

Abstract
The extracellular region of the transmembrane neural cell adhesion molecule (NCAM-EC) is shed as a soluble fragment at elevated levels in the schizophrenic brain. A novel transgenic mouse line was generated to identify consequences on cortical development and function of expressing soluble NCAM-EC from the neuron-specific enolase promoter in the developing and mature neocortex and hippocampus. NCAM-EC transgenic mice exhibited a striking reduction in synaptic puncta of GABAergic interneurons in the cingulate, frontal association cortex, and amygdala but not hippocampus, as shown by decreased immunolabeling of glutamic acid decarboxylase-65 (GAD65), GAD67, and GABA transporter 1. Interneuron cell density was unaltered in the transgenic mice. Affected subpopulations of interneurons included basket interneurons evident in NCAM-EC transgenic mice intercrossed with a reporter line expressing green fluorescent protein and by parvalbumin staining. In addition, there appeared to be a reduction in excitatory synapses, as revealed by synaptophysin staining and apical dendritic spine density of cortical pyramidal cells. Behavioral analyses demonstrated higher basal locomotor activity of NCAM-EC mice and enhanced responses to amphetamine and (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate compared with wild-type controls. Transgenic mice were deficient in prepulse inhibition, which was restored by clozapine but not by haloperidol. Additionally, NCAM-EC mice were impaired in contextual and cued fear conditioning. These results suggested that elevated shedding of NCAM perturbs synaptic connectivity of GABAergic interneurons and produces abnormal behaviors that may be relevant to schizophrenia and other neuropsychiatric disorders.
Keywords: neural cell adhesion molecule, NCAM, interneuron, GABAergic, cingulate, prepulse inhibition, schizophrenia
Introduction
The neural cell adhesion molecule (NCAM) controls diverse aspects of axon and dendritic development, synaptogenesis, and plasticity. Intercellular adhesive events are regulated through homophilic and heterophilic binding interactions mediated by the NCAM extracellular region (NCAM-EC), which consists of five immunoglobulin-like and two fibronectin III domains (Panicker et al., 2003). Alternative splicing of the NCAM gene generates three principal variants that differ only in the cytoplasmic domain: 180 and 140 kDa transmembrane isoforms found in neurons and other cells and a 120 kDa glycophosphatidyl inositol-linked form present mostly in glia. NCAM140 is preferentially expressed in developing neurons and regulates hippocampal-based learning and migration of olfactory granule cell precursors (Stork et al., 2000). In contrast, NCAM180 is restricted primarily to postsynaptic densities of mature neurons, where it is involved in synaptic functions (Persohn and Schachner, 1990; Dityatev et al., 2000), including those regulating sensory gating (Wood et al., 1998) and fear conditioning (Stork et al., 2000). The extracellular region of all NCAM isoforms is subject to posttranslational polysialylation, which modulates cell migration, axon growth, and activity-dependent synaptic plasticity (Muller et al., 1996; Rutishauser and Landmesser, 1996). NCAM knock-out (KO) mice display deficits in learning, memory, hippocampal long-term potentiation (LTP) (Cremer et al., 1994; Bukalo et al., 2004), impaired migration of olfactory neurons (Cremer et al., 2000), decreased sensory gating (Wood et al., 1998), and increased anxiety, aggression, and hyperactivity (Stork et al., 1999, 2000).
Recent evidence has revealed a dysregulation of NCAM associated with schizophrenia (Vawter, 2000). A soluble NCAM fragment consisting of most of its extracellular region is present at elevated levels in postmortem brain and in CSF (Poltorak et al., 1995; Honer et al., 1997; van Kammen et al., 1998; Vawter et al., 1998a,b, 2001), correlating with the severity and duration of schizophrenia (Vawter et al., 1998b). Soluble NCAM-EC in the schizophrenic brain does not arise from overexpression of a minor secreted splice variant of the NCAM extracellular region (Vawter et al., 2000) but appears to be derived from proteolytic cleavage (shedding) of transmembrane NCAM isoforms (Vawter et al., 2001). Soluble NCAM has been widely documented in the normal brain as well as neuronal cell cultures (Walmod et al., 2004), but the protease responsible for NCAM cleavage has not been identified. Soluble NCAM has the potential to act as a dominant-negative inhibitor of intact NCAM interactions, and regulation of shedding could be important in the mechanism of neuronal process outgrowth and synaptogenesis.
To study the effect of overproduction of soluble NCAM-EC in the brain, we generated transgenic mice that express the NCAM-EC fragment from the neuron-specific enolase (NSE) promoter. This promoter is induced late in neuronal differentiation and attains maximal activation in adults (Forss-Petter et al., 1990). This strategy circumvented the early embryonic lethality of mice expressing secreted NCAM generated by a genetic knock-in approach (Rabinowitz et al., 1996). These mice may reflect the brain expression of NCAM in schizophrenia, in which transmembrane and soluble forms of NCAM are present, and the level of the NCAM-EC fragment is ∼50% that of NCAM180 and NCAM140 (Vawter et al., 1998a). NCAM-EC transgenic mice exhibited a striking decrease in synaptic terminals of subpopulations of GABAergic interneurons in frontal cortical areas and the amygdala. The mice also displayed abnormal behaviors that included deficits in sensory gating and emotional memory as well as alterations in motor performance. These results suggested that abnormal expression of secreted NCAM may perturb frontal cortical and amygdala circuitry by interfering with presynaptic function of GABAergic interneurons and possibly other excitatory neurons associated with rodent models of schizophrenia.
Materials and Methods
Construction of plasmids. The pNSE-NCAM plasmid was constructed using the pUC19-based pMG2 expression plasmid (B. Popko, University of Chicago, Chicago, IL). A hemagglutinin (HA) tag and BamH1 site were added by PCR to the 3′-end of the cDNA sequence of the rat NCAM extracellular region (lacking the variable alternative splice exon). This resulted in a 2.2 kb HindIII-BamH1 fragment that represented the entire open-reading frame of the NCAM extracellular cDNA sequence and HA tag. The 1.8 kb NSE promoter was removed from pNSElacZ (G. Sutcliffe, The Scripps Research Institute, La Jolla, CA) and subcloned 5′ to the 2.2 kb NCAM-HA fragment into pMG2. β-Globulin splice sites and poly(A) signal sequences from the pMG2 vector were retained 3′ to the NCAM-HA coding region. Digestion with NotI released a 5.7 kb linear construct free of plasmid DNA for transgene injection.
Transgenic mice and genotyping. Transgenic mice expressing the pNSE-NCAM cDNA (NCAM-EC) were generated at the University of North Carolina Chapel Hill Transgenic Facility. The linearized 5.7 kb pNSE-NCAM construct was microinjected into fertilized C57BL/6 ova. Eggs were transferred to the oviducts of pseudopregnant females of the same strain. Injection of 100 eggs resulted in the birth of 42 pups. Genomic DNA was extracted from tail biopsies from 11 transgenic mice and characterized for transgene insertion by PCR and Southern blotting. Specific PCR primers (NCAM 1964, GCAGTGACCACGTCATGCTC; EXON3R, GGTGAATTCTTTGCCAAAGTGA) were designed for the transgenic construct so that endogenous NCAM DNA was not amplified. For Southern blotting, DNA was digested with EcoRI and probed with a 1.1 kb fragment hybridizing to the HA and β-globulin sequences. Behavioral studies were conducted with wild-type (WT) and NCAM-EC transgenic mice when they were 4-6 months of age; approximately equal numbers of males and females were tested from each genotype with 7-10 mice/genotype/experiment/condition. Founder males (3237, 3250, and 3272) were used to establish separate transgenic lines and were backcrossed to C57BL/6 for five to eight generations. Line 3237 was used for behavioral studies.
Transgenic C57BL/6 harboring a bacterial artificial chromosome (BAC) construct expressing enhanced green fluorescent protein (EGFP)-WT in a subpopulation of basket interneurons driven by the glutamic acid decarboxylase-67 (GAD67) promoter were generated as described previously (Chattopadhyaya et al., 2004). These mice were intercrossed with NCAM-EC transgenic mice (EGFP-NCAM) for five generations for analysis of the effect of the transgene on basket interneurons.
Western blot analysis and Nissl staining. Extracts were prepared from cerebral hemispheres of adult NCAM-EC transgenic mice and WT littermates in radioimmunoprecipitation assay lysis buffer, clarified by centrifugation, and electrophoresed (100 μg of protein) on 7.5% SDS-polyacrylamide gels. Proteins were transferred to nitrocellulose filters, which were blocked with 2% bovine serum albumin and incubated with monoclonal anti-HA antibodies (1:1000) (BabCo, Richmond, CA) or monoclonal anti-OB11 antibody (1:500) against the NCAM cytoplasmic domain (Sigma, St. Louis, MO) for 2 h. Antibody binding was detected by enhanced chemiluminescence.
For fractionation, brains were homogenized in 5 ml of ice-cold PBS with 0.1 mg/ml phenylmethylsulfonyl fluoride, 2 mg/ml pepstatin A, 2 mg/ml aprotinin, and 2 mg/ml leupeptin. Homogenates were centrifuged at 42,000 × g for 30 min at 4°C, and supernatants were collected to yield the soluble fraction. For Nissl staining, NCAM-EC transgenic mice and WT littermates were anesthetized and perfused, and brains were postfixed and processed for Nissl staining as described previously (Demyanenko et al., 1999).
Immunofluorescence and cell-density analysis. Adult transgenic and WT littermate control mice were processed for cryostat sectioning, and brains were sectioned coronally (12 μm) (Demyanenko et al., 1999). For immunofluorescence staining, sections were treated with 1% NP-40/PBS and blocked in 5% milk/5% normal goat or donkey serum. Sections were washed in PBS and incubated for 1 h in 1% serum containing one the following primary antibodies: 5 μg/ml mouse anti-GAD65 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), 2-4 μg/ml rabbit anti-GAD67 (AB108; Chemicon, Temecula, CA), 6 μg/ml mouse anti-parvalbumin (P-3171; Sigma), mouse anti-calretinin [monoclonal antibody (MAB) 1568; Chemicon] at a dilution of 1:2000, 1.6 μg/ml rabbit anti-GABA transporter 1 (GAT-1), 5 μg/ml rabbit anti-NCAM polyclonal antibody (AB5032; Chemicon), or 3 μg/ml mouse anti-synaptophysin (MAB 5258; Chemicon). Sections were washed in 1% NP-40/PBS and incubated for 30 min in 1% serum containing FITC-conjugated secondary antibodies, 1:500 for Alexa 488 (Molecular Probes, Eugene, OR) or 1:150 for tetramethylrhodamine isothiocyanate (TRITC)-conjugated goat anti-rabbit IgG and TRITC-conjugated goat anti mouse IgG (Jackson ImmunoResearch, West Grove, PA). Sections were rinsed, mounted in Vectashield (Vector Laboratories, Burlingame, CA), and imaged by confocal microscopy (Olympus FV500; Olympus, Tokyo, Japan) with a 60× objective at the University of North Carolina Microscopy Facility (Dr. Robert Bagnell, Director, Department of Pathology, University of North Carolina School of Medicine). For EGFP-transgenic mice, brains were sectioned at 100 μm and analyzed directly. Images were acquired using laser excitation (488 nm), laser power (10%), and aperture size (102 μm) and analyzed using Scion (Frederick, MD) Image software. For each image, the perisomatic signals (puncta rings) or neuropil area from at least six randomly selected target neurons were delineated and then pixel density within the delineated area was calculated and averaged as described previously (Huang et al., 1999). Four or more sections from each of three to seven mice of each genotype were analyzed. A total of at least 200 cells were analyzed for each genotype. NIH Image J software was used for analysis of total neuropil area (4 mice/genotype; 16 images per genotype). Data from at least 20 images from each genotype were averaged to derive the pixel density of puncta ring and/or neuropil.
