caspases (for review, see Hengartner, 2000).
Attempts to discriminate whether the mode of cell death in AD is necrotic or apoptotic have yielded mixed results, with features of each pathway having been detected via a variety of biochemical and histological approaches (for review, see Behl, 2000). A number of studies have detected DNA fragmentation, a hallmark of apoptosis, in AD tissue sections using the terminal deoxynucleotide transferase mediated dUTP nick-end labeling (TUNEL) method (Gavrieli et al., 1992; Sgonc and Wick, 1994) to identify 3′-OH ends of broken DNA (Su et al., 1994; Cotman and Anderson, 1995; Lassmann et al., 1995; Troncoso et al., 1996). However, of these studies, some groups have detected morphological changes consistent with apoptosis (Su et al., 1994; Cotman and Anderson, 1995), whereas other groups have not (Lassmann et al., 1995; Troncoso et al., 1996). Still other groups have found evidence in AD tissue, suggesting that the mode of cell death is necrotic (Lucassen et al., 1997; Stadelmann et al., 1998). Thus, categorizing the cell-death mode in AD has proven equivocal, perhaps in part because of the lack of convenient animal models that exhibit all of the pathological hallmarks of the disease (NFTs, β-amyloid deposits, and neuronal death), which would allow for greater experimental accessibility and thus a chance to “catch” the neurons in the act of dying.
To address the potential connection between nonmutant tau pathology, NFT formation, and modes of cell death, we examined aged htau mice, which express nonmutant human tau isoforms but no isoforms of mouse tau and have been previously determined to form aggregated, hyperphosphorylated tau by 9 months of age (Andorfer et al., 2003). We observed that the htau mice older than 15 months of age developed thioflavine-S-positive NFTs, DNA fragmentation (based on TUNEL staining), and a quantitative loss of neurons.
We additionally show that the tau-initiated cell death in the htau mice was accompanied by expression of cell-cycle regulatory proteins, similar to what has been shown in human AD brain in regions in which cell death is extensive (Lew et al., 1995; Busser et al., 1998; Yang et al., 2001, 2003; Herrup and Arendt, 2002). We therefore propose that the connection between nonmutant tau pathology and cell death in the htau mice may occur via changes in cell-cycle control and abnormal, incomplete, cell-cycle reentry.
Materials and Methods
Generation of htau mice
htau mice were generated as described previously (Andorfer et al., 2003) by crossing mice that express a tau transgene derived from a human PAC, H1 haplotype, termed 8c mice (Duff et al., 2000), with tau knock-out (KO) mice that have a targeted disruption of exon one of tau (Tucker et al., 2001). The F1 generation of mice that contained the human tau gene was backcrossed to the KO mice to obtain a population of mice that are homozygous for the mouse tau disruption but that also carry and express the human tau transgene.
Immunohistochemistry
Abnormal phosphorylation and conformational changes on tau can be detected with monoclonal antibodies, including the following: (1) CP13, reactive with phosphorylation on serine 202 on tau in pretangles (Kimura et al., 1996) and later-stage NFTs (Augustinack et al., 2002); (2) MC1, recognizing a conformation (Jicha et al., 1997a,b, 1999) that is specific to abnormal tau (Weaver et al., 2000); and (3) PHF1 (paired helical filament), specific for phosphorylation at serines 396/404 (Ps396/404), reacting with later-stage NFTs (Otvos et al., 1994; Jicha et al., 1997b).
Immunohistochemistry was performed using these antibodies on htau mouse brain serial coronal vibratome sections (50 μm) collected from multiple mice, of both sexes, aged 1, 8, 12, 14, 16, and 18 months. Mouse brains were immunostained using standard streptavidin biotin peroxidase methods as described previously (Duff et al., 2000) and developed with 3,3′-diaminobenzidine (DAB). The following monoclonal antibodies were used to stain the free-floating brain sections: CP13 (1:25) (Ps202), MC1 (1:10) (detecting a conformational abnormality of tau), and PHF1 (1:25) (Ps396/404). Sections were also immunostained with antibodies to neuronal nuclei (NeuN) (Chemicon, Temecula, CA).
Electron microscopy
Tissue sections. Electron microscopy (EM) on tissue sections was performed as described previously (Andorfer et al., 2003) on mice aged 15, 18, 22, and 26 months stained with uranyl acetate (5% in 50% ethanol).
Sarkosyl-insoluble filaments. Isolation of Sarkosyl-insoluble tau filaments was performed as described previously (Andorfer et al., 2003). Ultracentrifugation and Sarkosyl extraction (30 min incubation in 1% Sarkosyl) was performed according to Greenberg and Davies (1990) to obtain insoluble fractions of tau from htau mice aged 15 months. Insoluble fractions (pellets) were washed one time with 1% Sarkosyl and resuspended in 100 μl of 1× TBS. A 6 μl aliquot of the suspension of the insoluble material was placed on each carbon-coated Formvar grid (Electron Microscopy Sciences, Fort Washington, PA) for 10 min, rinsed in distilled water for 10 min, placed in 1× TBS for 2 min, and then drained. Grids were blocked for 10 min with a 1× TBS solution containing 0.1% Tween and 0.1% gelatin from coldwater fish skin (Sigma, St. Louis, MO) and then drained. Primary antibodies (CP13 and PHF1) were diluted 1:100 in the blocking solution above, added to grids for 1 h, and then washed off in 1× TBS three times, 5 min each. Immunogold-conjugated (10 nm) goat anti-mouse IgG (heavy and light chain) (Ted Pella, Redding, CA) was diluted 1:50 in the blocking solution, placed on grids for 1 h, and then drained and washed three times for 5 min each in 1× TBS. Grids were then fixed in 2% glutaraldehyde (diluted in 1× TBS) for 5 min, washed three times for 5 min each in distilled water, stained for 15 min in uranyl acetate (5% in 50% ethanol), washed in distilled water, and allowed to dry.
Thioflavine-S staining
Briefly, free-floating vibratome sections were washed in distilled water for 1 min and then incubated for 10 min in 0.1% thioflavine-S (Sigma) made in distilled water. Sections were then dipped in 70% ethanol two times and incubated in 100 mm sodium citrate, pH 4.0, for 4 min. Sections were mounted (in the sodium citrate solution, pH 4.0) on glass slides, coverslipped in 80% glycerol at pH 4.0, and examined using a Bio-Rad (Hercules, CA) 2000 scanning laser confocal microscope. Incubation and mounting were performed at pH 4.0 to quench enhanced green fluorescent protein fluorescence (Nakanishi et al., 2001). Quenching was confirmed by examination of KO mice.
Comparison of cortical thickness
Vibratome sections (50 μm) collected from several ages of htau mice (1, 10, and 14 months) were stained with NeuN (Chemicon), an antibody that specifically labels neuronal nuclei. Cortical thickness and density of neuronal nuclei were qualitatively compared between closely matched sections and brain regions from htau mice of each of the ages.
