

Abstract
Emerging evidence suggests that mitochondrial dysfunction contributes to the pathophysiology of epilepsy. Recurrent mitochondrial Ca2+ ion load during seizures might act on mitochondrial membrane potential (ΔΨm) and proton motive force. By using electrophysiology and confocal laser-scanning microscopy, we investigated the effects of epileptiform activity, as induced by low-Mg2+ ion perfusion in hippocampal slice cultures, on changes in ΔΨm and in mitochondrial Ca2+ ion concentration ([Ca2+]m).
The mitochondrial compartment was identified by monitoring ΔΨm in the soma and dendrites of patched CA3 pyramidal cells using the mitochondria-specific voltage-sensitive dye rhodamine-123 (Rh-123). Interictal activity was accompanied by localized mitochondrial depolarization that was restricted to a few mitochondria in small dendrites. In contrast, robust Rh-123 release into the cytosol was observed during seizure-like events (SLEs), indicating simultaneous depolarization of mitochondria. This was critically dependent on Ca2+ ion uptake and extrusion, because inhibition of the mitochondrial Ca2+ ion uniporter by Ru360 and the mitochondrial Na+/Ca2+ ion exchanger by 7-chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one but not the inhibitor of mitochondrial permeability transition pore, cyclosporin A, decreased the SLE-associated mitochondrial depolarization.
The Ca2+ ion dependence of simultaneous mitochondrial depolarization suggested enhanced Ca2+ ion cycling across mitochondrial membranes during epileptiform activity. Indeed, [Ca2+]m fluctuated during interictal activity in single dendrites, and these fluctuations spread over the entire mitochondrial compartment during SLEs, as revealed using mitochondria-specific dyes (rhod-2 and rhod-ff) and spatial frequency-based image analysis. These findings strengthen the hypothesis that epileptic activity results in Ca2+ ion-dependent changes in mitochondrial function that might contribute to the neuronal injury during epilepsy.
Keywords: mitochondria, epilepsy, hippocampus, calcium, patch clamp, imaging
Introduction
Epileptic activity is accompanied by large ionic shifts (Heinemann et al., 1986; Lux et al., 1986) that increase the demand for ATP to restore transmembrane ion gradients. Mitochondrial ATP synthesis depends on the proton motive force and consequently on the mitochondrial membrane potential (ΔΨm) (Mitchell, 1966). Neuronal activity attributable to electrical stimulation (Bindokas et al., 1998), seizure-like events (SLEs) (Kovács et al., 2001, 2002), and spreading depression (Bahar et al., 2000) results in changes of ΔΨm in brain tissue. Substantial ΔΨm loss may be a common feature of injurious processes, because it favors the opening of the mitochondrial permeability transition pore and the release of proapoptotic factors (Zoratti and Szabó, 1995). Moreover, ATP deficiency and disruption of the ionic gradients induce cell swelling and necrotic cell death (Ward et al., 2000; Krieger and Duchen, 2002). Therefore, seizure-induced changes in ΔΨm might contribute to cell death during and after status epilepticus.
The presence of Ca2+ ion-activated large-capacity mitochondrial Ca2+ ion-uptake pathways, the steep electrochemical gradient for Ca2+ ion, and the large Ca2+ ion-binding capacity of the mitochondrial matrix enable Ca2+ ion load of mitochondria, given that the free intracellular Ca2+ ion concentration ([Ca2+]i) is elevated above a set point of 150-300 nm (Babcock et al., 1997; Pitter et al., 2002). Such [Ca2+]i transients have been observed in the vicinity of plasmalemmal voltage-gated calcium channels or at the release sites of intracellular Ca2+ ion stores (Rizzuto et al., 1999; Szalai et al., 2000). Ca2+ ion cycling across mitochondrial membranes alters the kinetics of intracellular Ca2+ ion signaling (White and Reynolds, 1995; Colegrove et al., 2000a,b; Duchen, 2000; Nicholls and Budd, 2000) and allows sensitive regulation of oxidative ATP synthesis as a result of Ca2+ ion-dependent activation of mitochondrial dehydrogenases (McCormack and Denton, 1993; Robb-Gaspers et al., 1998; Jouaville et al., 1999; Kann et al., 2003a,b). However, extensive mitochondrial Ca2+ ion uptake and extrusion might dissipate ΔΨm (Nicholls and Budd, 2000; Scanlon et al., 2000), thereby decreasing the proton motive force and reducing ATP synthesis. Indeed, robust influx of Ca2+ ion into mitochondria, accompanied by a gradual loss of ΔΨm and a decrease in oxidized nicotinamide-adenine dinucleotide (NAD+) reduction, was observed during recurrent SLEs (Kovács et al., 2002). Because of the use of spatially averaged measures of ΔΨm and mitochondrial Ca2+ ion concentration ([Ca2+]m), the contribution of individual neurons to the signal and its subcellular localization remained unidentified.