Interneuron cell numbers within layer II/III were counted blind after immunostaining by epifluorescence microscopy in serial coronal sections matched for rostrocaudal level by bregma distance throughout the cingulate cortex in WT and NCAM-EC transgenic littermates. Sections were stained for either parvalbumin or calretinin and examined by indirect immunofluorescence or scored directly from brain sections of EGFP-expressing mice. The total cell number was then divided by the area of the image (7000 μm2) to obtain cell density. The density was reported as cells per square millimeter. In all cases, three to five mice of each genotype (two to six sections per mouse) were analyzed, and 150-200 cells per genotype were scored.
Analysis of spine densities on apical dendrites of pyramidal neurons. NCAM-EC mice were intercrossed with Thy1/yellow fluorescent protein (YFP) line H mice (Feng et al., 2000), in which layer V pyramidal cells are fully labeled with enhanced YFP. Apical dendritic branches located in layer II/III of the cingulate cortex were imaged by confocal microscopy, and Z-stacked projections were used for measurement of spine density. At least 30 dendritic segments from 16 sections were measured from wild-type and mutant mice (two mice of each genotype). The number and density of the spines were measured using the Neurolucida software as described previously for the Thy1/YFP line H mice (Demyanenko et al., 2004).
General neurophysiological status. The neurophysiological screen consisted of tests that involved an initial evaluation as well as assessments of orienting and reflexive behavior, postural and righting reflexes, paw strength, coordination and balance, and anxiety-like behavior (Ribar et al., 2000; Pogorelov et al., 2005). All measures, except where noted, were scored on a multipoint system. Normal responses were scored as 0, negative responses (-1 to -6) denoted deficiencies, and positive scores (+1 to + 6) reflected more proficient or exaggerated responses relative to normal or 0 scores. In the initial evaluation, animals were examined for evidence of changes in skin color, body tone, lacrimation or palperbral closure, exophthalmus, convulsions or tremor, heart or respiration rates, tail elevation or body posture, and barbering or hair loss. Assessments of orientation and reflexive behavior included visual orientation to an object (ability to orient to a dental mirror and follow its movement through the visual field), visual placement (propensity of the mouse to extend its forepaws just before contact with a flat surface), and whisker, eye, and ear reflexes (orientation to whisker touch, eye blink in response to touch, and ear twitch in response to touch with a dowel). Analyses of postural and righting reflexes involved testing the ability of the mouse to maintain its balance when its cage was shaken in a vertical or horizontal plane and for the animal to right itself when placed on its back. Paw strength, coordination, and balance were examined by several tests. Evaluation of forepaw and rearpaw strength was determined by the ability of the mouse to grasp a rod with its forepaws or rearpaws and its tenacity to hold the rod when it was pulled away. The wire-hang task examined forepaw strength and coordination by testing the ability of the mouse to remain suspended on a vertical wire by its forepaws for 90 s. Coordination, balance, and motor strength were studied by observing the time it took the mouse to climb down and up a vertical pole and to traverse a horizontal pole. Mice were also held by their tails for 60 s to ascertain whether they would display abnormal crossing of hindlimbs or body contortion. Finally, anxiety-like behaviors were evaluated in a zero maze for 5 min (Pogorelov et al., 2005). Behavior was videotaped with the investigator hidden from the view of the mouse. Percentage of time in the open arms of the maze was subsequently scored by an investigator who was unfamiliar with the genotypes of the mice.
Open-field activity. Spontaneous activity was evaluated in an automated Omnitech Digiscan apparatus (AccuScan Instruments, Columbus, OH) as described previously (Gainetdinov et al., 1999). WT and NCAM-EC transgenic mice were placed into the open field, and activity was summated at 5 min intervals over a 1 h period. Locomotion was measured as the total distance covered in centimeters (horizontal activity), rearing was evaluated as the total number of vertical beam breaks (vertical activity), and stereotypy was quantitated in terms of repetitive breaks of a given beam with intervals of <1 s. Pharmacological responses were also assessed after 60 min in the open field for baseline activity followed immediately by treatment with vehicle (water), amphetamine (2 or 4 mg/kg), or (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801) (0.2, 0.3, or 0.4 mg/kg, i.p.).
Prepulse inhibition of acoustic startle. Prepulse inhibition (PPI) of acoustic startle was measured in a Med Associates (St. Albans, VT) apparatus in the presence of a 64 dB white-noise background. Mice were given three different types of trials. One trial type consisted of a 40 ms 120 dB white-noise startle stimulus. In a second, the startle stimulus was preceded by a 20 ms prepulse stimulus that was 4, 8, or 12 dB above the white-noise background. In a third trial type, no auditory stimulus was presented, and these null trials served as control for background movements by the animal. During testing, mice were acclimated to the apparatus for 5 min and then given 74 test trials. In 30 of the trials, the 120 dB stimulus was given alone (startle-only trials), whereas eight additional trials served as null trials. For the remaining 36 trials, the prepulse stimulus preceded the startle stimulus by 100 ms. Testing began with 10 startle-only trials, followed by combinations of the 36 prepulse trials, 10 startle-only trials and the eight null trials in a pseudorandom order; testing was completed with 10 startle-only trials. The behavioral responses were measured as the peak startle response for each trial between 35 and 65 ms after the onset of the startle stimulus. Responses that fell outside of this range or that were at a magnitude below that of null trials were discarded. Inhibition of the startle response was calculated for each intensity of prepulse as the ratio of prepulse trials to startle-only trials subtracted from 1 and expressed as a percentage inhibition of response, where %PPI = [1 - (prepulse trials/startle-only trials)]*100. For pharmacological studies, NCAM-EC mice and WT littermates were injected 30 min before PPI testing with vehicle (saline), haloperidol (0.05 or 0.2 mg/kg), or clozapine (1 or 3 mg/kg, i.p.).
Fear conditioning. Fear conditioning was conducted in a mouse fear-conditioning apparatus (Med Associates). During the 3 min training session, WT littermates and NCAM-EC transgenic mice were placed into the apparatus, and after 2 min, a 72 dB tone was sounded for 30 s. Two seconds before offset of the tone, a 0.4 mA scrambled foot-shock was given, and the tone and shock terminated simultaneously. The mouse remained in the chamber for an additional 30 s and then was returned to its home cage. Twenty-four hours later, the animal was tested under one of two conditions. For context testing, the mouse was returned to the same chamber in which it was trained, and freezing behavior was scored every 5 s over a 5 min period; no tone or shock was presented. Under cued testing, the animal was placed into a chamber with a different floor that appeared visually distinct from that used during training. After 2 min, the 72 dB tone was given for 3 min, but no shock was presented. All behaviors were videotaped during the training and test sessions. Freezing behaviors were scored subsequently over the 4 min period for training and the 5 min period for both test conditions by an observer who had no previous knowledge of the genotypes of the mice.
Statistics. Image analyses of puncta rings and/or neuropil and evaluation of interneuron cell density (cells per square millimeter) were statistically calculated for each genotype using Student's t test. Behavioral analyses were conducted with the SPSS-11 programs of statistical tests (SPSS, Chicago, IL), and all results are presented as means and SEM. Responses on the neurophysiological screen were analyzed by t tests. Spontaneous horizontal, vertical, and stereotypy activities were collapsed and analyzed over the first hour by t tests for the two genotypes. For amphetamine and MK-801 responses, the data were analyzed by univariate ANOVA for the main effects of genotype and drug treatment. Decomposition analyses of main effects and significant interaction terms were preformed using pairwise comparisons with Bonferroni corrections. For PPI, baseline startle responses to the 120 dB tone were analyzed by t tests for vehicle-treated WT and NCAM-EC mice. A univariate ANOVA examined startle responses within each genotype for the various drugs and their doses. PPI responses were evaluated with a repeated-measures ANOVA (RMANOVA), and decomposition analyses were used to determine differences in drug responses. The fear conditioning data were analyzed by RMANOVA with decomposition analyses to examine effects at specific times. In all cases, p < 0.05 was considered significant.
Results
Generation of NCAM-EC transgenic mice
To investigate potential neurological deficits of NCAM-EC overexpression, we generated transgenic mice secreting soluble NCAM-EC in the adult brain. A plasmid was generated (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material) in which expression of the entire NCAM-EC, including its five Ig-like and two fibronectin III domains with a C-terminal HA epitope tag, was placed under control of the NSE promoter. This promoter is activated at low levels in all neurons during terminal differentiation and reaches maximal levels in adult neurons (Forss-Petter et al., 1990). The transgene was detected in genomic DNA isolated from 11 of 42 transgenic mice by PCR and Southern blotting (supplemental Fig. S1A, representative examples, available at www.jneurosci.org as supplemental material). The copy number in different transgenic lines was estimated between 1 (3255) and 20 (3272) copies by comparison to known amounts of NCAM-EC plasmid. The 11 transgenic mice were bred to WT C57BL/6 mice, and all crosses showed germ-line transmission. Expected litter sizes with equal numbers of males and females indicated that there was no deleterious effect on the viability of embryos. The transgenic mice were long lived and had normal breeding capability.
