

Abstract
Footshock stress can reinstate cocaine-seeking behavior through a central action of the stress-associated neurohormone corticotropin-releasing factor (CRF). Here we report (1) that footshock stress releases CRF in the ventral tegmental area (VTA) of the rat brain, (2) that, in cocaine-experienced but not in cocaine-naive rats, this CRF acquires control over local glutamate release, (3) that CRF-induced glutamate release activates the mesocorticolimbic dopamine system, and (4) that, through this circuitry, footshock stress triggers relapse to drug seeking in cocaine-experienced animals. Thus, a long-lasting cocaine-induced neuroadaptation, presumably at the level of glutamate terminals in the VTA, appears to play an important role in stress-induced relapse to drug use. Similar neuroadaptations may be important for the comorbidity between addiction and other stress-related psychiatric disorders.
Keywords: stress, addiction, relapse, ventral tegmental area, corticotropin-releasing factor, cocaine
Introduction
Stress is one source of provocation to relapse in detoxified addicts (Kosten et al., 1986; Sinha, 2001). Animal studies suggest that stress reinstates drug seeking through an action of the neurohormone corticotropin-releasing factor (CRF) (Shaham et al., 2000; Sarnyai et al., 2001). CRF is released by cells of the paraventricular hypothalamic nucleus to activate the hypothalamic-pituitary-adrenal axis (Herman et al., 2003); it is also expressed as a presumed cotransmitter in various neurons in the brain (Swanson et al., 1983). CRF antagonists attenuate footshock-induced reinstatement of either cocaine or heroin seeking (Shaham et al., 1997, 1998; Erb et al., 1998), and CRF injections into the bed nucleus of the stria terminalis (BNST) reinstate cocaine seeking (Erb and Stewart, 1999). CRF injections into the ventral tegmental area (VTA), another site associated with stress-induced reinstatement of cocaine seeking (McFarland et al., 2004), stimulate locomotion and the mesocorticolimbic dopamine system (Kalivas et al., 1987), a system that is also implicated in drug reward (Wise and Bozarth, 1987) and drug-seeking behavior (Stewart, 1984).
The effects of CRF in the VTA are of particular interest because this neurohormone and several drugs of abuse cause synaptic neuroadaptations in VTA dopamine neurons (Ungless et al., 2001; Saal et al., 2003). It is well known that stress activates the mesocorticolimbic dopamine system (Thierry et al., 1976; Deutch et al., 1991), a system implicated broadly in stress-associated psychiatric disorders (Drevets, 2000; Moghaddam, 2002; Sesack et al., 2003). Repeated stress or repeated exposure to psychomotor stimulants can sensitize the mesocorticolimbic dopamine system to the activating (Antelman et al., 1980; Kalivas and Stewart, 1991) and rewarding (Lett, 1989; Piazza et al., 1990) effects of cocaine and other addictive drugs. Similarly, stress and five addictive drugs (amphetamine, cocaine, morphine, nicotine, and ethanol) sensitize VTA dopamine neurons to glutamatergic input (Saal et al., 2003), thought to be the major excitatory input to these neurons. Such sensitization can be induced by local application of CRF itself (Ungless et al., 2003).
We have focused on CRF as a possible link between stress mechanisms and the mesocorticolimbic dopamine system. We used in vivo microdialysis to assess the effects of footshock stress on cocaine seeking and on VTA levels of three neurotransmitters: CRF, glutamate, and dopamine. CRF in this region was of interest because of reports of CRF-immunoreactive varicose fibers in this region (Swanson et al., 1983) and because VTA dopamine neurons express CRF receptors (Van Pett et al., 2000). Glutamate input was of interest because of the sensitivity of glutamatergic transmission to stress and cocaine (Saal et al., 2003). Dopamine was of interest because glutamatergic activation of VTA dopamine neurons (Legault et al., 2000) is accompanied by dendritic dopamine release (Legault and Wise, 1999). Here we report that footshock stress causes VTA release of CRF in both cocaine-naive and cocaine-experienced rats and that CRF in the VTA of cocaine experienced but not cocaine-naive rats causes glutamate release, dopamine release, and reinstatement of cocaine-seeking behavior.
Materials and Methods
Subjects
Male Long-Evans rats (Charles River, Raleigh, NC), weighing 350-400 g at the time of the surgery, were used. They were housed individually under a reverse light/dark cycle (light on at 8:00 P.M.) with ad libitum access to food and water. The guidelines of the National Institutes of Health (publication 86-23, 1996) were followed, and all procedures were approved by the local Animal Care and Use Committee.
Surgery
The rats were implanted, under a mixture of pentobarbital (30 mg/kg, i.p.) and chloral hydrate (140 mg/kg, i.p.) anesthesia, with intravenous catheters and unilateral or bilateral guide cannulas for microdialysis probes (CMA/Microdialysis, North Chelmsford, MA). The coordinates for VTA guide cannulas were as follows: 5.6 mm posterior to bregma, 2.2 mm lateral at an angle of 12° toward the midline, and 6.7 mm ventral to the skull surface; the coordinates for substantia nigra (SN) guide cannulas were 1.2 mm lateral to those of the VTA. The cannulas were anchored to the skull with stainless-steel screws and dental cement. In each animal, an intravenous SILASTIC catheter (Dow Corning, Midland, MI) was inserted into the right external jugular vein. The catheter was inserted to just penetrate the right atrium and was secured to the vein with silk suture. The end of the catheter was fed subcutaneously around the back of the neck to exit at the back of the skull, where it was seated on a 22-gauge stainless-steel cannula (Plastics One, Roanoke, VA) that was fixed to the head assembly with dental cement. Each rat was given a prophylactic subcutaneous injection of 0.25 ml of 2.27% enrolfloxacine (Baytril) daily for 3 d. The catheters were flushed daily with 0.05 ml of gentomicin (4 mg/ml in sterile saline) and 0.05 ml of heparinized saline (30 U/ml in sterile saline) before testing.
Self-administration training
After recovery from surgery, each rat's catheter was connected by polyethylene tubing through a fluid swivel to a syringe in a microprocessor-controlled syringe pump (Razel Scientific Instruments, Stamford, CT). The polyethylene tubing was protected by a metal spring. The animal was placed in an operant chamber equipped with two levers, one fixed and one retractable, 9 cm above the chamber floor. Each rat was trained to press the retractable (“active”) lever for intravenous cocaine (1 mg/kg per injection, delivered in a volume of 0.13 ml over 4.5 s) on a fixed ratio-1 schedule of reinforcement; a white cue light above the lever was lighted for the duration of each injection. A timeout period of 20 s in which additional lever pressing was ineffective followed each drug injection. Extinction trials or a microdialysis experiment followed a minimum of 10 d of stable responding in daily 4 h sessions. The mean active and inactive lever presses and the injections in the last three sessions of self-administration training were 37.8 ± 5.2, 5.4 ± 2.9, and 32.2 ± 3.4, respectively (n = 87).
