

Abstract
Olfactory receptor neurons (ORNs) undergo caspase-mediated retrograde apoptosis after target removal (bulbectomy), in which axonal caspase-9 and caspase-3 activation leads to terminal apoptosis in ORN soma of the olfactory epithelium. Here, we show that caspase-8 can act as an initiator of ORN apoptosis after bulbectomy and also after synaptic instability is induced by NMDA-mediated excitotoxic death of ORN target neurons in the olfactory bulb. Caspase-8 and caspase-3 are sequentially activated within ORN presynaptic terminals, and caspase-8 complexes with dynactin p150Glued, (a retrograde motor protein) and is transported retrogradely, preceding axonal caspase-3 activation and apoptosis of ORN cell bodies. Focal in vivo inhibition of initiator caspase activation or microtubule-dependent transport (with Taxol) at the lesioned axon terminus results in a significant reduction in retrograde axonal caspase-8 and caspase-3 activation and inhibition of retrograde ORN death. Caspase-8 activation and retrograde transport after NMDA lesion is similarly reduced in mice null for p75, the low-affinity nerve growth factor receptor. The retrograde apoptosis of ORNs thus involves a novel mechanism that used p75 in the local activation of caspase-8. Once caspase-8 is maximally activated in the presynaptic terminal, it is transported retrogradely by the motor complex dynactin/dynein, a process that can be inhibited focally to inhibit ORN apoptosis after acute axonal lesion. These data have revealed a novel mechanism of retrograde apoptosis, in which caspase-8 complexes directly with axonal dynactin p150Glued to reveal a differential vulnerability of subpopulations of ORNs to undergo apoptosis after axonal damage and the loss of olfactory bulb target neurons.
Keywords: olfactory, bulbectomy, NMDA, p75(NGFR), neoepitope, TUNEL, coimmunoprecipitation
Introduction
Neuronal apoptosis is the primary mechanism of neuronal loss in neurodegenerative disease, ischemia, or trauma (Rink et al., 1995; Choi, 1996; Stefanis et al., 1997) (for review, see Oppenheim, 1991; Yuan et al., 2003). Developmental neuronal apoptosis, necessary for sculpting the nervous system, is thought to result from a “survival of the fittest” mechanism, whereby excess immature neurons compete for limiting neurotrophic factors (Burek and Oppenheim, 1999; Earnshaw et al., 1999; Roth and D'Sa, 2001; Salvesen, 2002), which can subsequently stimulate retrograde prosurvival signaling (for review, see Miller and Kaplan, 2001). Such prosurvival signaling must dominate over the local activation of caspases, a family of intracellular cysteine proteases (Earnshaw and Kaufmann, 1999; Salvesen, 2002), which drive neuronal apoptosis. The initiator caspase-9 and the effector caspase-3 are essential for developmental neuronal apoptosis (Hakem et al., 1998; Kuida et al., 1998) and are also implicated in trauma-induced secondary neuronal and glial apoptosis. The manner in which caspases can transmit apoptotic signals in vivo after axonal damage has yet to be established (Endres et al., 1998; Clark et al., 1999; Springer et al., 1999; Le et al., 2002).
Apoptosis of adult olfactory receptor neurons (ORNs) depends on the retrograde activation of caspase-3 and caspase-9, the proenzymes of which are distributed throughout their axons to the synaptic terminals (Cowan et al., 2001). Caspase-3 and caspase-9 proenzymes first appear in ORN axons during early embryonic axogenesis, when the local environment is trophic factor rich (Cowan and Roskams, 2004). Localized activation of caspase-3 in ORN synaptic terminals is not detected, however, until the first two postnatal weeks, the peak of ORN synaptic remodeling, and when bulb neurons undergo apoptosis based on changes in afferent input (Fiske and Brunjes, 2001a; Yan et al., 2001). Collectively, this suggests a role for synaptic caspase activation in synaptic stability in addition to target-driven activity-dependent apoptosis. Changes in afferent activity (too much or too little) could therefore enable trans-synaptic signaling from bulbar targets to stimulate ORN remodeling (apoptosis, followed by neurogenesis) and subsequent compensatory local changes in interneuron circuitry that occur in the olfactory bulb (OB) throughout maturity (Cummings et al., 1997; Rochefort et al., 2002; Carleton et al., 2003).
Before this mechanism can be tested, upstream signals that drive caspase-3 and caspase-9 activation at the synapse must first be identified and their role in initiating retrograde apoptosis in different ORN subpopulations delineated. Although bulbectomy-induced neurodegeneration is partially modulated by Bcl-2 and Bax (Jourdan et al., 1998; Conley et al., 2003), caspase-3 could also be activated by an extrinsic death receptor pathway in ORN presynaptic terminals (Budihardjo et al., 1999; Fumarola and Guidotti, 2004; Thorburn, 2004). To examine the extrinsic contribution to the initiation of ORN retrograde apoptosis, we used a novel lesion to remove ORN target neurons in the olfactory bulb by NMDA-induced excitotoxicity, leaving ORN presynaptic terminals intact. Here, we identify caspase-8 as an initiator of ORN apoptosis and the low-affinity nerve growth factor receptor (lNGFR) p75 as a potential upstream death receptor in this mechanism. In addition, we show that the same retrograde motor system that signals survival (Eaton et al., 2002; LaMonte et al., 2002; Puls et al., 2003) can carry activated apical caspases and propagate retrograde neuronal cell death in response to both deafferentation and target deprivation.
Materials and Methods
Lesions
Bulbectomies. Unilateral olfactory bulbectomies were performed on adult CD-1 mice as described previously (Roskams et al., 1996). Complete removal of the olfactory bulb was confirmed after the animal was killed. Animals were killed 0, 12, 24, or 36 h after surgery.
NMDA microinjections. Three-month-old mice were anesthetized with Xylaket [25% ketamine HCl (Bimeda-MTC Animal Health, Cambridge, Ontario, Canada), 2.5% xylazine (Bayer, Toronto, Ontario, Canada), 15% ethanol, and 0.55% NaCl]. The mouse was fixed to a stereotaxic frame, and a hole was drilled into the skull above 5.1 ± 0.2 mm anterior, 1.0 mm lateral to the midline, from bregma. NMDA (40 μm) was injected directly perpendicular to the plane of the skull, to 1.5 mm depth in a volume of 0.5 μl, over 10 min. Sham animals were injected with the same volume of carrier (0.1 m PBS). Animals were killed 1 or 4 d after surgery.
Inhibitor/Taxol treatment. Immediately after bulbectomy, the bulb cavity was filled with gelfoam and 2 μl of either 50 μm initiator caspase inhibitor L-826920 (Merck Frosst, Dorval, Quebec, Canada) or 10 μm Taxol (Molecular Probes, Eugene, OR) was injected into the gelfoam. Animals were sutured and allowed to recover for 24-48 h (n = 4-6 per time point). Retrograde transport was detected by addition of 3-4 μl of 5% Fast Blue (Sigma, St. Louis, MO) in DMSO to the anterior edge of the lesion site at the end of surgery.
Bromodeoxyuridine labeling. To assess both short-term incorporation (24 h) and long-term retention (14 d), wild-type (wt) C57BL/6 and p75-/- mice (exon III mutants; strain Ngfrtm1Jae; The Jackson Laboratory, Bar Harbor, ME) (Lee et al., 1992) (n = 4 per time point per genotype) were given three injections of bromodeoxyuridine (BrdU) (50 mg/kg, i.p.; Sigma), administered at 2 h intervals, and killed at 24 h and 14 d after BrdU.
Tissue preparation
Immunohistochemistry or terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling. Mice (n = 3-5 per time point or treatment group) were anesthetized with Xylaket and perfused with PBS, followed by 4% paraformaldehyde (PFA), and brains, olfactory bulbs, and olfactory epithelia (OEs) were dissected out and postfixed in 4% PFA (Cowan et al., 2001). Tissues were equilibrated in 10 and 30% sucrose, embedded in TissueTek (Sakura Finetek, Torrance, CA), and frozen in liquid nitrogen. Ten to 16 μm numbered coronal or transverse sections were prepared on an HM 500 cryostat (Microm Instruments, San Marcos, CA) and stored at -20°C. For protein preparation, mice were decapitated after anesthesia, and their olfactory tissue was dissected out without perfusion, snap frozen on dry ice, and homogenized in ice-cold modified radioimmunoprecipitation assay (RIPA) buffer containing the following: 50 mm Tris-HCl, pH 8, 150 mm NaCl, 1% Nonidet P-40 (NP-40), 0.5% deoxycholic acid, 0.1% SDS, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 100 μg/ml PMSF. Tissue from four to six animals was pooled for each time point or treatment group. At least two sets of experimental groups were prepared for analysis. Homogenates were stored at -80°C.
Antibodies
For immunoprecipitation (IP) and Western blotting, mouse monoclonal anti-dynactin p150Glued (BD Biosciences, Franklin Lake, NJ), mouse anti-dynein, cytoplasmic (74 kDa intermediate chains) monoclonal antibody (mAb) (Chemicon, Temecula, CA), and anti-Flag mAb (Sigma) were used. Mouse monoclonal antibodies against dynactin p150Glued coiled coil 1 (CC1), mAb 150.1 and dynactin p150Glued coiled coil 2 (CC2), and mAb 150.B were kindly provided by Dr. T. Schroer (Johns Hopkins University, Baltimore, MD) and were described previously (Steuer et al., 1990; Gaglio et al., 1996). Caspase-8 antibody used for Western blot analysis was a rabbit polyclonal antibody against human caspase-8 large subunit, (R#MF438). For immunohistochemistry, cleaved caspase-8 was recognized with affinity-pure rabbit polyclonal antibody against caspase-8 neoepitope [KLH-(C)GIPVETD] (MF-E0022804K) (used at 1:1000), and active caspase-3 was detected with a rabbit polyclonal antibody (67341A; used at 1:500; PharMingen, San Diego, CA). Goat polyclonal antibody to olfactory marker protein (OMP) (used at 1:5000) was a gift from F. L. Margolis (University of Maryland, Baltimore, MD), and neuron-specific class III β-tubulin (NST) monoclonal antibody (used at 1:500; BabCo, Richmond, CA), mouse monoclonal antibodies to neuronal-specific nuclear protein (NeuN) (1:100), growth-associated protein 43 (GAP43) (1:1250), and synaptophysin (1:50) as well as p75-specific rabbit polyclonal antibody (1:1000) were all obtained from Chemicon.
Bioinformatics
We blasted the pseudo-death effector domain (pDED) sequence from Huntingtin interacting protein protein interactor (HIPPI) using National Center for Biotechnology Information nucleotide-nucleotide Basic Longitudinal Alignment Sequence Tool [BLAST (blastn)] and protein-protein BLAST (blastp) using a number of different search criteria and obtained a number of hits that were then tested for proteins implicated in axonal transport with the highest identity to HIPPI. Additional motifs were analyzed using a combination of Swissprot databases.
DNA constructs
Expression constructs for dynactin p150Glued, CC1 (amino acids 217-548), and CC2 (amino acids 926-1049) were kindly provided by Dr. T. Schroer (Johns Hopkins University) and were described by Quintyne et al. (1999). Expression constructs for Flag-tagged procaspase-8 and Flag-tagged caspase-8 deletions were described previously (Gervais et al., 2002).