Western blotting with HA antibodies was used to evaluate the levels of expression of transgenic NCAM-EC protein in brain extracts isolated from F1 offspring. Different levels of HA-tagged NCAM-EC protein were expressed in transgenic lines (supplemental Fig. S1B, selected cases, available at www.jneurosci.org as supplemental material). Epitope-tagged NCAM-EC had an expected molecular size of ∼110 kDa and appeared as a diffuse band probably resulting from known carbohydrate modification of NCAM. The soluble fraction isolated from brains of NCAM-EC transgenic mice contained the majority of the HA-tagged NCAM-EC protein (∼75% of total NCAM-EC in the brain) estimated by immunoblotting (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). Western blotting of brain extracts with NCAM antibody OB-11, which recognizes a common determinant in the intracellular domain of both NCAM180 and NCAM140, showed that transgene expression of NCAM-EC did not alter the levels of endogenous NCAM isoforms in the brain of most transgenic lines (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material). Levels of NCAM-EC expression did not correlate with endogenous NCAM expression across lines. Three founder males (3237, 3250, and 3272) expressing elevated levels of NCAM-EC protein were chosen for establishing separate transgenic lines. These mice were backcrossed to C57BL/6 for five to eight generations for neuroanatomical and behavioral analyses. No differences in GABAergic marker expression were observed among the three lines analyzed. Line 3237 was chosen for behavioral analysis.
Expression and localization of NCAM-EC in brain
Nissl staining of adult brain sections showed no differences in the size of the olfactory bulb, hippocampus, cerebellum, thalamus, amygdala, or superior colliculus in NCAM-EC mice compared with WT littermates (Fig. 1A,B). Unlike NCAM180 KO mice (Wood et al., 1998), brain ventricles were not enlarged in NCAM-EC mice. The cerebral cortex had a normal thickness with all layers in the correct order. Golgi staining revealed typical cortical neurons, including pyramidal cells with normal morphology and dendritic orientation (data not shown). The expression and localization of the transgenic NCAM-EC fragment in the adult brain was analyzed by immunoperoxidase staining with HA antibodies. NCAM-EC was present in all layers of the transgenic neocortex where it was evident both in the neuropil and soma of subsets of neurons in layers II-VI, as shown in the cingulate cortex (Fig. 1C,E). A similar pattern of immunoreactivity was observed in the motor, somatosensory, and visual cortices. NCAM-EC was prominent in neuropil-rich areas of the hippocampus (CA1, CA2, and CA3) and dentate gyrus (Fig. 1F,H). NCAM-EC was also evident in the amygdala (Fig. 1I) and cerebellum (Fig. 1K). Nonspecific immunoreactivity with HA antibodies was not observed in the corresponding regions of WT brain (Fig. 1D,G,J). Because a large fraction of NCAM-EC was soluble, it was not possible to determine which cell types express the transgene and which cells are binding targets of NCAM-EC. Also, because antibodies against endogenous NCAM also recognize NCAM-EC, potential region-specific alteration in endogenous NCAM isoforms could not be evaluated.
Figure 1.

Nissl staining of NCAM-EC transgenic brain and immunocytochemical localization of NCAM-EC protein. A, B, Nissl staining of adult WT and NCAM-EC brain in a coronal section through the hippocampus (HC), thalamus (th), corpus callosum (cc), lateral ventricle (LV), and dorsal third ventricle (D3V). Scale bar, 500 μm. Immunoperoxidase staining with anti-HA antibodies in adult transgenic cingulate cortex (C) shows localization in layers I-VI and no staining in WT cortex (D). Scale bar, 100 μm. E, NCAM-EC immunoreactivity in the cingulate of transgenic mice is evident in the neuropil and soma of large pyramidal-like neurons (arrowhead) and nonpyramidal-like cells (arrow). Scale bar, 50 μm. F, NCAM-EC staining in the hippocampus of transgenic mice in CA1, CA2, and CA3 and the dentate gyrus (DG). G, No staining in WT hippocampus. Scale bar, 100 μm. H, NCAM-EC staining in the hippocampus and dentate gyrus is evident in the neuropil and cell bodies. Scale bar, 150 μm. I, NCAM-EC staining in the basolateral nucleus of amygdala of transgenic mice. J, No staining in WT amygdala. Scale bar, 100 μm. K, NCAM-EC staining in the cerebellum of transgenic mice. Scale bar, 100 μm. IGL, Internal granular layer; Mo, molecular layer.
Decreased GAD65, GAD67, and GAT-1 in GABAergic interneurons of NCAM-EC transgenic mice
Glutamic acid decarboxylase is the rate-limiting enzyme leading to the biosynthesis of GABA. GAD65 and GAD67, the major isoenzymes of GAD in brain, are distinct gene products with molecular weights of 65 and 67 kDa, respectively. GAD65 is enriched in nerve terminals of GABAergic inhibitory neurons, where it may be activated during high-GABA demand, whereas GAD67 is localized throughout the neuronal processes and soma, where it may maintain basal GABA levels (Feldblum et al., 1993; Esclapez et al., 1994). Because expression and/or activity of presynaptic markers of GABAergic interneurons are decreased in the prefrontal cortex in schizophrenia (Akbarian et al., 1995; Ohnuma et al., 1999; Volk et al., 2000, 2001; Blum and Mann, 2002; Hashimoto et al., 2003), the expression of GAD65 and GAD67 was analyzed in frontal cortical areas (cingulate and frontal association cortex) of the NCAM-EC cerebral cortex by immunofluorescence staining. In the cingulate and frontal association cortex of WT mice, prominent GAD65 immunofluorescence was concentrated in discrete puncta rings, which represented axon terminals and presynaptic boutons of GABAergic interneurons, surrounding nonimmunoreactive soma of postsynaptic neurons in cortical layers II-VI (Fig. 2, arrows). GAD65 immunofluorescent staining in WT frontal areas was generally weaker in neuropil but clearly above background (Fig. 2, inset; compare nonimmune IgG). At equivalent rostrocaudal levels in the NCAM-EC cingulate and frontal association cortex, GAD65 localization in puncta rings was significantly decreased to less than half that of WT (puncta rings in NCAM-EC cingulate and frontal association cortex was reduced by 62 and 59%, respectively) (Fig. 2). Punctate staining for GAD65 in the neuropil also was reduced in transgenic frontal cortical areas but reached significance only in the frontal association cortex (reduced by 62%). There was no decrease in GAD65 fluorescent puncta rings surrounding pyramidal cell soma in the hippocampus (CA1 or CA2) (data not shown).
Figure 2.
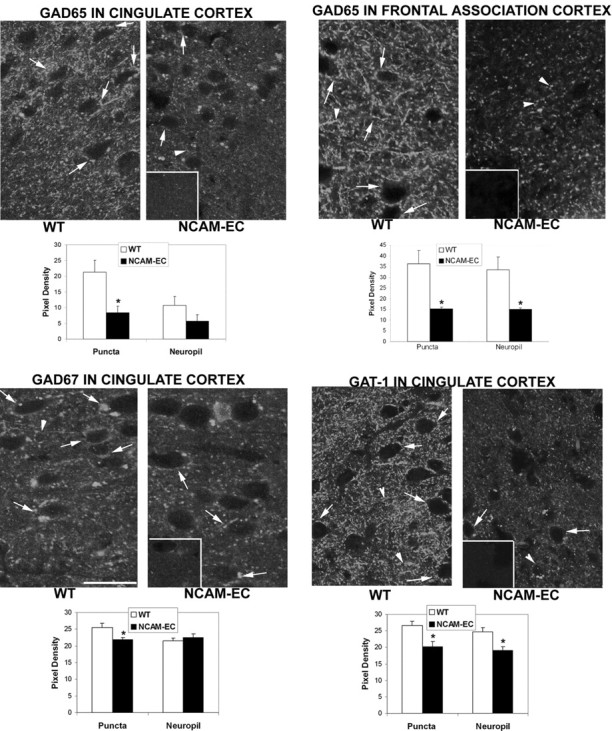
Immunofluorescence localization of GABAergic neuronal markers is decreased in synaptic puncta within the NCAM-EC cerebral cortex. Immunofluorescent staining for GAD65, GAD67, or GAT-1 was analyzed in the cingulate and frontal association cortex (layers II/III) of adult WT and NCAM-EC transgenic mice. Fluorescent puncta rings surrounding nonlabeled soma are shown in confocal images by arrows; punctate staining in the neuropil is shown by arrowheads. Control staining with nonimmune IgG is shown in the insets. Quantification of immunofluorescence in puncta rings and neuropil by mean pixel density from multiple images is depicted in bar graphs. *p < 0.05 from the WT control. Scale bar, 10 μm.
GAD67 immunofluorescence in the cingulate cortex of WT mice was present as puncta rings and as punctate staining in the neuropil (Fig. 2). In the NCAM-EC cingulate cortex, GAD67-labeled puncta rings showed a small but significant decrease of 16% compared with WT, whereas staining in the neuropil was not affected. GABA transporter GAT-1 is enriched in presynaptic nerve terminals of GABAergic interneurons in the cerebral cortex (Minelli et al., 1995). WT mice showed prominent GAT-1 localization in puncta rings surrounding immunonegative cell bodies as well as in the neuropil of cingulate cortex (Fig. 2). In contrast, punctate staining for GAT-1 in the NCAM-EC cingulate cortex was significantly decreased in puncta rings (24% reduction) and neuropil (21% reduction) (Fig. 2).
Decreased synaptic puncta of interneurons in NCAM-EC brain
Although immunofluorescence staining for GAD and GAT-1 was decreased in the transgenic cortex, an appreciable amount of labeling remained, suggesting that subpopulations of interneurons were specifically affected. Cortical GABAergic interneuron subpopulations display distinct electro-physiological characteristics and expression patterns of calcium-binding proteins such as parvalbumin. The parvalbumin-expressing subpopulation accounts for 40-50% of GABAergic interneurons in the rodent neocortex (Kubota et al., 1994). Parvalbumin+ interneurons include basket (wide arbor) and chandelier neurons (Conde et al., 1994).
The effect of NCAM-EC overexpression on basket interneurons was examined in NCAM-EC mice that were intercrossed with a BAC transgenic mouse line (EGFP-WT) expressing EGFP from the GAD67 promoter in subpopulations of basket interneurons (Chattopadhyaya et al., 2004). EGFP-fluorescent puncta rings surrounding cell soma were prominent in the cingulate cortex of EGFP-WT mice (Fig. 3A). NCAM-EC littermates (EGFP-NCAM) showed a significant decrease of 24% in fluorescent puncta rings compared with WT mice (Fig. 3A,B). Fluorescent puncta within the neuropil of EGFP-NCAM mice were not significantly decreased, consistent with the possibility that synaptic terminals were selectively disrupted. A significant decrease of 23% in fluorescent puncta rings was also observed in the amygdala of EGFP-NCAM mice (Fig. 3A,B). There was no significant reduction in EGFP-labeled puncta in the hippocampus (Fig. 3A,B). Endogenous NCAM was demonstrated to be localized in perisomatic synaptic puncta of basket interneurons within the cingulate cortex and amygdala, shown by immunostaining for NCAM in cortical sections from the EGFP-WT mice (Fig. 3C). NCAM immunostaining could be seen colocalized with EGFP-labeled synaptic puncta surrounding the cell soma, as well as in the neuropil.