Three groups of rats (n = 7-22 per group) that received saline self-administration opportunity rather than cocaine self-administration opportunity in the training phase were either used in the assigned experiments or subsequently given similar exposure to the extinction regimen and served as cocaine-naive controls in the reinstatement testing. The mean active and inactive lever presses and the injections in these rats in the last three sessions of saline self-administration were 4.9 ± 1.4, 5.2 ± 0.9, and 2.92 ± 0.7, respectively.
Extinction sessions
Immediately after the completion of cocaine self-administration training, five groups of rats (n = 6-13 per group) were subjected to extinction sessions in which saline (0.13 ml/injection) was substituted for cocaine. Extinction sessions were continued until active-lever response counts decreased to fewer than four responses per hour (16 responses per 4 h session), the baseline level of responding seen in control animals that were not trained with cocaine reinforcement (see Fig. 1 A). This criterion was reached in 14-25 sessions after the start of saline substitution.
Figure 1.

Footshock stress reinstated cocaine seeking (A) and increased extracellular levels of ventral tegmental CRF (B), glutamate (C), and dopamine (D). Footshock stress increased responding on the previously active lever, the lever previously associated with cocaine reinforcement, in rats with a history of cocaine (cocaine-experienced, n = 13) but not in rats with a history of saline (cocaine-naive, n = 7) self-administration. *p < 0.05, significant difference from all other experimental conditions (A). Footshock increased CRF release in the VTA of both cocaine-experienced and cocaine-naive rats. Footshock selectively increased glutamate and dopamine release in the VTA of rats with a history of cocaine, but not saline, self-administration. Footshock-induced increases in VTA CRF levels were blocked by VTA perfusion of TTX (0.1 μm; n = 6-7). Basal VTA CRF, glutamate, and dopamine levels in cocaine-experienced rats were 0.056 ± 0.018, 2277 ± 659, and 0.4 ± 0.1 and in cocaine-naive rats were 0.055 ± 0.013, 1489 ± 269, and 0.6 ± 0.2 nm, respectively. In rats with TTX perfusion, the drug was included in the perfusion medium throughout the experiment. Basal CRF levels in this group were 0.028 ± 0.004 nm.*p < 0.05, significant difference from preshock baseline values (B) or from drug-naive values (C, D).
Cocaine withdrawal
Three groups underwent microdialysis without being subjected to extinction training. One group (n = 7) was tested 24 h after the last self-administration session and was designated the 1-d-withdrawal group. The other two groups were returned to their home cages after completion of the cocaine self-administration training and remained there for 6-7 d (n = 8) or 21-23 d (n = 7) before the microdialysis system was connected. These two groups were defined as the 7- and 21-d-withdrawal groups, respectively.
Footshock-induced reinstatement and transmitter release
Five sets of footshock experiments were conducted after the completion of the extinction sessions.
Experiment 1: effects of footshock on reinstatement and VTA CRF release. After completion of the extinction phase, we used one group of cocaine-experienced and one group of cocaine-naive rats (n = 7 each) to determine the effect of footshock on lever pressing and on VTA CRF levels. Another group of cocaine-naive rats (n = 7) was used to test the effect of the sodium channel blocker tetrodotoxin (TTX) on footshock-induced release of CRF in the VTA. Blockers were removed from the guide cannulas, and bilateral VTA microdialysis probes (CMA/12 14/02; CMA/Microdialysis) were inserted through the guide cannulas and fixed in position. The microdialysis probes were then connected to the microdialysis pump (CMA/100) through the two channels of a three-channel swivel (Scipro, Sanborn, NY) with fluorinated ethylenepropylene tubing. The remaining channel was used to connect the intravenous catheter to its injection pump. Each rat was placed in its testing box and infused with artificial CSF (aCSF) (in mm: 148 NaCl, 2.7 KCl, 1.2 CaCl2, and 0.8 MgCl2, pH 7.4) at a flow rate of 0.4 μl/min overnight with ad libitum access to food and water. On the following day, the perfusion medium was replaced with aCSF containing 0.2% of bovine albumin serum (BSA) (Sigma, St. Louis, MO), and the flow rate was increased to 2 μl/min. After a 30 min equilibration period and the subsequent collection of five 25 min baseline samples, each animal was given a 20 min series of inescapable and unpredictable 0.5 s footshocks (Shaham and Stewart, 1995). Shock intensity was adjusted, for each animal, to a level below the threshold for freezing behaviors (0.3-0.6 mA), the night before reinstatement testing. During reinstatement testing, it was administered at random 40 ± 30 s intervals.
A microdialysis sample was collected at the end of each footshock series, and then reinstatement testing was initiated. The active lever was introduced to the chamber, and the house light was illuminated. The consequences of lever pressing during the reinstatement test were the same as during extinction. We recorded both active and inactive lever presses as well as number of saline injections for a period of 2 h. Six 25 min postshock microdialysis samples were collected. In rats with TTX perfusion, the drug was included in the perfusion medium at a concentration of 0.1 μm throughout the dialysis period. Samples from one side (left) were used to measure CRF levels by radioimmunoassay.
Experiment 2: effects of footshock on reinstatement and VTA glutamate and dopamine release. One group of cocaine-experienced and one group of cocaine-naive rats (n = 7 each) were used. The experimental design was the same as in experiment 1, except that the microdialysis probes were perfused with aCSF throughout the experiment and 20 min microdialysis samples were collected. Active and inactive lever presses and the number of earned saline infusions were recorded for a period of 2 h. Six 20 min postshock samples were collected before the end of the experiments. Samples from the left and right VTA were used for HPLC analysis of glutamate and dopamine, respectively.