To further explore the relationship between epileptiform activity, ΔΨm, and [Ca2+]m, we combined electrophysiology and confocal laser-scanning microscopy of mitochondria in the area CA3 of hippocampal slice cultures. ΔΨm was monitored using a voltage-sensitive mitochondrial dye, rhodamine-123 (Rh-123) (Schuchmann et al., 2000), whereas measurements of [Ca2+]m were performed using the mitochondria-specific Ca2+ ionsensitive dyes rhod-2 and rhod-ff (Kovács et al., 2001; Billups and Forsythe, 2002).
Materials and Methods
Hippocampal slice cultures. Organotypic hippocampal slice cultures were prepared and maintained as described previously (Stoppini et al., 1991; Kovács et al., 2001). In brief, 7- to 8-d-old Wistar rat pups were decapitated, and the brains were removed and rinsed in ice-cold gassed (95% O2, 5% CO2) minimal essential medium (MEM). Slices (400 μm thick) containing the entorhinal cortex and hippocampus were cut (McIllwain Tissue Chopper; Mickle Laboratory Engineering, Guildford, UK) and placed on a culture membrane (0.4 μm; Millicell-CM; Millipore, Eschborn, Germany) under sterile conditions. Slice cultures were used for experiments after 6-10 d in culture. One-half of the culture medium (containing 50% MEM, 25% HBSS, and 25% horse serum, pH 7.4; all from Invitrogen, Eggenstein, Germany) was replaced at every second day. Only slice cultures with intact anatomical structure were used for experiments.
Electrophysiology. A piece of culture membrane carrying a slice culture was placed in the recording chamber mounted on an epifluorescence microscope (BX50WI; Olympus, Budapest, Hungary) and perfused with gassed (95% O2, 5% CO2) artificial CSF (ACSF) containing the following (in mm): 129 NaCl, 3 KCl, 1.25 NaH2PO4, 1.8 MgSO4, 1.6 CaCl2, 21 NaHCO3, and 10 glucose, pH 7.4 (at ∼5 ml/min). Patch pipettes with 4-5 MΩ resistance were pulled by a vertical pipette puller (Narishige, Tokyo, Japan). Pipette solution contained the following (in mm): 135 potassium gluconate, 10 NaCl, 0.05 CaCl2, 1 EGTA, 10 HEPES, and 3 MgATP, pH 7.3 (270-290 mOsm). Visually identified CA3 pyramidal cells were recorded in the whole-cell mode with a patch-clamp amplifier, EPC 7, and WinTida 4.0 software (HEKA Elektronik, Lambrecht/Pfalz, Germany). After establishing the whole-cell configuration in voltage-clamp mode, we switched to current-clamp mode to allow the activation of voltage-gated calcium channels during epileptiform activity. Series resistance was 13 ± 1 MΩ (n = 36 cells). Series resistance and cell capacitance were not compensated. Additionally, an ACSF-filled patch pipette was placed either in the stratum radiatum or in the stratum oriens, in close proximity to the recorded cell to stimulate synaptic inputs. Input-output curves were obtained in normal ACSF with the same stimulation protocol (5 pulses at 100 Hz), which was also used for SLE induction. In normal ACSF, none of the measured cells showed epileptiform discharges spontaneously or after stimulation. Epileptiform activity was induced by switching the perfusion to ACSF that contained 5 mm KCl without added Mg2+ (low-[Mg2+] ACSF). In the case of [Ca2+]m measurements, an ACSF-filled recording electrode was put in the CA3 region, and field potential changes were simultaneously recorded with changes in the fluorescence of the mitochondria-specific Ca2+ ion-sensitive probes rhod-2 and rhod-ff. In the case of the whole-cell recordings, Ru360 (180 μm; Merck, Darmstadt, Germany) and 7-chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one (CGP-37157) (15 μm; Biotrend, Köln, Germany) were applied through the patch pipette, whereas carbonyl cyanide 3-chlorophenylhydrazone (CCCP) (10 μm in DMSO; final concentration, 0.1%), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) (10 μm in DMSO), and cyclosporin A (10 μm in DMSO) were added to the perfusion. In case of the field potential recording, slice cultures were preincubated with 50 μm Ru360 during the staining procedure, and 10 μm Ru360 was present in the perfusion during the entire experiment. Although this concentration already blocks mitochondrial Ca2+ ion uptake (Zhou and Bers, 2002), under whole-cell conditions, a higher Ru360 concentration was chosen to counterbalance the illumination-dependent decomposition of the ruthenium complex. If not noted otherwise, all chemicals were from Sigma (Deisenhofen, Germany).
