

Abstract
Regardless of proximal cause, photoreceptor injury or disease almost invariably leads to the activation of Muller cells, the principal glial cells in the retina. This observation implies the existence of signaling systems that inform Muller cells of the health status of photoreceptors. It further suggests that diverse types of photoreceptor damage elicit a limited range of biochemical responses. Using the mouse retina, we show by microarray, RNA blot, and in situ hybridization that the genomic responses to both light damage and inherited photoreceptor degeneration involve a relatively small number of genes and that the genes activated by these two insults overlap substantially with one another and with the genes activated by retinal detachment. Among the induced transcripts, those coding for endothelin2 (Edn2) are unusual in that they are localized to photoreceptors and are also highly induced in all of the tested models of photoreceptor disease or injury. Acute light damage also leads to a >10-fold increase in endothelin receptor B (Ednrb) in Muller cells 24 h after injury. These observations suggest that photoreceptor-derived EDN2 functions as a general stress signal, that EDN2 signals to Muller cells by binding to EDNRB, and that Muller cells can increase their sensitivity to EDN2 as part of the injury response.
Keywords: photoreceptor, retinal degeneration, retinal disease, endothelin, light damage, retinal detachment, microarray
Introduction
Photoreceptor dysfunction and/or loss is a central feature in a wide variety of retinal disorders, including retinitis pigmentosa, macular degeneration, retinal detachment, and solar retinitis. Despite the diversity of proximal causes that characterize these disorders, current evidence suggests that they converge on a relatively small number of molecular and cellular pathways leading to repair or to cell injury and death (Pacione et al., 2003). Similarly, current evidence suggests that some environmental risk factors, such as cigarette smoke and light exposure, may exacerbate diverse photoreceptor diseases, consistent with a sharing of pathogenic mechanisms of cellular damage (Young, 1988, 1992; Heckenlively et al., 1991; Taylor et al., 1992; Solberg et al., 1998; Tomany et al., 2004).
Regardless of the proximal causes, photoreceptor injury or disease almost invariably leads to the activation of Muller cells, the most abundant glial cells in the retina (Bringmann and Reichenbach, 2001; Garcia and Vecino, 2003). Glial activation is also observed in a variety of non-ocular CNS diseases and injuries, and the accumulation of glial fibrillary acidic protein (GFAP) in reactive Muller cells in the retina or in reactive astrocytes in the brain and spinal cord has become a widely used surrogate measure for neuronal disease or injury (Lewis and Fisher, 2003). Although glial activation presumably evolved to limit or repair neuronal damage, excessive activation can inhibit regeneration and functional recovery. In the retina, the contrast between the beneficial and detrimental effects of Muller cell activation can be seen by comparing the protective effects of potassium buffering, glutamate uptake, and glutathione-based antioxidant action by Muller cells with the damaging effect of proliferation and migration of Muller cells in the context of proliferative vitreoretinopathy and subretinal fibrosis (Bringmann and Reichenbach, 2001; Guidry, 2005).
The activation of Muller cells in response to diverse retinal disorders suggests that Muller cells continuously monitor the health status of retinal neurons and, in particular, that Muller cells might monitor the release of neuron-derived signaling molecules using specific receptors. In theory, such a system could be based on signaling molecules specifically released by healthy neurons or by unhealthy neurons, leading, respectively, to a decrease or increase in release after neuronal disease or injury. In considering the second of these possibilities (the release of signals from unhealthy neurons), it would be of interest to determine whether Muller cells simply sense the products of tissue injury or deranged metabolism or whether high-affinity neuron-derived ligands target Muller cell receptors in a dedicated paracrine signaling system. Regardless of which model proves to be correct, the identification of any signaling pathway between retinal neurons and Muller cells could be of practical interest because activating or inhibiting the pathway might alter the time course or severity of a wide variety of retinal disorders.
Based on studies in mice, we present evidence here that one such signaling pathway involves the production of endothelin-2 (EDN2) by photoreceptors subject to any of a wide variety of diseases or injuries and the sensing of this ligand by endothelin receptor B (EDNRB) on Muller cells. Moreover, we report that the genomic responses to light damage and to inherited photoreceptor degeneration involve a relatively small number of genes and that these gene sets show substantial overlap with one another and with the gene set activated by retinal detachment. These data strongly support the idea that diverse types of photoreceptor damage activate a small number of biochemical and cell biological responses.
Materials and Methods
cDNA probes. cDNA clones for mouse endothelin converting enzyme-1 (Ece1) (IMAGE identification number 6854311), Ece2 (IMAGE identification number 4502901), and Ednra (IMAGE identification number 2812426) were obtained from the American Type Culture Collection (Manassas, VA). The cDNA clone for Ednrb (IMAGE identification number 4971909) was obtained from Research Genetics (Carlsbad, CA). All other cDNA probes were PCR amplified using mouse retina cDNA or a developing mouse eye cDNA library as template. The identities of all probes were confirmed by sequencing.
Rapid amplification of cDNA ends. Total RNA was extracted from 1-month-old photoreceptor cadherin prCAD-/- (photoreceptor cadherin) mouse retina, and 5′ rapid amplification of cDNA ends (RACE) PCR amplification was performed with the Smart RACE cDNA amplification kit (BD Biosciences/Clontech, Franklin Lakes, NJ). Three sets of nested primer pairs, shown as arrowheads and labeled “1,” “2,” and “3” in Figure 2, were used in sequential 5′ RACE PCR reactions to amplify the presumptive full-length 4.5 kb Edn2 cDNA product. 5′ RACE PCR amplification with the extreme 5′ proximal primer pair (Fig. 2, “1”) failed to amplify additional sequences 5′ of those indicated by the boxed region in Figure 2.
Figure 2.

The Edn2 gene and its transcripts. Thin horizontal lines, Introns or 5′ and 3′ flanking DNA; black boxes, coding regions; open boxes, 5′ and 3′ noncoding sequences shared by the ∼1.4 kb and ∼4.5 kb transcripts; gray box, the extended 5′ noncoding region that accounts for the difference between the ∼1.4 and ∼4.5 kb transcripts; rightward arrows above the gene indicate the presumptive start sites of transcription for the ∼4.5 and ∼1.4 kb transcripts. Leftward arrowheads labeled “1,” “2,” and “3” indicate the locations of the three sets of nested RACE PCR primer pairs. Bottom, Summary of transcripts visualized by RNA blot hybridization of WT and prCAD-/- RNA with the three indicated probes. A, ApaI; B, BamHI; E, EcoRI; H, HindIII.
Microarray hybridization. Microarray experiments were performed using mouse genome MOE430 arrays (Affymetrix, Santa Clara, CA) with retina RNA probes obtained from either (1) ∼1-month-old prCAD-/- and wild-type (WT) 129 controls, or (2) BALB/c mice exposed with pupil dilation to 6 h of 6000 lux light, followed by 24 h in darkness and dark-adapted controls. For each comparison, data from three experiments (six independent RNA samples; 12 retinas per sample) were processed using robust multichip averaging (Irizarry et al., 2003). We note that the retinas are likely to have a low level of retinal pigment epithelium (RPE) contamination. The data were converted to a log2-transformed format, a p value was calculated for each transcript using t test statistics, and fold changes were calculated from the log2-transformed signal ratios. No scaling factor for variance stabilization or p value correction was used.
RNA preparation and blotting. Total RNA from retinas was extracted with Trizol (Invitrogen, Carlsbad, CA) and the RNeasy Mini kit (Qiagen, Valencia, CA). RNA blotting was performed using standard methods with 32P-labeled cDNA probes isolated by PCR amplification from a mouse retina cDNA library, as described above.
