

Abstract
δ-Opioid receptor (DOPr) activation fails to produce cellular physiological responses in many brain regions, including the periaqueductal gray (PAG), despite neural expression of high densities of the receptor. Previous histochemical studies have demonstrated that a variety of stimuli, including chronic morphine treatment, induce the translocation of DOPr from intracellular pools to the surface membrane of CNS neurons. PAG neurons in slices taken from untreated mice exhibited μ-opioid receptor (MOPr) but not DOPr-mediated presynaptic inhibition of GABAergic synaptic currents. In contrast, after 5-6 d of chronic morphine treatment, DOPr stimulation inhibited synaptic GABA release onto most neurons. Shorter exposure to morphine in vitro (upto 4 h) or in vivo (18 h) did not induce functional DOPr responses. DOPr-mediated presynaptic inhibition could not be induced in slices from untreated animals by increasing synaptic activity in vitro using high extracellular potassium concentrations or activation of protein kinase A. Induction of functional DOPr signaling by chronic morphine required MOPr expression, because no DOPr receptor responses were observed in MOPr knock-out mice. DOPr agonists also had no effect on miniature IPSCs in β-arrestin-2 knock-out mice after chronic morphine. These results suggest that induction of DOPr-mediated actions in PAG by chronic morphine requires prolonged MOPr stimulation and expression of β-arrestin-2.
Keywords: opioids, receptor trafficking, analgesia, patch clamp, presynaptic inhibition, synaptic terminal
Introduction
The actions of opioids are transduced via three receptor types: the μ-opioid (MOP), δ-opioid (DOP), and κ-opioid (KOP) receptors. All three opioid receptors are G-protein-coupled receptors (GPCRs) and have a high degree of sequence homology (Law et al., 2000). Most clinically used opioids act via the MOP receptor (MOPr) (Breivik, 2001), but DOP receptor (DOPr) agonists are also capable of producing antinociception, often with fewer negative side effects than MOPr agents (Cowan et al., 1988; Lazarus et al., 1999). Intrathecal or intracerebroventricular application of DOPr agonists also produces spinal and supraspinal antinociception in rodents (Heyman et al., 1987; Stewart and Hammond, 1993). Microinjection of DOPr agonists into discrete brain regions known to mediate antinociceptive opioid actions, such as the nucleus raphe magnus, the ventral medial medullary reticular formation, and the nucleus reticularis gigantocellularis pars α show that these regions can mediate modest DOPr-dependent antinociception (Rossi et al., 1994; Ossipov et al., 1995; Thorat and Hammond, 1997; Kovelowski et al., 1999). However, in another brain region critical for opioid antinociception, the periaqueductal gray (PAG), DOPr agonists have either little or no analgesic efficacy (Rossi et al., 1994; Ossipov et al., 1995). These behavioral studies are generally consistent with cellular physiological studies that have found that DOPr agonists do not affect GABAergic synaptic transmission of rat or mouse PAG neurons and have little (mouse PAG) or no (rat PAG) postsynaptic effect (Chieng and Christie, 1994; Vaughan and Christie, 1997; Connor and Christie, 1998; Connor et al., 1999; Vaughan et al., 2003).
The reason(s) for the lack of DOPr agonist actions in the PAG is unclear but may be explained by the subcellular distribution of DOPrs. Unlike MOPrs, which are primarily expressed on the cell surface, DOPrs, in many neurons, are almost exclusively intracellular (Arvidsson et al., 1995; Kalyuzhny et al., 1996; Kalyuzhny and Wessendorf, 1998; Cahill et al., 2001b; Commons et al., 2001). Recent work has shown that chronic treatment with morphine traffics intracellular DOPrs to the plasma membrane of neurons in the dorsal spinal cord of rats and mice (Cahill et al., 2001a; Morinville et al., 2003). This effect was correlated with enhanced DOPr-mediated antinociception, indicating that the number of functional DOPrs was increased in morphine-treated animals.
This study tested the hypothesis that chronic morphine treatment would induce efficacious actions of DOPr agonists in the PAG. Using patch-clamp techniques, we show that, after chronic morphine administration, DOPr agonists reduce vesicular GABA release in the PAG. The mechanism does not involve simple changes in activity of nerve terminals or hypertrophy of cAMP signaling. However, the mechanisms underlying this induction of DOPr function do require the expression of MOPrs and β-arrestin-2 (βarr2).
Materials and Methods
Experiments used adult male C57BL/6J mice (10-20 g) or knock-outs, as described below. All animal experiments were approved by the Royal North Shore Hospital/University of Technology Sydney Ethics Committee.
Knock-out mice. Wild-type mice, heterozygotes, and βarr2 knock-out mice were generated as littermates from crossing heterozygous βarr2 C57BL/6/129SvJ animals. Production and full phenotypic analysis were described previously (Bohn et al., 1999). Wild-type and knock-out mice homozygous for a deletion of exon 1 of the MOPr (MOPr-1) were used. Production and detailed initial phenotypic analysis of the MOPr knock-out mice have been described in detail previously (Schuller et al., 1999). Knock-out mice used in the present study were derived from matings of MOPr-/- mice and wild-type controls.
Treatment with morphine. Morphine was administered via subcutaneous injections of a sustained-release preparation of morphine base. Morphine base was suspended in a vehicle comprising mannide mono-oleate (Arlacel A; 10%) and light liquid paraffin (40%) in normal saline. Unless otherwise stated, chronic morphine treatment comprised three injections on alternate days over a period of 5 d. With each injection, mice were treated with 300 mg/kg morphine. These chronic treatment protocols have been shown previously to induce physical dependence in rats (Chieng and Christie, 1996; Ingram et al., 1998) and mice (Hack et al., 2003). Vehicle-treated mice were injected with only the vehicle solution. Injections were made while the animals were under light halothane anesthesia.