Figure 3.

Fluorescent synaptic puncta of basket interneurons are decreased in the NCAM-EC cerebral cortex and amygdala. A, Fluorescent synaptic puncta of GABAergic basket interneurons are decreased in the cingulate cortex (layers II/III) and amygdala (basolateral nucleus), but not the hippocampus (CA2 pyramidal cell layer shown), of adult NCAM-EC transgenic mice expressing EGFP specifically in basket cells. Arrows point to puncta rings. Scale bar, 10 μm. B, Quantification of fluorescence in puncta rings, expressed as mean pixel density, in WT and NCAM-EC cingulate cortex (layers II/III), amygdala, and hippocampus (CA2 pyramidal cell layer). *p < 0.05, from the WT mice. C, Colocalization of NCAM immunostaining with rhodamine-conjugated secondary antibodies (red) with EGFP-expressing basket interneurons (green) in adult WT cingulate cortex and amygdala. Arrows indicate colocalization of NCAM immunoreactivity in synaptic puncta surrounding cell bodies. D, Immunostaining for parvalbumin in cingulate cortex of EGFP-WT and EGFP-NCAM transgenic mice. Arrows show puncta rings. Insets, Control staining with nonimmune IgG. The graph shows quantification of immunofluorescence of parvalbumin+ puncta rings in EGFP-WT and EGFP-NCAM cingulate cortex (layers II/III).
The decrease in immunofluorescence of GABAergic neuronal markers in synaptic puncta rings in the NCAM-EC frontal cortex may be a result of fewer presynaptic GABAergic terminals or overall loss of neuropil. To examine this distinction further, the area occupied by neuropil excluding soma was measured in confocal images of EGFP-WT and EGFP-NCAM cortex matched for rostrocaudal level. The average total area occupied by neuropil for EGFP-WT mice (6601 ± 26 μm2 per image) was not significantly different from EGFP-NCAM mice (6533 ± 212 μm2). These results were consistent with a decrease in the number of synaptic terminals rather than an overall loss of neuropil. In accord with this, fluorescent puncta rings of neurons immunostained for parvalbumin in the cingulate cortex were reduced by ∼90% in NCAM-EC compared with WT mice (Fig. 3D). Because ∼80% of cortical parvalbumin-positive neurons are GABAergic (Powell et al., 2003), it is reasonable to conclude that synaptic terminals of parvalbumin-expressing GABAergic interneurons are decreased in NCAM-EC transgenic mice.
Interestingly, there was a significant reduction in immunofluorescence staining for synaptophysin in the NCAM-EC cingulate cortex. Approximately 40% reduction was observed both in puncta rings (pixel density: WT, 135 ± 6; NCAM, 81 ± 8; p < 0.05) and in neuropil (pixel density: WT, 159 ± 10; NCAM, 98 ± 9; p < 0.05) (Fig. 4A). Synaptophysin staining should be fairly proportional to synapse density. Because there are more excitatory synapses in the cortex than inhibitory synapses (DeFelipe et al., 2002), the substantial reduction of synaptophysin immunoreactivity in the transgenic cingulate cortex suggested that synapses other than those derived from GABAergic interneurons were also disrupted. To determine whether excitatory synapses were also decreased in the NCAM-EC brain, NCAM-EC transgenic mice were intercrossed with reporter mice (Thy1/YFP line H) that express YFP from Thy-1 promoter in layer V pyramidal neurons throughout the cell soma, axons, dendrites, and dendritic spines (Feng et al., 2000; Demyanenko et al., 2004). Spine density on branches of apical dendrites found in layer II/III of the cingulate cortex in NCAM-EC/Thy1 mice (1.7 ± 0.2 spines/μm) was reduced by ∼40% when compared with wild-type Thy1 mice (2.8 ± 0.3 spines/μm) (Fig. 4B). Thus, excitatory synapses also appeared to be reduced in the transgenic cortex.
Figure 4.

Synaptophysin immunofluorescence and dendrite spine density are decreased in NCAM-EC cingulate cortex. A, Immunofluorescent staining for synaptophysin was analyzed in cingulate cortex (layers II/III) of adult WT and NCAM-EC transgenic mice. Fluorescent puncta rings surrounding nonlabeled soma are shown in confocal images by arrows. Control staining with nonimmune IgG is shown in the inset. B, Spines on apical dendritic branches in the cingulate cortex (layers II/III) of adult WT (Thy1/YFP line H) and transgenic mice (NCAM-EC/Thy1) were calculated. Arrows indicate some of the spines on the dendrite. *p < 0.05, from the WT control. Scale bar, 10 μm.
Because a decrease in synaptic connectivity might lead to loss of activity-dependent cell survival, we measured the density of interneurons labeled by immunofluorescence staining for parvalbumin or calretinin in NCAM-EC and WT cortex. The density of parvalbumin+ cells was equivalent to WT in the NCAM-EC cingulate cortex (supplemental Fig. S2, available at www.jneurosci.org as supplemental material) as well as in the frontal association cortex, reticulothalamic nucleus, and striatum (data not shown). The density of calretinin+ cells was also unchanged in cingulate cortex (supplemental Fig. S2, available at www.jneurosci.org as supplemental material) or frontal association cortex of transgenic mice (data not shown). Furthermore, the density of basket interneurons in the cingulate cortex of EGFP-NCAM mice was equivalent to that in EGFP-WT mice. These results indicated that there was no loss of neurons expressing parvalbumin or calretinin nor was there loss of EGFP-expressing basket cell subpopulations in frontal cortical areas of NCAM-EC transgenic mice.
Gross sensory and motor function appears normal in NCAM-EC mice
Gross neurophysiological status was compared between WT and NCAM-EC transgenic mice (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Animals were evaluated on appearance, orientation and reflexive behaviors, postural and righting reflexes, paw strength, coordination, and balance (Ribar et al., 2000). No differences were distinguished among genotypes except in the latency to begin walking across a vertical pole. Despite this fact, WT and mutant mice responded similarly on the neurophysiological screen and did not appear to posses any gross features that might compromise subsequent behavioral testing. Importantly, in the zero maze, WT and NCAM-EC mice spent similar amounts of time in the open arms (Fig. S3, available at www.jneurosci.org as supplemental material). Hence, these findings suggest that the transgenic mice do not present an anxiety-like phenotype (Shepherd et al., 1994).
NCAM-EC mice show increased basal activity and enhanced amphetamine and MK-801 responses
Increased locomotor activity is often used as an index for schizophrenia-like behavior in rodents (Lipska and Weinberger, 2000). Accordingly, spontaneous activity of WT and NCAM-EC mice was measured in the open field over 1 h (`Fig. 5A-C). Horizontal or locomotor activity was significantly higher for transgenic mice than for WT littermates (t(1,14) = 2.310; p < 0.037). However, vertical or rearing activity was not distinguished by genotype. In contrast, stereotypy or repetitive beam breaks (Fig. 5C) were found to be significantly increased in NCAM-EC mice compared with WT controls (t(1,14) = 2.306; p < 0.037). These data show that NCAM-EC mice display a significant increase in spontaneous locomotor and stereotypical activities compared with WT animals.
Figure 5.

NCAM-EC mice show enhanced basal activity and locomotion to amphetamine and MK-801. A-C, Mice were placed in the open field, and spontaneous horizontal (A), vertical (B), and stereotypical (C) activities were monitored at 5 min intervals over 1 h (n = 7-9 mice/genotype; *p < 0.05 from WT controls). D-F, Locomotor, rearing, and stereotypy of mice injected with vehicle or 2 or 4 mg/kg, i.p. amphetamine (AMPH) are displayed. G-I, Locomotion, rearing, and stereotypy of mice given vehicle or 0.2, 0.3, or 0.4 mg/kg, i.p. MK-801 [n = 7-10 mice/genotype; #p < 0.01; +p < 0.005 from the WT controls given the same dose of AMPH/MK-801 as the NCAM-EC transgenic (Tg) mice].
Because schizophrenic patients show enhanced sensitivity to psychostimulants (Lieberman et al., 1987) and these drugs can exacerbate psychotic symptoms (Janowsky et al., 1973), we evaluated the responses of WT and NCAM-EC mice to amphetamine in the open field (`Fig. 5D-F). Animals were placed into the open field for 60 min to acclimate them to the apparatus and then were injected with water or 2 or 4 mg/kg amphetamine and immediately returned to the open field for an additional hour. A univariate ANOVA for locomotor activity demonstrated significant effects of genotype (F(1, 44) = 12.627; p < 0.001) and drug (F(2,44) = 43.297; p < 0.001) as well as a significant genotype by drug interaction (F(2,44) = 3.093; p < 0.05). Bonferroni corrected pairwise comparisons revealed that locomotion in NCAM-EC mice was stimulated by both doses of amphetamine (p < 0.006) compared with the vehicle control, whereas WT animals were responsive only to the highest dose (p < 0.015). Moreover, 4 mg/kg amphetamine stimulated higher levels of locomotion in transgenic mice than in WT littermates (Fig. 5D). For rearing, significant drug effects were noted (F(2,44) = 6.694; p < 0.003) in which 2 and 4 mg/kg amphetamine suppressed activity (p < 0.015) but only in WT mice (Fig. 5E). With respect to stereotypy, significant genotype (F(1,44) = 12.698; p < 0.001) and drug effects were obtained (F(2,44) = 44.953; p < 0.001). Decomposition analyses revealed that stereotypy in NCAM-EC mice was responsive to both doses of amphetamine (p < 0.001), whereas WT animals were only activated by the highest dose (p < 0.001) (Fig. 5F). Hence, these data show that locomotion and stereotypy in NCAM-EC mice are more responsive to amphetamine than in the WT littermates.