Mice. prCAD-/- mice are described by Rattner et al. (2001). rds-/- (retinal degeneration slow) and rd7 (retinal degeneration 7) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). The 8- to 15-week-old albino BALB/c and pigmented 129/SVJ or C57BL/6 females used for light damage and retinal detachment experiments were obtained from the National Cancer Institute (Bethesda, MD).
Light damage. For light damage, mice were exposed to cool white fluorescent lights at 6000 lux intensity for the indicated times and then allowed to recover in the dark for 24 h unless otherwise noted. Cyclic light conditions consisted of 12 hr light in 10 or 250 lux cool white fluorescent light/12 h in the dark. Dark adaptation consisted of housing in complete darkness for at least 1 week. When indicated, pupils were dilated 15 min before light exposure with 1% cyclopentolate and 5% phenylephrine in physiological saline at neutral pH. Light intensities were measured with a Cal-Light 400 photometer (Cooke Corporation, Romulus, MI).
Retinal detachment. Retinal detachments (Nour et al., 2003) were performed with C57BL/6 mice after induction of deep anesthesia with intra-peritoneal ketamine and xylazine and corneal application of 5 μl of 10% lidocaine in PBS. A 30 gauge needle was gently inserted at the limbus and withdrawn, and a blunt 33 gauge needle was inserted through the 30 gauge hole and advanced until it touched the back wall of the eye. To detach the retina, ∼1 μl of physiological saline was slowly expelled through the 33 gauge needle into the subretinal space. After the procedure, erythromycin ophthalmic ointment was applied to the corneal surface. Control mice underwent the same procedure except that the 33 gauge needle was not inserted.
Histology and immunocytochemistry. Eyes were fresh frozen in OCT compound (Tissue-Tek, Miles, Elkhart, IN), and 10 μm sections were cut and postfixed in PBS with 4% paraformaldehyde. In situ hybridization was performed essentially as described previously (Schaeren-Wiemers and Gerfin-Moser, 1993) using digoxygenin-labeled riboprobes transcribed from cloned PCR products. Antibodies were obtained from the following sources: rabbit anti-EDNRB antibody (Alomone Labs, Jerusalem, Israel) and monoclonal anti-GFAP antibody (G-3893; Sigma, St. Louis, MO). For immunostaining with anti-EDNRB antibody (Peters et al., 2003), fresh frozen sections (10 μm) were fixed for 5 min in ice-cold acetone, washed three times with PBS, blocked for 1 h with 10% normal goat serum in PBS with 0.3% Triton X-100, and incubated overnight at 4°C with primary antibody at 1:100 dilution in blocking solution. The specificity of this antibody was confirmed by comparing the immunostaining signal of untransfected COS cells versus COS cells transfected with a mouse Ednrb cDNA clone. Omission of the primary antibody leads to an absence of the immunostaining signal in the retina.
Endothelin binding to retinal membranes. Retinas were homogenized in 50 mm HEPES, pH 7.0, 150 mm NaCl, 5 mm KCl, and 3 mm MgCl2, with protease inhibitors (H buffer), nuclei were removed by centrifugation at 600 × g for 1 min, and the membranes pelleted by centrifugation at 15,000 × g for 30 min at 4°C. Membranes were resuspended in H buffer, recovered again by centrifugation, resuspended in H buffer, adjusted to identical protein concentrations (typically 0.8 mg/ml), and stored at -80°C. Binding reactions were performed in 200 μl of H buffer with 0.1% BSA in the presence of 60 pm 125I-EDN1 (Amersham Biosciences, Piscataway, NJ) with gentle rotation at room temperature for 3 h. Unlabeled competitors (EDN2, BQ123, or BQ788; Sigma) were used at a final concentration of 1 μm. Under these conditions, the linear range of the binding assay with respect to EDNR concentration is observed with membranes from ∼10% of a light-damaged retina per tube. Bound and free ligands were separated by centrifuging the sample at 15,000 × g for 30 min at 4°C, resuspending the membrane pellet in H buffer, and repeating the centrifugation.
Endothelin binding to tissue sections. Sections (10 μm) of fresh frozen eyes were air dried and stored at -80°C, hydrated with H buffer for 10 min, and then incubated with H buffer containing 30 pm 125I-EDN1 with or without one of the unlabeled competitors at 1 μm. After a 1 h incubation at room temperature, the sections were washed five times for 5 min each with H buffer, once with PBS with 1 mm MgCl2, and then fixed in PBS with 1 mm MgCl2 with fresh 4% paraformaldehyde for 30 min at room temperature. Air-dried slides were covered with Saran Wrap, and the 125I signal detected with a PhosphorImager (Amersham Biosciences). The slides were then stained with toluidine blue.
Results
Induction of Edn2 transcripts in mutant retinas with photoreceptor degeneration
In an initial experiment aimed at identifying transcripts that exhibit changes in abundance in the context of retinal degeneration, we performed microarray hybridization with RNA from adult WT and prCAD-/- mouse retinas. prCAD (protocadherin 21) is an unusual cadherin that is required for the proper assembly and/or structural integrity of the photoreceptor outer segment (Rattner et al., 2001). Targeted disruption of both copies of the prCAD gene leads to the slow death of photoreceptors. Given the severity of the mutant phenotype, the microarray analysis revealed surprisingly few transcripts with substantial alterations in abundance in prCAD-/- retinas: three independent microarray hybridizations show only five transcripts that are induced by at least fivefold and only 27 additional transcripts that are induced by at least two-fold, with p values <0.05 (Fig. 1A) (supplemental Table 1, available at www.jneurosci.org as supplemental material). Among the several transcripts with a large induction in the prCAD-/- retina was that coding for EDN2. RNA blot analysis confirmed this induction and also revealed a large increase in EDN2 transcripts in retinas homozygous for the rds mutation and a smaller increase in retinas homozygous for the rd7 mutation (Fig. 1B). The rds mutation eliminates a structural protein in the photoreceptor outer segment, and the rd7 mutation eliminates a rod photoreceptor-specific transcription factor (Travis et al., 1989; Akhmedov et al., 2000; Chen et al., 2005). By in situ hybridization to prCAD-/- and rds-/- retinas, Edn2 transcripts localize to photoreceptors (Fig. 1C). Homozygous rd7 retinas are characterized by small regions of folding and detachment (Akhmedov et al., 2000; Haider et al., 2001), and Edn2 transcripts accumulate at these sites (Fig. 1C). Thus, the modest increase in Edn2 transcripts in rd7 retinas observed by RNA blotting reflects the spatially heterogeneous nature of the retinal pathology.
Figure 1.

Increase in Edn2 transcript abundance in diverse mouse models of photoreceptor disease. A, Scatter plot comparing the ratio of prCAD-/-/WT retina RNA hybridization signals in two independent microarray experiments. Hybridizations were performed with the Affymetrix mouse genome MOE430 chip, and the ratios are plotted on a log2 scale. Points representing Prcad, Edn2, and noncoding transcript AK018172 are labeled. As expected, the transcript with the greatest reduction in abundance in the prCAD-/- retina is that coding for prCAD itself. B, RNA blot showing Edn2 transcripts in WT, prCAD-/-, rds-/-, and rd7 retinas. Arrows to the right of this and subsequent blots indicate the high abundance ∼4.5 kb and the lower abundance ∼1.4 kb Edn2 transcripts. Probing the blot with Gapdh in this and all subsequent RNA blots controls for equal loading of RNA. C, In situ hybridization showing the accumulation of Edn2 transcripts in photoreceptor cells in prCAD-/-, rds-/-, and rd7 retinas. Specific Edn2 hybridization is undetectable in the control WT retina (left). In the rd7 retina, Edn2 transcripts accumulate only at the sites of retinal detachment (vertical arrows). ONL, Outer nuclear layer.