Preparation of brain slices and electrophysiology. Midbrain PAG slices (250 μm) were cut in ice-cold artificial CSF (ACSF). Slices were maintained at 34°C in a submerged chamber containing ACSF equilibrated with 95% O2 and 5% CO2. The slices were then transferred to a recording chamber and superfused continuously (2 ml/min) with ACSF of the following composition (in mm): 126 NaCl, 2.5 KCl, 1.4 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 11 glucose, and 25 NaHCO3. Slices from morphine-treated mice were maintained either in ACSF containing 5 μm morphine or in normal morphine-free ACSF (for 1-6 h). All experiments were conducted on neurons located in the ventrolateral PAG. PAG neurons were visualized on an upright microscope (BX50WI; Olympus Optical, Tokyo, Japan) using infrared Nomarski optics. Whole-cell voltage-clamp recordings of synaptic currents were made using patch electrodes (2-5 MΩ) containing the following (in mm): 140 CsCl, 10 EGTA, 5 HEPES, 2 CaCl2, and 2 MgATP, pH 7.3 (osmolarity, 270-290 mOsm/L). Whole-cell voltage-clamp recordings of cell body membrane currents (holding potential, -62 mV) were made using an internal solution comprising the following (mm): 95 K-gluconate, 30 KCl, 15 NaCl, 2 MgCl2, 10 HEPES, 11 EGTA, 2 MgATP, and 0.25 NaGTP. Series resistance (≤12 MΩ) was compensated by 80% and continuously monitored during experiments.
Spontaneous GABAA-mediated miniature IPSCs (mIPSCs) were obtained in the presence of CNQX (5 μm), strychnine (3 μm), and TTX (300 nm). Miniature events were filtered (1 kHz low-pass filter) and sampled at 5 kHz for on-line and later off-line analysis (Axograph 4.6; Axon Instruments, Union City, CA). Spontaneous miniature events above a preset threshold (5 SDs above baseline noise) were automatically detected by a sliding-template algorithm and then manually checked off-line. Events were counted in 20-60 s epochs to construct time plots of the mIPSC rate and ranked by amplitude to construct cumulative probability functions. Electrically evoked IPSCs (eIPSCs) were elicited via bipolar tungsten stimulating electrodes (FHC, Bowdoinham, ME) placed 50-100 μm from the recording electrode (rate, 0.03-0.05 Hz; stimuli, 2-70 V, 20-200 μs) in the presence of CNQX (5 μm). Neurons were voltage clamped at -70 mV. eIPSCs were quantified by averaging 4-10 consecutive responses for each condition (Axograph 4.6). Experiments were conducted in the presence of the adenosine A1 receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) (1 μm) to relieve inhibitory adenosine tone present after chronic morphine treatment in mouse PAG neurons (Hack et al., 2003).
Drugs, reagents, and solutions. d-Phe-Cys-Tyr-d-Trp-Arg-Pen-Thr-NH2 (CTAP), Tyr-d-Ala-Gly-N-Me-Phe-Gly-ol enkephalin (DAMGO) and [d-Ala2]deltorphin II (DELT) were obtained from Auspep (Parkville, Victoria, Australia). DPCPX, DAMGO, forskolin, and Arlacel A were from Sigma (Sydney, New South Wales, Australia). TTX was from Alomone Laboratories (Jerusalem, Israel). N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide dihydrochloride (H-89) was from Biomol (Plymouth, PA). CNQX was from Tocris Cookson (Bristol, UK). Morphine base was obtained from GlaxoSmithKline (Boronia, Victoria, Australia). Naloxone hydrochloride was obtained from Research Biochemicals (Natick, MA). Stock solutions of all drugs were diluted to working concentrations in the extracellular solution immediately before use and applied by superfusion. Forskolin, H-89, and DPCPX were dissolved in dimethylsulfoxide (DMSO) stock to achieve a DMSO concentration of 0.01% in superfusion solutions. Stock solutions of all other drugs were made in distilled water.
Data analysis. All pooled values are expressed as mean ± SEM. Statistical tests between treatment groups were made using an unpaired t test. Within-group comparisons were made using paired t tests. Differences among proportions was analyzed using χ2 tests. To analyze miniature event data, cumulative amplitude and interevent interval histograms were compared using the Kolmogorov-Smirnov (K-S) test. Significance was accepted at p < 0.05.
Results
DOPr agonists inhibit GABAergic synaptic transmission only after chronic morphine treatment
DOPr agonists do not produce presynaptic inhibition of GABAergic synaptic currents in PAG from drug-naive mice (Vaughan et al., 2003) or rats (Vaughan and Christie, 1997). In the presence of TTX (300 nm), CNQX (5 μm), and strychnine (3 μm), spontaneous mIPSCs were readily observed in PAG neurons. These currents were totally abolished by picrotoxin (100 μm; data not shown), as reported previously (Hack et al., 2003; Vaughan et al., 2003). In slices taken from vehicle-treated mice, DELT (300 nm to 1 μm) failed to affect the mIPSC rate or amplitude (n = 10 cells) (data summarized in Fig. 1D) in any PAG neurons. In slices from morphine-treated mice maintained in morphine (5 μm) and DPCPX (1 μm), superfusion of CTAP (1 μm) increased the mIPSC rate (105 ± 9.9% increase; n = 8 cells), as reported previously (Hack et al., 2003). Similarly, the mIPSC rate was elevated in slices spontaneously withdrawn from morphine for 1 h in vitro (Fig. 1D). In contrast to vehicle-treated animals, in slices from morphine-treated mice, DELT (300 nm) caused a significant reduction in the rate of mIPSCs (p < 0.05; K-S test) (Fig. 1) in 16 of 22 cells (73%; p < 0.0001 vs vehicle treatment; χ2 test). DELT-mediated inhibition was similar when withdrawal was induced by either brief (<5 min) superfusion of CTAP (5 of 6 cells) (Fig. 1A) or washing of morphine from slices for 1 h (11 of 16 cells), suggesting that the duration of withdrawal in vitro did not influence DOPr responses. In all subsequent experiments, withdrawal was induced by washing morphine from slices for 1 h unless otherwise noted. DELT (300 nm) did not affect mIPSC amplitude or amplitude distributions in these cells (p > 0.05; K-S test) (Fig. 1C, D). DELT-mediated inhibition of mIPSCs was reversed by the DOPr antagonist N, N-diallyl-Tyr-Aib-Aib-Phe-Leu-OH (ICI 174864) (1 μm) (Fig. 1A, B). In all cases in which DELT did not inhibit the mIPSC rate, either the MOPr agonist DAMGO (1 μm; n = 4) or the κ-receptor agonist (5α,7α,8α)-(+)-N-methyl-N-[7-(pyrrolidinyl)-1-oxaspiro[4,5]dec-8-yl]-benzeneacetamide (U69593) (300 nm, n = 5) was used as a positive control and produced inhibition, as reported previously (Vaughan et al., 2003). A decrease in miniature synaptic current frequency, in the absence of a change in amplitude distribution, reflects a decrease in neurotransmitter release probability (Redman, 1990).