Another model of schizophrenia-like behaviors in rodents involves the use of NMDA receptor antagonists. In fact, phencyclidine elicits a wide range of psychotomimetic effects in humans (Luby et al., 1962), and MK-801 or dizocilpine mimics some of the features of schizophrenia in humans (Carlsson et al., 2001). To determine whether the NCAM-EC mice may be more responsive to MK-801 than WT controls, animals were tested with the same protocol as that described for amphetamine above. When the effects of MK-801 were examined on locomotor activity (Fig. 5G), significant genotype (F(1,65) = 6.536; p < 0.013), drug (F(3,65) = 31.239; p < 0.001), and a genotype by drug interaction were obtained (F(3,65) = 2.928; p < 0.048). Bonferroni-corrected pairwise comparisons revealed that activity of WT and NCAM-EC mice was significantly stimulated (p < 0.003) by 0.3 and 0.4 mg/kg MK-801 compared with the respective vehicle controls. The pairwise analyses also showed that although no significant differences in response between the 0.3 and 0.4 mg/kg doses for WT mice, locomotion in transgenic animals was stimulated to a greater extent by 0.4 mg/kg MK-801 (p < 0.001). Moreover, NCAM-EC animals were more responsive to the highest dose of MK-801 (p < 0.003) than the WT controls (Fig. 5G). Rearing was suppressed to equal extents in both genotypes by all doses of MK-801 (F(3,65) = 14.039; p < 0.001) (Fig. 5H). Stereotypy to MK-801 mimicked virtually the same effects as that described for locomotion above (genotype: F(1,65) = 6.290; p < 0.015; drug: F(3,65) = 37.750; p < 0.001; genotype by drug interaction: F(3,65) = 2.796; p < 0.056). Decomposition analyses demonstrated that all three doses of MK-801 were effective in stimulating activity in both WT and transgenic mice (p < 0.05). Once again, the 0.3 and 0.4 mg/kg comparison was only significant for NCAM-EC animals (p < 0.016), and the WT and transgenic animals only differed from each other (p < 0.001) at the 0.4 mg/kg dose (Fig. 5I). Thus, NCAM-EC transgenic mice are more sensitive to the activity-stimulating effects of amphetamine and MK-801 than WT littermates, suggesting that overexpression and secretion of NCAM-EC may influence dopaminergic and glutaminergic neurotransmission and, therefore, be relevant to schizophrenia and other psychiatric disorders associated with dysfunction of these systems.
NCAM-EC transgenic mice are impaired in sensory gating
NCAM-EC transgenic mice were tested for sensory gating deficits in PPI. Before testing, amplitudes of the startle response to the 120 dB stimulus were found to be equivalent in WT and NCAM-EC mice (Fig. 6A, inset). When RMANOVA was applied to the PPI results, significant prepulse intensity effects (F(2,38) = 12.763; p < 0.001) and a genotype by prepulse intensity interaction (F(2,38) = 3.164; p < 0.054) were noted. Decomposition analyses demonstrated that transgenic mice displayed significantly lower inhibition of the startle response (p < 0.006) than WT controls at each of three prepulse intensities (Fig. 6A). Examination of the interaction revealed that WT mice could distinguish among each prepulse intensity (p < 0.022), whereas the NCAM-EC mice did not appear capable of differentiating among the stimuli. These data show that transgenic mice are deficient both in the magnitude of inhibition of PPI responses and in gating among the acoustic prepulse stimuli.
Figure 6.

Prepulse inhibition of acoustic startle is decreased in NCAM-EC transgenic mice and is restored with clozapine. A, Percentage of PPI responses of WT and NCAM-EC transgenic (Tg) mice. Inset, Startle response to the 120 dB tone. *p < 0.05, from the WT control. B, Startle responses of WT and Tg mice administered vehicle, haloperidol, orclozapine (intraperitoneally). *p < 0.05, from the respective vehicle control within genotype. C, D, PPI of WT and Tg mice given vehicle or 0.05 or 0.2 mg/kg haloperidol (intraperitoneally) (#p < 0.05, vehicle and 0.05 mg/kg haloperidol vs 0.2 mg/kg heloperidol). E, F, PPI of WT mice and Tg mice administered vehicle or 1 or 3 mg/kg, i.p. clozapine (n = 8-12 mice/genotype/drug/dose; *p < 0.05, vehicle vs 1 mg/kg clozapine; +p < 0.05, vehicle vs 3 mg/kg clozapine; @p < 0.05, 1 mg/kg vs 3 mg/kg clozapine).
To determine whether sensory gating by the transgenic mice was improved by treatment with antipsychotics, startle and PPI responses were evaluated for mice given haloperidol or clozapine. Before PPI testing, the amplitudes of the startle responses to the 120 dB startle stimulus were compared between WT and NCAM-EC transgenic mice given vehicle or each of the drugs (Fig. 6B). For WT and NCAM-EC mice, significant treatment effects for startle responses were obtained for both haloperidol (WT: F(1,32) = 10.617; p < 0.001; NCAM-EC: F(1,29) = 27.260; p < 0.001) and clozapine (WT: F(1,32) = 3.42; p < 0.045; NCAM-EC: F(1,23) = 10.487; p < 0.001). In all cases, 0.05 and 0.2 mg/kg haloperidol and 1 and 3 mg/kg clozapine enhanced the magnitude of the startle response to the 120 dB stimulus over that of the respective vehicle controls (WT, p < 0.045; NCAM-EC, p < 0.003).
Effects of haloperidol and clozapine were evaluated separately within each genotype. RMANOVA for WT mice given haloperidol identified a significant main effect of prepulse intensity (F(2,72) = 74.510; p < 0.001) and a significant prepulse by treatment interaction (F(4,72) = 14.001; p < 0.001) (Fig. 6C,D). PPI responses at 4 and 8 dB were lower for the vehicle controls and animals given 0.05 mg/kg haloperidol (p < 0.007) than for WT animals given 0.2 mg/kg haloperidol (Fig. 6C). When responses under each drug condition were analyzed, it was found that mice given vehicle or 0.05 mg/kg haloperidol could still sensory gate (p < 0.014), whereas those administered the higher dose of haloperidol could not differentiate among the prepulses. Hence, PPI is enhanced by haloperidol; however, 0.2 mg/kg, but not the 0.05 mg/kg, dose disrupts sensory gating in the WT mice. In comparison, in NCAM-EC animals, haloperidol did not alter PPI performance over that of the vehicle control (Fig. 6D). Thus, transgenic mice did not respond significantly to haloperidol.
In addition to evaluating effects of haloperidol on PPI, we also studied responses to clozapine (Fig. 6E,F). RMANOVA of WT responses revealed a significant effect of prepulse intensity (F(2,64) = 35.655; p = 0.001) (Fig. 6E). These data show that WT mice could still differentiate among the prepulses regardless of vehicle-drug assignment. An examination of NCAM-EC responses gave a significant main effect of prepulse intensity (F(2,46) = 16.373; p = 0.001) and a significant prepulse by treatment interaction (F(4,46) = 2.656; p < 0.045). Decomposition analyses demonstrated that NCAM-EC animals given 1 mg/kg clozapine had higher PPI responses at 4 dB than the vehicle controls (p < 0.001) and the animals given 3 mg/kg clozapine (Fig. 6F). Responses of the vehicle controls were also lower (p < 0.001) than those given either dose of clozapine at prepulse intensities of 8 or 12 dB. Additional comparisons confirmed that NCAM-EC mice could not differentiate among the different prepulses, whereas animals given 1 or 3 mg/kg clozapine successfully gated the stimuli. Collectively, these findings showed that the atypical antipsychotic clozapine, but not haloperidol, could normalize PPI in NCAM-EC mice.
Impaired fear conditioning in NCAM-EC transgenic mice
To test the associative learning capability of NCAM-EC transgenic mice, animals were tested for cued- and contextual-dependent fear conditioning. Parenthetically, cued fear conditioning is disrupted by lesions of the amygdala but not the hippocampus (Phillips and LeDoux, 1992; Kim and Davis, 1993), whereas contextual fear conditioning is mediated by the hippocampus, amygdala, and periaqueductal gray in addition to cingulate and frontal cortex (Fendt and Fanselow, 1999). During conditioning, freezing behaviors were not statistically different between the two genotypes over the first 2 min of training, during presentation of the tone shock, or after the footshock (Fig. 7A). As might be anticipated, however, an RMANOVA found significant main effects of test period (F(2,30) = 59.188; p < 0.001). Bonferroni-corrected pairwise comparisons showed that this effect was a result of a progressive increase in freezing behavior from acclimatization to tone shock and from tone shock to the postshock periods for both genotypes of mice. In comparison, freezing behaviors were less pronounced in transgenic mice than in WT controls during contextual fear conditioning (F(4,60) = 2.556; p < 0.048) (Fig. 7B). In fact, NCAM-EC transgenic mice displayed significantly lower levels of freezing behavior in the contextual test than the WT littermates at all times examined (p < 0.011). In the cued test, significant time (F(4,60) = 48.519; p < 0.001) and a genotype by time interaction were found (F(4,60) = 17.383; p < 0.001). Freezing responses were low and were not distinguished by genotype before presentation of the tone (Fig. 7C). However, when the tone was sounded, freezing behavior immediately increased for WT animals while it remained low for the NCAM-EC transgenic mice throughout testing (p < 0.001). Despite this fact, there was some increment in freezing behavior within the final block of testing for transgenic mice (minutes 1 and 2 vs 5; p < 0.01). These findings suggest that emotional learning and memory were impaired in NCAM-EC transgenic mice.
Figure 7.
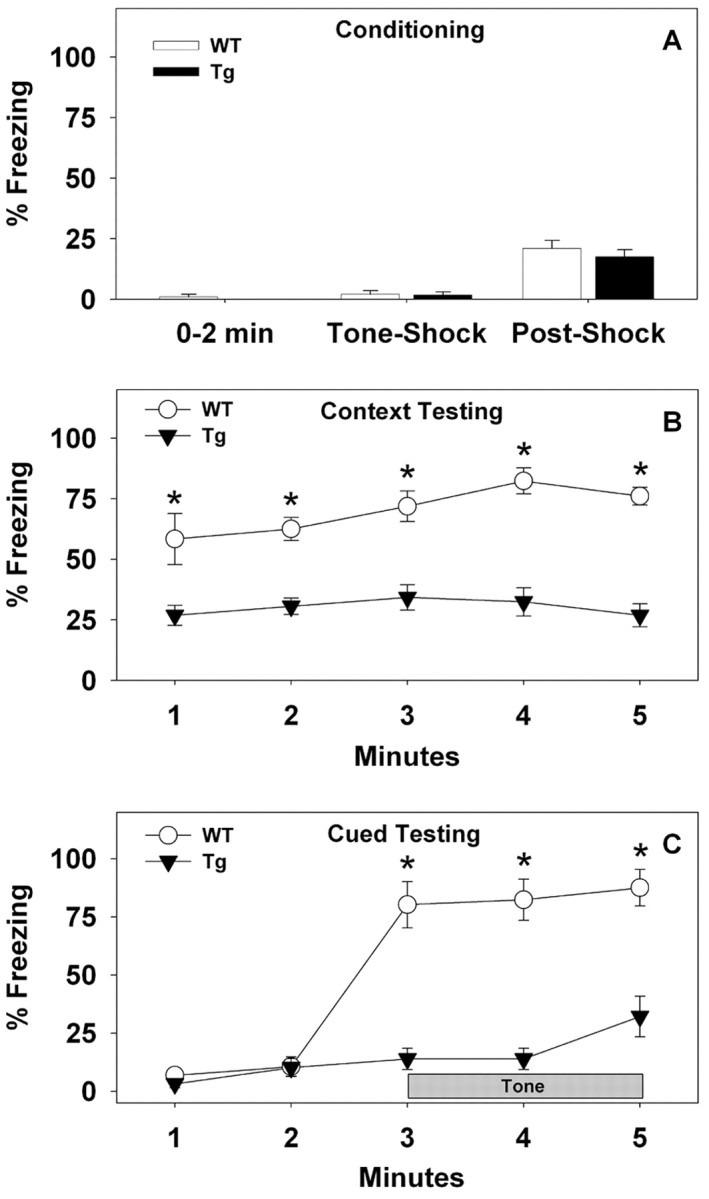
Fear conditioning is decreased in NCAM-EC transgenic mice. A, Freezing behavior of WT and NCAM-EC transgenic (Tg) mice during the first 2 min of conditioning, during the 30 s tone-shock interval, and 30 s after tone shock. B, Percentage of freezing responses of WT and Tg mice during each minute of the 5 min context test. C, Freezing behavior of WT and Tg mice before and during tone presentation (cue) over the 5 min test. n = 8-9 mice/genotype; *p < 0.05 from the transgenic mice.