Previous work has defined an ∼1.4 kb mature Edn2 transcript (Saida et al., 1989), consistent with the smaller of the two hybridizing bands in Figure 1B. The more abundant ∼4.5 kb species seen in Figure 1B has not been described previously. Its structure (as defined by sequencing of reverse transcriptase-PCR products and by RNA blot hybridization with probes from exon 1, intron 4, or exon 5) is summarized in Figure 2. The ∼4.5 kb transcript appears to differ from the previously characterized ∼1.4 kb transcript simply by the addition of ∼3 kb of 5′ untranslated sequences derived from genomic sequences that reside immediately upstream of the 5′ end of the smaller transcript. In preliminary experiments with transiently transfected HEK293 cells, the 5′ untranslated region of the ∼4.5 kb Edn2 message was found to significantly inhibit translation when placed 5′ of a β-galactosidase open reading frame (data not shown), suggesting the possibility that, in photoreceptor cells, the ∼4.5 kb transcript could be subject to translational regulation.
The genomic response to nongenetic photoreceptor damage
To explore the range of retinal degenerative processes that elicit Edn2 transcript induction, microarray analyses were performed on retinas damaged by acute exposure to visible light, a well characterized environmental insult that leads, in a dose-dependent manner, to photoreceptor death (Organisciak and Winkler, 1994). For this experiment, albino BALB/c mice were dark adapted for 1-2 weeks, exposed with fully dilated pupils to 6000 lux from cool white fluorescent lights for 6 h, and then allowed to recover for 24 h in the dark. By way of comparison, the light intensity at the horizon at midday is ∼7000 lux with an overcast sky and ∼12,000 lux with a blue sky; the light intensity at eye level is ∼1000 lux with standard indoor fluorescent ceiling lights. A 6 h light exposure and 24 h recovery period was chosen for the microarray analyses because an initial analysis of Edn2 transcript levels (described below) showed that maximal induction occurred after at least 1 h of light exposure, followed by at least 12 h of recovery.
Three independent microarray comparisons of control versus light-damaged retinas revealed 25 transcripts that are light induced by at least fivefold and an additional 256 transcripts that are light induced by at least twofold, with p values <0.05 (supplemental Table 2, available at www.jneurosci.org as supplemental material). These data are highly reproducible as judged by a scatter plot comparison of two of these independent experiments (Fig. 3A) and by RNA blot hybridization with probes derived from 12 of the most highly induced transcripts (Figs. 3C, 4A) (supplemental Table 3, available at www.jneurosci.org as supplemental material). Among the transcripts strongly induced by light damage, we were intrigued to find those coding for EDN2 and for one of two endothelin receptors, EDNRB. A cross comparison of the average fold induction among transcripts in prCAD-/- versus WT retinas and in light-damaged versus control retinas shows that Edn2 transcripts accumulate to an unusually high level under both conditions (Fig. 3B). Although many of the transcripts induced by light damage are not appreciably altered in prCAD-/- retinas as judged by a cross comparison of the microarray data sets, 8 of the 12 transcripts that are highly induced by light damage and that we analyzed by RNA blotting show an induction of 2.5-fold or greater in at least one of the three genetic models of photoreceptor degeneration tested (Figs. 1B, 3C; Table 1) (supplemental Table 3, available at www.jneurosci.org as supplemental material). Together, the microarray and RNA blot hybridization data suggest a pattern of substantially shared genomic responses to diverse photoreceptor diseases or injuries.
Figure 3.
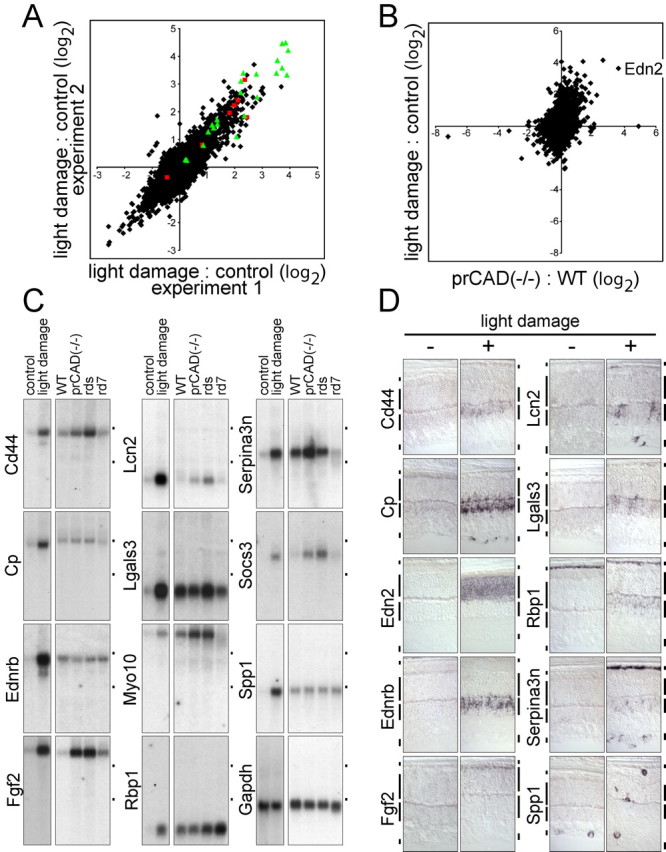
Changes in transcript levels in response to light damage. A, Scatter plot comparing the ratio of light-damaged/control retina RNA hybridization signals in two independent microarray experiments. Hybridizations were performed with the Affymetrix mouse genome MOE430 chip, and the ratios are plotted on a log2 scale. On the MOE430 chip, some of the transcripts are represented by groups of oligonucleotide hybridization targets that are listed as separate entries in compilations of transcript abundances. As an extreme example, ceruloplasmin transcripts are represented by seven different entries; these are indicated by red squares in the scatter plot. Green triangles indicate the 12 transcripts tested by RNA blotting and in situ hybridization (including Edn2 and Ednrb). For the light-damage samples, dark-adapted BALB/c mice were dilated, light exposed for 6 h, and then placed in darkness for 24 h. Control BALB/c mice were dark adapted. B, Scatter plot comparing the averaged prCAD-/-/WT microarray hybridization signals (Fig. 1A) to the averaged light-damaged/control microarray hybridization signals (A). The point representing Edn2 hybridization is labeled. C, RNA blot hybridization validates the transcript inductions predicted by microarray hybridization for 11 of 11 transcripts tested and shows different relative levels of induction in light-damaged and genetically based retinopathies. The WT control mice used for comparison with the genetic retinopathies was 129 maintained in a standard 12 h light/dark cycle. Fgf2 represents an example in which microarray hybridization significantly under estimates the fold induction: little or no induction was predicted by microarray hybridization with prCAD-/- versus WT retina RNA, but RNA blotting shows an ∼10-fold induction (supplemental Table 3, available at www.jneurosci.org as supplemental material). Cp, Ceruloplasmin; Lcn2, lipocalin2; Lgals3, soluble galactose binding lectin 3; Myo10, myosin X; Rbp1, retinoid binding protein 1; Socs3, suppressor of cytokine signaling 3; Spp1, secreted phosphoprotein 1/osteopontin. Dots to the right of each blot indicate the locations of the 28 and 18S ribosomal RNAs. D, Cellular localization of transcripts induced by light damage as determined by in situ hybridization. Vertical bars adjacent to each retinal section demarcate (from top to bottom) RPE, the outer nuclear layer ONL, INL, and GCL.
Figure 4.