Stereology
Tissue preparation. Brains were extracted from htau mice aged 8 and 17 months (five animals per age group) and immediately immersion fixed in 4% paraformaldehyde overnight at 4°C. Brains were cut in the coronal plane into 50-μm-thick sections using a vibratome. Sections were collected in order, mounted on slides, dried overnight at room temperature, stained with cresyl violet, dehydrated, and coverslipped.
Estimates of neuron loss. The optical fractionator (West et al., 1991) was used to obtain quantitative estimates of neuronal loss with age in the htau mice. After random selection of a starting point, six to eight sections, at equal intervals (800 μm), were collected through the length of the piriform cortex in each animal. We used an Axiophot IIc photomicroscope (Zeiss, Oberkochen, Germany) equipped with an ASI motorized stage, high-resolution Dage digital camera (Dage-MTI, Michigan City, IN), and StereoInvestigator software program (MicroBrightField, Wiliston, VT) running on a personal computer. The boundaries of the piriform cortex were delimited based on a mouse brain atlas (Hof, 2000). Neurons were identified using morphological criteria for nuclei and systematically and randomly sampled within counting frames of 20 × 20 μm, using a dissector height of 10 μm, across the entire extent of the piriform cortex. The average postprocessing thickness of sections was 18 μm. On average, 1000 neurons per animals were sampled. The coefficient of error was calculated according to Schmitz and Hof (2000). Volume estimates were calculated based on the outline of the region. Statistical analysis was performed with a Student's t test.
Immunoblotting
Immunoblotting was performed as described previously (Andorfer and Davies, 2000). Briefly, brains were extracted, weighed, and placed into a volume (in microliters) of homogenizing buffer (TBS, pH 7.4, containing 1 μm phenylmethylsulfonyl fluoride, 1 mm sodium vanadate, 2 mm EGTA, and 10 mm sodium fluoride) equal to five times brain mass (in milligrams). Samples were homogenized using a Polytron and stored at -80°C. Samples from a time course of htau brain homogenates (2, 5, 10, 13, 16, and 17 months of age) and an aged wild-type mouse control (20 months of age) were separated using 10% SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked for 1 h in 5% milk/1× TBS and probed with antibodies to caspase-3, -7, and -8 (Cell Signaling Technology, Beverly, MA) and caspase-6, -9, and -10 (Stressgen, Victoria, British Columbia, Canada), diluted at 1:1000 in 5% milk/1× TBS. Membranes were additionally probed with an antibody to SNAP-25 [synaptosome-associated protein of 25 kDa (SP-14)] (Honer et al., 1992) diluted 1:20 in 5% milk/1× TBS.
Paraffin sections
Antigen retrieval method. Sagittal brain hemispheres were incubated in 4% paraformaldehyde, embedded in paraffin, sectioned, and mounted on glass slides. Sections were deparaffinized and rehydrated, and endogenous peroxidase activity was quenched via serial incubation in xylene (two times for 10 min each), 100% Etoh (two times for 10 min each), 95% methanol/5% H2O2 (30 min), 95% Etoh (two times for 5 min each), and 80% Etoh (two times for 5 min each). Antigen retrieval was then performed by incubating sections in sodium citrate buffer, pH 6.0, for 20 min in a vegetable steamer (Black and Decker, Towson, MD). Sections were allowed to cool at room temperature for 30 min and then blocked in 5% milk/1× TBS for 1 h before antibody application.
Antibodies. Antibodies to the following cell-cycle regulatory molecules were used on the paraffin sections: proliferating cell nuclear antigen (PCNA) (1:100), Cdk5 (cyclin-dependent kinase 5) (1:1000), cyclin D1 (1:1000) (Santa Cruz Biotechnology, Santa Cruz, CA), cyclin B1 (1:1000), Cdk1/cdc2 (cell division cycle 2) (1:1000) (Upstate Biotechnology, Lake Placid, NY), ki67 (1:2000) (Vector Laboratories, Burlingame, CA), and cdc-2 (1:1000) (Cell Signaling Technologies). DAB was used as the chromogen, and, when double labeling was performed, reactivity was detected using the Vector Laboratories VIP substrate kit for peroxidase.
TUNEL assay. Detection of 3′-OH termini of DNA strand breaks was performed on paraffin sections of wild-type and htau mice aged 1, 8, 15, and 18 months using the ApopTag peroxidase kit (Intergen, Purchase, NY) following the recommendations of the manufacturer. Labeling was performed with DAB as the chromagen. Sections were counterstained with toluene blue.
Bromodeoxyuridine incorporation assay
To test for DNA synthesis, 12- and 18-month-old wild-type and htau mice were given water in a foil-wrapped bottle containing 0.5 mg/l of the thymidine analog 5-bromo-2′-deoxyuridine (BrdU) (Sigma) for either 4 or 7 d. Three animals per age group and treatment condition were killed. Brains were extracted and cut in the sagittal plane, placed in 4% paraformaldehyde overnight at 4°C, embedded in paraffin, sectioned, and mounted on slides. Antigen retrieval was performed as described above, and then sections were treated with 3N hydrochloric acid for 1 h after the 30 min cool-down phase. Sections were washed two times with 1× TBS (3 min each) to neutralize the acid and then blocked as above. Detection of BrdU was performed using an anti-BrdU monoclonal antibody (Roche Diagnostics, Indianapolis, IN) diluted 1:50 in 5% milk/1× TBS.
Results
Paired helical filaments and neurofibrillary tangles in htau mice aged 15 months and older
Sequential changes in tau phosphorylation, filament accumulation, and cell morphology occur as tau pathology progresses from early (pretangle stage), to more advanced intracellular, and ultimately, extracellular NFT stages (Kimura et al., 1996; Augustinack et al., 2002). We demonstrated previously that htau mice develop pretangles and early NFT-like pathology using monoclonal antibodies to show an age-dependent somatodendritic accumulation of phosphorylated tau that aggregates as paired helical filaments (Andorfer et al., 2003).
Here we examined tau accumulation in htau mice older than 15 months of age. Reactivity with antibodies to tau phosphorylated at serine 202 in 16-month-old htau mouse neocortex (Fig. 1A) and hippocampus (Fig. 1B) resembled reactivity of these antibodies to NFTs from human brain (Fig. 1C). Aging htau mice also developed a somatodendritic accumulation of tau in an abnormal conformation (detected with MC1, an AD conformation-specific antibody) within the piriform cortex (Fig. 1D) and hippocampus (Fig. 1E) and stained heavily with a marker for later-stage NFTs (PHF1, an antibody specific to tau phosphorylated at serine 396/404) in the neocortex (Fig. 1F) and hippocampus (Fig. 1G) in a manner that was similar to that of NFTs in human AD hippocampal neurons (data not shown). Advanced NFTs were detected with the classic fluorescent histochemical stain thioflavine-S. When applied to htau mouse brains, thioflavine-S revealed intensely labeled neurons in the cortex (Fig. 1I) and hippocampus (Fig. 1J) by 15 months of age. No NFTs were detected in age-matched control mice (Fig. 1H).