Discussion
Here, we describe a novel transgenic mouse strain (NCAM-EC) that expresses a soluble fragment of NCAM consisting of its extracellular domain in the brain. NCAM-EC transgenic mice displayed impaired synaptic connectivity involving GABAergic interneurons evident by a decrease in perisomatic synaptic puncta in the cingulate and frontal association cortex as well as the amygdala. Defective sensory gating, impaired emotional memory, and increased locomotor activities in NCAM-EC transgenic mice may be a result of defective circuitry in these regions. The results suggest that elevated shedding of the NCAM extracellular region may perturb NCAM180-dependent neuronal recognition affecting development or maintenance of inhibitory synapses in the cerebral cortex and amygdala.
The similarities in behavioral phenotypes of NCAM-EC mice and NCAM null mutants suggest that the neurological defects of the transgenic mice result from inhibition of NCAM function. Both strains exhibit impaired PPI performance, deficiencies in contextual and cued fear conditioning, and hyperlocomotion (Wood et al., 1998; Stork et al., 2000). Behavioral deficits in NCAM-EC mice probably arise from perturbation of NCAM180 function rather than NCAM140, because defective PPI (Wood et al., 1998) and fear conditioning (Stork et al., 2000) result from loss of NCAM180 specifically. Moreover, the olfactory bulb of NCAM-EC transgenic mice was normal in size, whereas that of NCAM140-mice is small because of impaired migration of granule neurons (Stork et al., 2000). Perturbation of NCAM180 function in NCAM-EC mice is likely to occur at the postnatal stage, when endogenous NCAM180 is upregulated and the NSE promoter is maximally activated (Forss-Petter et al., 1990).
Decreased numbers of presynaptic terminals of GABAergic neurons in the frontal cortex and amygdala of NCAM-EC mice were indicated by the reduction in perisomatic puncta rings immunoreactive for four different markers (GAD65, GAD67, GAT-1, and parvalbumin) and in EGFP-labeled basket cells. There is no loss of major interneuron subpopulations in the cingulate cortex of transgenic mice, as shown for parvalbumin+ interneurons, which account for 40-50% of GABAergic interneurons in the rodent neocortex (Kubota et al., 1994), for calretinin+ interneurons, which account for 12% of GABAergic interneurons in the mouse parietal cortex (Powell et al., 2003), or for the EGFP+ basket cell subpopulation. These results suggested that elevated expression of the soluble NCAM fragment perturbs the development or maintenance of presynaptic axon terminals of GABA interneurons. This finding is consistent with the known function of NCAM in regulating synapse number (Schuster et al., 1998; Dityatev et al., 2000), LTP (Luthl et al., 1994), and synaptic maturation (Landmesser et al., 1988). By disturbing neuronal connectivity, it is also possible that NCAM-EC might depress the expression of these markers. For example, activity-dependent regulation of GAD67 has been reported in interneurons (Hendry and Jones, 1988; Benson et al., 1994). This effect might be mediated by excitatory input from pyramidal cells (Melchitzky and Lewis, 2003), although activity-dependent regulation of GAD65 and GAT-1 has been noted less consistently (Patz et al., 2003). The finding that synaptophysin-labeled puncta were also decreased in the neuropil as well as perisomatic sites in the cerebral cortex of NCAM-EC transgenic mice suggested that connectivity of classes of neurons other than GABAergic interneurons may be perturbed, and that effects on interneurons could be indirect results of defects in glutamatergic or dopaminergic neurotransmission. Indeed, along with striking reductions in inhibitory synapses, a decrease in synaptophysin immunoreactivity and pyramidal cell dendrite spine density in the cingulate cortex of NCAM-EC mice suggests that there are decreases in excitatory synapses. Furthermore, synapses within other brain regions and cortical areas may be perturbed, because both the NCAM-EC transgene and endogenous NCAM are widely expressed in the brain.
Schizophrenia-related alteration in expression of genes involved in GABA neurotransmission has been repeatedly documented in the prefrontal cortex of human patients (Blum and Mann, 2002). Decreased expression of GAD67 (Akbarian et al., 1995; Volk et al., 2000) and GAT-1 (Pierri et al., 1999; Volk et al., 2001) in subpopulations of interneurons is observed consistently (Woo et al., 1998; Lewis et al., 2001), along with changes in GABAA receptors (Benes et al., 1996; Volk et al., 2002). Disturbances in chandelier axon terminals (Lewis et al., 2001) and basket interneurons have been implicated in working memory deficits in schizophrenia (Hashimoto et al., 2003). GABAergic dysfunction in the prefrontal cortex may perturb various cognitive functions, because interneurons are involved in information processing (Constantinidis et al., 2002). Although there was a reduction in GABAergic synaptic puncta in frontal cortex and amygdala of NCAM-EC mice, there was no reduction in GAD65-, GAD67-, or EGFP-labeled puncta in the hippocampus. Similarly, little or no reduction in GAD65 and GAD67 expression is evident in the hippocampus in schizophrenia (Todtenkopf and Benes, 1998; Heckers et al., 2002). Interestingly, a recent meta-analysis of 20 genome scans for schizophrenia susceptibility showed human chromosome region 11q22.3-q24.1, which encompasses the NCAM gene at 11q23.1, as the fourth-ranked candidate locus (Lewis et al., 2003).
Schizophrenia has been postulated to represent abnormalities in dopaminergic and/or glutaminergic neurotransmission (Carlsson et al., 2001). To assess responses of these systems, spontaneous activity was monitored in the open field. Under basal conditions, locomotor and stereotypical activities are higher in the NCAM-EC mice than in the WT controls. It is noteworthy that NCAM null mutants also show a similar increase in activity (Stork et al., 1999). The enhancement in baseline activity suggests that the NCAM-EC mice may possess alterations within the basal ganglia-thalamocortical circuitry. Additional support for this idea derives from observations with amphetamine and MK-801 treatments. In both cases, the NCAM-EC mice are more responsive to the locomotor-stimulating effects of these drugs than the WT controls. These findings suggest that neurons within the basal ganglia-thalamocortical circuit of NCAM-EC transgenic mice are more sensitive to the locomotor-stimulating effects of these agents than WT controls. Because GABAergic interneurons in frontal cortical regions and other brain areas can exert profound effects on this circuit (Sesack et al., 1995; Karler et al., 1998), deficits in GABAergic terminals may underlie the hyperactivity phenotype of the NCAM-EC animals and their enhanced responsivity to amphetamine and MK-801.
A decrease in GABAergic nerve terminals in frontal cortex and amygdala of NCAM-EC mice may disrupt the inhibitory limb of the PPI circuit and impair sensory gating. PPI of the acoustic response is mediated by a startle circuit that involves the auditory cortex, superior colliculus, and pons, which integrates with an inhibitory circuit comprised of inputs to the nucleus accumbens from the medial prefrontal cortex, amygdala, and hippocampus (Swerdlow et al., 2001). GABAergic neurotransmission in the forebrain is important in the response, because PPI is disrupted after infusion of the GABA antagonist picrotoxin into these brain regions (Fendt et al., 2000). Treatment of NCAM-EC transgenic mice with the atypical antipsychotic drug clozapine, but not the dopamine D2 receptor antagonist haloperidol, improved the ability of NCAM-EC transgenic mice to gate the auditory stimulus. Interestingly, deficits in PPI in GAD65 KO mice are also reversed by clozapine (Heldt et al., 2004). Clozapine is a broad-spectrum antipsychotic that has weak affinity for dopamine D1, D2, D3, and D5 receptors and substantial affinities for dopamine D4, serotonergic, α-adrenergic, muscarinic, and histaminergic receptors (Ashby and Wang, 1996). The inability of haloperidol to rescue PPI suggests that dopaminergic receptor signaling may be relatively normal in the forebrain of the NCAM-EC mice. In contrast, normalization of PPI responses by clozapine indicates that serotonergic neurotransmission, perhaps mediated through 2A receptors, may be abnormal. It is interesting that serotonin turnover and release in frontal cortex are stimulated by NMDA receptor antagonists such as MK-801 (Martin et al., 1998) and that in the open field, NCAM-EC mice are more responsive to this drug than WT animals. Because the raphe nucleus sends projections to the frontal cortex that synapse on GABA interneurons and pyramidal cells (Smiley and Goldman-Rakic, 1996), alterations in GABA function in this brain region could disrupt PPI performance in NCAM-EC mice and become normalized by clozapine treatment
In addition to deficits in PPI, NCAM-EC mice are also deficient in fear conditioning. It should be emphasized that the impairment in fear conditioning was most likely a result of abnormalities in learning and memory rather than increased locomotion, because activities of WT and NCAM-EC mice were very similar during the training phase of fear conditioning, and the mice did not show evidence of impulsive behavior in the neurophysiological screen. Because the transgenic mice are impaired in both contextual and cued fear conditioning, the results suggest that emotional memory in extrahippocampal regions or the amygdala is disrupted in these mutants (Phillips and LeDoux, 1992; Kim and Davis, 1993). In accord with this possibility, synaptic terminals of labeled GABAergic interneurons are diminished in the amygdala but not in the hippocampus of NCAM-EC transgenic mice. Moreover, NCAM is expressed on synaptic puncta of basket cells in the basolateral nucleus of the amygdala (shown here) as well as on interneuron-like cells in almost all amygdaloid nuclei in the rat (Nacher et al., 2002). In addition to the amygdala, GABAergic dysfunction in frontal cortical regions of NCAM-EC mice may also contribute to the deficits in fear conditioning, because these areas are known to regulate amygdala function in affective conditioning in rodents (Rosenkranz et al., 2003). In this regard, it is interesting that GAD65 null mutant mice are also deficient in fear conditioning (Stork et al., 2003).