Experiment 3: effects of VTA perfusion of a CRF antagonist on footshock-induced reinstatement and VTA levels of glutamate and dopamine. The effect of VTA CRF on footshock-induced reinstatement and VTA glutamate and dopamine release were assessed with bilateral VTA perfusion of the CRF antagonist α-helical CRF (1 μm) or aCSF. We tested each animal twice in each of two conditions: one in which footshock was given and one in which the shock was withheld. The two conditions were tested in each of 2 consecutive days; order of conditions was counterbalanced. The two tests each day were separated by at least 2 h, during which time aCSF was perfused through the probes. The footshock parameters were as described previously. The no-shock condition was the same as the shock condition, except that the wires from the shock generator were not connected to the test box. When α-helical CRF was perfused, it was included in the perfusion medium 20 min before the microdialysis baseline collection and maintained until the reinstatement test was completed. We collected microdialysis samples at 20 min intervals until the end of the reinstatement test. We recorded lever presses for each lever as well as the number of saline infusions earned in a 2 h test period. The collected samples from the left and right VTA were used for HPLC analysis of glutamate and dopamine, respectively.
Experiment 4: effects of SN perfusion of a CRF antagonist on footshock-induced reinstatement and SN levels of glutamate. Two groups of animals with bilateral SN microdialysis cannulas (n = 6 each) were used to test the effect of VTA α-helical CRF (1 and 10 μm) on footshock-induced reinstatement and SN glutamate release. The test procedures were the same as in experiment 3.
Experiment 5: effects of VTA perfusion of a glutamate antagonist on footshock-induced reinstatement and VTA levels of glutamate and dopamine. The effects of VTA perfusion of the ionotropic glutamate antagonist kynurenic acid (Kyn) were tested in a similar manner. Kyn (1 mm) or aCSF was perfused in footshock or no-shock conditions. Each condition was tested twice with 20 min of footshock and twice with no shock preceding the test. Details were the same as in experiment 3.
Effects of VTA CRF perfusion on local glutamate release
The effects of VTA infusion of CRF were assessed on behavior and neurotransmitter release in three experiments.
Experiment 1: effects of VTA CRF on local glutamate release in cocaine-naive and cocaine-experienced rats. Glutamate responses to VTA CRF were determined in one group of animals previously trained to self-administer cocaine and in one group similarly experienced but “rewarded” with saline rather than cocaine infusions during the training phase (n = 7 each). The animals were connected to the microdialysis systems immediately after the completion of the cocaine or saline self-administration training, and the probes were perfused with aCSF at a flow rate of 0.4 μl/min overnight, as described previously. On the next day, we increased the flow rate to 2 μl/min. After a 30 min equilibration period and subsequent collections of five 20 min baseline samples, CRF (10 μm) was added to the perfusion medium for 40 min. We terminated microdialysis after collecting seven post-CRF samples. Glutamate levels in the collected samples were analyzed by HPLC.
Experiment 2: effects of VTA CRF on local glutamate release in rats after various cocaine withdrawal periods. We assessed the effect of VTA infusions of CRF on local glutamate release in two additional groups 7 d (n = 7) or 21 d (n = 8) after the last cocaine self-administration training session. On the day before the completion of the assigned withdrawal period, the rats were connected to the microdialysis systems for testing as described above.
Experiment 3: effects of VTA perfusion of a CRF antagonist or a sodium channel blocker on CRF-induced VTA glutamate release in cocaine-experienced rats. Two groups were used (n = 7 each). After the last self-administration training session, the rats were connected to the microdialysis systems as described previously. On the next day, baseline samples were collected, and then α-helical CRF (1 μm) was added to the perfusion medium of one group and TTX (0.1 μm) was added to the perfusion medium of the other. Forty or 60 min later, respectively, CRF (10 μm) was introduced to the perfusion medium. CRF was kept in the medium for 40 min; the α-helical CRF or TTX was kept in the perfusion medium until the end of the experiment. Twenty minute samples were collected, and glutamate levels were analyzed by HPLC.
Effects of VTA CRF perfusion on reinstatement
Two experiments dealt with the effects of CRF on reinstatement of cocaine seeking.
Experiment 1: effects of VTA perfusion of a CRF antagonist on VTA CRF-induced reinstatement. After reaching the extinction criterion, one group of rats (n = 6) was connected to the microdialysis systems and perfused overnight as described previously. On the next day, the microdialysis flow rate was increased to 2 μl/min and the VTA was perfused with aCSF for 100 min before changing to drug treatments. After this equilibration period, the VTA perfusion was either continued for 3 more hours with aCSF (vehicle) or with CRF (10 μm), α-helical CRF (1 μm), or CRF plus α-helical CRF. We began tests for reinstatement during the last 2 h of each test session (20 min into the second perfusion period). We tested each animal under each of the four conditions, in counterbalanced order, two conditions per day, on consecutive days. The two tests of each day were separated by at least 2 h, during which the probe was perfused with aCSF.
Experiment 2: effects of VTA perfusion of a glutamate antagonist on VTA CRF-induced reinstatement. Another group (n = 7) was tested in the same manner, but CRF-induced reinstatement was challenged with Kyn rather than α-helical CRF. After reaching the extinction criterion, the rats were connected to the microdialysis system and perfused with aCSF overnight as described previously. On the test day, the VTA was first perfused with aCSF for 100 min. After this equilibration period, perfusion was continued either with aCSF (vehicle) or with CRF (10 μm), Kyn (1 mm), or CRF plus Kyn (10 μm and 1 mm, respectively) for 3 more hours. Tests for reinstatement commenced during the last 2 h of each test session, as above.
Biochemical analysis of microdialysis samples
A commercially available radioimmunoassay kit (RK-019-06; Phoenix Pharmaceuticals, Mountain View, CA), with minor modifications, was used to measure CRF levels in microdialysis samples. Briefly, 50 μl samples were mixed in 1.5 ml Eppendorf tubes with 50 μl of assay buffer used to dissolve the standard and other assay reagents. To balance the chemical components between the standard and assay samples, we added 50 μl of CRF microdialysis medium to tubes containing 50 μl of standard solutions at various concentrations. We first incubated (20 h) the samples and standards with 100 μl of 125I-CRF. Then we added the diluted CRF antibody and shook the tubes with a vortex shaker. We incubated at 4°C for another 20 h before separating the antibody-bound and antibody-free labeled CRF. Samples without CRF antibody (nonspecific binding) or CRF (total binding) and containing only 100 μ l of 125I-CRF [total count (TC)] were also incubated. The antibody-bound 125I-CRF in all except the TC tubes was incubated for 2 h with 100 μl of diluted goat anti-rabbit IgG serum and 100 μl of normal rabbit serum and then separated by centrifugation at 10,000 × g and subsequent aspiration of supernatant from the tubes. The test tubes were then loaded on a five-channel gamma counter (PerkinElmer Life Sciences, Boston, MA), and the radioactivity in each tube was counted for a period of 5 min. The TC tubes were loaded directly on the gamma counter after incubation. The CRF antibody in the assay kit showed no cross-reaction with urocortin, another stress peptide, or with other peptides such as adrenocorticotropin, Arg8-vasopressin, pituitary adenylate cyclase activating polypeptide, and leutinizing hormone-releasing hormone. The detection limit of the kit is 1-2 pg/tube.