Figure 1.

Pathology in aging htau mice was characterized by cell-body accumulation of phosphorylated and conformationally altered tau that aggregated into classic paired helical filaments and neurofibrillary tangles. An antibody to tau phosphorylated at serine 202 (CP13) labeled neurons in 16-month-old htau mouse piriform cortex (A) and hippocampus (B) in a manner that resembled that in neurofibrillary tangles of human brain (C). Neurons were also positive for MC1 (an AD conformation-specific antibody) in the piriform cortex (D) and hippocampus (E) of 16-month-old htau mice. PHF1 (Ps396/404) antibodies labeled distorted cortical neurons in the neocortex (F) and hippocampus (G) of 16-month-old htau mice. Thioflavine-S, a fluorescent histochemical stain commonly used to detect neurofibrillary tangles, did not detect any in control mice (H) but did reveal intensely labeled neurons in htau mice shown here in the piriform cortex (I) and hippocampus (J) of 15-month-old htau mice. Scale bars, 10 μm. Electron microscopy of Sarkosyl-insoluble material isolated from 16-month-old htau mice that were immunogold labeled with antibodies specific for phosphorylations on tau at serine 202 (CP13; K) and serines 396/404 (PHF1; L) confirmed the presence of paired helical filaments. Very similar results are found in mice aged 16-22 months, and representative data are shown.
Filaments isolated from these brains had an average period length of 44.7 nm and were comparable with that of PHF isolated from human AD brain. Immunogold labeling of the filaments revealed that they were composed of tau phosphorylated on serine 202 (Fig. 1K) and on serines 396/404 (Fig. 1L).
Cell loss with advanced age in htau mice
Morphologic abnormalities indicative of cellular injury were evident in some neurons in the older htau mice that were also positive for cell-body accumulation of tau phosphorylated at serine 202 (CP13) and for the abnormal MC1 conformational change, particularly within the piriform cortex. Predominantly evident were enlarged vacuoles, irregularly shaped membranes, and distorted processes (Fig. 2A-C). Additionally, some neurons appeared ballooned (Fig. 2D), and still others had the character of a so-called “ghost tangle” or a late-stage tangle (Fig. 2E).
Figure 2.
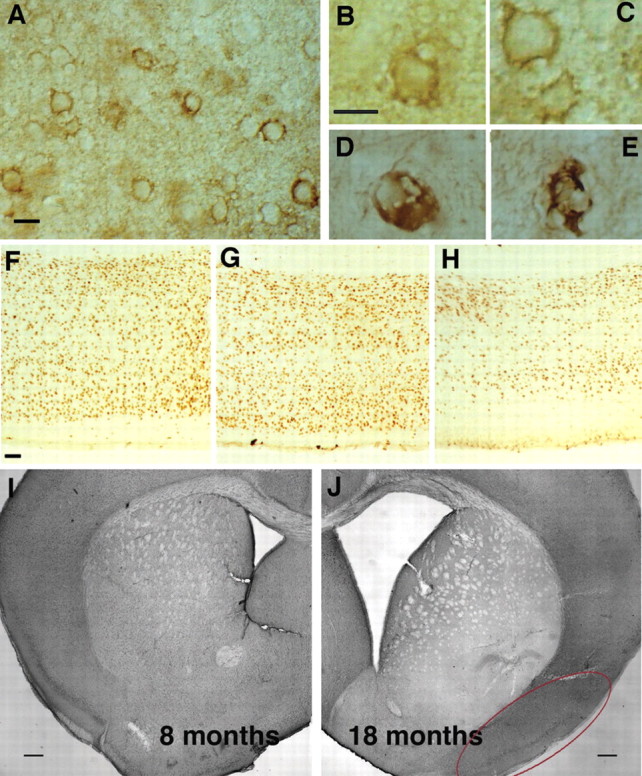
Gross changes in old htau brains, including abnormal neuronal morphology, a decrease in cortical thickness, and a dramatic enlargement of ventricle size. Irregularly shaped neurons were detected with CP13 (A-C) and MC1 (D, E). Note the presence of enlarged vacuoles, irregularly shaped membranes, and distorted processes (A-C). Additionally, some neurons appeared ballooned (D), and still others had the character of late-stage tangles (E). Comparison of cortical thickness from matched brain regions of 1-month-old (F), 10-month-old (G), and 14-month-old (H) htau mice indicated a decrease in thickness and an obvious reduction in the number of detectable nuclei with age. A comparison of ventricle size in level-matched coronal sections between htau mice aged 8 months (I) and 18 months (J) revealed a dramatic increase in the older mice. The red oval delineates the piriform cortex. Scale bars: A-E, 10 μm; F-H, 50 μm; I, J, 250 μm.
Gross morphological analysis of aging htau mouse brains compared with age-matched wild-type mice and younger htau mice revealed striking qualitative evidence of cell loss. Comparison of cortical thickness from level matched vibratome sections of 1-month-old (Fig. 2F), 10-month-old (Fig. 2G), and 14-month-old (Fig. 2H) htau mice stained with an antibody specifically reactive to neuronal nuclei indicated a decrease in thickness and a significant reduction in the number of detectable nuclei with age. Additionally, a comparison of ventricle size in level-matched coronal sections between htau mice aged 8 months (Fig. 2I) and 18 months (Fig. 2J) revealed a dramatic increase in the older htau mice, as well as a dramatic decrease in the thickness of the corpus callosum, both indicative of significant cell loss. This was not observed in aging wild-type control mice (data not shown). Considering these striking qualitative observations and the fact that neuronal injury and loss can be important features of diseases associated with NFT formation, quantitative stereologic evidence of cell loss in the htau mice was sought using the optical fractionator method.
Quantification of cell death in htau mice
We focused our examination of cell loss in the piriform cortex (Fig. 2J, surrounded by the red oval), a region in which older htau mice grossly display extensive accumulation of somatodendritic tau, morphologically abnormal cells, and a gross loss of cortical organization. The total number of neurons in the piriform cortex was estimated in 10 individual htau mice, aged 8 and 17 months. Three males and two females were examined per group, and the results were compiled in Figure 3A and presented graphically in Figure 3B. A significant reduction in the total number of neurons occurred between 8 and 17 months, from an average of 565,000 to 242,000, a decrease of 67.2% (p < 0.009). There was also a 23% reduction (p < 0.002) of the estimated volume of the piriform cortex between these age groups, consistent with the dramatic decrease in neuronal number and the gross brain observations described above.