Transfections and immunoprecipitations
Human embryonic kidney 293T (HEK293T) cells were cultured in DMEM supplemented with 10% fetal bovine serum. Transient transfections were performed using Lipofectamine Plus reagent (Invitrogen, San Diego, CA). HEK293T cells were transfected with the catalytically inactive Flag-tagged procaspase-8 C-A, Flag-tagged caspase-8 deletion constructs, Dynactin p150Glued CC1 (amino acids 217-548), or CC2 (amino acids 926-1049). At 40 h after transfection, HEK293T cells were washed in PBS, harvested, and lysed on ice for 30 min in buffer B150 (20 mm Tris-HCl, pH 8.0, 150 mm KCl, 10% glycerol, 5 mm MgCl2, 0.1% NP-40, and protease inhibitors). Supernatants were incubated for 2 h with M2 agarose beads (Sigma) at 4°C followed by three washes of the beads with lysis buffer. Immunoprecipitates were eluted from the beads using Flag peptides (Sigma) and were processed for Western blot analysis using anti-dynactin p150Glued antibodies or anti-Flag antibodies. For the co-IP of dynactin p150Glued and caspase-8 from tissue extracts of the olfactory bulb and epithelium, tissues were pooled from three to five CD1 adult mice and were homogenized in RIPA buffer. For IP, 400 μg extracts were incubated with 4 μg of anti-dynactin p150Glued antibodies for 2 h at 4°C followed by the addition of 30 μl of pre-equilibrated protein-G Sepharose beads for 18 h at 4°C. The beads were then washed as above, and the immunoprecipitates were eluted in 2× SDS-PAGE sample buffer and processed for Western blot analysis with anti-caspase-8 MF438 rabbit polyclonal antibodies or with anti-dynactin p150Glued antibodies. For the co-IP of dynactin p150Glued and procaspase-8 using in vitro translated proteins, dynactin p150Glued and procaspase-8 were transcribed and translated in vitro using TNT-coupled reticulocyte lysates (Promega, Madison, WI). Dynactin p150Glued was immunoprecipitated with anti-dynactin p150Glued antibodies on protein-G beads as above, and the co-IP of 35S-labeled procaspase-8 was detected by autoradiography.
Immunoblotting
Aliquots of tissue homogenates (25 μg of protein) were subjected to SDS-PAGE and transferred to Immobilon membrane (Millipore, Bedford, MA). Equal loading of lanes was confirmed by Ponceau S staining. Membranes were blocked for 1 h at room temperature with 5% nonfat milk in Tris-buffered saline (TBS), incubated for 12-20 h at 4°C in primary antibody in 2% milk/TBS, washed three times for 5 min each in 0.1% Tween 20 in TBS, and incubated for 1 h at room temperature in peroxidase-coupled goat anti-rabbit IgG (Bio-Rad, Hercules, CA) diluted in 2% milk/TBS. Signals were detected with chemiluminescence reagents (Pierce, Rockford, IL).
Immunohistochemistry and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling
Sections were rehydrated in PBS and then permeabilized in 0.1% Triton X-100 (Pierce) for 30 min, blocked with 4% normal serum, incubated at 4°C for 12-20 h with primary antibody, and rinsed twice in PBS plus 0.2% Tween 20. For immunohistochemistry, sections were incubated for 1 h at room temperature in biotin-conjugated polyclonal goat anti-rabbit antibody, and binding was visualized with the Vectastain ABC peroxidase and VIP chromogen kits (all from Vector Laboratories, Burlingame, CA). For double immunofluorescence, sections were incubated sequentially with Alexa 488-conjugated donkey anti-rabbit and either Alexa 594-conjugated donkey anti-goat or donkey anti-mouse polyclonal antibodies (all from Molecular Probes) for 1 h at room temperature each and then counterstained with 4′,6′-diamidino-2-phenylindole and mounted in Vectashield (Vector Laboratories). Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) was performed using both the TACS TdT Blue Label kit (R&D Systems, Minneapolis, MN) in accordance with the instructions of the manufacturer and as described previously (Cowan et al., 2001).
Quantitation of active caspase-8, TUNEL, and BrdU
Caspase-8 neoepitope-positive cells present in the OE were quantified in blinded samples as the number of positive cells per 200 μm OE in three sample areas (see Fig. 1 F). For knock-out mouse analysis, comparative active caspase-8 immunoreactivity in the glomerular layer of the bulb was quantified after confocal image capture from matched areas [glomerular and inner nerve fiber layer (NFL) combined] of lesioned and unlesioned olfactory bulb. Sections at three different caudorostral levels of the olfactory bulb from four lesioned mice of each genotype were examined and directly compared with the exact same area on the unlesioned contralateral side of the same mouse. Pseudoconfocal (stacked) images were captured from blinded samples using fixed settings for all sections and analyzed with NIH Image J, and the comparative (lesioned-unlesioned) results within each mouse were normalized to an area of 100,000 pixels.
Figure 1.
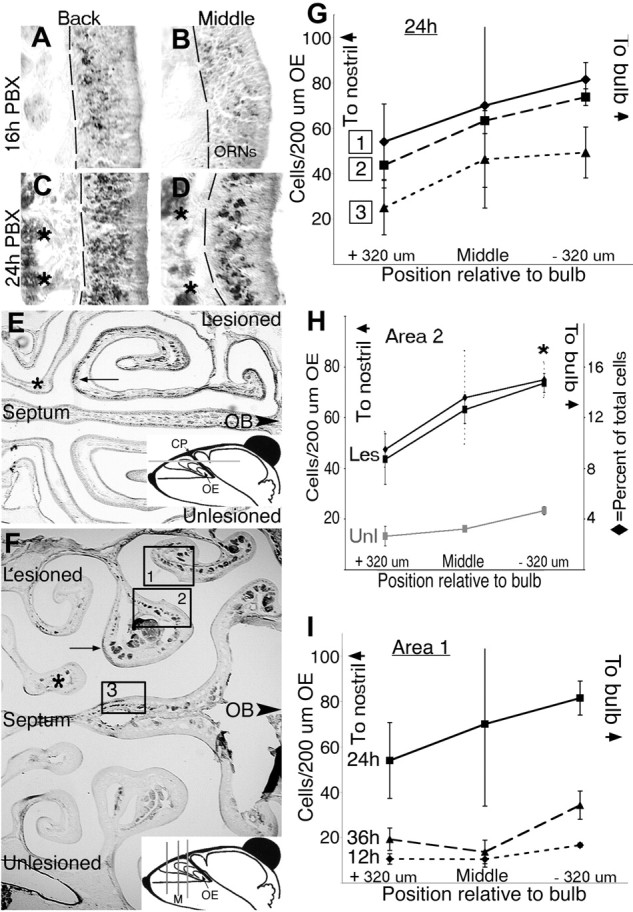
Caspase-8 is activated in a caudorostral gradient in olfactory receptor neurons after bulbectomy-activated caspase-8 at 16 h postbulbectomy (PBX) (A, B) was first detected in axon bundles and ORN soma (A) at the back of the OE, close to the olfactory bulb, at a time when it was barely detectable (B) in middle sections (F, inset, M) 1.5 mm closer to the nostril. By 24 h PBX, activated caspase-8 was detected in a much more widespread population of both ORNs and axon bundles (C; asterisks), close to the bulb, and was now readily detectable in subpopulations of axons and ORNs in middle sections (D) matched to those in B. In single transverse (E) and coronal (F) planes (as indicated in the insets), detection of activated caspase-8 at 24 h postbulbectomy in ORNs and axon bundles was most pronounced proximal to the olfactory bulb (arrows) and absent from axons and neurons more distal from the OB (asterisks). G, Activated caspase-8 present in the OE was quantified as the number of positive cells/200 μm OE in the three sample areas designated in F, at three distinct distances from the bulb in each animal, in a typical middle section (depicted in F), and 320 μm both rostral and caudal to this initial section. H, The number of cells containing activated caspase-8 was quantified for the unlesioned (Unl) side and also expressed as a percentage of total cells for sample area 2 at 24 h PBX. *p = 0.05 relative to +320 μm position, for counts/200 μm; Student's t test. I, The number of cells containing activated caspase-8 was quantified at sample area 1 in matched sections from mice allowed to survive for 12, 24, and 36 h PBX. A caudorostral pattern of active caspase-8 was recorded in all three sample areas at 24 h PBX (G, H), which was also the peak of signal intensity (I, sample area 1 only). In A-D, the dashed line indicates basal lamina. In G: ♦, sample area 1, endoturbinate IIa; ▪, sample area 2, endoturbinate IIb; ▴, sample area 3, septum. In H: ♦, lesioned (Les) cells as percentage of total; ▪, lesioned counts per 200 μm OE;  , unlesioned counts per 200 μm OE. In I: ♦, 12 h PBX; ▪, 24 h PBX; ▴, 36 h PBX. For all graphs, n = 3-5 per time point or position. Error bars represent SEM. CP, Cribriform plate.
, unlesioned counts per 200 μm OE. In I: ♦, 12 h PBX; ▪, 24 h PBX; ▴, 36 h PBX. For all graphs, n = 3-5 per time point or position. Error bars represent SEM. CP, Cribriform plate.
Cell death in the OE was assessed in blinded samples at sample area 2 (see Fig. 1 F) as the number of TUNEL-positive neurons per 200 μm OE. BrdU-positive cells were assessed in blinded samples at all three sample areas (see Fig. 1 F) over an extended distance (total, 6.5-8.5 mm/animal) and quantified as number per millimeter of OE. Cell counts and image intensities were tabulated and analyzed statistically for mean ± SEM, Student's t test, and one-way ANOVA using Microsoft (Seattle, WA) Excel.
Image capture
All images were visualized with an Axioplan 2 Imaging microscope (Zeiss, Jena, Germany) using a Retiga 1350EX camera (Quantitative Imaging Corporation, Burnaby, British Columbia, Canada) with Northern Eclipse software (Empix Imaging, Mississauga, ON) and were compiled using Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA). Confocal microscopy was performed using a Zeiss Axiovert S100 TV microscope fitted with Bio-Rad Radiance Plus confocal hardware and LaserSharp software running on a Dell Pentium II personal computer (Dell Computer Company, Round Rock, TX). Confocal Z-series were processed using NIH Image J software and imported into Adobe PhotoShop 6/7 (Adobe Systems) for colorization and determination of signal colocalization.