Figure 1.

DELT inhibits GABAergic release in PAG neurons from chronically morphine-treated mice. A, Representative current traces (Vh =-60 mV) from a PAG neuron from a morphine-dependent mouse showing typical mIPSCs during withdrawal induced by CTAP in control ACSF (containing 5 μm CNQX, 300 nm TTX, 3 μm strychnine, 1 μm DPCPX, and 1 μm CTAP). Superfusion of the DOPr agonist DELT (300 nm) inhibits the mIPSC rate, and the inhibition is reversed by the DOPr antagonist ICI 174864. B, Time course of mIPSC rate for the neuron shown in A during superfusion of DELT and ICI 174864. C, Cumulative distribution plots of mIPSC interevent interval and amplitude before (continuous line) and during (dashed line) DELT application for the neuron shown in A and B. Cumulative plots show that DELT induces a rightward shift in the distribution of interevent intervals (p < 0.001; K-S test), indicating a decrease in mIPSC frequency, whereas it does not induce a significant variation in mIPSC amplitude (p > 0.1; K-S test). D, Histogram illustrating the effects of DELT on mIPSC parameters in PAG neurons from vehicle- and morphine-treated mice [cells that responded to DELT after withdrawal induced by either CTAP (n = 5) or washout of morphine for 1 h (n = 11)]. The rate of mIPSCs was significantly higher in dependent neurons compared with vehicle (p < 0.01). DELT (300 nm) significantly inhibited the mIPSC rate in neurons from dependent animals (p < 0.01) without affecting amplitude (ampl.) (p = 0.26). DELT did not affect the mIPSC rate or amplitude in neurons from vehicle-treated animals. **p < 0.01 versus vehicle; #p < 0.01 versus control.
DOPr agonists fail to inhibit eIPSCs in neurons from untreated mice (Vaughan et al., 2003). DELT (300 nm) was without effect on eIPSCs in slices from vehicle-treated mice (n = 6; data not shown). In contrast, DELT (300 nm) inhibited eIPSC amplitude by 42 ± 9% in four of six neurons from chronically morphine-treated mice (p < 0.0001 vs vehicle treated; χ2 test) (Fig. 2) after withdrawal induced using CTAP (1 μm). The effects of DELT were reversed by the selective DOPr antagonist ICI 174864 (1 μm) in all cases (Fig. 2). Regardless of the response to DELT, eIPSC amplitudes in all cells were reduced by U69593 (300 nm), which is effective in the inhibition of eIPSCs in all mouse PAG neurons (Vaughan et al., 2003). These experiments demonstrate that chronic morphine treatment induces the expression of functional DOPrs in GABAergic nerve terminals in PAG.
Figure 2.

DELT inhibits electrically evoked GABAergic synaptic currents in neurons from morphine-treated mice. Top, Time course of evoked GABAergic IPSC amplitude during applications of DELT (300 nm) and ICI 174864 (1 μm) in a PAG neuron taken from a chronically morphine-treated mouse after washout of morphine for 1 h. Bottom, Traces averaged from 8-12 eIPSCs taken at the time points indicated in the top panel. The amplitude of eIPSCs was reduced by application of DELT, and this inhibition was reversed by the application of ICI 174864. The eIPSCs were elicited every 15 s at Vh =-70 mV in the presence of CTAP (1 μm), CNQX (5 μm), strychnine (3 μm), and DPCPX (1 μm).
Prolonged morphine treatment is required to induce DOPr-mediated presynaptic inhibition
Previous histochemical studies found that chronic treatment with morphine must persist for at least 48 h to observe increased surface expression of DOPr in neurons (Morinville et al., 2003). In contrast, stressful stimuli as brief as 3 min have been shown to induce translocation of DOPr to the surface membrane of PAG nerve terminals in previous studies (Commons, 2003). Slices of PAG from control mice were incubated in morphine (5 μm) in vitro for 1-3 h, followed by superfusion of CTAP (1 μm). Under these conditions, superfusion of DELT (1 μm) produced no inhibition of the mIPSC rate (97.6 ± 3.1% of control; p > 0.05; n = 7). In another experiment, animals were treated with morphine for 18 h as described in Materials and Methods. All PAG neurons taken from animals exposed to morphine for 18 h failed to respond to DELT (1 μm), as assessed by its ability to reduce mIPSC frequency (96.1 ± 5.6% of control; p > 0.05; n = 8 cells). All neurons tested responded to the application of DAMGO (1 μm; data not shown).
DOPr responses are not increased in the soma of PAG neurons after chronic morphine treatment
DOPr agonists induce a G-protein-coupled inwardly rectifying K+ current (GIRK) in ∼25% of PAG neurons from normal (i.e., drug naive) mice (Vaughan et al., 2003). In slices from chronically morphine-treated mice, DOPr agonist-induced GIRK currents were seen in 22% of cells studied (two of nine cells). The incidence (22% of neurons) and amplitude (∼12 pA) of DELT-mediated postsynaptic GIRK currents was similar to that reported in normal C57BL/6J mice (Vaughan et al., 2003). These results suggest that DOPr actions emerge on GABAergic nerve terminals in PAG after chronic morphine treatment without a substantial change in postsynaptic coupling of DOPr to GIRK channels in the cell body.