Deficits in sensory gating, emotional memory, and locomotor activities of NCAM-EC transgenic mice have been observed in other rodent models of schizophrenia-like behavior, including calcineurin conditional knockouts (Miyakawa et al., 2003), dopamine transporter mutants (Gainetdinov et al., 2001), NMDA receptor hypomorphs (Mohn et al., 1999), and mice with ventral hippocampal lesions (Lipska and Weinberger, 2000). However, NCAM dysregulation has been linked with other diseases, including bipolar disorder (Vawter, 2000; Arai et al., 2004) and Alzheimer's dementia (Todaro et al., 2004). Thus, the NCAM-EC transgenic mouse line provides a new model for investigating a subset of behavioral and neuropathogenic features that may provide unique insights into basic mechanisms that underlie certain neuropsychiatric disorders.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants MH064065 (P.F.M.), N626620 (P.F.M.), and EY13564 (J.Z.H.), by a National Alliance for Research on Schizophrenia and Depression (NARSAD) Distinguished Investigator award (P.F.M.), by an NARSAD Established Investigator award and unrestricted funds (W.C.W.), by NIH Postdoctoral Fellowship T32-HD40127 (N.P.-N.), by an NARSAD Young Investigator award (A.K.P.), by an American Psychological Association postdoctoral fellowship (R.M.R.), by the Whitehall Foundation, and by an NIH Pew scholarship (J.Z.H.). We gratefully acknowledge the contributions of Stacey Foti to the immunofluorescence protocol, Leann Hinkle to parvalbumin analysis, and James R. Stackhouse to the neurophysiological screen.
Correspondence should be addressed to Dr. Patricia F. Maness, Department of Biochemistry and Biophysics, University of North Carolina School of Medicine, Chapel Hill, NC 27599-7260. E-mail: srclab@med.unc.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254659-13$15.00/0
References
- Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney Jr WE, Jones EG (1995) Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry 52: 258-266. [DOI] [PubMed] [Google Scholar]
- Arai M, Itokawa M, Yamada K, Toyota T, Haga S, Ujike H, Sora I, Ikeda K, Yoshikawa T (2004) Association of neural cell adhesion molecule 1 gene polymorphisms with bipolar affective disorder in Japanese individuals. Biol Psychiatry 55: 804-810. [DOI] [PubMed] [Google Scholar]
- Ashby Jr CR, Wang RY (1996) Pharmacological actions of the atypical antipsychotic drug clozapine: a review. Synapse 24: 349-394. [DOI] [PubMed] [Google Scholar]
- Benes FM, Vincent SL, Marie A, Khan Y (1996) Up-regulation of GABAA receptor binding on neurons of the prefrontal cortex in schizophrenic subjects. Neuroscience 75: 1021-1031. [DOI] [PubMed] [Google Scholar]
- Benson DL, Huntsman MM, Jones EG (1994) Activity-dependent changes in GAD and preprotachykinin mRNAs in visual cortex of adult monkeys. Cereb Cortex 4: 40-51. [DOI] [PubMed] [Google Scholar]
- Blum BP, Mann JJ (2002) The GABAergic system in schizophrenia. Int J Neuropsychopharmacol 5: 159-179. [DOI] [PubMed] [Google Scholar]
- Bukalo O, Fentrop N, Lee AY, Salmen B, Law JW, Wotjak CT, Schweizer M, Dityatev A, Schachner M (2004) Conditional ablation of the neural cell adhesion molecule reduces precision of spatial learning, long-term potentiation, and depression in the CA1 subfield of mouse hippocampus. J Neurosci 24: 1565-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A, Waters N, Holm-Waters S, Tedroff J, Nilsson M, Carlsson ML (2001) Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Annu Rev Pharmacol Toxicol 41: 237-260. [DOI] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ (2004) Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci 24: 9598-9611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde F, Lund JS, Jacobowitz DM, Baimbridge KG, Lewis DA (1994) Local circuit neurons immunoreactive for calretinin, calbindin D-28k or parvalbumin in monkey prefrontal cortex: distribution and morphology. J Comp Neurol 341: 95-116. [DOI] [PubMed] [Google Scholar]
- Constantinidis C, Williams GV, Goldman-Rakic PS (2002) A role for inhibition in shaping the temporal flow of information in prefrontal cortex. Nat Neurosci 5: 175-180. [DOI] [PubMed] [Google Scholar]
- Cremer H, Lange R, Christoph A, Plomann M, Vopper G, Roes J, Brown R, Baldwin S, Kraemer P, Scheff S, Barthels D, Rajewsky K, Wille W (1994) Inactivation of the N-CAM gene in mice results in size reduction of the olfactory bulb and deficits in spatial learning. Nature 367: 455-459. [DOI] [PubMed] [Google Scholar]
- Cremer H, Chazal G, Lledo PM, Rougon G, Montaron MF, Mayo W, Le Moal M, Abrous DN (2000) PSA-NCAM: an important regulator of hippocampal plasticity. Int J Dev Neurosci 18: 213-220. [DOI] [PubMed] [Google Scholar]
- DeFelipe J, Alonso-Nanclares L, Arellano JI (2002) Microstructure of the neocortex: comparative aspects. J Neurocytol 31: 299-316. [DOI] [PubMed] [Google Scholar]
- Demyanenko GP, Tsai AY, Maness PF (1999) Abnormalities in neuronal process extension, hippocampal development, and the ventricular system of L1 knockout mice. J Neurosci 19: 4907-4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demyanenko GP, Schachner M, Anton E, Schmid R, Feng G, Sanes J, Maness PF (2004) Close homolog of L1 modulates area-specific neuronal positioning and dendrite orientation in the cerebral cortex. Neuron 44: 423-437. [DOI] [PubMed] [Google Scholar]
- Dityatev A, Dityateva G, Schachner M (2000) Synaptic strength as a function of post- versus presynaptic expression of the neural cell adhesion molecule NCAM. Neuron 26: 207-217. [DOI] [PubMed] [Google Scholar]
- Esclapez M, Tillakaratne NJ, Kaufman DL, Tobin AJ, Houser CR (1994) Comparative localization of two forms of glutamic acid decarboxylase and their mRNAs in rat brain supports the concept of functional differences between the forms. J Neurosci 14: 1834-1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldblum S, Erlander MG, Tobin AJ (1993) Different distributions of GAD65 and GAD67 mRNAs suggest that the two glutamate decarboxylases play distinctive functional roles. J Neurosci Res 34: 689-706. [DOI] [PubMed] [Google Scholar]
- Fendt M, Fanselow MS (1999) The neuroanatomical and neurochemical basis of conditioned fear. Neurosci Biobehav Rev 23: 743-760. [DOI] [PubMed] [Google Scholar]
- Fendt M, Schwienbacher I, Koch M (2000) Amygdaloid N-methyl-d-aspartate and gamma-aminobutyric acid(A) receptors regulate sensorimotor gating in a dopamine-dependent way in rats. Neuroscience 98: 55-60. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR (2000) Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28: 41-51. [DOI] [PubMed] [Google Scholar]
- Forss-Petter S, Danielson PE, Catsicas S, Battenberg E, Price J, Nerenberg M, Sutcliffe JG (1990) Transgenic mice expressing beta-galactosidase in mature neurons under neuron-specific enolase promoter control. Neuron 5: 187-197. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Wetsel WC, Jones SR, Levin ED, Jaber M, Caron MG (1999) Role of serotonin in the paradoxical calming effect of psychostimulants on hyperactivity. Science 283: 397-401. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Mohn AR, Bohn LM, Caron MG (2001) Glutamatergic modulation of hyperactivity in mice lacking the dopamine transporter. Proc Natl Acad Sci USA 98: 11047-11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA (2003) Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci 23: 6315-6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckers S, Stone D, Walsh J, Shick J, Koul P, Benes FM (2002) Differential hippocampal expression of glutamic acid decarboxylase 65 and 67 messenger RNA in bipolar disorder and schizophrenia. Arch Gen Psychiatry 59: 521-529. [DOI] [PubMed] [Google Scholar]
- Heldt SA, Green A, Ressler KJ (2004) Prepulse inhibition deficits in GAD65 knockout mice and the effect of antipsychotic treatment. Neuropsychopharmacology 29: 1610-1619. [DOI] [PubMed] [Google Scholar]
- Hendry SH, Jones EG (1988) Activity-dependent regulation of GABA expression in the visual cortex of adult monkeys. Neuron 1: 701-712. [DOI] [PubMed] [Google Scholar]
- Honer WG, Falkai P, Young C, Wang T, Xie J, Bonner J, Hu L, Boulianne GL, Luo Z, Trimble WS (1997) Cingulate cortex synaptic terminal proteins and neural cell adhesion molecule in schizophrenia. Neuroscience 78: 99-110. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S (1999) BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98: 739-755. [DOI] [PubMed] [Google Scholar]
- Janowsky DS, el-Yousel MK, Davis JM, Sekerke HJ (1973) Provocation of schizophrenic symptoms by intravenous administration of methylphenidate. Arch Gen Psychiatry 28: 185-191. [DOI] [PubMed] [Google Scholar]
- Karler R, Calder LD, Thai DK, Bedingfield JB (1998) The role of dopamine and GABA in the frontal cortex of mice in modulating a motor-stimulant effect of amphetamine and cocaine. Pharmacol Biochem Behav 60: 237-244. [DOI] [PubMed] [Google Scholar]
- Kim M, Davis M (1993) Electrolytic lesions of the amygdala block acquisition and expression of fear-potentiated startle even with extensive training but do not prevent reacquisition. Behav Neurosci 107: 580-595. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Hattori R, Yui Y (1994) Three distinct subpopulations of GABAergic neurons in rat frontal agranular cortex. Brain Res 649: 159-173. [DOI] [PubMed] [Google Scholar]
- Landmesser L, Dahm L, Schultz K, Rutishauser U (1988) Distinct roles for adhesion molecules during innervation of embryonic chick muscle. Dev Biol 130: 645-670. [DOI] [PubMed] [Google Scholar]
- Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, Williams NM, Schwab SG, Pulver AE, Faraone SV, Brzustowicz LM, Kaufmann CA, Garver DL, Gurling HM, Lindholm E, Coon H, Moises HW, Byerley W, Shaw SH, Mesen A, et al. (2003) Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: schizophrenia. Am J Hum Genet 73: 34-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Cruz DA, Melchitzky DS, Pierri JN (2001) Lamina-specific deficits in parvalbumin-immunoreactive varicosities in the prefrontal cortex of subjects with schizophrenia: evidence for fewer projections from the thalamus. Am J Psychiatry 158: 1411-1422. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Kane JM, Alvir J (1987) Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacology (Berl) 91: 415-433. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR (2000) To model a psychiatric disorder in animals: schizophrenia as a reality test. Neuropsychopharmacology 23: 223-239. [DOI] [PubMed] [Google Scholar]