Results
Caspase-8 is activated at the onset of ORN retrograde apoptosis
Caspase-9 and caspase-3 are activated retrogradely in ORNs in response to bulbectomy (Cowan et al., 2001); however, what initiates and propagates this proapoptotic caspase signaling is not known. We assayed for expression of a number of caspase-3 activators and detected procaspase-8 in both the developing and adult OE (data not shown), suggesting that it could act as an initiator caspase and activator of caspase-3. We then generated caspase-8 neoepitope antibodies that specifically recognize the auto-cleaved form of caspase-8 as a tool to immunohistochemically track axonal caspase-8 activation after bulbectomy, the same lesion used to demonstrate retrograde axonal activation of caspase-3 and caspase-9. The caspase-8 neoepitope was detected as early as 12 h after bulbectomy in a few ORN axons and cell bodies situated close to the olfactory bulb (Fig. 1I). At 16 h after bulbectomy (before we previously detected caspase-3 activation), we detected activated caspase-8 in ORN axons and cell bodies closest to the bulb cavity (Fig. 1A) but significantly lower levels further away (Fig. 1B). By 24 h, identical areas of the OE demonstrated much more widespread activation of caspase-8 both close to and more distant from (Fig. 1C,D) the bulb. This pattern was also reflected in transverse OE at 24 h (Fig. 1E). To measure spatial retrograde activation of caspase-8 at different distances from the lesioned target, we assayed the number of caspase-8 neoepitope-positive cell bodies per millimeter on coronal sections along three individual turbinates (pinpointed on Fig. 1F), sampling at three 320 μm caudorostral intervals from the cribriform plate 24 h after lesion. To test for temporal activation, we also assayed a single turbinate (Fig. 1F, sample area 1) at the same middle (Fig. 1F, M) distance from the olfactory bulb cavity and at three different time points (12, 24, and 36 h) after bulbectomy (Fig. 1G-I). Activated caspase-8 detection peaked at 12-24 h after bulbectomy (Fig. 1I) and, by 24 h, had accumulated in all turbinates at sites closest to the olfactory bulb cavity (Fig. 1G), reflecting the pattern seen in transverse sections of OE (Fig. 1E). These data demonstrated a caudorostral sequence of caspase-8 activation but did not relate this to the activation of executioner caspases. We then examined successive adjacent sections to test the relationship between the activation of caspase-8 and activation of caspase-3 (both polyclonal neoepitope antibodies). The detection of caspase-8 neoepitope was greater than that of caspase-3 neoepitope at both 12 h (Fig. 2A,C) and 24 h (Fig. 2B,D) after bulbectomy in some of the same (and some adjacent) ORN populations (active C3/active C8 overlap) (Fig. 2E,F). ORN cell body processing of caspase-8, however, was not restricted to the OMP-positive mature ORN population (the major ORN population that undergoes apoptosis after bulbectomy) (Fig. 2G) but was also found in GAP43-positive immature ORNs (Fig. 2H). At 24 h after bulbectomy, OMP-positive neurons represented 49% of the detectable activated caspase-8 signal, whereas 40% of active caspase-8 was consistently detected in GAP 43-positive immature ORNs.
Figure 2.
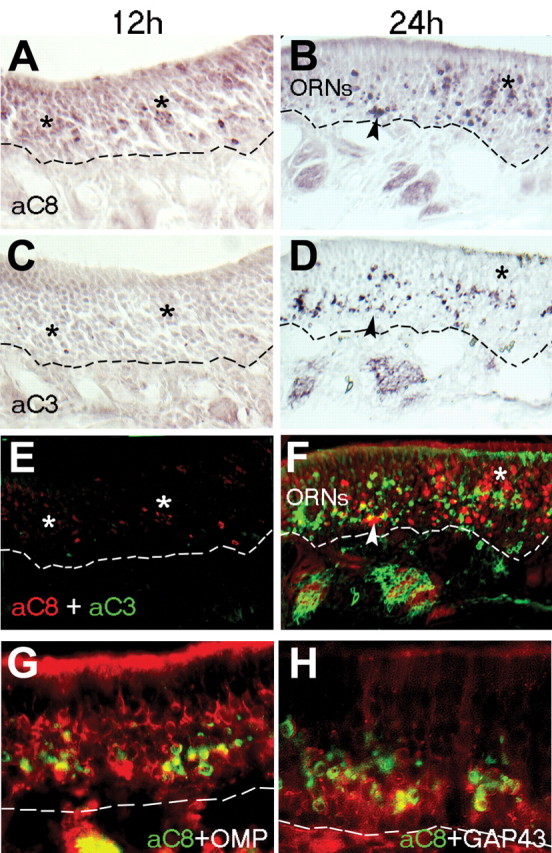
Caspase-8 activation peaks ahead of, and is more widespread than, active caspase-3 after bulbectomy. A, C, E, At 12 h postbulbectomy (12 h), active caspase-8 (A; aC8) is detected in a greater proportion of cells (asterisks) in the OE than active caspase-3 (aC3) on an adjacent section (C), a pattern emphasized when both images are overlaid (E; red, active caspase-8; green, active caspase-3). B, D, F, At 24 h postbulbectomy (24 h), some ORNs shared active caspase-8 (B; F, red) and caspase-3 (D; F, green) in common (arrowheads), but a greater proportion of ORNs in the more mature, apical layers of OE contain active caspase-8 alone (asterisks). G, H, In immunolabeled coronal sections of OE at 24 h postbulbectomy, active caspase-8 (green) is detected in both OMP-positive mature ORNs (G, red) and immature neurons expressing GAP43 (H, red). ORNs, Mature neuronal layers of olfactory epithelium. The dashed line indicates basal lamina.
ORN target neurons in the olfactory bulb die after excitotoxic lesion
A number of studies have highlighted the inter-relationship between afferent ORN stimulation, olfactory learning, and the regulation of apoptosis and neurogenesis in both the OE and the OB (Cummings et al., 1997; Rochefort et al., 2002; Carleton et al., 2003). Despite its usefulness in stimulating widespread ORN degeneration and subsequent regeneration, the olfactory bulbectomy is not just a target deprivation but also an axotomy and, therefore, has limited usefulness in examining the initiation of synaptic apoptosis. To investigate whether postsynaptic target neurons in the olfactory bulb may directly regulate ORN survival signaling at the glomerular synapse of mature ORNs, we took advantage of the fact that ORNs use an NMDA receptor-containing synapse. The ORN postsynaptic targets (mitral and tufted cells) contain combinations of both NMDA and metabotropic glutamate receptors (Ennis et al., 1998; Montague and Greer, 1999; Salin et al., 2001) and are therefore susceptible to undergo excitotoxic cell death at high levels of stimulation. We first used immunocytochemistry and reverse transcription-PCR for the NR1 subunit of the NMDA receptor to rule out expression of NMDA receptors on ORNs themselves (data not shown). When NMDA was infused unilaterally into the olfactory bulb, TUNEL detection of apoptotic nuclei indicated that, as early as 24 h after NMDA administration, many mitral and tufted cells in the lesioned olfactory bulb had been stimulated to undergo apoptosis (Fig. 3A,B). NeuN staining revealed that neuronal populations within the unlesioned olfactory bulb maintained their concentric layered structure, whereas neuronal layering was disrupted by NMDA infusion already at 24 h (Fig. 3E,F). By 4 d after lesion, TUNEL positivity appeared restricted primarily to the granule and periglomerular cell layers, and NeuN immunohistochemistry revealed that many different olfactory bulb neurons had been lost by this time (Fig. 3G,H). Holes were apparent where mitral cell bodies had died (Fig. 3D,H), and sampling every eighth section through the olfactory bulb revealed that 89-95% of mitral cells were lost by 4 d after NMDA lesion, with the lower percentages at the most rostral and caudal tips of the bulb. Twenty-four hours after NMDA treatment, there was no corresponding increase in TUNEL-positive ORNs in the OE projecting to the NMDA-lesioned bulb (Fig. 3I,J). However, at 4 d, there were TUNEL-positive cells in the more superficial and mature layers of the OE of septal and endoturbinates, resulting in a 22-38% increase in the number of TUNEL-positive ORNs in restricted focal areas of the OE compared with matched regions on contralateral, unlesioned OE (Fig. 3K,L). These data suggest that a subpopulation of ORNs undergoes delayed apoptosis after loss of their postsynaptic target in a similar time window (48-72 h after target death) to that reported after bulbectomy (Michel et al., 1997; Cowan et al., 2001), but that most mature ORNs were in fact mostly resistant to the loss of target-derived trophic support.
Figure 3.
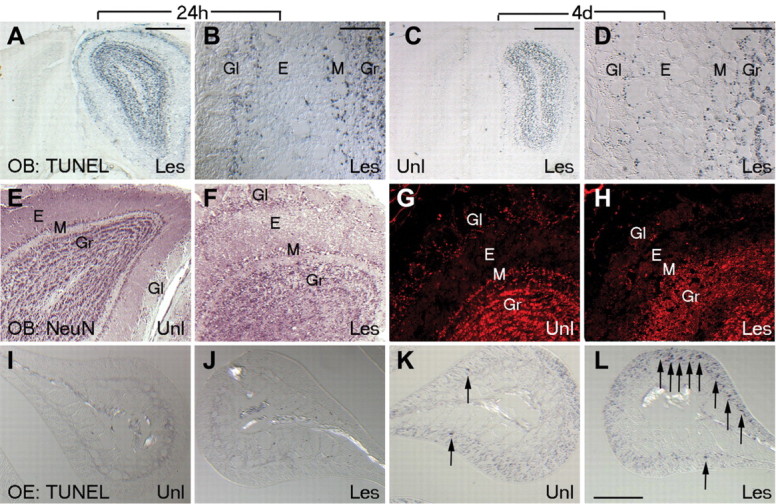
Olfactory bulb and epithelial neurons undergo apoptosis after NMDA lesion. A-D, Coronal sections of unilaterally NMDA-lesioned olfactory bulb show TUNEL-positive apoptotic cells restricted to the lesioned side only at 24 h (A, B) and 4 d (C, D) after NMDA infusion. TUNEL-positive neurons in several neuronal layers of the olfactory bulb (periglomerular, mitral, and granule) can be seen on the lesioned side but are relatively absent from the unlesioned side. E-H, Coronal sections of unilaterally NMDA-lesioned bulb at 24 h (E, F) and 4 d (G, H) immunolabeled for NeuN show disruption of neuronal layering in olfactory bulb at 24 h and disappearance of many bulb cells, notably in the mitral layer by 4 d. I-L, TUNEL labeling in the OE (shown on endoturbinate Iib; sample area 2 in Fig. 1 B) at 24 h and 4 d after NMDA lesion. Lesion-induced TUNEL-positive ORNs seen at 4 d (K, L) are not seen in the unlesioned side or in sham animals. E, External plexiform; Gl, glomerular; Gr, granule layers; Les, lesioned side; M, mitral/tufted; Unl, unlesioned side.