Induction of functional DOPrs is not attributable to short-term increases in nerve terminal excitability
The mechanism of induction of functional DOPrs in PAG nerve terminals could involve depolarization or increased synaptic activity during opioid withdrawal. Membrane depolarization can cause κ-opioid receptor translocation from the cell interior to the membrane (Shuster et al., 1999). The effects of DELT on mIPSCs in PAG from control animals were therefore examined after increasing the concentration of KCl in the extracellular solution ([K+]o) by 15 mm (from 2.5 to 17.5 mm) for 10 min in the presence of TTX to depolarize the presynaptic membrane and activate Ca2+ channels (Fig. 3). Elevation of [K+]o increased the frequency of mIPSCs (Fig. 3). Under these conditions, DELT (1 μm) failed to modulate mIPSC frequency or amplitude (Fig. 3). To confirm that PAG neurons expressed functional MOPr responses under these conditions, the nonselective opioid agonist Met-enkephalin (10 μm) was applied. Application of Metenkephalin (10 μm) in the presence of high [K+]o caused a robust and reversible reduction in mIPSC frequency without affecting mIPSC amplitude (Fig. 3). These results indicate that short-term depolarization does not induce the expression of functional DOPr responses in nerve terminals in PAG.
Figure 3.
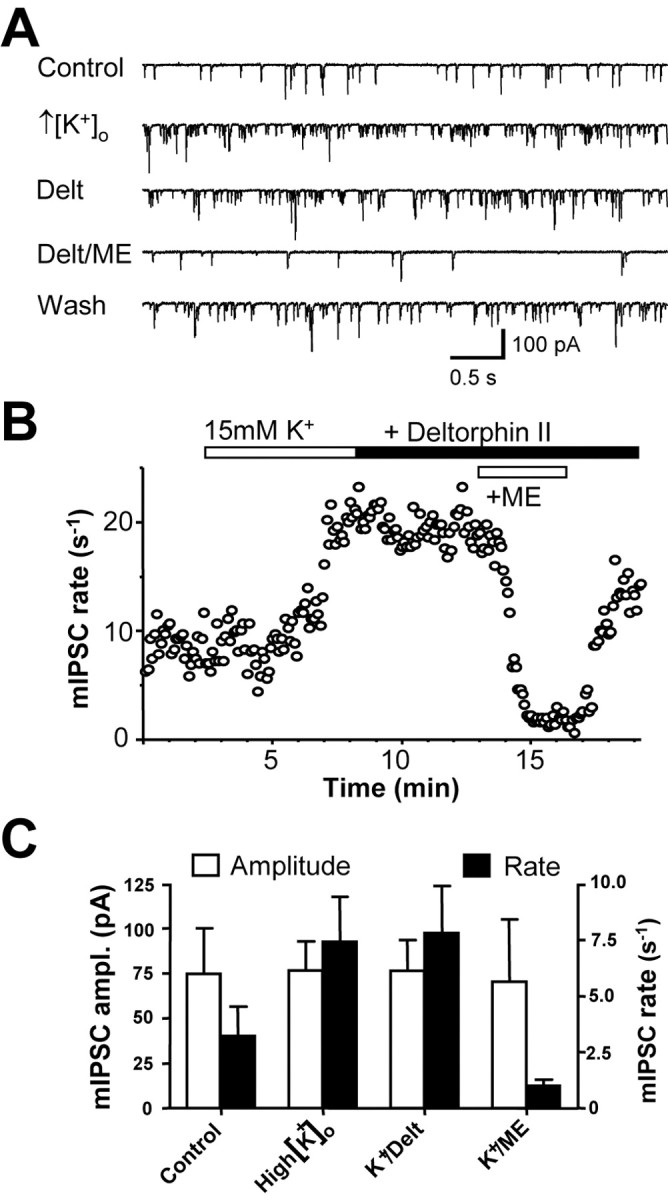
Depolarization does not induce DOPr activity in morphine-naive PAG neurons. A, Representative pharmacologically isolated mIPSC traces (Vh =-60 mV) recorded in normal ACSF ([K+]o = 2.5 mm) and after superfusions of high [K+]o ACSF (+15 mm), DELT (1 μm), Met-enkephalin (ME; 10 μm), and washout (Wash) of Met-enkephalin. B, Time course of mIPSCs for the neuron shown in A. Elevation of [K+]o increased the rate of mIPSCs. Application of DELT in the presence of 17.5 mm [K+]o does not affect the mIPSC rate, but subsequent application of Met-enkephalin reversibly reduces the mIPSC rate. C, Histogram illustrating the effects of high [K+]o ACSF and applications of DELT (1 μm) and Met-enkephalin (10 μm) on the mIPSC rate and amplitude (ampl.).
Withdrawal from chronic morphine treatment causes cAMP/protein kinase A (PKA)-dependent depolarization and enhanced GABAergic neurotransmission in PAG neurons (Chieng et al., 1996; Ingram et al., 1998; Hack et al., 2003). Upregulated cAMP signaling can induce translocation of other GPCRs (e.g., D1 receptors) (Brismar et al., 1998) from the cytoplasm to the surface in other cell types. The ability of DELT to modulate the mIPSC rate after chronic morphine treatment persisted despite the pre-incubation of slices with the PKA inhibitor H-89 (10 μm) (Fig. 4A). This concentration of H-89 was sufficient to completely prevent withdrawal-induced hyperexcitation of mIPSCs (Hack et al., 2003). The effective block of PKA by H-89 (10 μm) was also confirmed by its ability to block the increase in the mIPSC rate induced by forskolin (n = 4 cells) (Fig. 4B).
Figure 4.

Upregulation of cAMP/PKA-mediated signal transduction during morphine withdrawal is not responsible for inducing DOPr function. A, mIPSC rate and amplitude in the absence and presence of H-89. PAG slices from morphine-treated mice were preincubated in the absence of morphine for 1 h and in the presence of the PKA inhibitor H-89 (10 μm) for 30 min before commencement and throughout the course of the experiment. DELT- (300 nm) mediated inhibition of mIPSC frequency persisted in the presence of H-89 (**p < 0.01; *p < 0.05; n = 5; paired t tests in the absence and presence of DELT). B, Histogram showing the augmentation of mIPSC frequency in morphine-naive PAG neurons by forskolin (1 μm; n = 4; ***p < 0.001; paired t test). Subsequent application of DELT (1 μm) in the continued presence of forskolin did not affect the mIPSC rate, but Met-enkephalin (ME; 10 μm) did reduce the mIPSC rate. The forskolin-induced increase in mIPSC rate was abolished by previous treatment with H-89 (10 μm; p < 0.001; unpaired t test; n = 5). ICI, ICI 174864.