Figure 3.

Quantification of age-related neuronal loss in the piriform cortex of htau mice was performed using the optical fractionator method of stereology. A, Neuronal counts and region volume estimates in the piriform cortex provided for the individual htau mouse examined, aged 8 months (n = 5) or 17 months (n = 5). CE, Coefficient of error. B, Comparison of estimated total of neurons between the two ages of htau mice revealed a dramatic reduction in the total number of neurons between 8 and 18 months, from an average of 565,000 to 242,000, a decrease of 67.2% (p < 0.009). There was also a 23% reduction (p < 0.002) in the estimated volume of the piriform cortex between these age groups.
Electron microscopy of dying htau neurons
Ultrastructural morphological criteria are often used to discern the mode of cell death, particularly with respect to nuclear morphology (for review, see Leist and Jaattela, 2001). When examined by EM, neurons in the older htau mice display morphological evidence that is not easily classified into a single cell-death category. A number of dying neurons in the htau mice displayed features of apoptosis, including nuclear breakdown, chromatin condensation, membrane blebbing, and cytoplasmic shrinkage (Fig. 4A,B). Other neurons showed chromatin condensation into dark dispersed bodies, but there was no sign of cell shrinkage or cell fragmentation (Fig. 4C,D), suggesting that these cells were not dying by a classic apoptotic cell death. Further morphological diversity was seen in neurons that displayed no prominent chromatin condensation but extensive organelle swelling and cytoplasmic vacuolization more suggestive of necrotic cell lysis (Fig. 4E,G). Interestingly, the majority of the dying neurons with nuclear abnormalities in the old htau mice did not have significant accumulations of filamentous tau (Fig. 4A-E) (Andorfer et al., 2003), whereas many neurons with tau aggregates displayed intact nuclei (Fig. 4F). Local damage, swelling, and demyelination were evident in many of the axonal processes (Fig. 4H,I).
Figure 4.
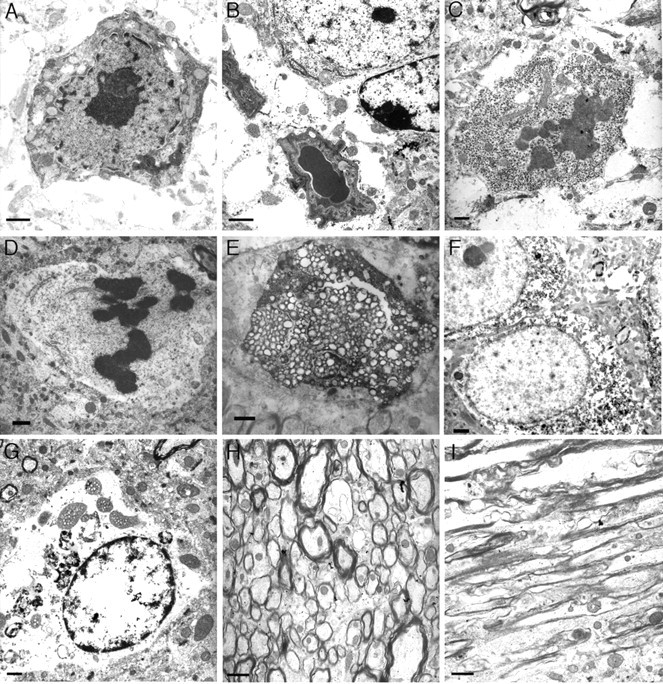
Nuclear abnormalities in neurons in the older htau mice detected by electron microscopy suggest multiple pathways of cell death. A, B, Features of apoptosis, including nuclear breakdown, chromatin condensation, membrane blebbing, and cytoplasmic shrinkage in a 22-month-old htau mouse cortex (A) and striatum (B). Chromatin condensation into dark dispersed bodies in the absence of cell shrinkage or cell fragmentation was also detected in 22-month-old htau hippocampus (C) and cortex (D). Other neurons in the cortex (E) and thalamus (G) of htau mice displayed no prominent chromatin condensation but extensive organelle swelling and cytoplasmic vacuolization suggestive of cell lysis. Local damage, swelling, vacuolization, and demyelination were visible in axonal processes (H, I), and many of these processes contained filamentous aggregates. Neurons were also detected in the 22-month-old htau brain that had little damage (F) and displayed dispersed chromatin and intact organelles but that had accumulated aggregates. Note that the neurons that displayed nuclear breakdown did not have significant accumulations of filamentous tau (A-F). Scale bars: A-G, 2 μm; H, I, 5 μm.
Cells in htau brains show evidence of DNA fragmentation but no activation of caspases
DNA fragmentation can be detected using the TUNEL method (Gavrieli et al., 1992; Sgonc and Wick, 1994) to label broken 3′-OH ends of DNA. When we used this method on 15-month-old wild-type mouse brain paraffin sections, we did not detect any TUNEL-positive cells (Fig. 5A), whereas age-matched htau mouse brain sections were TUNEL-positive in the hippocampus (Fig. 5B) and piriform cortex (Fig. 6C). Numbers of positive cells were low, with two to three positive cells per section. TUNEL has been classically used as evidence of caspase driven apoptosis; however, TUNEL has also been detected independently of caspase activation. We therefore looked for more direct biochemical evidence of caspase activation in Western blots of a time course of htau brain homogenates (2, 5, 10, 13, 16, 17, and 20 months of age) and an aged wild-type mouse control (20 months old) using antibodies to cleaved caspase-3, -6, -7, -8, -9, and -10. Caspases become active when cleaved (Hengartner, 2000), but, consistent with the small numbers of TUNEL-positive cells detected, we found no evidence of any caspase cleavage at the gross level in the htau mice, as shown in Figure 6A for caspase-3 (cleavage fragment would appear at 17/19 kDa). Similar results were obtained for other members of the caspase family, including caspase-6, -7, -8, -9, and -10 (data not shown).
Figure 5.

DNA fragmentation was detected in the htau mice using TUNEL on paraffin sections. No TUNEL reactivity was detected in a 15-month-old control mouse brain (A), whereas TUNEL-positive cells were detected in an age-matched htau mouse hippocampus (B) and piriform cortex (C). Numbers of positive cells were low: two to three positive cells per section in mice aged 15-18 months. Scale bars, 10 μm.
Figure 6.

Cell death in htau mice did not occur through a classic caspase-dependent apoptotic pathway. A Western blot of brain homogenates of a time course of htau mice (ht) (2, 5, 10, 13, 16, and 17 months of age) and an old aged wild-type (wt) mouse (20 months) probed with an antibody that detects both full length and cleaved (active) forms of caspase-3 revealed no cleavage of caspase-3 (A). Active caspase-3 would be detected as a band at 17/19 kDa. Similar negative results were obtained for other members of the caspase family, including caspase-6, -7, -8, -9, and -10 (data not shown). B, Blots were also probed with SP-14 and antibody that recognizes SNAP-25 to control for loading.