The stress-induced form of caspase-8 is upregulated and synaptically activated when ORN target neurons are lost
To assess caspase-8 proenzyme expression and activation during the initiation of ORN synaptic apoptosis, protein extracts from unlesioned and NMDA-treated mouse OE and OB were examined by immunoblotting using rabbit polyclonal antibodies (MF438) directed against the large catalytic subunit (p17) of human caspase-8 (Fig. 4A). During the first 24 h after NMDA lesion, increased expression of the long stress-induced proform of caspase-8 was observed on the lesioned side but not on the contralateral side (Fig. 4B, arrow), coupled with proteolytic cleavage of caspase-8 and corresponding activation of caspase-3 only in lesioned tissue (Fig. 4B, arrowheads). Active caspase-8 was first detected in the glomeruli (containing ORN termini) and NFL (containing ORN axons) of the NMDA-treated olfactory bulb but was absent from glomeruli of the unlesioned bulb (Fig. 4C,D). In adjacent sections of olfactory bulb, caspase-3 was activated in glomeruli and the NFL in a similar manner to caspase-8 (Fig. 4F,G). It is also important to note that other neuronal layers of the OB did not appear to activate caspase-3 and caspase-8 during NMDA-induced excitotoxic cell death (at least not at the levels detected by the neoepitope). However, we have detected apoptosis inducing factor nuclear translocation in subsets of periglomerular neurons after NMDA treatment, suggesting that alternative death pathways may be used in some OB interneurons targeted for replacement (data not shown). By 4 d after NMDA treatment, many ORN axons still remained in the NFL, and a persistent activation of both caspase-3 and caspase-8 was detected in the glomerular layer of only the NMDA-treated OB (Fig. 4E,H).
Figure 4.
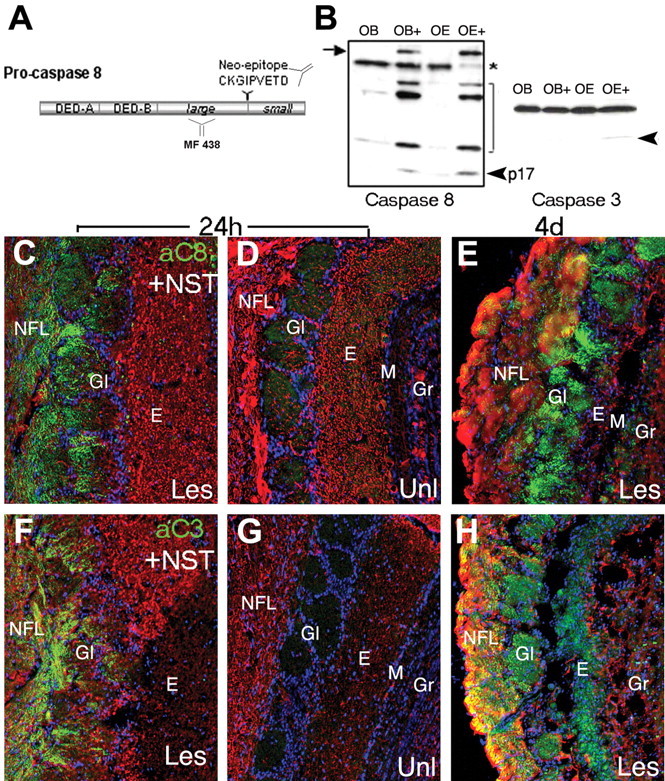
Caspase-8 proenzyme expression and activation after NMDA-induced excitotoxic death of ORN target neurons. A, A rabbit polyclonal antibody directed against the p17 large subunit of caspase-8 (MF 438) was used in Western blot analysis of protein extracts from unilaterally NMDA-lesioned olfactory bulb and epithelium. MF-E0022804K directed against caspase-8 neoepitope (CKGIPVETD) was used to detect in vivo caspase-8 cleavage by immunohistochemistry. B, On Western blots of microdissected tissue extracts prepared at 4 d after NMDA infusion, the stress-induced caspase-8 long proform (arrow) was induced only in samples from the NMDA-lesioned tissue (OE+, OB+ lanes), was absent in the unlesioned bulb and epithelium (OE-, OB- lanes), and was accompanied by the concomitant detection of caspase-8 (p17, arrowhead) and caspase-3 (arrowhead) cleavage products. C-H, Coronal sections of OB 24 h (C, D, F, G) and 4 d (E, H) after unilateral NMDA lesion showing caspase-8 activation (C-E, green) compared with axonal and dendritic NST (red) in the presynaptic glomerular compartment of the lesioned (C, E) but not unlesioned (D) OB. A similar pattern is seen for active caspase-3 (F-H, green), which is absent in the unlesioned OB (G). E, External plexiform; Gl, glomerular; Gr, granule layers; Les, lesioned side; M, mitral/tufted; Unl, unlesioned side.
Caspase-8 interacts with the p150Glued subunit of dynactin
Collectively, the preceding results suggested that caspase-8 activation may catalyze an initiating apoptotic event that occurs first at the ORN synapse and leads to the stimulation of a retrograde apoptotic program in the ORNs. In an effort to identify synaptic and axonal proteins that could interact with caspase-8 and potentially integrate the balance between synaptic remodeling and retrograde apoptotic signaling, we used a combination of bioinformatic approaches to identify neuronal proteins with structural similarity to the DED of caspase-8. For an initial bioinformatics approach, we searched for proteins with sequence and structural similarities to the pDED of the protein HIPPI, a known interactor with caspase-8 (Gervais et al., 2002). We then searched through candidates for proteins that may be implicated in axonal transport. The p150Glued subunit of Dynactin came back as a lead candidate containing two potential DED domains. The DED of p150Glued lies in its first coiled coil domain (amino acids 217-548) and is most highly similar to the pDED of HIPPI (Gervais et al., 2002) (Fig. 5A). Because DEDs represent homotypic interaction modules, and because HIPPI was shown previously to interact with caspase-8 through its pDED (Gervais et al., 2002), the presence of a pDED in CC1 prompted us to determine a direct biochemical interaction between caspase-8 and dynactin p150Glued.
Figure 5.

Interaction of caspase-8 and dynactin p150Glued is mediated by the dynactin CC1 domain. A, Dynactin p150Glued contains a pDED within its first coiled-coil domain (CC1), which could form the basis for an interaction with the DEDs of procaspase-8. The first and last amino acids of the pDED are indicated on the left and right of the sequence, whereas the region corresponding to the fifth helix (α5) of the pDED containing the critical lysine (K) residue that differentiates a pDED from a classical DED, which would have a hydrophobic residue at the equivalent position, is underlined. B, Transfected, mutated (Cys-Ala in catalytic domain) flag-tagged procaspase-8 (ProC8) (C-A) was immunoprecipitated and found to be complexed with endogenous dynactin p150Glued from extracts of HEK293T cells (lane 2). The flag-tagged procaspase-8 (C-A):dynactin p150Glued interaction was reduced (arrow) by co-overexpression of the dynactin CC1 domain but not the CC2 domain (lanes 3, 4; B, beads alone control). Western blots confirm expression of flagged caspase-8 in whole-cell extracts (WCE) of HEK293 cells. C, An anti-dynactin mAb immunoprecipitated cold (unlabeled) in vitro translated dynactin p150Glued with 35S-labeled in vitro translated procaspase-8 (proC8) (Mock, mock caspase-8 transfection; B, beads alone control). D, Flag-tagged procaspase-8 (ProC8) (C-A) will coimmunoprecipitate with the dynactin CC1 domain but not the CC2 domain in HEK293T cells. E, Binding of dynactin p150Glued (p150) or the CC1 domain to caspase-8 did not require the prodomain. Immunoprecipitation of flag-tagged caspase-8 lacking its prodomain (caspase-8 Δprodomain) coimmunoprecipitated both full-length dynactin p150Glued or CC1 but not CC2 in overexpressing HEK293T cell extracts.
The p150Glued subunit of dynactin is a required activator of neuronal retrograde transport, is expressed in multiple neurons, including ORNs (Melloni et al., 1995; Tokito et al., 1996; Waterman-Storer et al., 1997; Shah et al., 2000), and has been implicated in developmental axonal guidance and retrograde signaling (Dillman et al., 1996; McCabe et al., 2003). Genetic disruption of the retrograde motor complex dynein/dynactin resulted in motor neuron degeneration, presumably because of interruption of retrograde survival signals on which these cells depend (LaMonte et al., 2002; Puls et al., 2003). No direct mechanistic link has yet been made between dynactin and the cellular apoptotic machinery. To do this, we first performed coimmunoprecipitation experiments using whole-cell extracts from HEK293T cells transfected with a flag-tagged catalytically inactive caspase-8 (mutated C-A), or in vitro translated 35S-labeled procaspase-8 and “cold” in vitro translated dynactin p150Glued. Immunoprecipitation of flag-tagged caspase-8 (C-A) coimmunoprecipitated endogenous dynactin p150Glued from HEK293T extracts (Fig. 5B, left). Similarly, doing the reverse IP with in vitro translated proteins showed that immunoprecipitation of cold in vitro translated dynactin p150Glued coimmunoprecipitated in vitro translated 35S-labeled procaspase-8 (Fig. 5C). Overexpression of the CC1 but not the CC2 domain interfered with the caspase-8 - endogenous dynactin p150Glued interaction (Fig. 5B, right), suggesting that within dynactin p150Glued, CC1 contains the binding site for caspase-8. To further support this, we immunoprecipitated mutated flag-tagged caspase-8 (Cys-Asp mutation in active site) and showed, using specific antibodies directed against either CC1 or CC2, that caspase-8 interacted with CC1 but not CC2 when overexpressed independently (Fig. 5D). We next mapped the domains in caspase-8 that mediate its interaction with dynactin p150Glued and showed that the interaction did not require the prodomain but was mediated through caspase-8 catalytic domain (caspase-8 Δprodomain) (Fig. 5E). These data indicated that both full-length and cleaved caspase-8 could interact with dynactin p150Glued if they were localized in the same subcellular compartment. We next investigated whether this interaction could be an initiating event associated with the propagation of retrograde ORN apoptosis.
Caspase-8 becomes processed and progressively retrogradely associated with dynactin p150Glued during ORN retrograde apoptosis
To examine whether the interaction between caspase-8 and dynactin p150Glued may contribute to a mechanism-driving retrograde neuronal apoptosis in vivo, we tested the following: (1) whether caspase-8 (either full-length or activated) and the p150Glued dynactin subunit form a complex in ORNs, (2) whether their interaction is altered after NMDA lesion, and (3) whether this interaction may change in a retrograde pattern. At 24, 48, and 96 h after NMDA lesion, both lesioned and unlesioned OB and OE (Fig. 3) were dissected into four segments representing proximal and distal segments of the olfactory nerve in the manner indicated in Figure 6A. In the OE, proximal axons (OE2) were separated from more distal axons (OE1). Similarly, the OB was dissected into rostral and caudal segments (OB1, OB2) containing microdissected NFL and glomerular layers. This allowed us to examine potential biochemical interactions of p150Glued and caspase-8 in enriched synaptic extracts and axons at different levels of the neuraxis at two key times during the initiation and progression of the retrograde apoptotic signal (24-48 h). We prepared microdissected extracts, pooled from four to six animals per group, to generate sufficient tissue from each segment for analysis. Samples were coded, and the immunoprecipitations were performed blind on two experimental sets of mice. Extracts were immunoprecipitated with anti-dynactin p150Glued antibodies, and the coimmunoprecipitation of caspase-8 from lesioned and unlesioned sides of the same mice was examined by Western blot analysis using polyclonal antibodies (MF438) directed against caspase-8 large subunit (p17) (Fig. 6B). p150Glued did coimmunoprecipitate with full-length caspase-8 after lesion (on both lesioned an unlesioned sides), but only in lesioned tissue did it form a complex with the activated p17 catalytic subunit (Fig. 6 B). In addition, immunoprecipitation with MF438 (which would recognize both activated and full-length caspase-8) demonstrated that caspase-8 was present in both the OE and OB at this same time point in a complex containing both p150Glued and the intermediate chain of dynein (Dynein IC) (Fig. 6C).