Conversely, to test whether DOPr function was induced after acute upregulation of PKA signaling in untreated mice, slices were superfused with the adenylyl cyclase activator forskolin (1 μm) and subsequently exposed to DELT (1 μm). Application of forskolin (1 μm) increased the mIPSC rate to an extent similar to that produced by opioid withdrawal (n = 5 cells) (Fig. 4B), without affecting mIPSC amplitude. However, DELT (1 μm) did not alter the rate or amplitude of mIPSCs in any cells tested in the presence of forskolin (n = 5) (Fig. 4B). These studies suggest that the emergence of functional DOPr responses in GABAergic terminals after chronic morphine is not attributable to a short-term increase in nerve terminal excitability induced by either direct depolarization or enhanced PKA activity.
Induction of DOPr function is dependent on the expression of MOPrs and β-arrestin-2
Previous studies in the spinal cord have demonstrated that the translocation of DOPrs to the cell surface after chronic morphine treatment (Morinville et al., 2003) or inflammation (Cahill et al., 2003; Morinville et al., 2004) is dependent on the activation of MOPrs. In MOPr knock-out mice (MOPr-/-), DELT (0.3-1 μm) failed to inhibit mIPSCs in any cells tested (0 of 7 cells) after chronic morphine treatment (Fig. 5). DELT (300 nm) did inhibit the mIPSC rate in cells from chronically morphine-treated wild-type (MOPr+/+) littermates (6 of 10 cells; p < 0.001 vs MOPr-/- mice; χ2 test; 33.2 ± 5.8% inhibition) (Fig. 5). In cells that were not responsive to DELT, the κ-opioid agonist U69593 (300 nm) was used as a positive control (Vaughan et al., 2003) and inhibited the mIPSC rate in all neurons (n = 10; data not shown). As reported previously (Vaughan et al., 2003), basal mIPSC rates and amplitudes of MOR-/- did not differ from MOR+/+ mice (data not shown).
Figure 5.

Induction of DOPr function by long-term morphine treatment requires MOPrs and β-arrestin-2. Neurons from MOPr-/- or βarr2-/- do not develop DOPr-mediated presynaptic inhibition after chronic treatment with morphine. Knock-out mice and their wild-type littermates were treated with the standard morphine treatment protocol and probed for DOPr-mediated presynaptic inhibition of GABA release after washout of morphine for 1 h in vitro. Six of 10 neurons tested from wild-type mice expressing MOPr responded to DELT, but no PAG neurons tested from MOPr-/- mice responded to DELT. Wild-type littermates expressing βarr2 (βarr2+/+) demonstrated DOPr-mediated inhibition of GABA release in five of nine neurons after morphine treatment, but no neurons tested from βarr2-/- mice responded to DELT (0 of 9; p < 0.05 vs wild-type littermates; χ2 test). Data from vehicle and morphine-treated C57BL/6J mice are shown for comparison. Numbers within the bars indicate the number of neurons that responded or did not respond to DELT application.
βarr2 is necessary for rapid endocytosis of MOPrs and influences the development of morphine tolerance but is not required for the expression of opioid withdrawal (Bohn et al., 1999). To further explore the mechanisms of the requirement for MOPr expression to produce expression of functional DOPrs, slices from βarr2 knock-out mice (βarr2-/-) were examined after chronic treatment with morphine. Spontaneous mIPSCs were not affected by the application of DELT (300 nm and up to 3 μm was without effect) in any PAG neurons from morphine-treated βarr2-/- mice (Fig. 5). In contrast, 56% of PAG neurons from morphine-treated wild-type littermates responded to DELT (p < 0.05 vs βarr2-/- mice; χ2 test) (Fig. 5). As in wild-type mice (Hack et al., 2003), spontaneous mIPSC rate (1.1 ± 0.2 s-1 before withdrawal; n = 9) was enhanced by morphine withdrawal in βarr2-/- mice (105 ± 32% increase vs 88 ± 10% in βarr2+/+ littermates), consistent with the persistence of behavioral signs of opioid withdrawal in βarr2-/- mice. Basal mIPSC rates and amplitudes in vehicle-treated βarr2-/- mice (1.3 ± 0.5 s-1;41 ± 6 pA; n = 9) did not differ from βarr2+/+ mice (1.2 ± 0.4 s-1;43 ± 10 pA).
To confirm that neurons from βarr2-/- were able to express DOPrs on the cell surface, we examined DELT-activated GIRK currents in cell bodies of PAG neurons from βarr2-/- mice. DELT (1 μm) agonist-induced GIRK currents (8 ± 2 pA) reversed by ICI 174864 were seen in 33% of the cells studied (4 of 12 cells), similar to that reported in normal C57BL/6J mice (Vaughan et al., 2003). In the same cells, DAMGO (3 μm) also produced GIRK currents (25 ± 11 pA). These results indicate that functional DOPrs can be expressed on the surface of PAG neurons in the absence of βarr2 expression.
Discussion
We established that chronic treatment with morphine induces functional DOPr actions in GABAergic nerve terminals in the PAG. After chronic morphine treatment, DELT inhibited mIPSCs and eIPSCs, impinging on 60-70% of neurons in the ventrolateral PAG. The presynaptic actions of DOPr agonists only on a subpopulation of neurons could reflect heterogeneity of GABAergic projections onto different PAG neurons, as reported previously for another GPCR, the ORL1 receptor (Vaughan et al., 1997). The present results provide evidence for the physiological significance of recent reports that prolonged MOPr agonist exposure induces translocation of DOPrs from the cytosol to the plasma membrane (Cahill et al., 2001a; Morinville et al., 2003). In the PAG of untreated animals, the DOPr is primarily localized in axon terminals (Arvidsson et al., 1995; Kalyuzhny et al., 1996; Kalyuzhny and Wessendorf, 1998) Under these conditions, DOPrs in the PAG are rarely observed on the plasma membrane but are primarily associated with intracellular membranes (Commons et al., 2001; Commons, 2003). The intracellular localization of DOPrs is consistent with previous studies suggesting that DOPr agonists have zero or low efficacy in the PAG region at the cellular and systems level in untreated animals (Rossi et al., 1994; Ossipov et al., 1995; Vaughan et al., 2003).