Glutamate concentrations were determined by HPLC. We used an ESA 582 pump (ESA, Chelmsford, MA), a CMA/260 degasser, a CMA/200 refrigerated microsampler, a phase II ODS column (3 μm particle size, 3.2 × 100 mm; Bioanalytical Systems, West Lafayette, IN), a CMA/280 fluorescence detector, and an ESA model 501 data station. The CMA/280 is a fixed-wavelength fluorescence detector operating at a maximal excitation of 330-365 nm and emission of 440-530 nm. We performed precolumn derivation of glutamate with an o-phthalaldehyde/mercaptoethanol reagent (in m: 0.4 borate, 0.04 phthalaldehyde, and 0.4 2-mercaptoethanol, pH 10.4). Briefly, 10 μl of the reagent was added to and mixed with the samples by the microsampler. After a 60 s reaction period at 6°C in the microsampler, 20 μl of the mixture was injected onto the column. The elution of glutamate was achieved with a mobile phase consisting of 0.15 m sodium acetate, 10% methanol, and 1.5% tetrahydrofuran at a flow rate of 0.6 ml/min. After the appearance of the glutamate peak on the chromatogram, an injection of 20 μl of 100% methanol was made by the microsampler before the end of the chromatogram to accelerate the elution of the residuals on the column. The detection limit was 0.2 pmol/injection.
Dopamine was measured with an HPLC coupled to an ESA Coulo-chem II Detector (model 5200) with a dual-electrode microdialysis cell and an ESA model 501 data station. Samples were manually injected onto the column (3 μm particle size, 3 × 150 mm; Analytical MD-150; ESA). The mobile phase for dopamine separation consisted of 75 mm NaH2PO4, 1.5 mm N-1-octanesulfonic acid, 10 μm EDTA, and 8% acetonitrile, pH 3.0 adjusted with H3PO4. Dopamine was quantified on both reducing (-250 mV) and oxidizing (350 mV) electrodes. The limit of detection for dopamine was ∼5 fmol/injection.
Drugs
Cocaine hydrochloride and the anesthetics used in the surgery were obtained from the pharmacy department within the National Institute on Drug Abuse. CRF (rat/human CRF), α-helical CRF (CRF 9-41), Kyn, BSA, and TTX were purchased from Sigma. Kyn was dissolved first with a small aliquot of 5N NaOH and diluted with aCSF to the final concentration. Other drugs were dissolved directly in saline or aCSF when appropriate. Each drug solution was adjusted to pH 7.4 before use.
Histology
After the completion of the microdialysis experiments, the rats were decapitated under anesthesia, and their brains were removed and fixed in a 10% Formalin solution. After at least 7 d of fixation, the brains were frozen and 50 μm coronal sections were taken. Probe placement was determined under low magnification after wetting the tissue to differentiate fibers from cell masses.
Statistical analysis
VTA glutamate and dopamine levels were expressed as the concentrations in the perfusate (means ± SEM). Basal values refer to those obtained before the drug was added into the perfusion medium or before footshock was given. When data were expressed as percentage of baseline values, the mean concentration of the three samples preceding the drug administration was defined as 100%. Data were analyzed with mixed ANOVAs using group or antagonist as the between-subjects factor and time as the within-subjects factor. Significant differences (α = 0.05) were followed by Fisher's PLSD tests.
The total responses on the active or inactive levers between the various test conditions in each group of rats were analyzed separately with two-way repeated-measures ANOVA using the within-subjects factors of footshock/CRF (no shock/vehicle, shock/CRF) and antagonist (vehicle/drug).
Results
Effects of footshock
Footshock reinstated lever pressing and elevated VTA levels of endogenous CRF, glutamate, and dopamine (Fig. 1) in rats that had been previously trained to lever press for intravenous cocaine but that had subsequently extinguished the habit when lever pressing resulted in saline rather than cocaine injections. Foot-shock caused such animals to return to, and press selectively, a lever that had been previously (but not recently) associated with rewarding cocaine injections (the “active” lever). Footshock did not cause increased pressing of an identical lever that had never been associated with cocaine injections (the “inactive” lever). Footshock had no effect on lever responding in a comparison group of cocaine-naive rats with a history of lever pressing for saline rather than cocaine. This was confirmed by a significant shock × training (cocaine vs saline) interaction for active (F(1,18) = 8.8; p < 0.01), but not inactive (p > 0.1), lever responding. Footshock caused responding on the active lever in the cocaine-experienced rats, and this responding was significantly different from the levels of responding in all other groups and conditions (Fig. 1A). Footshock caused equal elevations of VTA CRF in rats that had previously self-administered cocaine and rats that had previously self-administered saline (main effect of time, F(9,108) = 7.3, p < 0.01 with no time × training interaction). In contrast, footshock elevated glutamate and dopamine levels only in the cocaine-experienced animals (time × training interactions, F(9,108) = 5.0, p < 0.01 and F(9,99) = 2.9, p < 0.01, respectively). Footshock did not cause VTA release of CRF when the VTA was infused with a sodium channel-blocking concentration of TTX (Fig. 1B).
The response-reinstating effect of foot-shock was significantly attenuated by VTA perfusion (via the dialysis probe) of the CRF antagonist α-helical CRF [shock × antagonist interaction for active (F(1,5) = 25.1; p < 0.01), but not inactive (p > 0.1), lever responding] (Fig. 2), a finding that confirms a local action of CRF. Perfusion of α-helical CRF into the adjacent SN had no effect on footshock-induced reinstatement of responding [the effect of shock (F(1,10) = 26.6; p < 0.01) was significant, but the shock × antagonist interaction (p > 0.1) was not] (Fig. 3). The elevations in VTA glutamate and dopamine were also attenuated by VTA perfusion of α-helical CRF, implicating an action at local CRF1 (Van Pett et al., 2000) or CRF2 (Ungless et al., 2003) receptors (time × antagonist interactions, F(27,180) = 3.3, p < 0.01 and F(27,180) = 3.5, p < 0.01, respectively). Footshock did not elevate glutamate levels in the SN (p > 0.1) (Fig. 3).
Figure 2.