Figure 6.

Interaction of caspase-8 with dynactin p150Glued and the dynein intermediate chain after NMDA lesion. A, NMDA was administered unilaterally into the olfactory bulb, and dissection of the olfactory neuraxis into four regions spanning the caudal end of the olfactory bulb to the rostral end of the OE was performed. B, Dynactin p150Glued was immunoprecipitated from OB and OE protein extracts containing pooled, microdissected regions of the NMDA-lesioned and unlesioned olfactory neuraxis. The interaction with caspase-8 (α-casp8) was analyzed by Western blots using rabbit polyclonal antibodies recognizing the p17 subunit (MF438). Dynactin p150Glued (α-p150Glued) coimmunoprecipitated with full-length caspase-8 (on both lesioned and unlesioned sides), but it was seen only on the lesion side to complex with the p17 large catalytic subunit of cleaved caspase-8 (arrow). prG, Protein-G beads only. C, Dynactin p150Glued and dynein intermediate chain (IC) were readily detected in total cell lysates of the olfactory epithelium (TOE) or olfactory bulb (TOB). When caspase-8 (α-casp8) was immunoprecipitated from these lysates, it was found to be complexed with both dynactin p150Glued and dynein IC (top two panels, OE and OB lanes). Similarly, immunoprecipitates of dynein IC (bottom panel, OE and OB lanes) coimmunoprecipitated dynactin p150Glued. prG, Protein-G beads only. D, Dynactin p150Glued immunoprecipitates were probed by Western blot analysis for the p17 subunit, and active caspase-8 associated with dynactin p150Glued was quantified in the four sample areas specified in A by densitometric scanning (n = 4-5). The complex of active caspase-8 with dynactin p150Glued increased dramatically (compared with contralateral, unlesioned side) at 24 h after NMDA treatment, and this complex shifted from the bulb to the periphery between 24 and 48 h after NMDA treatment. co-IPed, Coimmunoprecipitated. Concomitantly, at 24 h (E), active caspase-8 immunolabeling could be detected in ORN axons in both the NFL of the bulb and the OE turbinates in the periphery. E, External plexiform; Gl, glomerular.
Densitometric analysis of Western blots was used to quantify the relative proportion of dynactin-associated activated caspase-8 p17 subunit that could be coimmunoprecipitated (compared with a standard IP product of dynactin p150Glued) from pooled, microdissected samples of different parts of the olfactory neuraxis (collected as indicated in Fig. 6A). This analysis demonstrated that, as early as 24 h after NMDA, the interaction between activated caspase-8 and dynactin was significantly enhanced fourfold to fivefold in the OB (over unlesioned OB, pooled samples from the same mice) (Fig. 6D), with little difference between lesioned and unlesioned OE samples. By 48 h, however, activated caspase complexed with dynactin was only two-fold higher in the OB (compared with unlesioned), and the majority of the dynactin p150Glued-complexed activated caspase-8 p17 subunit was now distributed in a retrograde pattern throughout the olfactory nerve (Fig. 6D). A similar retrograde pattern was observed in coronal sections of the olfactory system at 24 h after NMDA treatment (Fig. 6E), in which active caspase-8 was detected in both ORN termini in the OB and in proximal axon bundles extending only partially into the OE turbinates.
Focal inhibition of retrograde transport and caspase activation in ORN axon termini inhibits retrograde caspase-8 activation and ORN apoptosis
The preceding results suggested a role for caspase-8 activation and the p150Glued subunit of dynactin in the retrograde apoptosis of mature ORNs after loss of their target population. To test whether disrupting retrograde transport could inhibit this retrograde apoptotic signaling, we first performed unilateral bulbectomy to generate a maximal retrograde apoptotic response in a tightly defined time window (Cowan et al., 2001) and then combined this with focal inhibition of either microtubule transport or localized caspase activation at axon termini. After bulbectomy, we immediately introduced the microtubule stabilizing agent, Taxol, and a retrograde tracer, Fast Blue (Choi et al., 2002), (in gelfoam) into the cavity created after removal of the olfactory bulb, where it could directly impact microtubule-dynein-dynactin interactions at axotomized ORN axon terminals. In parallel experiments, we injected a specific small-molecule inhibitor (L-826920; Merck Frosst) to block upstream caspase signaling at the axotomized termini. Twenty-four hours later, normally the peak of active caspase-8 detection (Fig. 1I), mice were killed and examined for axonal and ORN cell body activation of caspase-8. In control animals (+Bulbectomy, no Taxol), retrograde tracer could be detected in axons and cell bodies all the way back to the ORNs of the olfactory epithelium (Fig. 7A,B). In some regions of the OE, tracer extended beyond the ORN population and may reflect the labeling of cells to which ORNs are junctionally coupled. In contrast, in Taxol-treated animals, tracer was excluded primarily from the ORN cell bodies (Fig. 7C,D), with residual tracer detected in some axons (Fig. 7D). In bulbectomized mice that did not receive Taxol, axonal and ORN soma caspase-8 neoepitope was readily detectable (Fig. 7F) but was significantly less in mice that had received Taxol (Fig. 7G), where ORN cell bodies were not positive for Fast Blue (Fig. 7C,D). Activation of caspase-8 was demonstrated by two independent methods, by the presence of neoepitope, and also by an in situ reaction, in which caspase-8 was first immunocaptured from OE extracts from different treatment groups and IP products were then analyzed for their ability to catalyze the cleavage of caspase-3. When either Taxol or the small molecular caspase inhibitor L-826920 was added to the ORN axon termini, a 6- to 10-fold reduction in the lesion-specific ORN active caspase-8 neoepitope immunoreactivity (ratio, lesioned:unlesioned) was found 24 h after bulbectomy (Fig. 7K). Similarly, a threefold to fourfold reduction in activated, immunocaptured caspase-8 in OE extracts was obtained in the caspase-3 cleavage assay, in which Taxol-treated extracts were compared with bulbectomy alone (data not shown). Both L-826920 and Taxol-treated animals demonstrated similar twofold to threefold decreases in TUNEL-positive cells in the OE (lesioned:unlesioned) (Fig. 7L). Because active caspase-8 was found with comparable frequency in GAP43-positive ORNs in both Taxol-treated and unlesioned animals (Fig. 7H-J), this decrease in TUNEL positivity is accounted for predominantly by the OMP-positive mature ORN population. These results indicate that active caspase-8 at the lesion site needs to engage with dynactin and axon-based retrograde motors for the maximal propagation of the retrograde proapoptotic program in ORNs.
Figure 7.
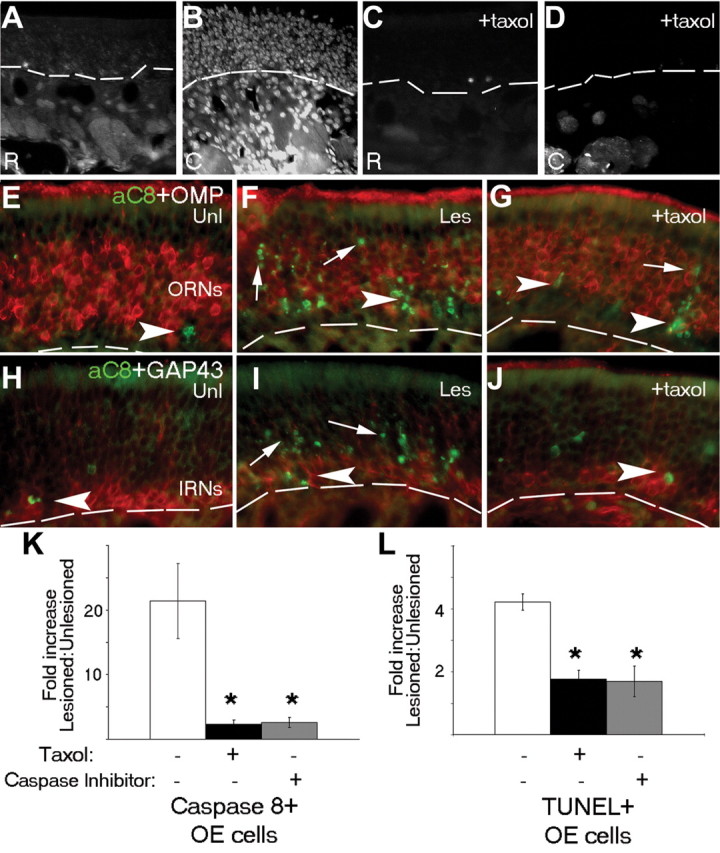
Disruption of microtubule transport inhibits retrograde apoptosis induced by bulbectomy. A-J, Coronal sections of the olfactory epithelium at 24 h after unilateral bulbectomy, in which the retrograde tracer Fast Blue was added to the gelfoam at the lesion site in the OB, show accumulation of the tracer in axon bundles in the distal OE (A) and ORN nuclei at points closer to the bulb (B). When the microtubule-stabilizing agent Taxol was added along with the tracer at the time of the bulbectomy, nuclear accumulation of Fast Blue was significantly reduced in ORN cell bodies (C, D). Activation of caspase-8 (E-J, green) was readily detected in both apical, more mature neuronal layers of the OE (arrows) expressing OMP (E-G, red) and immature neuronal layers (arrows) expressing GAP43 (H-J, red), specifically on the lesioned side of bulbectomized animals (E, F, H, I). In Taxol-treated animals, residual caspase-8 activation was also noted in the immature neuronal compartment (G, J, arrowheads). The ratio of caspase-8 neoepitope-positive (K) or TUNEL-positive (L) cells/200 μm OE, compared with unlesioned contralateral OE (on endoturbinate IIb, sample area 2 in Fig. 1B), was significantly reduced after bulbectomy in mice that received either Taxol (black; to focally inhibit microtubule transport) or a small molecular inhibitor of initiator caspases (gray; L-826920) (to focally inhibit caspase activity) compared with no additional treatment with bulbectomy (white) at the 24 h time point. In K and L, error bars represent SEM (*p < 0.003). The dashed line indicates basal lamina. aC8, Active caspase-8; C, caudal; Les, lesioned side; R, rostral; Unl, unlesioned side.