The mechanisms responsible for the emergence of functional DOPrs in PAG nerve terminals after chronic morphine treatment remain unresolved; however, the present results, as in the histochemical study by Morinville et al. (2003), suggest that several days of exposure to morphine are required. Neither acute exposure to morphine for several hours in vitro nor continuous exposure for 18 h in vivo produced functional DOPr responses in PAG nerve terminals in the present study. The prolonged time course needed for morphine treatment to upregulate DOPrs is considerably longer than that reported previously for other stimuli that induce the surface expression of DOPrs (see below), suggesting that the underlying mechanisms may differ.
Conditions known to induce translocation of DOPrs to the plasma membrane, such as depolarization, do not appear to account for the functional expression of DOPrs after chronic morphine treatment. Brief depolarization increases the membrane expression of a number of GPCRs, including KOPrs (Shuster et al., 1999) and DOPrs (Bao et al., 2003), as do brief physiological stimuli, such as swim stress (Commons, 2003). Insertion of DOPrs into the membrane could therefore be triggered by membrane depolarization or the increased nerve terminal excitability occurring in the PAG during opioid withdrawal in vitro (Chieng and Christie, 1996; Ingram et al., 1998; Hack et al., 2003). However, in the present study, membrane depolarization produced by elevating [K+]o for up to 10 min in PAG neurons from morphine-naive mice failed to cause DOPr-mediated inhibition of GABA release, nor did exposure to a concentration of forskolin sufficient to increase mIPSC frequency severalfold. Furthermore, no induction of DOPr function was observed in PAG nerve terminals from βarr2-/- mice, despite the occurrence of a robust increase in mIPSC frequency during opioid withdrawal in the PAG. These results indicate that the depolarization of nerve terminals or increased synaptic activity does not induce functional DOPrs in PAG nerve terminals. The mechanisms of the induction of functional DOPrs by chronic morphine therefore appear to differ from the rapid activity-dependent translocation described in other models.
Other adaptations associated with chronic morphine treatment, such as enhanced PKA signaling during withdrawal, also fail to explain the induction of functional DOPrs after chronic morphine treatment. Upregulated PKA signaling can increase the expression of receptors at the plasma membrane (Meyer-Franke et al., 1998) and induce functional receptor sensitization of MOPrs and A1 receptors (Ingram et al., 1998; Hack et al., 2003). Morphine withdrawal induces PKA-dependent hyperexcitation in PAG GABAergic nerve terminals (Ingram et al., 1998; Hack et al., 2003) and might therefore be responsible for induction of functional DOPrs. The present study ruled this out, because block of PKA activity failed to prevent DOPr function in GABA-containing nerve terminals. Furthermore, direct activation of the cAMP/PKA cascade with forskolin in PAG slices from untreated animals failed to mimic the induction of DOPr responses. It is therefore very unlikely that activity-dependent or rebound increases in PKA signaling occurring during opioid withdrawal produced functional DOPr responses after chronic morphine treatment. Consistent with this interpretation, we observed a similar expression of DOPr actions after withdrawal induced briefly with CTAP (<5 min) or 1 h of washout of morphine. Similarly, Cahill et al. (2001a) reported that the level of DOPr expression on the cell surface after chronic morphine administration was not different 8 h after the final morphine treatment compared with 1 h after treatment, suggesting that the effect was not mediated by opioid withdrawal.
Induction of DOPr receptor responses in the PAG after chronic morphine treatment requires expression of MOPrs, because the effect was abolished in MOPr-/- mice. Similarly, the translocation of DOPr to the surface membrane in spinal cord neurons by either chronic morphine treatment (Morinville et al., 2003) or inflammation (Morinville et al., 2004) is absent in MOPr-/-. These findings provide important confirmation that morphine induces functional DOPr activity via selective stimulation of MOPr and not via direct interaction of morphine with DOPr or other targets. Previous studies in MOPr knock-out mice have reported no evidence for compensatory changes in DOPr expression (Kitchen et al., 1997; Chen et al., 2000) or subcellular distribution (Morinville et al., 2003).
The mechanism underlying the requirement for MOPr expression to produce DOPr responses after chronic morphine treatment is unknown. One possibility is the involvement of MOPr trafficking and/or a functional interaction between MOPrs and DOPrs on synaptic terminals in the PAG region. Morphine treatment failed to induce DOPr function in βarr2-/- mice, suggesting that this protein plays a crucial role in the induction of surface expression of DOPrs in the PAG. β-Arrestins are cytosolic proteins that regulate the uncoupling of activated receptors from their G-protein-mediated signaling pathways, mediate receptor endocytosis via interaction with clathrin-coated pits, and act as signaling molecules in their own right (for review, see Claing et al., 2002). Given that MOPr stimulation is necessary to induce DOPr function, one possibility for the increased surface expression of DOPrs is that excessive stimulation of MOPrs during chronic morphine treatment promotes their internalization and that, during subsequent redistribution to the surface membrane, MOPrs induce DOPr translocation (possibly as heterodimers) from intracellular compartments to the surface. Both MOPrs and DOPrs are able to form heterodimers when coexpressed in isolated cell lines (Gomes et al., 2000) and spinal cord membranes (Gomes et al., 2004). In these models, treatment with MOPr agonists increases the surface expression of DOPrs and vice versa. If analogous mechanisms exist in PAG neurons, it would explain the involvement of both MOPrs and βarr2 in the induction of functional DOPrs after chronic morphine, as reported here.
The prolonged morphine treatment period required to induce DOPr actions in the current study and others (Cahill et al., 2001a; Morinville et al., 2003) suggests that upregulation of gene products and/or proteins linked to cell-surface recruitment may play a role in DOPr translocation. A number of accessory proteins have been linked to cell-surface targeting of GPCRs (Bouvier, 2001). It was proposed recently that intracellular chaperoning of DOPrs from the endoplasmic reticulum to the plasma membrane by opioid receptor agonists and antagonists may rescue improperly configured receptor proteins, resulting in increased agonist binding (Morello et al., 2000; Petaja-Repo et al., 2002). Therefore, prolonged stimulation with an MOPr agonist could lead to the expression of gene product(s) involved in the folding and translocation of DOPrs, perhaps as a consequence of β-arrestin binding and trafficking.