Parameters affecting Edn2 and Ednrb transcript induction in response to light damage. A-E, RNA blots showing retinal Edn2 and Ednrb transcript accumulation as follows: A, BALB/c in response to 1 h bright light exposure with pupil dilation, followed by the indicated number of hours of recovery in darkness; B, BALB/c in response to 10 h bright light exposure without pupil dilation, followed by 24 h of recovery in darkness; C, pigmented 129/SVJ in response to the indicated times of bright light exposure with pupil dilation, followed by 24 h of recovery in darkness; D, BALB/c in response to 5, 15, 30, or 60 min bright light exposure with pupil dilation, followed by 24 h of recovery in darkness; E, BALB/c either preconditioned for 10 d in constant light (CL; 250 lux) or maintained in darkness (DA, dark adapted) and then exposed for 1 h to 6000 lux with pupil dilation, followed in each case by 24 h of recovery in darkness. Also shown are Spp1 transcripts, a marker for invading microglia and/or macrophages, and Prcad transcripts, a photoreceptor-specific marker. With respect to the blots in A and B, additional experiments show that, for BALB/c mice, a 1 h exposure with pupil dilation produces a transcript induction response equivalent to that of a 6 h exposure without dilation (data not shown).
Table 1.
Summary of the disease or damage responses and retinal localization(s) for transcripts from 12 genes
|
Gene |
Induction with light exposure |
Induction with retinal detachment |
Induction in one or more genetic retinopathies |
Cell type in which the RNA is induced |
|---|---|---|---|---|
| Cd44 | + | − | + | Muller cells |
| Cp | + | + | − | Muller cells, astrocytes |
| Ednrb | + | − | − | Muller cells |
| Edn2 | + | + | + | Photoreceptors |
| Fgf2 | + | +/− | + | Photoreceptors |
| Lcn2 | + | + | + | Muller cells, astrocytes |
| Lgals3 | + | − | +/− | Muller cells |
| Myo10 | + | ND | + | ND |
| Rbp1 | + | − | + | Muller cells |
| Serpina3n | + | + | + | Muller cells, astrocytes, RPE |
| Socs3 | + | ND | + | ND |
|
Spp1.
|
+ |
− |
− |
Microglia, macrophages |
+/−, Weak induction; ND, not determined. The assignments of cell type are based on transcript localization in differential interference contrast images; they have not been confirmed by double labeling with cell type-specific markers. In the pigmented C57BL/6 mouse used for retinal detachment, any hybridization signals in the RPE would be obscured by melanin.
By hybridization to RNA blots identical to the ones shown in Figure 3C, we observe no effect of light exposure or genetically based retinopathies on the levels of transcripts coding for the two proteases involved in endothelin maturation, ECE1 and ECE2 (data not shown). Transcripts coding for EDN1, EDN3, and EDNRA were below the limit of detection by RNA blot hybridization (data not shown).
The transcripts induced by light damage can be divided into distinct classes based on the cell types in which they accumulate (Fig. 3D; Table 1). Only 2 of the 10 transcripts chosen for in situ hybridization based on their high level of induction by light damage, those coding for EDN2 and basic fibroblast growth factor (FGF2), accumulate in photoreceptors, the principal site of light-induced cell damage. Induction of Fgf2 after any of a variety of retinal insults has been described by Wen et al. (1995) and Gao and Hollyfield (1996). Both proteins presumably act as paracrine messengers. Several transcripts, including those coding for CD44, ceruloplasmin (previously reported by Chen et al., 2003), EDNRB, lipocalin2, Lgals3, retinol binding protein 1 (RBP1), and serpin A3N accumulate in the inner nuclear layer (INL), a distribution consistent with accumulation in Muller cell bodies. We note that this analysis does not rule out the possibility that one or more of these transcripts accumulates in other cells within the INL. Ceruloplasmin, lipocalin2, and serpin A3N transcripts also accumulate in the ganglion cell layer (GCL) in light-damaged eyes, possibly in astrocytes; RBP1 transcripts also accumulate in the RPE in both control and light-damaged eyes; and serpin A3N transcripts also accumulate in the RPE in light-damaged eyes. Spp1 (secreted phosphoprotein 1/osteopontin) transcripts are found in the GCL in both control and light-damaged eyes and appear to mark macrophages or microglia that have invaded the damaged retina. In the skin, Spp1 has been implicated in the wound-healing response based on defects observed in Spp1-/- mice (Liaw et al., 1998).
Parameters affecting the induction of Edn2 and Ednrb transcripts by light
In light-damaged albino BALB/c retinas, the accumulation of both Edn2 and Ednrb transcripts follows a delayed time course, with maximal accumulation occurring 12-24 h after a 1 h light exposure with pupil dilation (Fig. 4A). Based on this observation, all light-damage experiments described in the text that follows were performed with a 24 h recovery period in the dark. Edn2 and Ednrb transcripts also accumulate, albeit to lower levels, in albino BALB/c mice exposed without pupil dilation to 6000 lux for 10 h (Fig. 4B) and in pigmented 129/SVJ mice exposed with pupil dilation to 6000 lux for 2 or more hours (Fig. 4C). Pigmented 129/SVJ mice exposed without pupil dilation to 6000 lux for 6 h show little or no induction of either transcript (data not shown).
BALB/c mice with fully dilated pupils display a remarkable sensitivity to light exposure as judged by the induction of Edn2 and Ednrb transcripts after as little as 15 min of exposure to 6000 lux (Fig. 4D). Indeed, control experiments to determine whether pupil dilation alone leads to an accumulation of Edn2 transcripts demonstrated that exposure to standard indoor lighting during the several minutes required for transport and instillation of eye drops is sufficient to induce a detectable accumulation of Edn2 transcripts (data not shown). Only if the mice are maintained in nearly complete darkness do we observe no effect of pupil dilation on Edn2 transcript levels.
Many organisms exhibit enhanced resistance to environmental stress if preconditioned with a low level of the same stress. For example, bacteria exposed to low levels of oxidants or ultraviolet light develop resistance to a subsequent challenge with oxidants or ultraviolet light, respectively (Wick and Egli, 2004). This preconditioning phenomenon has also been observed with light exposure in the mammalian retina (Liu et al., 1998; Li et al., 2003). For example, Li et al. (2003) observed enhanced photoreceptor survival after a bright light challenge in albino rats maintained in moderate-intensity cyclic light (12 h at 400 lux/12 h in darkness) compared with albino rats maintained in very-low-intensity cyclic light (12 h at 5 lux/12 h in darkness).
Figure 4E shows that Edn2 and Ednrb transcript levels are significantly affected by preconditioning with light. Maintaining albino BALB/c mice in moderate-intensity constant light, using standard animal room lighting (250 lux), for 10 d leads to a significant induction of Edn2, but not Ednrb or Spp1, transcripts. After this preconditioning regimen, a 6000 lux exposure for 1 h with pupil dilation produces little or no further induction of Edn2 transcripts, although the level of Edn2 transcripts elicited by the preconditioning regimen is approximately twofold lower than the level attained when dark-adapted mice are identically exposed to damaging light. The preconditioning regimen also eliminates the light-damage-dependent induction of Ednrb and Spp1 transcripts.
The photoreceptor localization of Edn2 transcripts (Fig. 3D) is consistent with the possibility that cellular damage during preconditioning in constant light might somehow block further Edn2 transcript induction. However, this mechanism cannot account for the absence of Ednrb transcript induction in the INL or the lack of invasion by microglia/macrophages as indicated by the unaltered level of Spp1 transcripts. The continued presence of normal Prcad transcript levels in all of the retina samples (Fig. 4E) also argues against this model. In contrast to preconditioning in moderate intensity constant light, preconditioning with a regimen of dim or moderate cyclic light (12 h at 10 or 250 lux, respectively/12 h in darkness) produced, respectively, no or only a small effect on basal levels of Edn2 transcripts and on light-damage induction of Edn2 and Ednrb transcripts relative to that seen with mice maintained in complete darkness (data not shown). This experiment demonstrates that light exposure history strongly influences the genomic response of the photoreceptor to damaging light.