The low-affinity nerve growth factor receptor, p75, contributes to the initiation of synaptic apoptotic signaling in ORNs
The previous data suggest that there is an initiating signal at the presynaptic terminal of ORNs that can stimulate apoptosis at the level of the synapse. That only a subpopulation of ORNs die after NMDA lesion also indicates either that this signal is not evenly distributed across ORNs and their glomeruli or that there are a number of pathways balanced to prevent this apoptosis signal in different ORN subpopulations. The low-affinity nerve growth factor receptor, p75, has recently emerged as a prodeath-stimulating pathway that could interact with multiple proapoptotic pathways (Barker, 2004; Kaplan and Miller, 2004). It also has a unique and striking distribution in rat ORNs. Only after glomeruli have formed mature synapses does p75 become discretely localized to this compartment, where it colocalizes with the olfactory marker protein (Roskams et al., 1996). This suggests that synaptic p75 serves a function that is predominantly restricted to subpopulations of mature ORNs and could be involved in balancing prosurvival/apoptotic signals at the mature synapse. We first confirmed that p75 is also restricted to the same glomerular synaptic compartment in C57BL/6 wild-type mice by showing its coincidence with synaptophysin (Fig. 8A) and exclusion from either axonal (ORN) or dendritic (mitral cell) β-III NST (Fig. 8B). It was also clear that p75 was not evenly distributed, because some synaptophysin-positive glomeruli exhibited strong p75 expression, whereas in others, it was barely detectable (Fig. 8A). Despite their apparent formation of synaptophysin-positive functioning glomeruli, we confirmed that p75 is entirely absent in glomeruli of p75 null mice (exon III mutants) (Fig. 8C) (Lee et al., 1992). To test whether p75 may be involved in initiating synaptic apoptosis in ORNs, we first performed NMDA lesions on a set of p75 null mice and, 24 h later, found TUNEL-positive neurons in the olfactory bulb similar to that seen in wild-type mice (Fig. 3). Unlike wild-type mice, however (Fig. 4), activated caspase-8 was barely detectable in the synaptic terminals and terminal axonal compartments of ORNs at 24 h after lesion. Because the detectable signal in p75 null mice was minimal, we sampled large areas of glomeruli (from lesioned and unlesioned olfactory bulb) within individual mice to compare caspase-8 activation 24 h after NMDA administration and found that there was a ∼90% reduction in caspase-8 activation in the glomerular and nerve fiber layers after NMDA lesion in the absence of glomerular p75 (Fig. 8D). Although this suggests a role for p75 in initiating synaptic apoptosis in this model, it does not tell us whether this is part of “normal” ORN turnover. We first established that, using immunofluorescence, the p75 null mice contained similar baseline percentages of GAP43-positive/OMP-positive olfactory neurons (data not shown). The baseline number of TUNEL-positive ORNs in the OE is extremely low in a normal, young adult mouse and contains a significant number of immature ORNs and basal cells (Cowan et al., 2001, 2004). This low number and high background (∼50% of baseline apoptotic cells in the OE are not yet mature ORNs) made it hard to accurately measure shifts in TUNEL-positive cells in only mature ORNs (our population of interest) in the steady state. As an alternative (and a measure of longevity), to test whether ORNs survive longer in the absence of p75, we first labeled adult, unlesioned mice with BrdU and found that there are equivalent numbers of mitotic, BrdU-labeled olfactory progenitors in p75 null mice and wild-type mice 24 h after injection (Fig. 8E,F,I). When these labeled mice were allowed to survive for 14 d, progeny of the original BrdU-labeled cells were found throughout the progenitor and differentiated immature and mature neuronal layers of the OE of both null and wild-type mice (Fig. 8G,H). When we counted the differentiated (nonbasal) cells that retained BrdU since labeling, however, we found that, even within as short a time period as 2 weeks, p75 null mice retained twice the number of differentiated neurons in the superficial layers of the OE, suggesting that in the absence of p75, newly generated ORNs exhibit greater longevity and enhanced survival (Fig. 8I). These results suggest a role for p75 in the induction of apoptosis in ORNs both after lesion and during normal turnover and that, in the absence of p75, ORNs survive longer.
Figure 8.
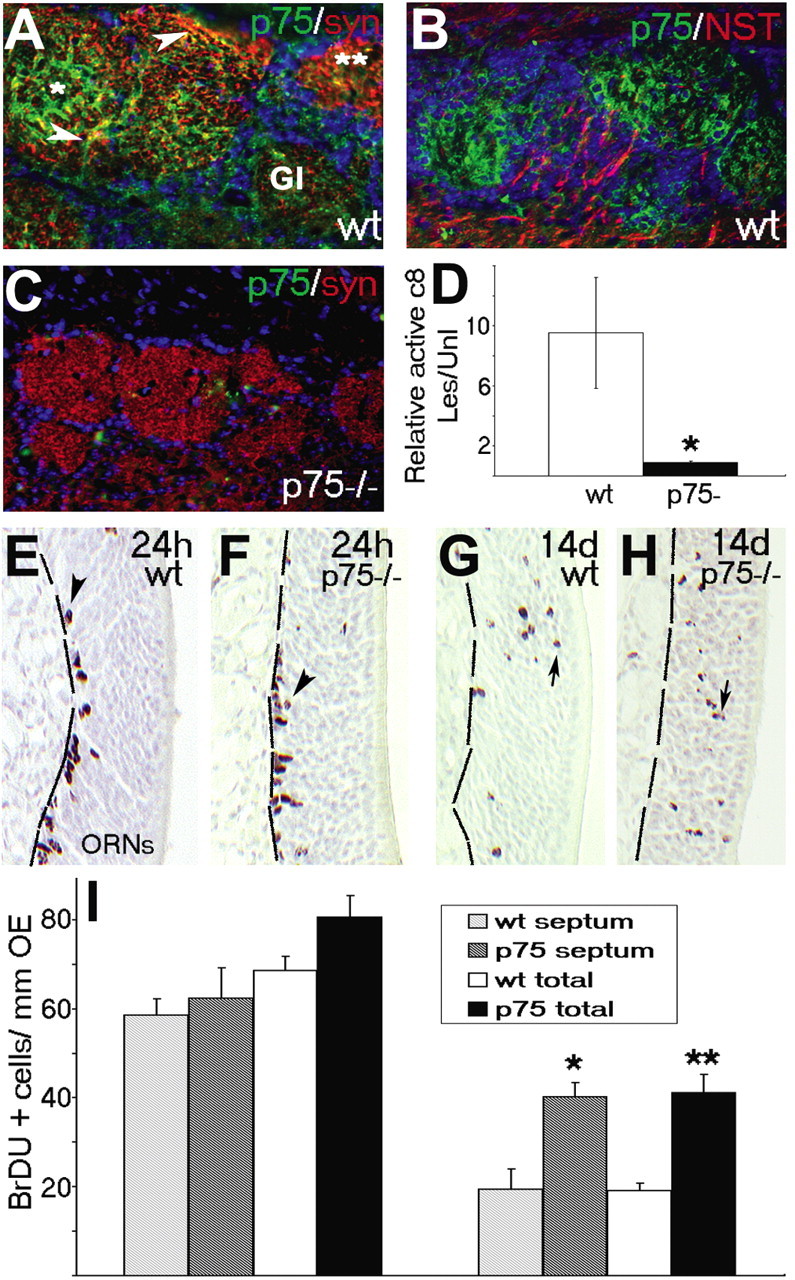
Lesion-induced activation of caspase-8 and ORN turnover are significantly reduced in p75 (lNGFR) null mice. Using immunofluorescence, p75 low-affinity nerve growth factor receptor (green) marks ORN axon termini in the glomeruli of the olfactory bulb of wt mice (A, B) but is not detected in p75 null mice (C). A, p75 immunoreactivity partially overlapped (green) with synaptophysin (red) and was clearly expressed in glomeruli that were distinct from ORN or mitral axon tracts labeled with βIII NST (B; red). Twenty-four hours after NMDA, lesion-specific active caspase-8 could be detected in the glomeruli of wt mice (signal normalized for area) but not p75 null mice (D). E, F, BrdU was readily detected 24 h after IP injection in basal progenitors in the OE of both wild-type (E) and p75 null (F) mice. G, H, Long-term retention of the signal (indicative of ORN longevity and turnover) assessed 14 d after injection was detected in cells throughout progenitor, immature, and mature neuronal layers of the OE in both wild-type (G) and p75-/- (H) animals. I, Quantitation of the number of BrdU-labeled cells in the OE within the same sample areas delineated in Figure 1 demonstrated no difference either in the septum (area 3; patterned bars) or in the total counts (solid bars) at 24 h, but retention of BrdU-labeled ORNs was increased in p75 knock-out mice by 14 d after labeling. *p < 0.05; **p < 0.01; Student's paired t test. Gl, Glomerular; Les, lesioned side; syn, synaptophysin; Unl, unlesioned side.
Discussion
ORNs undergo apoptosis after bulbectomy by upregulating procaspase-3 and procaspase-9 and then inducing their activation retrogradely from ORN axons to soma (Cowan et al., 2001). How caspase-3 can be activated synaptically to produce injury-induced retrograde ORN apoptosis (Cowan et al., 2001), yet developmentally stimulate localized synaptic remodeling without concurrent apoptosis (Mattson and Duan, 1999; Yan et al., 2001), is not understood. Here, we show that caspase-8, once activated, can undergo retrograde transport and stimulate ORN apoptosis by complexing directly with the DED domain of Dynactin p150Glued. Despite widespread activation of synaptic caspase-8 after NMDA-induced loss of target neurons, however, only a vulnerable subset of mature ORNs subsequently undergo apoptosis. p75, which is highly localized to olfactory bulb glomeruli, is implicated in the initiation of apoptosis at the ORN synapse, resulting in synaptic caspase-8 activation, followed by retrograde activation of caspase-3 (and caspase-9), as the p17-activated subunit of caspase-8 is transported from presynaptic terminals back down axons. Localized inhibition of initiator caspase activity or microtubule-dependent retrograde transport at the lesioned afferent terminal can inhibit both the retrograde activation of caspase-8 at the ORN soma and subsequent caspase-3-mediated ORN apoptosis. Collectively, these data have led us to propose a model (Fig. 9) whereby ORNs at different developmental and functional stages differentially interpret bulb-derived signals to maintain a balance between apoptosis and survival.
Figure 9.

Opposing prosurvival and proapoptosis pathways in ORNs as they shift in a developmental state. A, Summary indicating the representative frequencies of OE-based ORN populations and the frequency with which ORNs of different stages undergo apoptosis in the OE. Newly generated and molecularly senescent ORNs are the most vulnerable, and immature and mature ORNs stabilized by activity are the most resistant. Markers of populations used in this study (GAP43, OMP) are also indicated. B, Pathways of apoptotic vulnerability at the synapse and how they may inhibit, or lead to, caspase-8 translocation to dynactin. Prosurvival signals are in gray, and proapoptotic signals are in black. Dyn p150, p150Glued, Dynactin subunit; ECM, extracellular matrix; JNK, Jun kinase; M/T, mitral/tufted cells; nNOS, neuronal nitric oxide synthase; NO*, nitric oxide; p75, low-affinity nerve growth factor receptor, p75; PG, periglomerular cells; TrkB, kinase active; TrkB-, truncated TrkB.
Using an antibody recognizing a neoepitope revealed after activation of caspase-8, we demonstrated retrograde activation of axonal and then ORN soma caspase-8 activation after bulbectomy (Fig. 1). The caspase-8 neoepitope was also consistently detected in a subpopulation of immature (GAP43-positive) ORNs in lesioned and unlesioned OE (Fig. 2). This suggests that OE-residing death receptor stimuli (potentially FasL or tumor necrosis factor α) (Farbman et al., 1999; Suzuki and Farbman, 2000) could stimulate caspase-8 activation and developmental apoptosis in immature ORNs using alternative receptor-based pathways from those present at the mature ORN presynaptic terminal.