Expression of cell-cycle markers in htau mice
In light of the connection that has been drawn between abnormal expression of cell-cycle molecules and cell death in human tauopathies (for review, see Vincent et al., 2003), we hypothesized that a similar reexpression of some of these regulatory molecules might also occur in the htau mice. To test this, we performed immunohistochemistry on paraffin sections from htau mouse brains and wild-type control mice aged 12, 18, and 22 months with a series of antibodies to cell-cycle molecules that have been implicated in the human disease, including PCNA, cyclin D1, ki67, cyclin B1, and cdc2.
We found that the htau brains were positive for the cell-cycle regulating proteins cyclin D1, ki67, and PCNA, but brains from age-matched wild-type mice were not reactive with antibodies to these proteins (Figs. 7, 8). The amount of ki67 is normally tightly regulated (Scholzen and Gerdes, 2000), and, although its exact function in the cell cycle is not known, it has been determined to be essential for cell proliferation (Schluter et al., 1993) and is aberrantly expressed in AD (Nagy et al., 1997). Select populations of neurons in htau cortex (Fig. 7B), thalamus (Fig. 7C), and hippocampus (data not shown) were positive for expression of ki67. The appearance of ki67 in these cells suggested a loss of regulation of ki67 expression, or degradation, and a possible reentry of these normally postmitotic cells into the cell cycle. Cyclin D1 is another cell-cycle regulating molecule that is involved in controlling the progression of the cell cycle through G1-phase to the boundary of the S-phase (for review, see Coqueret, 2002). Although cyclin D1 is not normally expressed in postmitotic neurons, it has been detected in AD brains (Busser et al., 1998; Yang et al., 2003). Cyclin D1-positive cells were detected in the htau mice, shown here in the cortex (Fig. 7E) and thalamus (Fig. 7F) of a 22-month-old mouse.
Figure 7.

The S-phase associated cell-cycle molecules ki67 and cyclin D1 are detectable in htau neurons. Brains from age-matched wild-type mice were not reactive with antibodies to ki67 (A), but sections from htau mice showed numerous positive neurons in the cortex (B) and thalamus (C). In wild-type mice, there was no reactivity with cyclin D antibodies (D), and select populations of neurons in htau mice were positive, shown here in the cortex (E) and thalamus (F) of a 22-month-old mouse. Scale bars, 10 μm. Very similar results have been obtained in 12- and 18-month-old htau mice.
Figure 8.

Cell cycling and DNA synthesis in the htau mouse brain was detected with antibodies to PCNA and ki67 and by incorporation of the thiamine analog BrdU. No reactivity with the PCNA antibody was detected in wild-type mice (A) shown here at 14 months, but reactivity was detected in an age-matched htau mouse brain (B). Double labeling with PCNA (C, brown) and NeuN (C, purple) (a neuronal marker) showed that both neurons and glial cells expressed PCNA. Most cells that were positive for PCNA (D, brown staining) were not positive for CP13, reactive to phospho-serine 202 on tau (D, purple staining), with some rare exceptions. Incorporation of the thiamine analog BrdU (E, F, H, I), indicating DNA synthesis, was detected in some postmitotic brain regions of 12- and 18-month-old htau mice, including the somatosensory cortex (H, brown). Double labeling with antibodies to BrdU and ki67 (H, purple) revealed some cells that were positive for either marker and other cells that were positive for both (I).
Paraffin sections of htau brains were also reactive with an antibody to PCNA (Fig. 8B). PCNA is a cell-cycle regulatory molecule that appears to play a coordinating role in a number of interactions at the DNA replication fork but has also been shown to be involved in DNA repair (for review, see Maga and Hubscher, 2003). To determine which brain cell types were reactive to the PCNA antibody, we performed double labeling (Fig. 8C) with PCNA and NeuN, a neuronal marker. With this double labeling, we noted that both neurons and glial cells expressed PCNA, shown for a 22-month-old htau brain. Additionally, to determine whether there was any colocalization between PCNA and the abnormal tau phosphorylation in the htau brains, we performed double labeling with the PCNA antibody and CP13 (reactive with tau phosphorylated at serine 202). Additionally, we noted that most cells that were positive for PCNA were not positive for CP13 (Fig. 8D).
The cell-cycle regulators that we detected in the htau mice were for the most part associated with G1-/S-phases of the cell cycle. Normally, the cell-cycle progresses in a highly stereotyped manner beginning with entry into G1/S in which DNA replication occurs, progressing through mitosis, and ultimately ending in cell division. For mitosis to occur, cells must progress through the G2/M transition that is initiated after the assembly of the maturation-promoting factor, composed of cyclin B and cdc2. We did not detect these mitosis-associated molecules cyclin B1, and Cdk1 (cdc2) in the htau brains (data not shown), suggesting that the changes in cell-cycle regulation fall short of mitosis and cell division.
Bromodeoxyuridine incorporation in cortical neurons in htau mice
Cells that are actively synthesizing DNA can be detected by looking for the incorporation of the thiamine analog BrdU. To confirm the cell-cycle reentry of htau neurons, we provided a set of htau and wild-type mice (12 and 18 months old) with BrdU in their drinking water for 4 and 7 d before they were killed (three mice per age per treatment length) and looked for incorporation using an antibody specific for BrdU. Neurogenesis has been shown to continue through adult life in specific brain regions, including the subventricular zone of the lateral ventricles and the subgranular zone of the dentate gyrus of the hippocampus (Eriksson et al., 1998; Gould et al., 1998), but it does not normally occur in most regions with large populations of postmitotic neurons. In both htau and age-matched wild-type mice, two to four small cells per section were BrdU positive in the dentate gyrus, presumably reflecting ongoing neurogenesis. We detected BrdU incorporation in the htau mice after both treatment lengths in several postmitotic regions, including the somatosensory cortex (Fig. 8E,F) of 12-month-old htau mice treated for 4 and 7 d, respectively. In both 12- and 24-month-old htau mice, an average of 23 cells per section (range of 12-39) were BrdU positive (excluding small cells in the dentate gyrus or associated with blood vessels). We additionally performed double labeling on this set of mouse brain paraffin sections using the BrdU antibody in conjunction with an antibody to ki67. No reactivity with either antibody was detected in the somatosensory cortex (Fig. 8G) of wild-type mice, but reactivity with both BrdU and ki67 antibodies was detected in the htau mice (Fig. 8H). A number of these cells were positive for both markers of DNA synthesis, as shown in Figure 8I. Sections stained with the BrdU antibody were also double labeled with CP13, a phosphotau antibody that labels neurons at all stages of tangle formation in humans. In 12 sections from six htau mice (three each of 12 and 18 months), only 2 of 275 BrdU-labeled cells were also positive for CP13 (0.72%)
Discussion
Using our htau model, we have shown that NFT formation and cell death occur with expression of nonmutant tau, although overexpression of tau and an abnormal ratio of tau isoforms are probably important factors. NFT-like formation and cell death from tau with no mutations has been shown in a Drosophila model combining overexpression of wild-type human tau with manipulation of GSK3β (glycogen synthase kinase-3β) activation (Jackson et al., 2002). Mice expressing a cDNA for the shortest 3R isoform of tau (Ishihara et al., 1999) also develop tau pathology at high age (2 years), although the pathology is not obviously accompanied by neurodegeneration. The expression of human tau in the htau mouse is approximately four times the expression of mouse tau in wild-type animals, and there is an excess of 3R isoforms over 4R tau (Andorfer et al., 2003). It is currently unclear whether overexpression or the isoform imbalance are responsible for the tangle formation and neuronal death or whether the loss of mouse tau is critical, as suggested by the lack of pathology in the 8c mouse (Duff et al., 2000), from which the htau was derived.