Bilateral VTA perfusions of a CRF receptor antagonist attenuated stress-induced reinstatement of cocaine seeking (A) and the local release of glutamate (B) and dopamine (C). When the CRF antagonist α-helical CRF (α-H-CRF; 1 μm) was infused via bilateral dialysis probes 20 min before footshock, it attenuated footshock stress-induced reinstatement of responding on the previously active lever (n = 6). Perfusions of α-helical CRF attenuated footshock-induced increases in VTA glutamate and dopamine release. Basal glutamate and dopamine levels were 1747 ± 577 and 0.5 ± 0.2 nm, respectively. Perfusions of α-helical CRF had no significant effects on the levels of either of the two neurotransmitters. *p < 0.05, significant difference from the other experimental conditions.
Figure 3.

Footshock did not increase glutamate levels in the substantia nigra, and perfusions of α-helical CRF into this region had no effect on footshock-induced reinstatement of cocaine seeking. Basal glutamate levels were 1726 ± 430 nm in this region. Perfusions of α-helical CRF (A, 1 μm; B, 10 μm) had no effect on basal glutamate levels (n = 6 per group). *p < 0.05, significant differences from the other experimental conditions.
VTA perfusion of the ionotropic glutamate receptor antagonist kynurenic acid also blocked both reinstatement of lever pressing [shock × antagonist interaction for active (F(1,6) = 8.6; p < 0.01) but not inactive (p > 0.1) lever pressing] and elevation of dopamine levels (time × antagonist interaction, F(27,216) = 5.2; p < 0.01) (Fig. 4). VTA perfusion with kynurenic acid had no effect on the ability of footshock to elevate VTA glutamate levels (time × antagonist interaction, p > 0.1). This finding indicates that CRF acts directly on glutamate terminals or on local nondopamine elements that control glutamate release at the level of the glutamate terminal. The possibility that CRF first acts to release dopamine which then augments glutamate release (Kalivas and Duffy, 1995) at D1 dopamine receptors on glutamate terminals (Dewar et al., 1997) appears to be ruled out, because footshock did not significantly elevate dopamine levels when glutamate receptors were blocked (time × antagonist interaction, F(27,216) = 5.2; p < 0.01).
Figure 4.
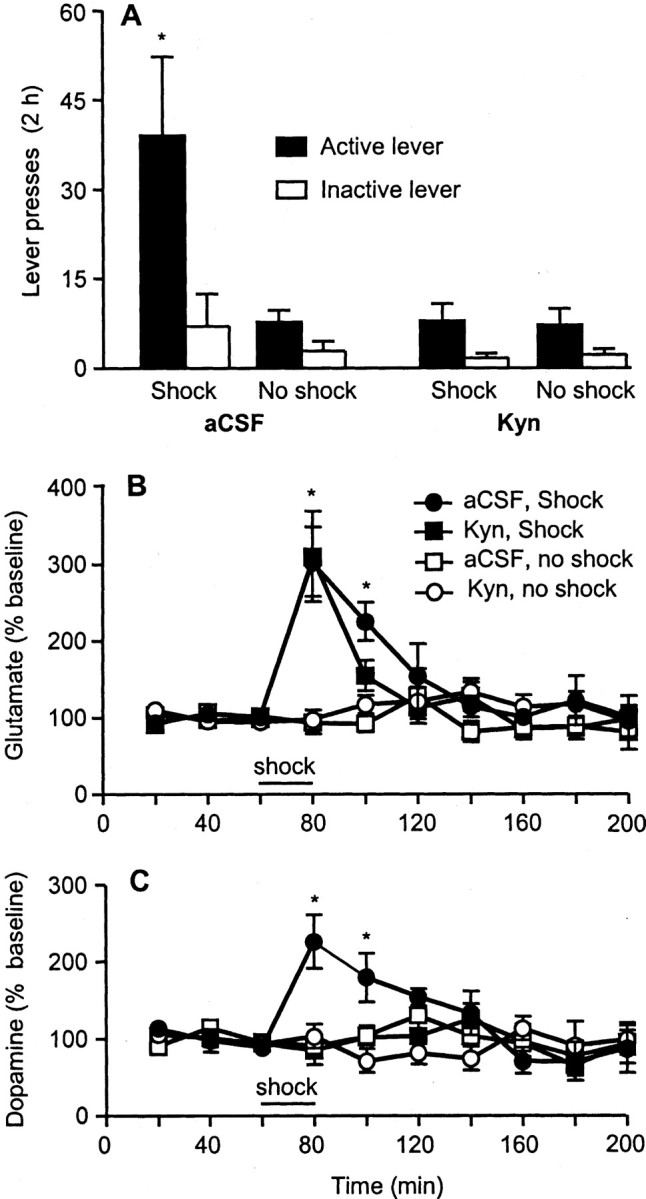
Bilateral VTA perfusions of an ionotropic glutamate receptor antagonist attenuated stress-induced reinstatement of cocaine seeking and stress-induced release of local dopamine. Basal glutamate and dopamine levels were 2444 ± 681 and 0.4 ± 0.1 nm, respectively. Kyn (1 mm infused via the dialysis probe) attenuated footshock stress-induced reinstatement of responding on the previously active lever (n = 7). *p < 0.05, significant difference from the other experimental conditions. Kyn attenuated footshock-induced increases in dopamine, but not glutamate, release in the ventral tegmental area. *p < 0.05, significant difference from the aCSF condition.
Effects of VTA perfusion of CRF
To explore these possibilities further, we assessed the effects of direct perfusion of exogenous CRF into the VTA. Perfusion of CRF through the VTA dialysis probe of cocaine-trained rats reinstated responding on the previously active but not the previously inactive lever (Fig. 5). CRF was ineffective when coperfused with the CRF antagonist α-helical CRF or with the glutamate antagonist kynurenic acid. Perfusion of aCSF, α-helical CRF, or kynurenic acid alone had no significant effect on the behavior. ANOVAs revealed significant CRF × α-helical CRF and CRF × kynurenic acid interactions for active (F(1,5) = 23.5, p < 0.01 and F(1,6) = 42.3, p < 0.01, respectively), but not inactive (p values >0.1), lever responding.
Figure 5.

Intra-VTA perfusions of CRF reinstated cocaine seeking, and this effect was attenuated by receptor antagonists of CRF and glutamate. Perfusions of α-helical CRF (α-H-CRF; 1 μm infused via the dialysis probe) attenuated CRF (10 μm)-induced reinstatement of responding on the previously active lever (n = 6). Kyn (1 mm infused via the dialysis probe) also attenuated CRF-induced reinstatement of responding on the previously active lever (n = 7). CRF, Kyn, and α-helical CRF were added to the perfusion medium 1 h before the tests commenced and kept in the perfusion medium until the end of the reinstatement testing. *p < 0.05, significant difference from the other experimental conditions.