Infusing NMDA into the olfactory bulb allowed us to examine, first, how target neurons in the bulb respond to excess NMDA (mimicking overstimulation by ORNs) and, second, which apoptosis-stimulating events may occur in ORN terminals after their main synaptic targets are destroyed. Multiple OB neurons died after NMDA infusion (not just those with NMDA receptors), but the OB remained relatively intact macroscopically, including the persistence of many ORN axon terminals (Fig. 3). This suggests that olfactory bulb neuronal death and remodeling can occur with excessive stimulation of NMDA receptors, in addition to too little stimulation (Fiske and Brunjes, 2001a,b). Like procaspase-3 and procaspase-9, the stress-induced form of caspase-8 was specifically upregulated in the OE during the initiation of apoptosis induced by OB NMDA (Fig. 4). Activated caspase-8 was detected in ORN presynaptic terminals immediately after NMDA and later in some ORN axons. However, only small subpopulations of TUNEL-positive mature ORNs (20-30% in focal locations within the OE) were detected, peaking at 4 d after NMDA lesion.
Despite mounting evidence linking dysfunction of dynein-regulated cellular transport mechanisms with neurodegeneration (Hayward, 2003), this is the first evidence directly linking synaptic activation of initiator caspases with the molecular motors that power retrograde transport and signaling. By mapping the domains of caspase-8 that preferentially bind dynactin p150Glued (Fig. 5), we have shown that both full-length and activated caspase-8 can complex with dynactin and the intermediate chain of dynein in ORNs, the postsynaptic targets of which have been eradicated, but that activated caspase-8 dynactin is the preferential cargo at the leading edge of a wave of retrograde apoptotic signaling (Fig. 6) (Cowan et al., 2001). By then inhibiting either microtubule transport or caspase-8 cleavage at the axon terminal, we were able to stall retrograde apoptosis in its tracks and reduce both axonal caspase-8 activation (and subsequent caspase-3 activation) and inhibit ORN apoptosis (Fig. 7). The motor complex dynein requires dynactin for optimal retrograde axonal transport, including retrograde survival signaling (Heerssen et al., 2004), and directly opposes kinesin-driven anterograde transport, which is essential for axonal outgrowth (Waterman-Storer et al., 1997; Shah et al., 2000) (Martin et al., 1999). Intriguingly, mutations of both dynactin p150Glued (Puls et al., 2003) and its neuron-specific interactors (Liu et al., 2003) have already been implicated in progressive sensory and motor neurodegeneration disorders in mice, effects caused by interrupted retrograde survival signaling. Our results raise the possibility that the late onset and relatively mild phenotype described for some of these mutants could result from simultaneous interruption of retrograde proapoptotic signals.
Here, p75, the low-affinity nerve growth factor receptor, emerged as a candidate death receptor restricted only to mature glomeruli, yet varying in intensity from one glomerulus to the next (Figs. 8, 9) (Roskams et al., 1996). At the synapse, p75 could differentially respond to either processed or full-length pro-BDNF and/or pro-NGF, which could be synthesized in a variety of bulb target neurons, including periglomerular neurons, which persist after NMDA lesion (Guthrie and Gall, 1991; Carter and Roskams, 2002; Barker, 2004). p75 has already been implicated in glomerular formation or persistence by the appearance of ectopic, disorganized glomeruli in the p75 null mouse (Tisay et al., 2000). Here, we have shown that the absence of p75 reduces ORN synaptic activation of caspase-8 after NMDA lesion and significantly increases the longevity of differentiated ORNs (Fig. 8). Although this strongly suggests a role for p75 in both lesion-induced and normal ORN apoptosis, it is not likely to be the sole determining factor in the synaptic balance between ORN life and death. For example, mature ORNs (which downregulate neuronal NOS after synaptogenesis) could also generate reactive oxygen species in response to nitric oxide released from periglomerular neurons (Fig. 9). The summation of these independent but related proapoptotic events could collectively result in the translocation of activated caspase-8 onto dynactin and the induction of retrograde apoptosis.
Given the widespread loss of bulb neurons, synaptic activation of caspase-8 after NMDA, and a dogma suggesting that ORN survival is dependent on trophic support from the bulb, it was remarkable that several days after lesion, the majority of ORN axons persisted. Collectively, these data lead to a model whereby the glomerular synapse is developmentally dynamic and contains multiple, opposing proapoptotic and prosurvival forces, delicately balancing state-dependent changes in ORN survival signaling. Extracellular stimuli from olfactory bulb neurons in single glomeruli could use a single effector to stimulate highly localized apoptosis in some ORN presynaptic terminals while maintaining survival of others, depending on how well matched their input is with adjacent neurons and glomeruli. Aberrantly firing ORNs could thus be distinguished from more stable neighbors and selected for apoptosis, thereby initiating their own replacement and stimulating turnover first in ORNs and then in the OB interneurons that will adapt to new ORN input (summarized in Fig. 9).
So, how does this model help to distinguish the ORNs that die from those that persist? The majority of persistent axons are GAP43/NST positive, with a concomitant reduction in OMP-positive ORNs, suggesting differential vulnerability to caspase-8 dynactin-mediated retrograde apoptosis as ORNs age (Fig. 9). First, immature ORN terminals could resist induction of retrograde apoptosis, because full-length presynaptic tyrosine receptor kinase B (TrkB) can bind target-derived BDNF and signal retrograde survival, endogenous neuronal nitric oxide synthase can S-nitrosylate (and inhibit) caspase-9 (Roskams et al., 1994, 1996), and postsynaptic extracellular matrix can indirectly inhibit local caspase-3 activation. This, coupled with the sporadic activity essential for synaptogenesis, could maintain immature axons even at a “compromised” target (Yu et al., 2004). Second, OMP-positive ORNs with functional, reinforced synapses are less dependent on target-derived trophic support, can use truncated TrkB to protect from p75 stimulation, and exhibit a greater dependence on afferent odor stimulation (Watt et al., 2004) and support from olfactory ensheathing cells (OECs) of the nerve fiber layer (Woodhall et al., 2001; Lipson et al., 2003). In the absence of proapoptotic signals, persistent OMP-positive axons represent the most stable ORNs able to combine activity and OEC-derived signals to remain and form new targets, similar to many central neurons after injury (Snider et al., 2002). Third, the final subpopulation of ORNs likely represents a malfunctioning (“molecularly senescent”) ORN population, with a higher potential to amplify localized synaptic caspase activation and initiate retrograde apoptosis. This compromised population is revealed in the excitotoxic lesion and likely represents a population primed for turnover. In these vulnerable mature ORNs, if the strong survival drive produced by evoked activity is not matched to stimulation of adjacent axons (Yu et al., 2004), synaptic contacts could become unstable, removing the final barrier to local inhibition of apoptosis.
Our data have now created a link between an activated protease capable of stimulating apoptosis, with a key regulatory protein for retrograde axonal transport. In both of the lesions used here, it appears that, once localized, activated caspase becomes coupled to axonal microtubular transport machinery, a summation of mitochondrial and nonmitochondrial caspase signaling initiates degeneration of both the axon and the cell body. These mechanisms may also be relevant in other neuronal populations, in which synaptic caspase activation does occur developmentally but does not usually result in widespread apoptosis (Mattson and Duan, 1999) in lesion-induced neuronal apoptosis or in understanding degenerative vulnerability after transient axonal damage in established neurodegenerative disorders with a significant olfactory component.
Footnotes
This work was supported by the National Institute on Deafness and Other Communication Disorders (5 RO1 DC04579-04), by a start-up grant from Merck Frosst of Canada (A.J.R.), by a Natural Sciences and Engineering Research Council of Canada Industrial Collaborative Fellowship (M.S.), and by a fellowship from the National Centres of Excellence Stem Cell Network (C.C.). We thank Erin Currie, David Sharon, and Teresa Wang for technical assistance, members of the Roskams laboratory for ideas and discussion, and Lynn Raymond and Wolfram Tetzlaff for comments on this manuscript. We also thank Kate Guthrie for her generous input and insights regarding NMDA lesion in rats.
Correspondence should be addressed to Dr. A. J. Roskams, Department of Zoology, University of British Columbia, 6270 University Boulevard, Vancouver, British Columbia, Canada V6T 1Z4. E-mail: roskams@zoology.ubc.ca.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256092-13$15.00/0
C.C. and M.S. contributed equally to this work.
References
- Barker PA (2004) p75NTR is positively promiscuous: novel partners and new insights. Neuron 42: 529-533. [DOI] [PubMed] [Google Scholar]
- Budihardjo I, Oliver H, Lutter M, Luo X, Wang X (1999) Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol 15: 269-290. [DOI] [PubMed] [Google Scholar]
- Burek MJ, Oppenheim RW (1999) Cellular interactions that regulate programmed cell death in the developing vertebrate nervous system. In: Cell death and diseases of the nervous system (Ratan VKAR, ed), pp 145-179. Ottawa: Humana.