The results of this study suggest that NFT toxicity may not be the only cause of the dramatic neuronal loss observed in this model. Whereas a number of phosphotau-positive cells displayed morphological abnormalities such as excess vacuolization and ballooning and even ghost tangles, other individual neurons within the htau brains appeared to die without the formation of NFTs. When examined by EM, these neurons displayed “dying” morphologies, such as chromatin condensation and vacuolization, but did not appear to have a significant load of tau filaments. Indeed, many of the cells in the htau mice that accumulated filamentous tau appeared “healthy” in terms of nuclear morphology. Neurodegeneration has been demonstrated to occur without NFT formation in a Drosophila model of tauopathy in which wild-type human tau is expressed (Wittmann et al., 2001), although the extent of neurodegeneration was much higher in flies expressing mutant (R406W) human tau. Moreover, there is evidence to suggest that some neurons in AD brain may die without forming NFTs (Gomez-Isla et al., 1997). Our data provide additional evidence that cell death may not result directly or primarily from NFT formation but rather may result from a loss of normal tau function or a gain of pathologic tau function.
Abnormal tau function in the htau mice could result from an altered tau isoform ratio that we demonstrated previously in the initial characterization of the htau mice (Andorfer et al., 2003). Tau isoforms can be separated into two groups, either 3R or 4R, depending on the inclusion or exclusion of exon 10 (Goedert et al., 1988, 1989; Lee et al., 1988; Himmler, 1989). Alterations in the ratio of 3R and 4R isoforms, normally equal in humans, have been linked to a number of neurodegenerative diseases that develop NFTs (Spillantini and Goedert, 1998; Hutton, 2001; Poorkaj et al., 2001). We demonstrated previously excess production of the 3R tau isoforms in htau mice. The 3R isoforms of tau have been shown to have a lower affinity for microtubules (Goode and Feinstein, 1994; Panda et al., 1995) and a differential ability to regulate microtubule dynamics (Panda et al., 2003; Bunker et al., 2004). It has been proposed that there may be a narrow window of acceptable microtubule dynamics (Bunker et al., 2004) because overstabilization (Jordan et al., 1996; Yvon et al., 1999) and destabilization (Jordan et al., 1996) have each been demonstrated to result in mitotic block and cell death. Given this, it is possible that the excess 3R tau in htau mice could result in more unstable microtubules, potentially disrupting their normal function and resulting in neuronal death.
We also demonstrated here that the neuronal death in the htau mice was morphologically diverse. It was also associated with the aberrant appearance of cell-cycle molecules and the initiation of DNA synthesis. Changes in microtubule dynamics could also be related to these observed changes in cell-cycle control because microtubules are vital not only for the maintenance of cell shape but also play important roles in cell signaling, cell division, and mitosis. Although the process of cell division and cell death would intuitively appear to be quite distinct, they may in fact be mechanistically related. Changes in microtubule behavior have been shown to activate programmed cell-death signals (Srivastava et al., 1998; Giannakakou et al., 2001; Ye et al., 2001) (for review, see Michaelis et al., 2002). It is therefore possible that disorganization of chromatin, potentially caused by microtubule instability, causes cells in the htau mice to develop imbalances that cannot be tolerated and thus set off a cascade of cell death. Such a cell-cycle-driven mechanism of neurodegeneration has been proposed to occur in AD and several other tauopathies (Herrup and Yang, 2001; Yang et al., 2001, 2003) (for review see, Vincent et al., 2003), but a direct link to nonmutant tau pathology had not been established so far. Involvement of cell-cycle mechanisms in the neuronal death that follows hypoxia-ischemia has also been suggested in two recent papers (Kuan et al., 2004; Wen et al., 2004).
Although the changes in cell-cycle control in the htau mice must be downstream of the abnormal tau function, these do not appear to occur exclusively as a direct result of NFT formation. Many of the cells that were observed to reenter the cell cycle and to exhibit catastrophic changes in nuclear morphology, indicative of cell death, did not form NFTs. However, significant NFT loads were noted in a number of axons that displayed signs of pathology, including swelling and demyelination. This axonal pathology could be related to a disruption of axonal transport, which has been shown to cause cell death in several animal models (Gunawardena and Goldstein, 2001; LaMonte et al., 2002; Gunawardena et al., 2003) and has been suggested as a potential cause of neurodegeneration in AD (Goldstein, 2003). It would appear that, in the htau mouse, both NFT formation and cell-cycle activation occur simultaneously in different populations of neurons in the piriform and probably other cortical regions. It seems likely that both processes result in neuronal death, although the time course of death may be somewhat different.
We detected mixed features of apoptosis, necrosis, and auto-phagy, indicating that there is diversity in the cell-death pathways of individual neurons in the htau mice that is reminiscent of the diversity that has been noted in human neurodegenerative dementias. Zehr et al. (2004) have reported evidence of apoptosis in oligodendrocytes of mutant human tau transgenics, and we cannot exclude the possibility that the TUNEL-positive cells detected in htau mice were glia rather than neurons. Additional work will be necessary to examine this possibility. Although a number of dying neurons in the htau mice display features of apoptosis, including nuclear breakdown, chromatin condensation, and membrane blebbing, other neurons display no prominent chromatin condensation but extensive organelle swelling and cytoplasmic vacuolization more suggestive of necrotic cell lysis. This morphological variation may reflect variation in affected cell types. The unique biochemical properties of different neuronal subpopulations might influence the kinetics of cell death and therefore underlie the particular manifestation of death within individual neurons in the htau mice. Although it is generally assumed in AD that neurons with less filament load are in earlier stages of the degenerative process, it is possible that this is related more to individual neurons dealing with the stresses in different ways.