Perfusion of CRF through the dialysis probe also elevated glutamate levels in the VTA of cocaine-experienced, but not cocaine-naive, animals (time × group interaction, F(33,297) = 2.4; p < 0.01) (Fig. 6). Glutamate was elevated equally in groups tested 1, 7, or 21 d after the last exposure to cocaine. The elevations were blocked by perfusions of the CRF antagonist α-helical CRF (time × antagonist interaction, F(11,154) = 3.5; p < 0.01) and also of sodium channel-blocking concentrations of tetrodotoxin (time × antagonist interaction, F(11,154) = 3.6; p < 0.01). Again, these findings suggest that CRF first causes local glutamate release, which, in turn, causes dopaminergic activation and associated dendritic dopamine release.
Figure 6.

Intra-VTA perfusions of CRF increased glutamate levels in cocaine-experienced rats but not in drug-naive rats (A); this effect was blocked by α-helical CRF (B) and by the sodium channel blocker TTX (C). Perfusion of CRF (10 μm) increased glutamate levels in cocaine-experienced rats, whether tested 1, 7, or 21 d after withdrawal from 14 to 18 d of cocaine self-administration (n = 7-8 per withdrawal day). This effect of CRF was not observed in rats with a history of saline self-administration (cocaine-naive groups) that were tested 1 d after their last training day (n = 7). Basal glutamate levels in the cocaine-experienced rats were 2493 ± 379, 3438 ± 921, and 2920 ± 547 nm for withdrawal days 1, 7, and 21, respectively. Basal levels in the cocaine-naive rats were 2019 ± 454 nm. *p < 0.05, significant difference from the cocaine-naive group (Fig. 5A). Coperfusions of α-helical CRF (1 μm) or TTX (0.1 μm) blocked the CRF-induced glutamate release. *p < 0.05, significant difference from the α-helical CRF plus CRF or the TTX plus CRF groups (B, C), which were tested in the 1-d-withdrawal condition.
Discussion
Our most important finding is that cocaine experience causes a long-lasting neuroadaptation that gives stress input access to mesolimbic circuitry and that predisposes stressed animals to relapse to cocaine seeking. Although cocaine experience is known to cause a variety of neuroadaptations in drug reward systems (Nestler and Aghajanian, 1997; White and Kalivas, 1998), most of these neuroadaptations are not permanent (most normalize in 1 week). In the present case, CRF infusions that failed to cause VTA glutamate release in cocaine-naive animals were as effective 3 weeks after cocaine as they were 1 d after cocaine in cocaine-experienced animals. Moreover, our findings implicate a cocaine-induced neuroadaptation in the brain circuitry responsible for initiation of drug seeking, not just for drug reinforcement.
Ours is not the only report of cocaine-induced changes in the glutamatergic response to CRF. One day after cocaine, amphetamine, morphine, nicotine, ethanol, or restraint stress, postsynaptic responses of dopamine neurons to glutamatergic input is augmented in a ventral tegmental slice preparation (Saal et al., 2003). This takes the form of an increase in the AMPA receptor-mediated synaptic currents to NMDA receptor-mediated currents in VTA dopamine neurons (Saal et al., 2003), and this increase is potentiated by CRF (Ungless et al., 2003). Whereas the change in the AMPA/NMDA receptor-mediated response to glutamate reflects a postsynaptic neuroadaptation influenced by CRF, our findings suggest an additional presynaptic neuroadaptation. Both neuroadaptations suggests important ways that the cocaine-experienced brain differs from the normal brain.
The present studies yielded three findings that clarify the interaction between stress- and reward-associated brain circuitry in the cocaine-experienced animal. First, the microdialysis findings identified the VTA as a stress-associated release site for endogenous CRF. CRF is important not just as a hypothalamic hormone; it is also expressed in cell groups that are widely distributed throughout the brain (Bale and Vale, 2004). It also acts as a neurotransmitter or neuromodulator in neurons of the central nucleus of the amygdala and the BNST (Swanson et al., 1983), regions implicated in stress-induced reinstatement of cocaine seeking (Erb et al., 1999; Shaham et al., 2000; Leri et al., 2002; McFarland et al., 2004). However, although CRF perfusion into the VTA is known to have behavioral effects (Kalivas et al., 1987), anatomical studies have not yet identified a CRF-containing projection from either of these structures to the VTA.
Second, the actions of CRF in this region are mediated by glutamate (presumably by glutamate that provides excitatory input to the VTA), causing glutamate release in cocaine-experienced but not in cocaine-naive rats. Neuroadaptations in glutamatergic control over VTA dopamine neurons have been postulated to contribute to the increasingly compulsive nature of cocaine use (Wolf, 1998; Nestler, 2001; Kalivas, 2004). Most cocaine-induced neuroadaptations in the VTA are short-lived, but some are known to last at least 21 d (Kalivas and Duffy, 1998; Lu et al., 2003), the period over which our animals' glutamate responses remained sensitive to local CRF. Interestingly, both cocaine and forced-swim stress sensitize VTA dopamine neurons to glutamatergic input (Saal et al., 2003), and cocaine sensitization augments VTA glutamate release (Kalivas and Duffy, 1998). In addition to previous findings of sensitization of glutamate receptors or within dopamine neurons themselves, our findings suggest sensitization of the local (presynaptic) control of glutamate release and suggest a mechanism by which a stress-associated transmitter can come to influence such release.
Finally, our findings implicate VTA actions of neuronal CRF in stress-induced relapse to drug seeking. Previous studies have suggested the involvement of CRF at two terminal fields of the mesocorticolimbic system: the BNST and the central nucleus of the amygdala (Erb and Stewart, 1999; Shaham et al., 2000; Erb et al., 2001). A VTA site of action for CRF was first suggested from the effects of VTA CRF on locomotion (Kalivas et al., 1987), a loose correlate of reward. Additional support for this site of action comes from the observation that reversible inactivation of this brain area attenuates stress-induced reinstatement of cocaine seeking (McFarland et al., 2004). The sequence of VTA CRF release triggering VTA glutamate release and, in turn, dopaminergic activation provides a substrate for such reinstatement. Our hypothesis is that distal elevation of extracellular dopamine levels, an effect shared with cocaine priming, induces the behavior: this hypothesis is suggested by the findings that dopamine antagonists injected into either nucleus accumbens (Xi et al., 2004) or the prefrontal cortex (Capriles et al., 2003; McFarland et al., 2004) block stress-induced reinstatement.