Confocal laser-scanning microscopy-based imaging. Measurements of Rh-123 fluorescence were made with a Noran OZ confocal laser-scanning microscope system (Prairie Technologies, Middleton, WI). For excitation of Rh-123, the 488 nm line of a Kr-Ar laser (Omnichrom 643; Melles Griot, Bensheim, Germany) was used. The maximal laser power was 10 μW, as measured under the objective, which was dimmed to 20-30%. At this laser intensity, several (up to 10) 10-min-long recording periods could be made without signs of phototoxic damage (i.e., “bleb-bing” of dendrites). The fluorescence was measured with a 60× water-immersion objective (numeric aperture, 0.9). The pinhole was set to 100 μm for somatic and to 50 μm for dendritic fluorescence recordings. Averaged (8-16 frame) eight-bit pictures were taken at 2-1 Hz by using the acquisition software Intervision running on a Silicon Graphics (Mountain View, CA) workstation under Irix 6.3. Rh-123 (10 μg/ml) solved in EtOH (final concentration, 0.025-0.1%) was added to the pipette solution before the recording. To fasten dye loading, 20 μg/ml dye was used for dendritic fluorescence measurements. Even at this concentration, Rh-123 did not exert any overt changes on passive and active electrophysiological properties of the cells. By using a Fluoview 300 (Olympus) confocal microscope for Rh-123 fluorescence monitoring, the dye concentration was decreased to 2.5 μg/ml to prevent illumination-dependent superoxide formation (Buckman and Reynolds, 2001) attributable to the higher laser power of that microscope. Acquisition of Rh-123 fluorescence time series was synchronized with the recording of the membrane potential changes. After recording of a given number of time series, a z-scan was taken from the recorded cell for representation purposes.
To study changes in [Ca2+]m, slice cultures were bulk loaded with the AM ester of either rhod-2 (5 μm; Kd = 570 nm) or rhod-ff (5 μm; Kd = 19 μm) at 36.5°C for 30 min in serum-free culture medium, followed by a subsequent 30 min period for hydrolysis of the dye. Both dyes, in AM-ester form, carry a positive charge, which allows accumulation in negatively charged mitochondria, in which the ester bond is hydrolyzed. Because the hydrolysis might also occur in the cytosol, colocalization with mitochondria-specific fluorescent marker (MitoTracker Green; Molecular Probes, Leiden, The Netherlands) was done for differentiation between mitochondrial and cytosolic rhod-2 fluorescence. After washout, slice cultures were transferred to the recording chamber mounted on an Olympus BX61WI equipped with a Fluoview 300 confocal laser-scanning system. MitoTracker Green (5 μm) was loaded simultaneously with the Ca2+ ion-sensitive probes (n = 2 slice cultures each), and colocalization of both dyes was studied by sequential scanning at 488 and 543 nm while monitoring fluorescence through a 60× objective at 515 and 600 nm for MitoTracker Green and rhod-2 (or rhod-ff), respectively. Image acquisition and three-dimensional (3-D) reconstruction were made with the software Tiempo for Fluoview 300 (Olympus). All fluorescent probes were purchased from Molecular Probes.