The appearance of key pathological hallmarks, cell death, and NFT formation within our single mammalian model provides an exciting opportunity to further explore the relationship between them and their connection to inappropriate cell-cycle activation and ultimately to understand their importance in human tauopathies, including AD.
Footnotes
This work was supported by National Institute of Mental Health Grants 38623 and AG022102 and Applied Neurosolutions (Vernon Hills, IL).
Correspondence should be addressed to Dr. Cathy Andorfer, Albert Einstein College of Medicine, Bronx, NY10461. E-mail: cathy.andorfer@sbri.org.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255446-09$15.00/0
References
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P (2003) Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem 86: 582-590. [DOI] [PubMed] [Google Scholar]
- Andorfer CA, Davies P (2000) PKA phosphorylations on tau: developmental studies in the mouse. Dev Neurosci 22: 303-309. [DOI] [PubMed] [Google Scholar]
- Augustinack JC, Schneider A, Mandelkow EM, Hyman BT (2002) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol (Berl) 103: 26-35. [DOI] [PubMed] [Google Scholar]
- Behl C (2000) Apoptosis and Alzheimer's disease. J Neural Transm 107: 1325-1344. [DOI] [PubMed] [Google Scholar]
- Bunker JM, Wilson L, Jordan MA, Feinstein SC (2004) Modulation of microtubule dynamics by tau in living cells: implications for development and neurodegeneration. Mol Biol Cell 15: 2720-2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busser J, Geldmacher DS, Herrup K (1998) Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci 18: 2801-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coqueret O (2002) Linking cyclins to transcriptional control. Gene 299: 35-55. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Anderson AJ (1995) A potential role for apoptosis in neurodegeneration and Alzheimer's disease. Mol Neurobiol 10: 19-45. [DOI] [PubMed] [Google Scholar]
- Duff K, Knight H, Refolo LM, Sanders S, Yu X, Picciano M, Malester B, Hutton M, Adamson J, Goedert M, Burki K, Davies P (2000) Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol Dis 7: 87-98. [DOI] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH (1998) Neurogenesis in the adult human hippocampus. Nat Med 4: 1313-1317. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA (1992) Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119: 493-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakakou P, Robey R, Fojo T, Blagosklonny MV (2001) Low concentrations of paclitaxel induce cell type-dependent p53, p21 and G1/G2 arrest instead of mitotic arrest: molecular determinants of paclitaxel-induced cytotoxicity. Oncogene 20: 3806-3813. [DOI] [PubMed] [Google Scholar]
- Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A (1988) Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci USA 85: 4051-4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA (1989) Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J 8: 393-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LS (2003) Do disorders of movement cause movement disorders and dementia? Neuron 40: 415-425. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT (1997) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol 41: 17-24. [DOI] [PubMed] [Google Scholar]
- Goode BL, Feinstein SC (1994) Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J Cell Biol 124: 769-782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, Tanapat P, McEwen BS, Flugge G, Fuchs E (1998) Proliferation of granule cell precursors in the dentate gyrus of adult monkeys is diminished by stress. Proc Natl Acad Sci USA 95: 3168-3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SG, Davies P (1990) A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA 87: 5827-5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS (2001) Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila Neuron 32: 389-401. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS (2003) Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila Neuron 40: 25-40. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA (1992) Alzheimer's disease: the amyloid cascade hypothesis. Science 256: 184-185. [DOI] [PubMed] [Google Scholar]
- Hengartner MO (2000) The biochemistry of apoptosis. Nature 407: 770-776. [DOI] [PubMed] [Google Scholar]
- Herrup K, Arendt T (2002) Re-expression of cell cycle proteins induces neuronal cell death during Alzheimer's disease. J Alzheimers Dis 4: 243-247. [DOI] [PubMed] [Google Scholar]
- Herrup K, Yang Y (2001) Pictures in molecular medicine: contemplating Alzheimer's disease as cancer: a loss of cell-cycle control. Trends Mol Med 7: 527. [DOI] [PubMed] [Google Scholar]
- Himmler A (1989) Structure of the bovine tau gene: alternatively spliced transcripts generate a protein family. Mol Cell Biol 9: 1389-1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof PR (2000) Comparative cytoarchitectonic atlas of the C57BL/6 and 129/Sv mouse brains. Amsterdam: Elsevier.
- Honer WG, Dickson DW, Gleeson J, Davies P (1992) Regional synaptic pathology in Alzheimer's disease. Neurobiol Aging 13: 375-382. [DOI] [PubMed] [Google Scholar]
- Hutton M (2001) Missense and splice site mutations in tau associated with FTDP-17: multiple pathogenic mechanisms. Neurology 56: S21-S25. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, et al. (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393: 702-705. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM (1999) Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron 24: 751-762. [DOI] [PubMed] [Google Scholar]
- Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, Geschwind DH (2002) Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila Neuron 34: 509-519. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Bowser R, Kazam IG, Davies P (1997a) Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res 48: 128-132. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Lane E, Vincent I, Otvos Jr L, Hoffmann R, Davies P (1997b) A conformation- and phosphorylation-dependent antibody recognizing the paired helical filaments of Alzheimer's disease. J Neurochem 69: 2087-2095. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Rockwood JM, Berenfeld B, Hutton M, Davies P (1999) Altered conformation of recombinant frontotemporal dementia-17 mutant tau proteins. Neurosci Lett 260: 153-156. [DOI] [PubMed] [Google Scholar]
- Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L (1996) Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res 56: 816-825. [PubMed] [Google Scholar]
- Kerr JF (1972) Shrinkage necrosis of adrenal cortical cells. J Pathol 107: 217-219. [DOI] [PubMed] [Google Scholar]