Localized transcript induction after retinal detachment
The induction of transcripts from a small set of genes in both genetically determined and light damage-induced degenerations suggests that this induction could represent a generic response to photoreceptor damage. To test this idea, we asked whether an experimental retinal detachment could elicit a similar response. In humans, retinal detachment is one of the most common ophthalmological emergencies. The resulting photoreceptor damage likely arises from the loss of intimate contact between photoreceptors and the underlying retinal pigment epithelium and from inefficient oxygenation of the photoreceptors by the choroidal circulation. In the experiments reported here, small detachments were induced in C57BL/6 mice by subretinal injection of ∼1 μl of physiological saline. Four days later, in situ hybridization revealed localized induction of Edn2 transcripts in photoreceptors, with the highest levels of accumulation centered at the detachment site (Fig. 5; Table 1). Altogether, 4 of 10 light-induced transcripts that we tested by in situ hybridization were clearly induced at the site of the detachment (Fig. 5; Table 1). In every instance, the induced transcripts accumulate in the same pattern and/or cell type(s) in which they accumulated after light damage. Interestingly, although the induction of ceruloplasmin, lipocalin 2, and serpin A3N transcripts in the INL is localized to the site of detachment, these transcripts are also induced in scattered cells across the inner face of the entire retina, indicating both local and global cell type-specific responses. These data suggest that the accumulation of Edn2 transcripts observed in or near the folds in rd7 retinas (Fig. 1C) represents a response to retinal detachment rather than a direct result of the loss of Nr2e3 (the photoreceptor transcription factor mutated in the rd7 mouse) (Akhmedov et al., 2000; Haider et al., 2001).
Figure 5.

In situ hybridization shows transcript accumulation 4 d after a localized retinal detachment in C57BL/6 mice. The probes are indicated for each panel, and the region of retinal detachment is indicated with a black bracket. Edn2 accumulates in photoreceptors; ceruloplasmin (Cp), lipocalin2 (Lcn2), and Serpina3n accumulate in the INL, presumably in Muller cells. ONL, Outer nuclear layer.
Increase in EDNRB in Muller glia after light exposure
Both EDNRA and EDNRB are normally produced in the mammalian retina, as determined previously by binding of radiolabeled endothelins and endothelin analogs to membrane homogenates or to tissue sections followed by in situ autoradiography (MacCumber and D'Anna, 1994; de Juan et al., 1995). Although the spatial resolution of in situ autoradiography is limited, the published data suggest that EDNRA is localized primarily to the retinal vasculature and EDNRB primarily to glia. Consistent with our in situ hybridization results assigning Muller cells as the primary cell type in which Ednrb transcripts accumulate (Fig. 3D), immunostaining with affinity-purified anti-EDNRB antibodies revealed a pattern of light damage-induced EDNRB that substantially overlaps the pattern of damage-induced GFAP (Fig. 6). Induced EDNRB immunoreactivity is especially prominent at the outer limiting “membrane,” the retinal layer defined by the most distal processes of Muller cells and the adjacent photoreceptor inner segments with which the Muller cell processes form adherens junctions (Cohen, 1992). Additional sites of EDNRB immunoreactivity in the ganglion cell layer may represent astrocytes.
Figure 6.
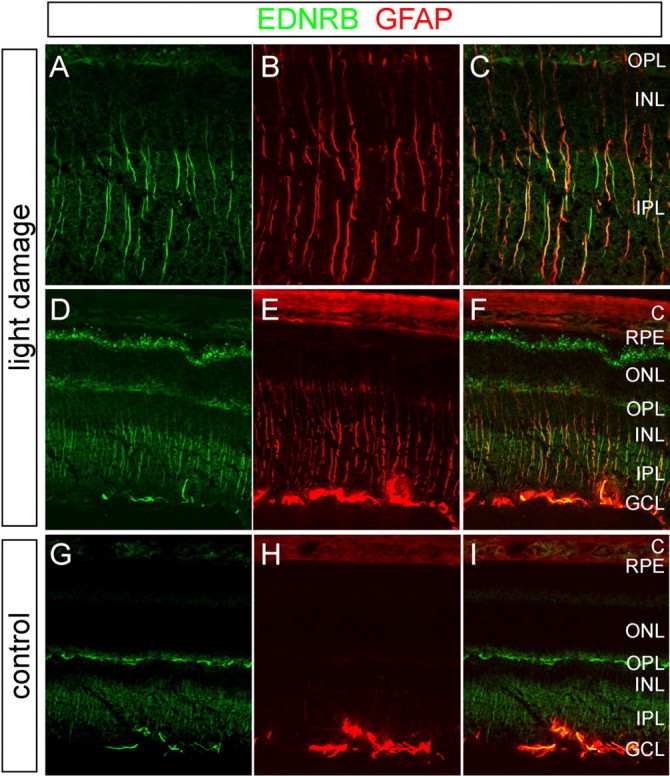
Accumulation of EDNRB and GFAP in Muller cells in light-damaged retinas as determined by immunostaining. A-F, BALB/c mice were exposed to 6 h of bright light with pupil dilation, followed by a 24 h recovery in darkness. A-C, Enlarged views of the central retina from D-F. G-I, Control BALB/c mice maintained in darkness. Light exposure leads to the accumulation of EDNRB and GFAP in Muller cells, characterized in the central retina by their radial fibers. EDNRB also accumulates in presumptive astrocytes in the GCL and at the outer tips of the Muller cells, which abut the photoreceptor inner segments between the outer nuclear layer and the RPE. Among Muller cells in the light-damaged retina, both EDNRB and GFAP show substantial cell-to-cell variability. C, Choroid; ONL, outer nuclear layer; OPL, outer plexiform layer; IPL, inner plexiform layer.
To assess the competence of the induced EDNRB to bind endothelin ligands and to quantitate the relative levels of EDNRA and EDNRB binding sites, 10 μm frozen sections of control or light-damaged BALB/c retinas were incubated with 30 pm 125I-EDN1 in either the absence of competing ligands or the presence of 1 μm unlabeled EDN2, 1 μm BQ123 (an EDNRA-specific antagonist), or 1 μm BQ788 (an EDNRB-specific antagonist) (Fig. 7A) (Davenport and Kuc, 2002). Both EDN1 and EDN2 bind EDNRA and EDNRB with subnanomolar affinities, and therefore competition with 1 μm EDN2 should eliminate all specific binding to EDN receptors. An analogous ligand-binding experiment was also performed with suspensions of control or light-damaged BALB/c retinal membranes (Fig. 7B). Both experiments reveal a large increase in the abundance of EDNRB in the light-damaged BALB/c retina with little change in the abundance of EDNRA; in Figure 7B, the increase in EDNRB is ∼12-fold. Together with the immunostaining data shown in Figure 6, these data predict that light damage in BALB/c mice leads to a large increase in EDN2 sensitivity in Muller cells (and perhaps other glia), which is presumably accompanied by increased production of EDN2 by photoreceptor cells. Interestingly, 125I-EDN1 binding experiments show that the abundance of EDNRB in dark-adapted C57BL/6 retinas is 2.5-fold higher than in dark-adapted BALB/c retinas, but the EDNRB level in C57BL/6 remains essentially unchanged after exposure with pupil dilation to the standard 6 h 6000 lux light damage regimen (data not shown). If EDNRB mediates a Muller cell repair response, as the present work suggests, then the greater resistance to light damage of C57BL/6 mice compared with BALB/c mice may arise not only from differences in iris and RPE pigmentation but also from differences in the intrinsic state of the dark-adapted retina.
Figure 7.