- Carleton A, Petreanu LT, Lansford R, Alvarez-Buylla A, Lledo PM (2003) Becoming a new neuron in the adult olfactory bulb. Nat Neurosci 6: 507-518. [DOI] [PubMed] [Google Scholar]
- Carter LA, Roskams AJ (2002) Neurotrophins and their receptors in the primary olfactory neuraxis. Microsc Res Tech 58: 189-196. [DOI] [PubMed] [Google Scholar]
- Choi D, Li D, Raisman G (2002) Fluorescent retrograde neuronal tracers that label the rat facial nucleus: a comparison of Fast Blue, Fluoro-ruby, Fluoro-emerald, Fluoro-Gold and DiI. J Neurosci Methods 117: 167-172. [DOI] [PubMed] [Google Scholar]
- Choi DW (1996) Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol 6: 667-672. [DOI] [PubMed] [Google Scholar]
- Clark RS, Kochanek PM, Chen M, Watkins SC, Marion DW, Chen J, Hamilton RL, Loeffert JE, Graham SH (1999) Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human. FASEB J 13: 813-821. [DOI] [PubMed] [Google Scholar]
- Conley DB, Robinson AM, Shinners MJ, Kern RC (2003) Age-related olfactory dysfunction: cellular and molecular characterization in the rat. Am J Rhinol 17: 169-175. [PubMed] [Google Scholar]
- Cowan CM, Roskams AJ (2004) Caspase-3 and caspase-9 mediate developmental apoptosis in the mouse olfactory system. J Comp Neurol 474: 136-148. [DOI] [PubMed] [Google Scholar]
- Cowan CM, Thai J, Krajewski S, Reed JC, Nicholson DW, Kaufmann SH, Roskams AJ (2001) Caspases 3 and 9 send a pro-apoptotic signal from synapse to cell body in olfactory receptor neurons. J Neurosci 21: 7099-7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings DM, Henning HE, Brunjes PC (1997) Olfactory bulb recovery after early sensory deprivation. J Neurosci 17: 7433-7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillman III JF, Dabney LP, Karki S, Paschal BM, Holzbaur EL, Pfister KK (1996) Functional analysis of dynactin and cytoplasmic dynein in slow axonal transport. J Neurosci 16: 6742-6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw WC, Martins LM, Kaufmann SH (1999) Mammalian caspases: structure, activation, substrates and function during apoptosis. Annu Rev Biochem 68: 383-424. [DOI] [PubMed] [Google Scholar]
- Eaton BA, Fetter RD, Davis GW (2002) Dynactin is necessary for synapse stabilization. Neuron 34: 729-741. [DOI] [PubMed] [Google Scholar]
- Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gomez-Isla T, Hyman BT, Moskowitz MA (1998) Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J Cereb Blood Flow Metab 18: 238-247. [DOI] [PubMed] [Google Scholar]
- Ennis M, Linster C, Aroniadou-Anderjaska V, Ciombor K, Shipley MT (1998) Glutamate and synaptic plasticity at mammalian primary olfactory synapses. Ann NY Acad Sci 855: 457-466. [DOI] [PubMed] [Google Scholar]
- Farbman AI, Buchholz JA, Suzuki Y, Coines A, Speert D (1999) A molecular basis of cell death in olfactory epithelium. J Comp Neurol 414: 306-314. [DOI] [PubMed] [Google Scholar]
- Fiske BK, Brunjes PC (2001a) Cell death in the developing and sensory-deprived rat olfactory bulb. J Comp Neurol 431: 311-319. [PubMed] [Google Scholar]
- Fiske BK, Brunjes PC (2001b) NMDA receptor regulation of cell death in the rat olfactory bulb. J Neurobiol 47: 223-232. [DOI] [PubMed] [Google Scholar]
- Fumarola C, Guidotti GG (2004) Stress-induced apoptosis: toward a symmetry with receptor-mediated cell death. Apoptosis 9: 77-82. [DOI] [PubMed] [Google Scholar]
- Gaglio T, Saredi A, Bingham JB, Hasbani MJ, Gill SR, Schroer TA, Compton DA (1996) Opposing motor activities are required for the organization of the mammalian mitotic spindle pole. J Cell Biol 135: 399-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervais FG, Singaraja R, Xanthoudakis S, Gutekunst CA, Leavitt BR, Metzler M, Hackam AS, Tam J, Vaillancourt JP, Houtzager V, Rasper DM, Roy S, Hayden MR, Nicholson DW (2002) Recruitment and activation of caspase-8 by the Huntingtin-interacting protein Hip-1 and a novel partner Hippi. Nat Cell Biol 4: 95-105. [DOI] [PubMed] [Google Scholar]
- Guthrie KM, Gall CM (1991) Differential expression of mRNAs for the NGF family of neurotrophic factors in the adult rat central olfactory system. J Comp Neurol 313: 95-102. [DOI] [PubMed] [Google Scholar]
- Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW (1998) Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 94: 339-352. [DOI] [PubMed] [Google Scholar]
- Hayward P (2003) Dysfunctional cellular transport causes neurodegeneration. Lancet Neurol 2: 266. [DOI] [PubMed] [Google Scholar]
- Heerssen HM, Pazyra MF, Segal RA (2004) Dynein motors transport activated Trks to promote survival of target-dependent neurons. Nat Neurosci 7: 596-604. [DOI] [PubMed] [Google Scholar]
- Jourdan F, Moyse E, De Bilbao F, Dubois-Dauphin M (1998) Olfactory neurons are protected from apoptosis in adult transgenic mice overexpressing the bcl-2 gene. NeuroReport 9: 921-926. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD (2004) Neurobiology: a move to sort life from death. Nature 427: 798-799. [DOI] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA (1998) Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 94: 325-337. [DOI] [PubMed] [Google Scholar]
- LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M, Van Winkle T, Howland DS, Holzbaur EL (2002) Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron 34: 715-727. [DOI] [PubMed] [Google Scholar]
- Le DA, Wu Y, Huang Z, Matsushita K, Plesnila N, Augustinack JC, Hyman BT, Yuan J, Kuida K, Flavell RA, Moskowitz MA (2002) Caspase activation and neuroprotection in caspase-3-deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Proc Natl Acad Sci USA 99: 15188-15193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R (1992) Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell 69: 737-749. [DOI] [PubMed] [Google Scholar]
- Lipson AC, Widenfalk J, Lindqvist E, Ebendal T, Olson L (2003) Neurotrophic properties of olfactory ensheathing glia. Exp Neurol 180: 167-171. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Ding J, Kowal AS, Nardine T, Allen E, Delcroix JD, Wu C, Mobley W, Fuchs E, Yang Y (2003) BPAG1n4 is essential for retrograde axonal transport in sensory neurons. J Cell Biol 163: 223-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Iyadurai SJ, Gassman A, Gindhart Jr JG, Hays TS, Saxton WM (1999) Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport. Mol Biol Cell 10: 3717-3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Duan W (1999) “Apoptotic” biochemical cascades in synaptic compartments: roles in adaptive plasticity and neurodegenerative disorders. J Neurosci Res 58: 152-166. [PubMed] [Google Scholar]
- McCabe BD, Marques G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O'Connor MB (2003) The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron 39: 241-254. [DOI] [PubMed] [Google Scholar]
- Melloni Jr RH, Tokito MK, Holzbaur EL (1995) Expression of the p150Glued component of the dynactin complex in developing and adult rat brain. J Comp Neurol 357: 15-24. [DOI] [PubMed] [Google Scholar]
- Michel D, Moyse E, Trembleau A, Jourdan F, Brun G (1997) Clusterin/ApoJ expression is associated with neuronal apoptosis in the olfactory mucosa of the adult mouse. J Cell Sci 110: 1635-1645. [DOI] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR (2001) Neurotrophin signalling pathways regulating neuronal apoptosis. Cell Mol Life Sci 58: 1045-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montague AA, Greer CA (1999) Differential distribution of ionotropic glutamate receptor subunits in the rat olfactory bulb. J Comp Neurol 405: 233-246. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW (1991) Cell death during development of the nervous system. Annu Rev Neurosci 14: 453-501. [DOI] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown Jr RH, Ludlow CL, Fischbeck KH (2003) Mutant dynactin in motor neuron disease. Nat Genet 33: 455-456. [DOI] [PubMed] [Google Scholar]
- Quintyne NJ, Gill SR, Eckley DM, Crego CL, Compton DA, Schroer TA (1999) Dynactin is required for microtubule anchoring at centrosomes. J Cell Biol 147: 321-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink A, Fung KM, Trojanowski JQ, Lee VM, Neugebauer E, McIntosh TK (1995) Evidence of apoptotic cell death after experimental traumatic brain. Am J Pathol 147: 1575-1583. [PMC free article] [PubMed] [Google Scholar]
- Rochefort C, Gheusi G, Vincent JD, Lledo PM (2002) Enriched odor exposure increases the number of newborn neurons in the adult olfactory bulb and improves odor memory. J Neurosci 22: 2679-2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskams AJ, Bredt DS, Dawson TM, Ronnett GV (1994) Nitric oxide mediates the formation of synaptic connections in developing and regenerating olfactory receptor neurons. Neuron 13: 289-299. [DOI] [PubMed] [Google Scholar]
- Roskams AJ, Bethel MA, Hurt KJ, Ronnett GV (1996) Sequential expression of Trks A, B, and C in the regenerating olfactory neuroepithelium. J Neurosci 16: 1294-1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth KA, D'Sa C (2001) Apoptosis and brain development. Ment Retard Dev Disabil Res Rev 7: 261-266. [DOI] [PubMed] [Google Scholar]
- Salin PA, Lledo PM, Vincent JD, Charpak S (2001) Dendritic glutamate autoreceptors modulate signal processing in rat mitral cells. J Neurophysiol 85: 1275-1282. [DOI] [PubMed] [Google Scholar]
- Salvesen GS (2002) Caspases and apoptosis. Essays Biochem 38: 9-19. [DOI] [PubMed] [Google Scholar]
- Shah JV, Flanagan LA, Janmey PA, Leterrier JF (2000) Bidirectional translocation of neurofilaments along microtubules mediated in part by dynein/dynactin. Mol Biol Cell 11: 3495-3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider WD, Zhou FQ, Zhong J, Markus A (2002) Signaling the pathway to regeneration. Neuron 35: 13-16. [DOI] [PubMed] [Google Scholar]
- Springer JE, Azbill RD, Knapp PE (1999) Activation of the caspase-3 apoptotic cascade in traumatic spinal cord. Nat Med 5: 943-946. [DOI] [PubMed] [Google Scholar]
- Stefanis L, Burke RE, Greene LA (1997) Apoptosis in neurodegenerative disorders. Curr Opin Neurol 10: 299-305. [DOI] [PubMed] [Google Scholar]
- Steuer ER, Wordeman L, Schroer TA, Sheetz MP (1990) Localization of cytoplasmic dynein to mitotic spindles and kinetochores. Nature 345: 266-268. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Farbman AI (2000) Tumor necrosis factor-alpha-induced apoptosis in olfactory epithelium in vitro: possible roles of caspase 1 (ICE), caspase 2 (ICH-1), and caspase 3 (CPP32). Exp Neurol 165: 35-45. [DOI] [PubMed] [Google Scholar]
- Thorburn A (2004) Death receptor-induced cell killing. Cell Signal 16: 139-144. [DOI] [PubMed] [Google Scholar]
- Tisay KT, Bartlett PF, Key B (2000) Primary olfactory axons form ectopic glomeruli in mice lacking p75NTR. J Comp Neurol 428: 656-670. [DOI] [PubMed] [Google Scholar]
- Tokito MK, Howland DS, Lee VM, Holzbaur EL (1996) Functionally distinct isoforms of dynactin are expressed in human neurons. Mol Biol Cell 7: 1167-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, Karki SB, Kuznetsov SA, Tabb JS, Weiss DG, Langford GM, Holzbaur EL (1997) The interaction between cytoplasmic dynein and dynactin is required for fast axonal transport. Proc Natl Acad Sci USA 94: 12180-12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt WC, Sakano H, Lee ZY, Reusch JE, Trinh K, Storm DR (2004) Odorant stimulation enhances survival of olfactory sensory neurons via MAPK and CREB. Neuron 41: 955-967. [DOI] [PubMed] [Google Scholar]
- Woodhall E, West AK, Chuah MI (2001) Cultured olfactory ensheathing cells express nerve growth factor, brain-derived neurotrophic factor, glia cell line-derived neurotrophic factor and their receptors. Brain Res Mol Brain Res 88: 203-213. [DOI] [PubMed] [Google Scholar]
- Yan XX, Najbauer J, Woo CC, Dashtipour K, Ribak CE, Leon M (2001) Expression of active caspase-3 in mitotic and postmitotic cells of the rat forebrain. J Comp Neurol 433: 4-22. [DOI] [PubMed] [Google Scholar]
- Yu CR, Power J, Barnea G, O'Donnell S, Brown HE, Osborne J, Axel R, Gogos JA (2004) Spontaneous neural activity is required for the establishment and maintenance of the olfactory sensory map. Neuron 42: 553-566. [DOI] [PubMed] [Google Scholar]
- Yuan J, Lipinski M, Degterev A (2003) Diversity in the mechanisms of neuronal cell death. Neuron 40: 401-413. [DOI] [PubMed] [Google Scholar]