- Kimura T, Ono T, Takamatsu J, Yamamoto H, Ikegami K, Kondo A, Hasegawa M, Ihara Y, Miyamoto E, Miyakawa T (1996) Sequential changes of tau-site-specific phosphorylation during development of paired helical filaments. Dementia 7: 177-181. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Schloemer AJ, Lu A, Burns KA, Weng WL, Williams MT, Strauss KI, Vorhees CV, Flavell RA, Davis RJ, Sharp FR, Rakic P (2004) Hypoxia-ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J Neurosci 24: 10763-10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M, Van Winkle T, Howland DS, Holzbaur EL (2002) Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron 34: 715-727. [DOI] [PubMed] [Google Scholar]
- Lassmann H, Bancher C, Breitschopf H, Wegiel J, Bobinski M, Jellinger K, Wisniewski HM (1995) Cell death in Alzheimer's disease evaluated by DNA fragmentation in situ. Acta Neuropathol (Berl) 89: 35-41. [DOI] [PubMed] [Google Scholar]
- Lee G, Cowan N, Kirschner M (1988) The primary structure and heterogeneity of tau protein from mouse brain. Science 239: 285-288. [DOI] [PubMed] [Google Scholar]
- Leist M, Jaattela M (2001) Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol 2: 589-598. [DOI] [PubMed] [Google Scholar]
- Lew J, Qi Z, Huang QQ, Paudel H, Matsuura I, Matsushita M, Zhu X, Wang JH (1995) Structure, function, and regulation of neuronal Cdc2-like protein kinase. Neurobiol Aging 16: 263-268, discussion 268-270. [DOI] [PubMed] [Google Scholar]
- Lucassen PJ, Chung WC, Kamphorst W, Swaab DF (1997) DNA damage distribution in the human brain as shown by in situ end labeling; area-specific differences in aging and Alzheimer disease in the absence of apoptotic morphology. J Neuropathol Exp Neurol 56: 887-900. [DOI] [PubMed] [Google Scholar]
- Maga G, Hubscher U (2003) Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci 116: 3051-3060. [DOI] [PubMed] [Google Scholar]
- Michaelis ML, Dobrowsky RT, Li G (2002) Tau neurofibrillary pathology and microtubule stability. J Mol Neurosci 19: 289-293. [DOI] [PubMed] [Google Scholar]
- Nagy Z, Esiri MM, Cato AM, Smith AD (1997) Cell cycle markers in the hippocampus in Alzheimer's disease. Acta Neuropathol (Berl) 94: 6-15. [DOI] [PubMed] [Google Scholar]
- Nakanishi T, Ikawa M, Yamada S, Toshimori K, Okabe M (2001) Alkalinization of acrosome measured by GFP as a pH indicator and its relation to sperm capacitation. Dev Biol 237: 222-231. [DOI] [PubMed] [Google Scholar]
- Otvos Jr L, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM (1994) Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res 39: 669-673. [DOI] [PubMed] [Google Scholar]
- Panda D, Goode BL, Feinstein SC, Wilson L (1995) Kinetic stabilization of microtubule dynamics at steady state by tau and microtubule-binding domains of tau. Biochemistry 34: 11117-11127. [DOI] [PubMed] [Google Scholar]
- Panda D, Samuel JC, Massie M, Feinstein SC, Wilson L (2003) Differential regulation of microtubule dynamics by three- and four-repeat tau: implications for the onset of neurodegenerative disease. Proc Natl Acad Sci USA 100: 9548-9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorkaj P, Grossman M, Steinbart E, Payami H, Sadovnick A, Nochlin D, Tabira T, Trojanowski JQ, Borson S, Galasko D, Reich S, Quinn B, Schellenberg G, Bird TD (2001) Frequency of tau gene mutations in familial and sporadic cases of non-Alzheimer dementia. Arch Neurol 58: 383-387. [DOI] [PubMed] [Google Scholar]
- Schluter C, Duchrow M, Wohlenberg C, Becker MH, Key G, Flad HD, Gerdes J (1993) The cell proliferation-associated antigen of antibody Ki-67: a very large, ubiquitous nuclear protein with numerous repeated elements, representing a new kind of cell cycle-maintaining proteins. J Cell Biol 123: 513-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz C, Hof PR (2000) Recommendations for straightforward and rigorous methods of counting neurons based on a computer simulation approach. J Chem Neuroanat 20: 93-114. [DOI] [PubMed] [Google Scholar]
- Scholzen T, Gerdes J (2000) The Ki-67 protein: from the known and the unknown. J Cell Physiol 182: 311-322. [DOI] [PubMed] [Google Scholar]
- Sgonc R, Wick G (1994) Methods for the detection of apoptosis. Int Arch Allergy Immunol 105: 327-332. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M (1998) Tau protein pathology in neurodegenerative diseases. Trends Neurosci 21: 428-433. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95: 7737-7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava RK, Srivastava AR, Korsmeyer SJ, Nesterova M, Cho-Chung YS, Longo DL (1998) Involvement of microtubules in the regulation of Bcl2 phosphorylation and apoptosis through cyclic AMP-dependent protein kinase. Mol Cell Biol 18: 3509-3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadelmann C, Bruck W, Bancher C, Jellinger K, Lassmann H (1998) Alzheimer disease: DNA fragmentation indicates increased neuronal vulnerability, but not apoptosis. J Neuropathol Exp Neurol 57: 456-464. [DOI] [PubMed] [Google Scholar]
- Su JH, Anderson AJ, Cummings BJ, Cotman CW (1994) Immunohistochemical evidence for apoptosis in Alzheimer's disease. NeuroReport 5: 2529-2533. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Hardy J, Fischbeck KH (2002) Toxic proteins in neurodegenerative disease. Science 296: 1991-1995. [DOI] [PubMed] [Google Scholar]
- Troncoso JC, Martin LJ, Dal Forno G, Kawas CH (1996) Neuropathology in controls and demented subjects from the Baltimore Longitudinal Study of Aging. Neurobiol Aging 17: 365-371. [DOI] [PubMed] [Google Scholar]
- Tucker KL, Meyer M, Barde YA (2001) Neurotrophins are required for nerve growth during development. Nat Neurosci 4: 29-37. [DOI] [PubMed] [Google Scholar]
- Vincent I, Pae CI, Hallows JL (2003) The cell cycle and human neurodegenerative disease. Prog Cell Cycle Res 5: 31-41. [PubMed] [Google Scholar]
- Weaver CL, Espinoza M, Kress Y, Davies P (2000) Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging 21: 719-727. [DOI] [PubMed] [Google Scholar]
- Wen Y, Yang S, Liu R, Brun-Zinkernagel AM, Koulen P, Simpkins JW (2004) Transient cerebral ischemia induces aberrant neuronal cell cycle re-entry and Alzheimer's disease-like tauopathy in female rats. J Biol Chem 279: 22684-22692. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ (1991) Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec 231: 482-497. [DOI] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293: 711-714. [DOI] [PubMed] [Google Scholar]
- Yang Y, Geldmacher DS, Herrup K (2001) DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci 21: 2661-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Mufson EJ, Herrup K (2003) Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J Neurosci 23: 2557-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Zhou J, Landen JW, Bradbury EM, Joshi HC (2001) Sustained activation of p34(cdc2) is required for noscapine-induced apoptosis. J Biol Chem 276: 46697-46700. [DOI] [PubMed] [Google Scholar]
- Yvon AM, Wadsworth P, Jordan MA (1999) Taxol suppresses dynamics of individual microtubules in living human tumor cells. Mol Biol Cell 10: 947-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehr C, Lewis J, McGowan E, Crook J, Lin WL, Godwin K, Knight J, Dickson DW, Hutton M (2004) Apoptosis in oligodendrocytes is associated with axonal degeneration in P301L tau mice. Neurobiol Dis 15: 553-562. [DOI] [PubMed] [Google Scholar]