Our interpretation of the present findings rests heavily on the pharmacological challenges that tie our observed actions of stress and CRF to the VTA. First, VTA (but not SN) perfusion of the CRF antagonist α-helical CRF blocked or greatly attenuated the ability of either footshock (Fig. 2) or CRF (Figs. 3, 5) to reinstate drug seeking or to elevate VTA glutamate or dopamine levels. Perfusions by reverse dialysis should diffuse on the order of 50 μm and should thus limit the agent to only a portion of the dopaminergic neurons and their afferents. mRNA for CRF receptors has been localized to many cell groups in the brain (Van Pett et al., 2000), including cells of the VTA (Sauvage and Steckler, 2001). Thus, it seems likely that the effects of footshock and the effects of VTA perfusion of CRF are mediated by CRF receptors within the VTA.
VTA perfusion of the glutamate antagonist kynurenic acid also blocked the ability of footshock (Fig. 4) to reinstate cocaine seeking or to cause dopamine release. Here too, the antagonist was given through the dialysis probe and thus most likely acted within the VTA. The blockade of these effects by kynurenic acid would appear to rule out a direct action of CRF on the dopamine neurons, because the ability of footshock stress to cause dopamine release (indicative of dopaminergic cell firing) was lost when glutamate input to the dopamine cells was blocked. Thus, the possibility that CRF acted on VTA dopamine cells to cause dendritic dopamine release that acts, in turn, on D1 dopamine receptors on glutamatergic terminals (Kalivas and Duffy, 1995) seems unlikely. Rather, our working hypothesis is that CRF acts directly on glutamate terminals to cause glutamate release. The possibility of CRF receptors on VTA glutamate terminals, however, awaits direct anatomical evidence.
Challenge with the sodium channel blocker TTX, routinely used in electrophysiological and microdialysis studies to block local synaptic transmission, suggests that synaptic release of glutamate, rather than, for example, reversal of the cystine-glutamate transporter (Baker et al., 2003), was responsible for the CRF-induced glutamate elevations. Perfusion of TTX also blocked footshock-induced CRF release. In addition, we infused KCl through the dialysis probe at the end of two of the CRF dialysis experiments; KCl, which induces neurotransmitter release from nerve terminals, elevated CRF levels in these animals (data not shown). These findings suggest that the CRF elevations, like the glutamate elevations, resulted from transmitter release from nerve terminals or varicosities.
Studies to further characterize the local mechanisms underlying the effects of footshock stress and of VTA CRF on behavior and local neurotransmitter release are in progress. Whatever the full mechanism proves to be, our observations to date reveal a brain alteration that is caused by cocaine experience and that sensitizes the mesocorticolimbic dopamine system to stress. That stress can interact with the dopamine system via CRF in the VTA suggests a link between stress mechanisms and drug reward mechanisms that has long been suspected but not, until now, identified. Because abnormal relationships between the mesocorticolimbic dopamine system and the medial prefrontal cortex are of current interest not only in relation to addiction but also in relation to schizophrenia, depression, and other stress-associated disorders (Drevets, 2000; Moghaddam, 2002; Sesack et al., 2003), these observations may prove to have very broad implications for biological psychiatry.
Footnotes
We thank Nobuyoshi Suto and Laurel Ecke for suggestions and help with the data collection in the final phases of this study.
Correspondence should be addressed to Roy A. Wise, National Institute on Drug Abuse Intramural Research Program, 5500 Nathan Shock Drive, Baltimore, MD 21224. E-mail: rwise@intra.nida.nih.gov.
D. Zitzman's present address: Department of Psychology, Tufts University, The Psychology Building, 490 Boston Avenue, Medford, MA 02155.
S. Azari's present address: University of Pennsylvania School of Medicine, 399 South 34th Street, Philadelphia, PA 19104.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255389-08$15.00/0
References
- Antelman SM, Eichler AJ, Black CA, Kocan D (1980) Interchangeability of stress and amphetamine in sensitization. Science 207: 329-331. [DOI] [PubMed] [Google Scholar]
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW (2003) Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci 6: 743-749. [DOI] [PubMed] [Google Scholar]
- Bale TL, Vale WW (2004) CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol 44: 525-557. [DOI] [PubMed] [Google Scholar]
- Capriles N, Rodaros D, Sorge RE, Stewart J (2003) A role for the prefrontal cortex in stress- and cocaine-induced reinstatement of cocaine seeking in rats. Psychopharmacology 168: 66-74. [DOI] [PubMed] [Google Scholar]
- Deutch AY, Lee MC, Gillham MH, Cameron DA, Goldstein M, Iadarola MJ (1991) Stress selectively increases fos protein in dopamine neurons innervating the prefrontal cortex. Cereb Cortex 1: 273-292. [DOI] [PubMed] [Google Scholar]
- Dewar KM, Rompré PP, Stewart J, Warren RA (1997) Excitotoxic lesions of the prefrontal cortex reduce dopamine D1-like receptors in the ventral tegmental area. Eur J Pharmacol 336: 155-158. [DOI] [PubMed] [Google Scholar]
- Drevets WC (2000) Functional anatomical abnormalities in limbic and prefrontal cortical structures in major depression. Prog Brain Res 126: 413-431. [DOI] [PubMed] [Google Scholar]
- Erb S, Stewart J (1999) A role for the bed nucleus of the stria terminalis, but not the amygdala, in the effects of corticotropin-releasing factor on stress-induced reinstatement of cocaine seeking. J Neurosci 19: RC35(1-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb S, Shaham Y, Stewart J (1998) The role of corticotropin-releasing factor and corticosterone in stress- and cocaine-induced relapse to cocaine seeking in rats. J Neurosci 18: 5529-5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb S, Salmaso N, Rodaros D, Stewart J (2001) A role for the CRF-containing pathway from central nucleus of the amygdala to bed nucleus of the stria terminalis in the stress-induced reinstatement of cocaine seeking in rats. Psychopharmacology 158: 360-365. [DOI] [PubMed] [Google Scholar]
- Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, Cullinan WE (2003) Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol 24: 151-180. [DOI] [PubMed] [Google Scholar]