Increase in EDNRB-specific binding sites in the light-damaged retina. A, Binding of 125I-EDN1 to fresh-frozen eye sections from control BALB/c mice maintained in darkness (left member of each pair of eyes) or BALB/c mice exposed to bright light for 6 h with pupil dilation, followed by 24 h of recovery in darkness (right member of each pair of eyes). Binding was performed in either the absence of competitor or the presence of 1 μm of the indicated competitor. The 125I signal is shown in the top row of panels, and the toluidine blue staining pattern is shown in the bottom row. Light exposure produces a large increase in 125I-EDN1 binding, almost all of which is blocked by BQ788 (an EDNRB antagonist) and unaffected by BQ123 (an EDNRA antagonist) and is therefore referable to EDNRB. B, Binding of 125I-EDN1 to retinal membranes from BALB/c mice exposed to bright light for 3 h with pupil dilation, followed by 24 h of recovery in darkness (left 4 bars), or from control BALB/c mice maintained in darkness (right 4 bars). Binding was performed in either the absence of competitor or the presence of 1 μm of the indicated competitor. The histogram shows the means and SDs for three independent experiments. In the dark-adapted retina, ∼70% of endothelin binding sites are EDNRB. With light damage, the number of EDNRB sites increases ∼12-fold; the number of EDNRA sites shows little change.
By binding 125I-EDN1 to a dilution series of retinal membranes, we observe ∼30 fmol (∼2 × 1010) EDNRB binding sites per light-damaged BALB/c retina (data not shown). Based on previous estimates of 6.4 × 105 Muller cells per mouse retina (Jeon et al., 1998) and with the assumption that approximately half of the binding sites are on Muller cells (Fig. 6), we calculate that, in the light-damaged BALB/c retina, each Muller cell normally contains ∼15,000 EDNRB binding sites. This is likely to represent a lower estimate because some EDNRB sites may be topologically unavailable for ligand binding in membrane suspensions or may already be occupied by endogenous endothelin. Thus far, we have been unable to detect endogenous EDN2 in the mouse retina, because its abundance appears to be below the ∼80 fmol/retina lower limit for detection by immunoblotting or by competitive binding with 125I-EDN1 and recombinant EDNRB. We note that, based on the measured abundance of EDNRB described above, only ∼30 fmol of EDN2 would be required to saturate all of the EDNRB sites per light-damaged BALB/c retina, and only ∼2.5 fmol of EDN2 would be required per dark-adapted BALB/c retina.
Discussion
The principal findings of this study are that (1) genetically based photoreceptor diseases and acute photoreceptor injuries lead to the induction of partially overlapping sets of transcripts in photoreceptors, Muller cells, astrocytes, and RPE cells, (2) Edn2 transcripts are unusual in that they are localized to photoreceptors and are also highly induced in all of the tested models of photoreceptor disease or injury, (3) acute light damage, but not the other genetic or nongenetic insults tested, induces Ednrb transcripts and EDNRB receptors in Muller cells, and (4) preconditioning with moderate-intensity constant light leads to a partial induction of Edn2 transcripts and an abrogation of further Edn2 transcript induction after intense light exposure. These observations strongly suggest that photoreceptor-derived EDN2 functions as a general stress signal, that EDN2 activates Muller cells by binding to EDNRB, that Muller cells can increase their sensitivity to EDN2 by upregulating EDNRB as part of the injury response, and that light exposure history influences the genomic response of the retina to damaging light. Critical tests of this model will require genetic and/or pharmacological manipulation of EDN signaling in the retina. An analogous induction of Fgf2 and Fgfr1 (fibroblast growth factor receptor 1) follows retinal injury, as first reported by Wen et al. (1995, 1998), but the cellular localization of Fgf2 transcripts may be more complex: Wen et al. (1995) observed primarily inner nuclear layer localization, whereas we observed primarily photoreceptor localization (Fig. 3D).
The general picture that photoreceptor disease or injury elicits a well defined program of transcriptional responses is in keeping with several recent surveys of retinal transcripts after light damage or mechanical trauma (Chen et al., 2004; Grewal et al., 2004; Roca et al., 2004; Vazquez-Chona et al., 2004). However, the present study is the first to compare the genomic response to multiple types of retinal diseases and injuries. The induced transcripts identified here encode protein products with diverse biochemical properties. For example, ceruloplasmin is an enzyme that converts Fe+2 to Fe+3, which presumably serves to decrease hydroxyl radical production (Osaki et al., 1966), lipocalin2 is a small extracellular protein that sequesters bacterial enterochelin and perhaps other ligands (Flo et al., 2004), CD44 is a cell surface protein that is concentrated within the retina at the outer limiting membrane (Chaitin et al., 1996), Spp1/osteopontin is a secreted glycoprotein involved in wound healing and tissue remodeling (Liaw et al., 1998), and Lgals3 is a galactose binding lectin (Cherayil et al., 1989). Additional biochemical diversity is apparent from supplemental Tables 1 and 2 (available at www.jneurosci.org as supplemental material). The in situ hybridization analysis reported here pointing to a prominent Muller cell genomic response to photoreceptor damage is consistent with the well known induction of GFAP in Muller cells and with previous work demonstrating phosphorylation of ERK (extracellular signal-regulated kinase) and accumulation of c-Fos in Muller cells after retinal detachment (Geller et al., 2001).
prCAD-/-, rd7 (Chen et al., 2005), light-damaged, and detached retinas show appreciable overlap in their transcript responses, but they also show substantial differences. These differences could reflect, at least in part, differences in the time course, severity, and spatial heterogeneity of photoreceptor stress. Despite these differences, the experiments reported here demonstrate that light damage in albino rodents represents a useful model for identifying a core set of molecular components involved in a broad array of photoreceptor injury and disease responses. This paradigm lends itself especially well to an experimental analysis of retinal damage and repair because of the extreme sensitivity of albino animals to light and because the light stimulus can be delivered in a quantitative, reproducible, and noninvasive manner (Organisciak and Winkler, 1994). The conceptually analogous use of cancer-prone lines of mice to study carcinogenesis has shown that sensitized animal models can provide numerous insights into pathogenic mechanisms that are relevant to nonsensitized animals, including humans (Pattengale et al., 1989).
The role proposed here for EDN2/EDNRB signaling in communicating photoreceptor stress or damage is reminiscent of the proposed role for endothelin signaling in response to mechanical trauma elsewhere in the CNS in various rodent models. For example, EDNRB is induced in astrocytes after spinal cord compression injury or optic nerve transection (Rogers et al., 1997; Peters et al., 2003), injection of EDNRB agonists into the striatum promotes a reactive astrocytosis that can be blocked by an EDNRB-specific antagonist (Ishikawa et al., 1997), and local administration of an EDNRB-specific antagonist decreases the number of reactive astrocytes near the site of mechanical trauma in the adult brain or optic nerve (Koyama et al., 1999; Rogers et al., 2003). However, the published data regarding endothelins and endothelin receptors in CNS trauma models differ in a number of respects from the data reported here. For example, after transection of the rabbit or rat optic nerve, a twofold elevation in EDNRB is first observed at 7 or 60 d after injury, respectively (Rogers et al., 1997). In contrast, light damage in the BALB/c retina induces a ∼12-fold elevation in retinal EDNRB within 1 d. One study of spinal cord compression injury in rats reported a 1.5-fold increase in tissue endothelin within 30 min, followed by a return to the baseline level over the next 48 h (Salzman et al., 1996). However, a different study of spinal cord transection in rats reported a 30-fold increase in tissue endothelin levels at 24 h, with accumulation beginning within several hours of injury (Uesugi et al., 1996). It is unlikely that the rapid endothelin increase reported in the first of these studies is mechanistically related to the Edn2 transcript accumulation that we observe in photoreceptors 12-24 h after light damage. Finally, we note that the published pharmacological experiments in living animals, although suggestive, generally do not resolve direct from indirect effects of endothelins.