- Luby ED, Gottlieb JS, Cohen BD, Rosenbaum G, Domino EF (1962) Model psychoses and schizophrenia. Am J Psychiatry 119: 61-67. [DOI] [PubMed] [Google Scholar]
- Luthl A, Laurent JP, Figurov A, Muller D, Schachner M (1994) Hippocampal long-term potentiation and neural cell adhesion molecules L1 and NCAM. Nature 372: 777-779. [DOI] [PubMed] [Google Scholar]
- Martin P, Carlsson ML, Hjorth S (1998) Systemic PCP treatment elevates brain extracellular 5-HT: a microdialysis study in awake rats. NeuroReport 9: 2985-2988. [DOI] [PubMed] [Google Scholar]
- Melchitzky DS, Lewis DA (2003) Pyramidal neuron local axon terminals in monkey prefrontal cortex: differential targeting of subclasses of GABA neurons. Cereb Cortex 13: 452-460. [DOI] [PubMed] [Google Scholar]
- Minelli A, Brecha NC, Karschin C, DeBiasi S, Conti F (1995) GAT-1, a high-affinity GABA plasma membrane transporter, is localized to neurons and astroglia in the cerebral cortex. J Neurosci 15: 7734-7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa T, Leiter LM, Gerber DJ, Gainetdinov RR, Sotnikova TD, Zeng H, Caron MG, Tonegawa S (2003) Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc Natl Acad Sci USA 100: 8987-8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn AR, Gainetdinov RR, Caron MG, Koller BH (1999) Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 98: 427-436. [DOI] [PubMed] [Google Scholar]
- Muller D, Wang C, Skibo G, Toni N, Cremer H, Calaora V, Rougon G, Kiss JZ (1996) PSA-NCAM is required for activity-induced synaptic plasticity. Neuron 17: 413-422. [DOI] [PubMed] [Google Scholar]
- Nacher J, Lanuza E, McEwen BS (2002) Distribution of PSA-NCAM expression in the amygdala of the adult rat. Neuroscience 113: 479-484. [DOI] [PubMed] [Google Scholar]
- Ohnuma T, Augood SJ, Arai H, McKenna PJ, Emson PC (1999) Measurement of GABAergic parameters in the prefrontal cortex in schizophrenia: focus on GABA content, GABA(A) receptor alpha-1 subunit messenger RNA and human GABA transporter-1 (HGAT-1) messenger RNA expression. Neuroscience 93: 441-448. [DOI] [PubMed] [Google Scholar]
- Panicker AK, Buhusi M, Thelen K, Maness PF (2003) Cellular signalling mechanisms of neural cell adhesion molecules. Front Biosci 8:d 900en]911. [DOI] [PubMed] [Google Scholar]
- Patz S, Wirth MJ, Gorba T, Klostermann O, Wahle P (2003) Neuronal activity and neurotrophic factors regulate GAD-65/67 mRNA and protein expression in organotypic cultures of rat visual cortex. Eur J Neurosci 18: 1-12. [DOI] [PubMed] [Google Scholar]
- Persohn E, Schachner M (1990) Immunohistological localization of the neural adhesion molecules L1 and N-CAM in the developing hippocampus of the mouse. J Neurocytol 19: 807-819. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE (1992) Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci 106: 274-285. [DOI] [PubMed] [Google Scholar]
- Pierri JN, Chaudry AS, Woo TU, Lewis DA (1999) Alterations in chandelier neuron axon terminals in the prefrontal cortex of schizophrenic subjects. Am J Psychiatry 156: 1709-1719. [DOI] [PubMed] [Google Scholar]
- Pogorelov VM, Rodriguiz RM, Insco ML, Caron MG, Wetsel WC (2005) Novelty seeking and stereotypic activation of behavior in mice with disruption of the Dat1 gene. Neuropsychopharmacology, in press. [DOI] [PubMed]
- Poltorak M, Khoja I, Hemperly JJ, Williams JR, el-Mallakh R, Freed WJ (1995) Disturbances in cell recognition molecules (N-CAM and L1 antigen) in the CSF of patients with schizophrenia. Exp Neurol 131: 266-272. [DOI] [PubMed] [Google Scholar]
- Powell EM, Campbell DB, Stanwood GD, Davis C, Noebels JL, Levitt P (2003) Genetic disruption of cortical interneuron development causes region- and GABA cell type-specific deficits, epilepsy, and behavioral dysfunction. J Neurosci 23: 622-631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JE, Rutishauser U, Magnuson T (1996) Targeted mutation of NCAM to produce a secreted molecule results in a dominant embryonic lethality. Proc Natl Acad Sci USA 93: 6421-6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribar TJ, Rodriguiz RM, Khiroug L, Wetsel WC, Augustine GJ, Means AR (2000) Cerebellar defects in Ca2+/calmodulin kinase IV-deficient mice. J Neurosci 20: RC107(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz JA, Moore H, Grace AA (2003) The prefrontal cortex regulates lateral amygdala neuronal plasticity and responses to previously conditioned stimuli. J Neurosci 23: 11054-11064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutishauser U, Landmesser L (1996) Polysialic acid in the vertebrate nervous system: a promoter of plasticity in cell-cell interactions. Trends Neurosci 19: 422-427. [DOI] [PubMed] [Google Scholar]
- Schuster T, Krug M, Hassan H, Schachner M (1998) Increase in proportion of hippocampal spine synapses expressing neural cell adhesion molecule NCAM180 following long-term potentiation. J Neurobiol 37: 359-372. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Snyder CL, Lewis DA (1995) Axon terminals immunolabeled for dopamine or tyrosine hydroxylase synapse on GABA-immunoreactive dendrites in rat and monkey cortex. J Comp Neurol 363: 264-280. [DOI] [PubMed] [Google Scholar]
- Shepherd JK, Grewal SS, Fletcher A, Bill DJ, Dourish CT (1994) Behavioural and pharmacological characterisation of the elevated “zero-maze” as an animal model of anxiety. Psychopharmacology (Berl) 116: 56-64. [DOI] [PubMed] [Google Scholar]
- Smiley JF, Goldman-Rakic PS (1996) Serotonergic axons in monkey prefrontal cerebral cortex synapse predominantly on interneurons as demonstrated by serial section electron microscopy. J Comp Neurol 367: 431-443. [DOI] [PubMed] [Google Scholar]
- Stork O, Welzl H, Wotjak CT, Hoyer D, Delling M, Cremer H, Schachner M (1999) Anxiety and increased 5-HT1A receptor response in NCAM null mutant mice. J Neurobiol 40: 343-355. [DOI] [PubMed] [Google Scholar]
- Stork O, Welzl H, Wolfer D, Schuster T, Mantei N, Stork S, Hoyer D, Lipp H, Obata K, Schachner M (2000) Recovery of emotional behaviour in neural cell adhesion molecule (NCAM) null mutant mice through transgenic expression of NCAM180. Eur J Neurosci 12: 3291-3306. [DOI] [PubMed] [Google Scholar]
- Stork O, Yamanaka H, Stork S, Kume N, Obata K (2003) Altered conditioned fear behavior in glutamate decarboxylase 65 null mutant mice. Genes Brain Behav 2: 65-70. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Braff DL (2001) Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology (Berl) 156: 194-215. [DOI] [PubMed] [Google Scholar]
- Todaro L, Puricelli L, Gioseffi H, Guadalupe Pallotta M, Lastiri J, Bal de Kier Joffe E, Varela M, Sacerdote de Lustig E (2004) Neural cell adhesion molecule in human serum. Increased levels in dementia of the Alzheimer type. Neurobiol Dis 15: 387-393. [DOI] [PubMed] [Google Scholar]
- Todtenkopf MS, Benes FM (1998) Distribution of glutamate decarboxylase65 immunoreactive puncta on pyramidal and nonpyramidal neurons in hippocampus of schizophrenic brain. Synapse 29: 323-332. [DOI] [PubMed] [Google Scholar]
- van Kammen DP, Poltorak M, Kelley ME, Yao JK, Gurklis JA, Peters JL, Hemperly JJ, Wright RD, Freed WJ (1998) Further studies of elevated cerebrospinal fluid neuronal cell adhesion molecule in schizophrenia. Biol Psychiatry 43: 680-686. [DOI] [PubMed] [Google Scholar]
- Vawter MP (2000) Dysregulation of the neural cell adhesion molecule and neuropsychiatric disorders. Eur J Pharmacol 405: 385-395. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Cannon-Spoor HE, Hemperly JJ, Hyde TM, VanderPutten DM, Kleinman JE, Freed WJ (1998a) Abnormal expression of cell recognition molecules in schizophrenia. Exp Neurol 149: 424-432. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Hemperly JJ, Freed WJ, Garver DL (1998b) CSF N-CAM in neuroleptic-naive first-episode patients with schizophrenia. Schizophr Res 34: 123-131. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Frye MA, Hemperly JJ, VanderPutten DM, Usen N, Doherty P, Saffell JL, Issa F, Post RM, Wyatt RJ, Freed WJ (2000) Elevated concentration of N-CAM VASE isoforms in schizophrenia. J Psychiatr Res 34: 25-34. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Usen N, Thatcher L, Ladenheim B, Zhang P, VanderPutten DM, Conant K, Herman MM, van Kammen DP, Sedvall G, Garver DL, Freed WJ (2001) Characterization of human cleaved N-CAM and association with schizophrenia. Exp Neurol 172: 29-46. [DOI] [PubMed] [Google Scholar]
- Volk D, Austin M, Pierri J, Sampson A, Lewis D (2001) GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons. Am J Psychiatry 158: 256-265. [DOI] [PubMed] [Google Scholar]
- Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA (2000) Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry 57: 237-245. [DOI] [PubMed] [Google Scholar]
- Volk DW, Pierri JN, Fritschy JM, Auh S, Sampson AR, Lewis DA (2002) Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb Cortex 12: 1063-1070. [DOI] [PubMed] [Google Scholar]
- Walmod PS, Kolkova K, Berezin V, Bock E (2004) Zippers make signals: NCAM-mediated molecular interactions and signal transduction. Neurochem Res 29: 2015-2035. [DOI] [PubMed] [Google Scholar]
- Woo TU, Whitehead RE, Melchitzky DS, Lewis DA (1998) A subclass of prefrontal gamma-aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc Natl Acad Sci USA 95: 5341-5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood GK, Tomasiewicz H, Rutishauser U, Magnuson T, Quirion R, Rochford J, Srivastava LK (1998) NCAM-180 knockout mice display increased lateral ventricle size and reduced prepulse inhibition of startle. NeuroReport 9: 461-466. [DOI] [PubMed] [Google Scholar]