Data evaluation. Each single-cell recording was performed in a different slice culture, and at least two batches prepared from four animals were used for each experimental set. Because of spatial limitations of the objective field, Rh-123 fluorescence measurement was performed either at the soma (31 cells) or at the dendrites (five cells). To compare somatic Rh-123 fluorescence changes, 12 cells were used for control, 9 for Ru360, 5 for CGP-57137, and 5 for cyclosporin A treatment. Rh-123 fluorescence time series were evaluated off-line by the NIH ImageJ 1.32 software. Mean fluorescence intensity was measured in selected regions of interest (ROIs) and is expressed after baseline correction as ΔF/F0 as a percentage. The ROIs were outlined by hand and enclosed either the brightly stained organelles identified as mitochondria or the entire soma, with the exception of the nucleus, thus representing both mitochondrial and cytosolic Rh-123 fluorescence. In the case of mitochondrial fluorescence monitoring, the ROI contained the mitochondrion and its immediate vicinity; therefore, dye release resulted in an increase in the mean fluorescence intensity. Rh-123 fluorescence changes obtained from the soma were chosen for statistical comparison, because changes in Rh-123 fluorescence in dendrites or in single mitochondria varied considerably, depending on baseline fluorescence level, length of dye loading, and distance from the culture surface. All values are given as mean ± SEM. For statistical comparison between groups, the Mann-Whitney U test was used. Z-direction movements (i.e., resulting from SLE-associated swelling of the tissue) were corrected on-line by refocusing. When necessary, focusing artifacts were removed from the ΔF/F0 traces off-line together with the values of previous out-of-focus frames. Speed of mitochondrial traffic was estimated by measuring the average time for crossing subsequent waypoints of the route.
For evaluation of epileptiform activity-associated changes in [Ca2+]m, seven and four slice cultures were used after bulk staining with either rhod-2 AM or rhod-ff AM, respectively. In contrast to Rh-123 fluorescence recordings, measurement of [Ca2+]m was complicated by the fact that rhod-ff and rhod-2 fluorescence originated partly from dye trapped in the cytosol and thereby reflected changes both in [Ca2+]i and [Ca2+]m. Therefore, we used the spatial frequency filtering-based method (Gerencsér and Ádám-Vizi, 2001) for differentiation of [Ca2+]i and [Ca2+]m signals. Colocalization of rhod-2 and rhod-ff with the mitochondria-specific probe MitoTracker Green revealed that the dotted rhod-2 and rhod-ff fluorescence pattern represented [Ca2+]m, whereas cytosolic fluorescence was homogeneous. Therefore, low spatial-frequency rhod-2 (or rhod-ff) fluorescence originated from dye trapped in the cytosol, whereas high spatial-frequency components represented mitochondria. The highest spatial-frequency pixel-to-pixel intensity variations, representing the photon shot noise superimposed on the image information, were also removed. Acquired images were transformed in the frequency domain by fast Fourier transform (FFT) macro of NIH ImageJ 1.32, multiplied with an appropriate filter function, and then reversed to the space domain by inverse FFT. The filter function was chosen empirically for each image stack, depending on the zoom factor. The remaining dotted fluorescence pattern represented [Ca2+]m. There was some unavoidable overlap between the two signals, i.e., out-of-focus blurred mitochondrial fluorescence, which may appear at low spatial frequencies, as well as cytosolic fluorescence from narrow dendrites and spines, which may appear at high spatial frequencies. To minimize signal cross talk, additional threshold masking was applied in some cases, which allowed the exclusion of any remaining cytosolic rhod-2 fluorescence. Changes in rhod-2 and rhod-ff fluorescence were measured in ROIs covering a single mitochondrion or small groups of mitochondria. For measurement of the amplitude and duration of the [Ca2+]m transients, 10-min-long periods of interictal activity or SLE were evaluated for each mitochondrion. The results are expressed as ΔF/F0 as a percentage. Because of the linear nature of FFT and inverse FFT, the relative amplitudes and time course of the fluorescence changes are not affected by the filtering procedure. In vitro calibration of the signal was not performed. Changes in [Ca2+]i were estimated by measuring fluorescence in ROIs placed over the nucleus, i.e., in areas in which cross talk between the two signals could not occur (Gerencsér and Ádám-Vizi, 2001).
Results
Epileptiform activity in CA3 pyramidal cells as induced by the low-[Mg2+] condition
Somatic whole-cell recordings were obtained in visually identified CA3 pyramidal cells (n = 36). Passive membrane properties (resting membrane potential, -65 ± 1.3 mV; input resistance, 171 ± 9 MΩ) and stimulus-induced responses (Fig. 1A) were determined before induction of epileptiform activity. After switching the perfusion to low-[Mg2+] ACSF, the membrane potential increased by 6.2 ± 0.4 mV (n = 36) within 10 min, and spontaneous epileptiform activity emerged (Fig. 1). Epileptiform activity was classified into three types, namely, interictal activity, SLEs, and late recurrent discharges, according to previous characterization of low-[Mg2+] ACSF-induced epileptiform activity (Mody et al., 1987; Dreier and Heinemann, 1991; Kovács et al., 1999, 2001).