With respect to previously reported ocular effects of endothelins, Prasanna et al. (2003) have suggested that endothelin signaling may play multiple roles in the pathogenesis of glaucoma. In particular, these workers report that EDN1 inhibits anterograde axonal transport along optic nerve fibers, induces optic nerve damage and reactive gliosis of the optic nerve head, and regulates aqueous humor dynamics. The extent to which these observations reflect the direct actions of EDN1 on retinal or anterior segment targets or are secondary to the vasoactivity of EDN1 remains to be determined. The work described here, together with that reported for traumatic CNS injury models, adds to the growing list of functions mediated by endothelins (Kedzierski and Yanagisawa, 2001).
The induction of Edn2 transcripts in response to brief exposures to bright light and to preconditioning with moderate-intensity light implies that even a modest light exposure can modulate damage response pathway(s). These observations are consistent with both the enhanced resistance to light damage afforded by preconditioning with moderate light (Liu et al., 1998; Li et al., 2003) and the suggestion that light exposure within the “normal” intensity range might be toxic to the retina if integrated over many years (Young, 1988, 1992). In support of the latter hypothesis, epidemiological evidence suggests that cumulative retinal light exposure, as determined by lifetime sunlight exposure and the density of iris pigmentation, may be one risk factor for age-related macular degeneration (Taylor et al., 1992; Tomany et al., 2004). Also consistent with this hypothesis are biochemical, cell culture, and animal studies on the light-dependent accumulation and phototoxic properties of ocular retinoid derivatives (Sparrow et al., 2003) and the enhanced susceptibility to light damage observed among animals carrying mutations in retinal disease genes (Wang et al., 1997; Vaughan et al., 2003). This last observation indicates that diverse disease processes can sensitize the retina to light toxicity.
The present work suggests a number of questions for future research. What is the role of the various induced and repressed gene products in the retinal disease or injury response? Does EDN induction also occur as a generic response to neuronal damage in the inner retina? Finally, can EDN agonists or antagonists delay the onset or decrease the severity of retinal disease or injury?
Footnotes
This work was supported by the National Eye Institute and the Howard Hughes Medical Institute. We thank Jichao Chen, Dr. Francisco Murillo, Dr. Charlotte Reme, Dr. Gabriele Ronnett, Leila Toulabi, Dr. Mark Tso, and Dr. Yanshu Wang for advice, materials, or assistance and Drs. Tudor Badea, Thomas Rotolo, and Donald Zack for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Amir Rattner, 805 Preclinical Teaching Building, 725 North Wolfe Street, Johns Hopkins University School of Medicine, Baltimore, MD 21205. E-mail: arattner@jhmi.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254540-10$15.00/0
References
- Akhmedov NB, Piriev NI, Chang B, Rapoport AL, Hawes NL, Nishina PM, Nusinowitz S, Heckenlively JR, Roderick TH, Kozak CA, Danciger M, Davisson MT, Farber DB (2000) A deletion in a photoreceptor-specific nuclear receptor mRNA causes retinal degeneration in the rd7 mouse. Proc Natl Acad Sci USA 97: 5551-5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann A, Reichenbach A (2001) Role of Muller cells in retinal degenerations. Front Biosci 6: 72-92. [DOI] [PubMed] [Google Scholar]
- Chaitin MH, Ankrum MT, Wortham HS (1996) Distribution of CD44 in the retina during development and the rds degeneration. Brain Res Dev Brain Res 94: 92-98. [DOI] [PubMed] [Google Scholar]
- Chen J, Rattner A, Nathans J (2005) The rod photoreceptor-specific nuclear receptor Nr2e3 represses transcription of multiple cone-specific genes. J Neurosci 25: 118-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Dentchev T, Wong R, Hahn P, Wen R, Bennett J, Dunaief JL (2003) Increased expression of ceruloplasmin in the retina following photic injury. Mol Vis 9: 151-158. [PubMed] [Google Scholar]
- Chen L, Wu W, Dentchev T, Zeng Y, Wang J, Tsui I, Tobias JW, Bennett J, Baldwin D, Dunaief JL (2004) Light damage induced changes in mouse retinal gene expression. Exp Eye Res 79: 239-247. [DOI] [PubMed] [Google Scholar]
- Cherayil BJ, Weiner SJ, Pillai S (1989) The Mac-2 antigen is a galactose-specific lectin that binds IgE. J Exp Med 170: 1959-1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AI (1992) The retina. In: Adler's physiology of the eye, Ed 9 (Hart WM, ed), pp 579-615. St. Louis: Mosby.
- Davenport AP, Kuc RE (2002) Radioligand binding assays and quantitative autoradiography of endothelin receptors. Methods Mol Biol 206: 45-70. [DOI] [PubMed] [Google Scholar]
- de Juan JA, Moya FJ, Fernandez-Cruz A, Fernandez-Durango R (1995) Identification of endothelin receptor subtypes in rat retina using subtype-selective ligands. Brain Res 690: 25-33. [DOI] [PubMed] [Google Scholar]
- Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A (2004) Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432: 917-921. [DOI] [PubMed] [Google Scholar]
- Gao H, Hollyfield JG (1996) Basic fibroblast growth factor: increased gene expression in inherited and light-induced photoreceptor degeneration. Exp Eye Res 62: 181-189. [DOI] [PubMed] [Google Scholar]
- Garcia M, Vecino E (2003) Role of Muller glia in neuroprotection and regeneration in the retina. Histol Histopathol 18: 1205-1218. [DOI] [PubMed] [Google Scholar]
- Geller SF, Lewis GP, Fisher SK (2001) FGFR1, signaling, and AP-1 expression after retinal detachment: reactive Muller and RPE cells. Invest Ophthalmol Vis Sci 42: 1363-1369. [PubMed] [Google Scholar]
- Grewal R, Stepczynski J, Kelln R, Erickson T, Darrow R, Barsalou L, Patterson M, Organisciak DT, Wong P (2004) Coordinated changes in classes of ribosomal protein gene expression is associated with light-induced retinal degeneration. Invest Ophthalmol Vis Sci 45: 3885-3895. [DOI] [PubMed] [Google Scholar]
- Guidry C (2005) The role of Muller cells in fibrocontractive retinal disorders. Prog Retin Eye Res 24: 75-86. [DOI] [PubMed] [Google Scholar]
- Haider NB, Naggert JK, Nishina PM (2001) Excess cone cell proliferation due to lack of a functional NR2E3 causes retinal dysplasia and degeneration in rd7/rd7 mice. Hum Mol Genet 10: 1619-1626. [DOI] [PubMed] [Google Scholar]
- Heckenlively JR, Rodriguez JA, Daiger SP (1991) Autosomal dominant sectoral retinitis pigmentosa. Two families with transversion mutation in codon 23 of rhodopsin. Arch Ophthalmol 109: 84-91. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP (2003) Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa N, Takemura M, Koyama Y, Shigenaga Y, Okada T, Baba A (1997) Endothelins promote the activation of astrocytes in rat neostriatum through ET(B) receptors. Eur J Neurosci 9: 895-901. [DOI] [PubMed] [Google Scholar]
- Jeon CJ, Strettoi E, Masland RH (1998) The major cell populations of the mouse retina. J Neurosci 18: 8936-8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedzierski RM, Yanagisawa M (2001) Endothelin system: the double-edged sword in health and disease. Annu Rev Pharmacol Toxicol 41: 851-876. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Takemura M, Fujiki K, Ishikawa N, Shigenaga Y, Baba A (1999) BQ788, an endothelin ET(B) receptor antagonist, attenuates stab wound injury-induced reactive astrocytes in rat brain. Glia 26: 268-271. [DOI] [PubMed] [Google Scholar]