In conclusion, the present study has demonstrated that prolonged and sustained treatment with morphine leads to the expression of functional DOPrs in GABAergic nerve terminals of the PAG. We showed, via the use of knock-out mice, that this effect is dependent on the MOPr and βarr2. These results may have important implications for the management of chronic pain and/or opioid dependence, because they suggest that chronic treatment with MOPr agonists increases the surface expression of DOPrs and thereby the potency of DOPr-selective agents, which have low efficacy under normal circumstances. Consistent with this possibility, chronic treatment with morphine has been reported to increase the behavioral effects of DOPr agonists (Melchiorri et al., 1992).
Footnotes
This work was supported by Grant 302087 from the National Health and Medical Research Council of Australia. Donations of μ-receptor knock-out animals by J. E. Pintar and β-arrestin-2 knock-out animals by R. J. Lefkowitz and M. G. Caron are gratefully acknowledged.
Correspondence should be addressed to MacDonald J. Christie, Pain Management Research Institute, L5 Main Block, Royal North Shore Hospital, St. Leonards, New South Wales 2065, Australia. E-mail: macc@med.usyd.edu.au.
S. P. Hack's present address: Allergan Pharmaceuticals, 77 Ridge Street, Gordon, New South Wales 2072, Australia.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253192-07$15.00/0
References
- Arvidsson U, Dado RJ, Riedl M, Lee JH, Law PY, Loh HH, Elde R, Wessendorf MW (1995) δ-Opioid receptor immunoreactivity: distribution in brainstem and spinal cord, and relationship to biogenic amines and enkephalin. J Neurosci 15: 1215-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Jin S-X, Zhang C, Wang L-H, Xu Z-Z, Zhang F-X, Wang L-C, Ning F-S, Cai H-J, Guan J-S, Xiao H-S, Xu Z-Q, He C, Hokfelt T, Zhou Z, Zhang X (2003) Activation of delta opioid receptors induces receptor insertion and neuropeptide secretion. Neuron 37: 121-133. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin F-T (1999) Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286: 2495-2498. [DOI] [PubMed] [Google Scholar]
- Bouvier M (2001) Oligomerization of G-protein-coupled transmitter receptors. Nat Rev Neurosci 2: 274-286. [DOI] [PubMed] [Google Scholar]
- Breivik H (2001) Opioids in cancer and chronic non-cancer pain therapy—indications and controversies. Acta Anaesthesiol Scand 45: 1059-1066. [DOI] [PubMed] [Google Scholar]
- Brismar H, Asghar M, Carey RM, Greengard P, Aperia A (1998) Dopamine-induced recruitment of dopamine D1 receptors to the plasma membrane. Proc Natl Acad Sci USA 95: 5573-5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill CM, Morinville A, Lee MC, Vincent JP, Collier B, Beaudet A (2001a) Prolonged morphine treatment targets δ opioid receptors to neuronal plasma membranes and enhances δ-mediated antinociception. J Neurosci 21: 7598-7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill CM, McClellan KA, Morinville A, Hoffert C, Hubatsch D, O'Donnell D, Beaudet A (2001b) Immunohistochemical distribution of delta opioid receptors in the rat central nervous system: evidence for somatodendritic labeling and antigen-specific cellular compartmentalization. J Comp Neurol 440: 65-84. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Morinville A, Hoffert C, O'Donnell D, Beaudet A (2003) Upregulation and trafficking of delta opioid receptor in a model of chronic inflammation: implications for pain control. Pain 101: 199-208. [DOI] [PubMed] [Google Scholar]
- Chen H, Seybold VS, Loh HH (2000) An autoradiographic study in muopioid receptor knockout mice. Brain Res Mol Brain Res 76: 170-172. [DOI] [PubMed] [Google Scholar]
- Chieng B, Christie MD (1996) Local opioid withdrawal in rat single periaqueductal gray neurons in vitro J Neurosci 16: 7128-7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieng B, Christie MJ (1994) Hyperpolarization by opioids acting on mureceptors of a sub-population of rat periaqueductal gray neurones in vitro. Br J Pharmacol 113: 121-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claing A, Laporte SA, Caron MG, Lefkowitz RJ (2002) Endocytosis of G protein-coupled receptors: roles of G protein-coupled receptor kinases and beta-arrestin proteins. Prog Neurobiol 66: 61-79. [DOI] [PubMed] [Google Scholar]
- Commons KG (2003) Translocation of presynaptic delta opioid receptors in the ventrolateral periaqueductal gray after swim stress. J Comp Neurol 464: 197-207. [DOI] [PubMed] [Google Scholar]
- Commons KG, Beck SG, Rudoy C, Van Bockstaele EJ (2001) Anatomical evidence for presynaptic modulation by the delta opioid receptor in the ventrolateral periaqueductal gray of the rat. J Comp Neurol 430: 200-208. [PubMed] [Google Scholar]
- Connor M, Christie MJ (1998) Modulation of Ca2+ channel currents of acutely dissociated rat periaqueductal grey neurons. J Physiol (Lond) 509: 47-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor M, Schuller A, Pintar JE, Christie MJ (1999) Mu-opioid receptor modulation of calcium channel current in periaqueductal grey neurons from C57B16/J mice and mutant mice lacking MOR-1. Br J Pharmacol 126: 1553-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan A, Zhu XZ, Mosberg HI, Omnaas JR, Porreca F (1988) Direct dependence studies in rats with agents selective for different types of opioid receptor. J Pharmacol Exp Ther 246: 950-955. [PubMed] [Google Scholar]