- Kalivas PW (2004) Glutamate systems in cocaine addiction. Curr Opin Pharmacol 4: 23-29. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Duffy P (1995) D1 receptors modulate glutamate transmission in the ventral tegmental area. J Neurosci 15: 5379-5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Duffy P (1998) Repeated cocaine administration alters extracellular glutamate in the ventral tegmental area. J Neurochem 70: 1497-1502. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Stewart J (1991) Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Rev 16: 223-244. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Duffy P, Latimer LG (1987) Neurochemical and behavioral effects of corticotropin-releasing factor in the ventral tegmental area of the rat. J Pharmacol Exp Ther 242: 757-763. [PubMed] [Google Scholar]
- Kosten TR, Rounsaville BJ, Kleber HD (1986) A 2.5-year follow-up of depression, life crises, and treatment effects on abstinence among opioid addicts. Arch Gen Psychiatry 43: 733-738. [DOI] [PubMed] [Google Scholar]
- Legault M, Wise RA (1999) Injections of N-methyl-d-aspartate into the ventral hippocampus increase extracellular dopamine in the ventral tegmental area and nucleus accumbens. Synapse 31: 241-249. [DOI] [PubMed] [Google Scholar]
- Legault M, Rompré P-P, Wise RA (2000) Chemical stimulation of the ventral hippocampus elevates nucleus accumbens dopamine by activating dopaminergic neurons of the ventral tegmental area. J Neurosci 20: 1635-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leri F, Flores J, Rodaros D, Stewart J (2002) Blockade of stress-induced, but not cocaine-induced reinstatement by infusion of noradrenergic antagonists into the bed nucleus of the stria terminalis or the central nucleus of the amygdala. J Neurosci 22: 5713-5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lett BT (1989) Repeated exposures intensify rather than diminish the rewarding effects of amphetamine, morphine, and cocaine. Psychopharmacology 98: 357-362. [DOI] [PubMed] [Google Scholar]
- Lu L, Grimm JW, Shaham Y, Hope BT (2003) Molecular neuroadaptations in the accumbens and ventral tegmental area during the first 90 days of forced abstinence from cocaine self-administration in rats. J Neurochem 85: 1604-1613. [DOI] [PubMed] [Google Scholar]
- McFarland K, Davidge SB, Lapish CC, Kalivas PW (2004) Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J Neurosci 24: 1551-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B (2002) Stress activation of glutamate neurotransmission in the prefrontal cortex: implications for dopamine-associated psychiatric disorders. Biol Psychiatry 51: 775-787. [DOI] [PubMed] [Google Scholar]
- Nestler EJ (2001) Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci 2: 119-128. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Aghajanian GK (1997) Molecular and cellular basis of addiction. Science 278: 58-63. [DOI] [PubMed] [Google Scholar]
- Piazza PV, Deminiere JM, Le Moal M, Simon H (1990) Stress- and pharmacologically-induced behavioral sensitization increases vulnerability to acquisition of amphetamine self-administration. Brain Res 514: 22-26. [DOI] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC (2003) Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37: 577-582. [DOI] [PubMed] [Google Scholar]
- Sarnyai Z, Shaham Y, Heinrichs SC (2001) The role of corticotropin-releasing factor in drug addiction. Pharmacol Rev 53: 209-243. [PubMed] [Google Scholar]
- Sauvage M, Steckler T (2001) Detection of corticotropin-releasing hormone receptor 1 immunoreactivity in cholinergic, dopaminergic and noradrenergic neurons of the murine basal forebrain and brainstem nuclei—potential implication for arousal and attention. Neuroscience 104: 643-645. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Carr DB, Omelchenko N, Pinto A (2003) Anatomical substrates for glutamate-dopamine interactions: evidence for specificity of connections and extrasynaptic actions. Ann NY Acad Sci 1003: 36-52. [DOI] [PubMed] [Google Scholar]
- Shaham Y, Stewart J (1995) Stress reinstates heroin-seeking in drug-free animals: an effect mimicking heroin, not withdrawal. Psychopharmacology 119: 334-341. [DOI] [PubMed] [Google Scholar]
- Shaham Y, Funk D, Erb S, Brown T, Walker C (1997) Corticotropin-releasing factor, but not corticosterone, is involved in stress-induced relapse to heroin-seeking in rats. J Neurosci 17: 2605-2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham Y, Erb S, Leung S, Buczek Y, Stewart J (1998) CP-154,526, a selective, non-peptide antagonist of the corticotropin-releasing factor1 receptor attenuates stress-induced relapse to drug seeking in cocaine- and heroin-trained rats. Psychopharmacology 137: 184-190. [DOI] [PubMed] [Google Scholar]
- Shaham Y, Erb S, Stewart J (2000) Stress-induced relapse to heroin and cocaine seeking in rats: a review. Brain Res Rev 33: 13-33. [DOI] [PubMed] [Google Scholar]
- Sinha R (2001) How does stress increase risk of drug abuse and relapse? Psychopharmacology 158: 343-359. [DOI] [PubMed] [Google Scholar]
- Stewart J (1984) Reinstatement of heroin and cocaine self-administration behavior in the rat by intracerebral application of morphine in the ventral tegmental area. Pharmacol Biochem Behav 20: 917-923. [DOI] [PubMed] [Google Scholar]
- Swanson L, Sawchenko P, Rivier J, Vale W (1983) Organization of bovine corticotropin releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology 36: 165-186. [DOI] [PubMed] [Google Scholar]
- Thierry AM, Tassin JP, Blanc G, Glowinski J (1976) Selective activation of the mesocortical DA system by stress. Nature 263: 242-244. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A (2001) Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411: 583-587. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Singh V, Crowder TL, Yaka R, Ron D, Bonci A (2003) Corticotropin-releasing factor requires CRF binding protein to potentiate NMDA receptors via CRF receptor 2 in dopamine neurons. Neuron 39: 401-407. [DOI] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE (2000) Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Com Neurol 428: 191-212. [DOI] [PubMed] [Google Scholar]
- White FJ, Kalivas PW (1998) Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend 51: 141-153. [DOI] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA (1987) A psychomotor stimulant theory of addiction. Psychol Rev 94: 469-492. [PubMed] [Google Scholar]
- Wolf ME (1998) The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol 54: 679-720. [DOI] [PubMed] [Google Scholar]
- Xi ZX, Gilbert J, Campos AC, Kline N, Ashby CRJ, Hagan JJ, Heidbreder CA, Gardner EL (2004) Blockade of mesolimbic dopamine D(3) receptors inhibits stress-induced reinstatement of cocaine-seeking in rats. Psychopharmacology 176: 57-65. [DOI] [PMC free article] [PubMed] [Google Scholar]