- Lewis GP, Fisher SK (2003) Up-regulation of glial fibrillary acidic protein in response to retinal injury: its potential role in glial remodeling and a comparison to vimentin expression. Int Rev Cytol 230: 263-290. [DOI] [PubMed] [Google Scholar]
- Li F, Cao W, Anderson RE (2003) Alleviation of constant-light-induced photoreceptor degeneration by adaptation of adult albino rat to bright cyclic light. Invest Ophthalmol Vis Sci 44: 4968-4975. [DOI] [PubMed] [Google Scholar]
- Liaw L, Birk DE, Ballas CB, Whitsitt JS, Davidson JM, Hogan BL (1998) Altered wound healing in mice lacking a functional osteopontin gene (spp1). J Clin Invest 101: 1468-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Peng M, Laties AM, Wen R (1998) Preconditioning with bright light evokes a protective response against light damage in the rat retina. J Neurosci 18: 1337-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCumber MW, D'Anna SA (1994) Endothelin receptor-binding subtypes in the human retina and choroid. Arch Ophthalmol 112: 1231-1235. [DOI] [PubMed] [Google Scholar]
- Nour M, Quiambao AB, Peterson WM, Al-Ubaidi MR, Naash MI (2003) P2Y(2) receptor agonist INS37217 enhances functional recovery after detachment caused by subretinal injection in normal and rds mice. Invest Ophthalmol Vis Sci 44: 4505-4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organisciak DT, Winkler BS (1994) Retinal light damage: practical and theoretical considerations. Prog Retinal Eye Res 13: 1-29. [Google Scholar]
- Osaki S, Johnson DA, Frieden E (1966) The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J Biol Chem 241: 2746-2751. [PubMed] [Google Scholar]
- Pacione LR, Szego MJ, Ikeda S, Nishina PM, McInnes RR (2003) Progress toward understanding the genetic and biochemical mechanisms of inherited photoreceptor degenerations. Annu Rev Neurosci 26: 657-700. [DOI] [PubMed] [Google Scholar]
- Pattengale PK, Stewart TA, Leder A, Sinn E, Muller W, Tepler I, Schmidt E, Leder P (1989) Animal models of human disease. Pathology and molecular biology of spontaneous neoplasms occurring in transgenic mice carrying and expressing activated cellular oncogenes. Am J Pathol 135: 39-61. [PMC free article] [PubMed] [Google Scholar]
- Peters CM, Rogers SD, Pomonis JD, Egnaczyk GF, Keyser CP, Schmidt JA, Ghilardi JR, Maggio JE, Mantyh PW (2003) Endothelin receptor expression in the normal and injured spinal cord: potential involvement in injury-induced ischemia and gliosis. Exp Neurol 180: 1-13. [DOI] [PubMed] [Google Scholar]
- Prasanna G, Narayan S, Krishnamoorthy RR, Yorio T (2003) Eyeing endothelins: a cellular perspective. Mol Cell Biochem 253: 71-88. [DOI] [PubMed] [Google Scholar]
- Rattner A, Smallwood PM, Williams J, Cooke C, Savchenko A, Lyubarsky A, Pugh EN, Nathans J (2001) A photoreceptor-specific cadherin is essential for the structural integrity of the outer segment and for photoreceptor survival. Neuron 32: 775-786. [DOI] [PubMed] [Google Scholar]
- Roca A, Shin KJ, Liu X, Simon MI, Chen J (2004) Comparative analysis of transcriptional profiles between two apoptotic pathways of light-induced retinal degeneration. Neuroscience 129: 779-790. [DOI] [PubMed] [Google Scholar]
- Rogers SD, Demaster E, Catton M, Ghilardi JR, Levin LA, Maggio JE, Mantyh PW (1997) Expression of endothelin-B receptors by glia in vivo is increased after CNS injury in rats, rabbits, and humans. Exp Neurol 145: 180-195. [DOI] [PubMed] [Google Scholar]
- Rogers SD, Peters CM, Pomonis JD, Hagiwara H, Ghilardi JR, Mantyh PW (2003) Endothelin B receptors are expressed by astrocytes and regulate astrocyte hypertrophy in the normal and injured CNS. Glia 41: 180-190. [DOI] [PubMed] [Google Scholar]
- Saida K, Mitsui Y, Ishida N (1989) A novel peptide, vasoactive intestinal contractor, of a new (endothelin) peptide family. Molecular cloning, expression, and biological activity. J Biol Chem 264: 14613-14616. [PubMed] [Google Scholar]
- Salzman SK, Acosta R, Beck G, Madden J, Boxer B, Ohlstein EH (1996) Spinal endothelin content is elevated after moderate local trauma in the rat to levels associated with locomotor dysfunction after intrathecal injection. J Neurotrauma 13: 93-101. [DOI] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A (1993) A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry 100: 431-440. [DOI] [PubMed] [Google Scholar]
- Solberg Y, Rosner M, Belkin M (1998) The association between cigarette smoking and ocular diseases. Surv Ophthalmol 42: 535-547. [DOI] [PubMed] [Google Scholar]
- Sparrow JR, Fishkin N, Zhou J, Cai B, Jang YP, Krane S, Itagaki Y, Nakanishi K (2003) A2E, a byproduct of the visual cycle. Vision Res 43: 2983-2990. [DOI] [PubMed] [Google Scholar]
- Taylor HR, West S, Munoz B, Rosenthal FS, Bressler SB, Bressler NM (1992) The long-term effects of visible light on the eye. Arch Ophthalmol 110: 99-104. [DOI] [PubMed] [Google Scholar]
- Tomany SC, Cruickshanks KJ, Klein R, Klein BE, Knudtson MD (2004) Sunlight and the 10-year incidence of age-related maculopathy: the Beaver Dam Eye Study. Arch Ophthalmol 122: 750-757. [DOI] [PubMed] [Google Scholar]
- Travis GH, Brennan MB, Danielson PE, Kozak CA, Sutcliffe JG (1989) Identification of a photoreceptor-specific mRNA encoded by the gene responsible for retinal degeneration slow (rds). Nature 338: 70-73. [DOI] [PubMed] [Google Scholar]
- Uesugi M, Kasuya Y, Hama H, Yamamoto M, Hayashi K, Masaki T, Goto K (1996) Endogenous endothelin-1 initiates astrocytic growth after spinal cord injury. Brain Res 728: 255-259. [DOI] [PubMed] [Google Scholar]
- Vaughan DK, Coulibaly SF, Darrow RM, Organisciak DT (2003) A morphometric study of light-induced damage in transgenic rat models of retinitis pigmentosa. Invest Ophthalmol Vis Sci 44: 848-855. [DOI] [PubMed] [Google Scholar]
- Vazquez-Chona F, Song BK, Geisert Jr EE (2004) Temporal changes in gene expression after injury in the rat retina. Invest Ophthalmol Vis Sci 45: 2737-2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Lam TT, Tso MO, Naash MI (1997) Expression of a mutant opsin gene increases the susceptibility of the retina to light damage. Vis Neurosci 14: 55-62. [DOI] [PubMed] [Google Scholar]
- Wen R, Song Y, Cheng T, Matthes MT, Yasumura D, LaVail MM, Steinberg RH (1995) Injury-induced upregulation of bFGF and CNTF mRNAs in the rat retina. J Neurosci 15: 7377-7385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen R, Cheng T, Song Y, Matthes MT, Yasumura D, LaVail MM, Steinberg RH (1998) Continuous exposure to bright light upregulates bFGF and CNTF expression in the rat retina. Curr Eye Res 17: 494-500. [DOI] [PubMed] [Google Scholar]
- Wick LM, Egli T (2004) Molecular components of physiological stress responses in Escherichia coli Adv Biochem Eng Biotechnol 89: 1-45. [DOI] [PubMed] [Google Scholar]
- Young RW (1988) Solar radiation and age-related macular degeneration. Surv Ophthalmol 32: 252-269. [DOI] [PubMed] [Google Scholar]
- Young RW (1992) Sunlight and age-related eye disease. J Natl Med Assoc 84: 353-358. [PMC free article] [PubMed] [Google Scholar]