- Gomes I, Jordan BA, Gupta A, Trapaidze N, Nagy V, Devi LA (2000) Heterodimerization of μ and δ opioid receptors: a role in opiate synergy. J Neurosci 20: RC110(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA (2004) A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci USA 101: 5135-5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack SP, Vaughan CW, Christie MJ (2003) Modulation of GABA release during morphine withdrawal in midbrain neurons in vitro. Neuropharmacology 45: 575-584. [DOI] [PubMed] [Google Scholar]
- Heyman JS, Mulvaney SA, Mosberg HI, Porreca F (1987) Opioid delta-receptor involvement in supraspinal and spinal antinociception in mice. Brain Res 420: 100-108. [DOI] [PubMed] [Google Scholar]
- Ingram SL, Vaughan CW, Bagley EE, Connor M, Christie MJ (1998) Enhanced opioid efficacy in opioid dependence is caused by an altered signal transduction pathway. J Neurosci 18: 10269-10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyuzhny AE, Wessendorf MW (1998) Relationship of mu- and delta-opioid receptors to GABAergic neurons in the central nervous system, including antinociceptive brainstem circuits. J Comp Neurol 392: 528-547. [PubMed] [Google Scholar]
- Kalyuzhny AE, Arvidsson U, Wu W, Wessendorf MW (1996) μ-Opioid and δ-opioid receptors are expressed in brainstem antinociceptive circuits: studies using immunocytochemistry and retrograde tract-tracing. J Neurosci 16: 6490-6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen I, Slowe SJ, Matthes HW, Kieffer B (1997) Quantitative autoradiographic mapping of mu-, delta- and kappa-opioid receptors in knockout mice lacking the mu-opioid receptor gene. Brain Res 778: 73-88. [DOI] [PubMed] [Google Scholar]
- Kovelowski CJ, Ossipov MH, Hruby VJ, Porreca F (1999) Lesions of the dorsolateral funiculus block supraspinal opioid delta receptor mediated antinociception in the rat. Pain 83: 115-122. [DOI] [PubMed] [Google Scholar]
- Law PY, Wong YH, Loh HH (2000) Molecular mechanisms and regulation of opioid receptor signaling. Annu Rev Pharmacol Toxicol 40: 389-430. [DOI] [PubMed] [Google Scholar]
- Lazarus LH, Bryant SD, Cooper PS, Salvadori S (1999) What peptides these deltorphins be. Prog Neurobiol 57: 377-420. [DOI] [PubMed] [Google Scholar]
- Melchiorri P, Maritati M, Negri L, Erspamer V (1992) Long-term sensitization to the activation of cerebral δ-opioid receptors by the deltorphin Tyr-D-Ala-Phe-Glu-Val-Val-Gly-NH2 in rats exposed to morphine. Proc Natl Acad Sci USA 89: 3696-3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson Jr MG, Reichardt LF, Barres BA (1998) Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 21: 681-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello JP, Petaja-Repo UE, Bichet DG, Bouvier M (2000) Pharmacological chaperones: a new twist on receptor folding. Trends Pharmacol Sci 21: 466-469. [DOI] [PubMed] [Google Scholar]
- Morinville A, Cahill CM, Esdaile MJ, Aibak H, Collier B, Kieffer BL, Beaudet A (2003) Regulation of δ-opioid receptor trafficking via μ-opioid receptor stimulation: evidence from μ-opioid receptor knock-out mice. J Neurosci 23: 4888-4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinville A, Cahill CM, Kieffer B, Collier B, Beaudet A (2004) Mu-opioid receptor knockout prevents changes in delta-opioid receptor trafficking induced by chronic inflammatory pain. Pain 109: 266-273. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Kovelowski CJ, Nichols ML, Hruby VJ, Porreca F (1995) Characterization of supraspinal antinociceptive actions of opioid delta agonists in the rat. Pain 62: 287-293. [DOI] [PubMed] [Google Scholar]
- Petaja-Repo UE, Hogue M, Bhalla S, Laperriere A, Morello JP, Bouvier M (2002) Ligands act as pharmacological chaperones and increase the efficiency of delta opioid receptor maturation. EMBO J 21: 1628-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman S (1990) Quantal analysis of synaptic potentials in neurons of the central nervous system. Physiol Rev 70: 165-198. [DOI] [PubMed] [Google Scholar]
- Rossi GC, Pasternak GW, Bodnar RJ (1994) mu and delta opioid synergy between the periaqueductal gray and the rostro-ventral medulla. Brain Res 665: 85-93. [DOI] [PubMed] [Google Scholar]
- Schuller AG, King MA, Zhang J, Bolan E, Pan YX, Morgan DJ, Chang A, Czick ME, Unterwald EM, Pasternak GW, Pintar JE (1999) Retention of heroin and morphine-6 beta-glucuronide analgesia in a new line of mice lacking exon 1 of MOR-1. Nat Neurosci 2: 151-156. [DOI] [PubMed] [Google Scholar]
- Shuster SJ, Riedl M, Li X, Vulchanova L, Elde R (1999) Stimulus-dependent translocation of κ opioid receptors to the plasma membrane. J Neurosci 19: 2658-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PE, Hammond DL (1993) Evidence for delta opioid receptor subtypes in rat spinal cord: studies with intrathecal naltriben, cyclic[D-Pen2, D-Pen5] enkephalin and [D-Ala2, Glu4]deltorphin. J Pharmacol Exp Ther 266: 820-828. [PubMed] [Google Scholar]
- Thorat SN, Hammond DL (1997) Modulation of nociception by microinjection of delta-1 and delta-2 opioid receptor ligands in the ventromedial medulla of the rat. J Pharmacol Exp Ther 283: 1185-1192. [PubMed] [Google Scholar]
- Vaughan CW, Christie MJ (1997) Presynaptic inhibitory action of opioids on synaptic transmission in the rat periaqueductal grey in vitro. J Physiol (Lond) 498: 463-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Christie MJ (1997) Actions of ORL1 receptor ligand, nociceptin, on membrane properties and synaptic transmission in rat periaqueductal gray neurons in vitro J Neurosci 17: 996-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Bagley EE, Drew GM, Schuller A, Pintar JE, Hack SP, Christie MJ (2003) Cellular actions of opioids on periaqueductal grey neurons from C57B16/J mice and mutant mice lacking MOR-1. Br J Pharmacol 139: 362-367. [DOI] [PMC free article] [PubMed] [Google Scholar]