Figure 1.

Stimulus-induced and spontaneous membrane potential changes in CA3 pyramidal cells. A, EPSP/IPSP sequence induced by a short stimulus train applied on the mossy fibers in normal (a) and in low-[Mg2+] (b) ACSF. Stimuli are indicated with a short bar below the traces, and the dotted line indicates the resting membrane potential of the cell. B, Interictal activity within a single CA3 neuron was represented by packets of synaptic inputs often resulting in a burst of action potentials. The discharge marked with an asterisk is seen to the right on a shorter time scale. C, SLEs consisted of an IBP, a TLP with high-frequency, small-amplitude oscillations (∼5 Hz) on a large depolarization shift, and a CLADP of variable length, while membrane potential slowly recovered to baseline or even hyperpolarized. D, The traces show the IBP of spontaneous (a) and a stimulus-induced (b) SLE (stimuli are indicated with a short bar below the traces). The traces on the right are from the TLP (c) and the CLADP (d) phases.
Interictal activity, as observed between SLEs, corresponded to spontaneous rhythmic synaptic activity consisting of short EPSP-IPSP sequences at a frequency of 0.2-0.7 Hz (Fig. 1B). Some of the EPSPs already reached spike threshold, and bursts of action potentials were superimposed on a short depolarization shift (Fig. 1B). SLEs represented the second major class of epileptiform activity (Fig. 1C). Although they also occurred spontaneously, SLEs were elicited by a short stimulus train applied to the neighboring neuropil, to minimize exposure to laser illumination. SLEs were characterized by three major phases: the initial bursting period (IBP), the tonic-like period (TLP), and clonic-like afterdischarges (CLADPs) (Kovács et al., 2001). The IBP started with a burst of action potentials during a slowly developing depolarization (Fig. 1Da,Db), which reached its maximum of 29.8 ± 0.8 mV (n = 90 SLE from 36 cells) during the TLP. Small oscillations occurred at this membrane potential, whereas action potentials were not present (Fig. 1Dc). In contrast to short depolarization shifts occurring during interictal activity, the depolarization during the TLP lasted 37.6 ± 2 s (n = 90 SLE from 36 cells). CLADPs consisted of a few to several hundred afterdischarges of decreasing amplitude and duration superimposed on a slowly developing repolarization of the plasma membrane (168 ± 21 s; n = 80 SLE from 31 cells) (Fig. 1C,Dd). Occasionally, the post-SLE hyperpolarization became reduced with a concomitant lengthening of the CLADPs of subsequent SLEs until transition to the late recurrent discharges occurred (data not shown) (but see Zhang et al., 1995; Kovács et al., 1999, 2002). Mitochondrial responses associated with late recurrent discharges were not evaluated because of the considerable variability in their onset.
The mitochondrial compartment of CA3 pyramidal cells
The mitochondria-specific voltage-sensitive dye Rh-123 was applied via the patch pipette. Within 5-10 min after establishing the whole-cell recording mode, the somata and the primary dendrites, but not the nuclear areas, became fluorescent, showing a bright, dotted staining pattern. After 15-25 min, fine dendritic structures appeared. Despite the fluorescence quenching within mitochondria (Nicholls and Ward, 2000) and the homogeneous cytosolic Rh-123 fluorescence, small (≤1 μm) mitochondria-like structures were well discernible both in the soma and in the dendrites (Figs. 2A-C, 6A,B). These structures might represent either a single mitochondrion (Collins et al., 2002) or small aggregates of several mitochondria (Müller et al., 2005). Rh-123 fluorescence intensity varied between mitochondria, apparently indicating differences in resting ΔΨm.
Figure 2.

Rh-123 staining pattern in CA3 pyramidal cells. A, A 3-D reconstruction of the mitochondrial compartment of an Rh-123-filled CA3 pyramidal cell after retraction of the patch pipette. The squares correspond to the images shown in B. B, A 3-D reconstruction of the apical dendrites (top) and the soma (bottom) of the same cell at higher magnification. Small mitochondria and larger mitochondrial aggregates were discernible in both the soma and the dendrites. C, A 3-D reconstruction of the mitochondrial compartment of another CA3 pyramidal cell containing rounded mitochondria.
Figure 6.
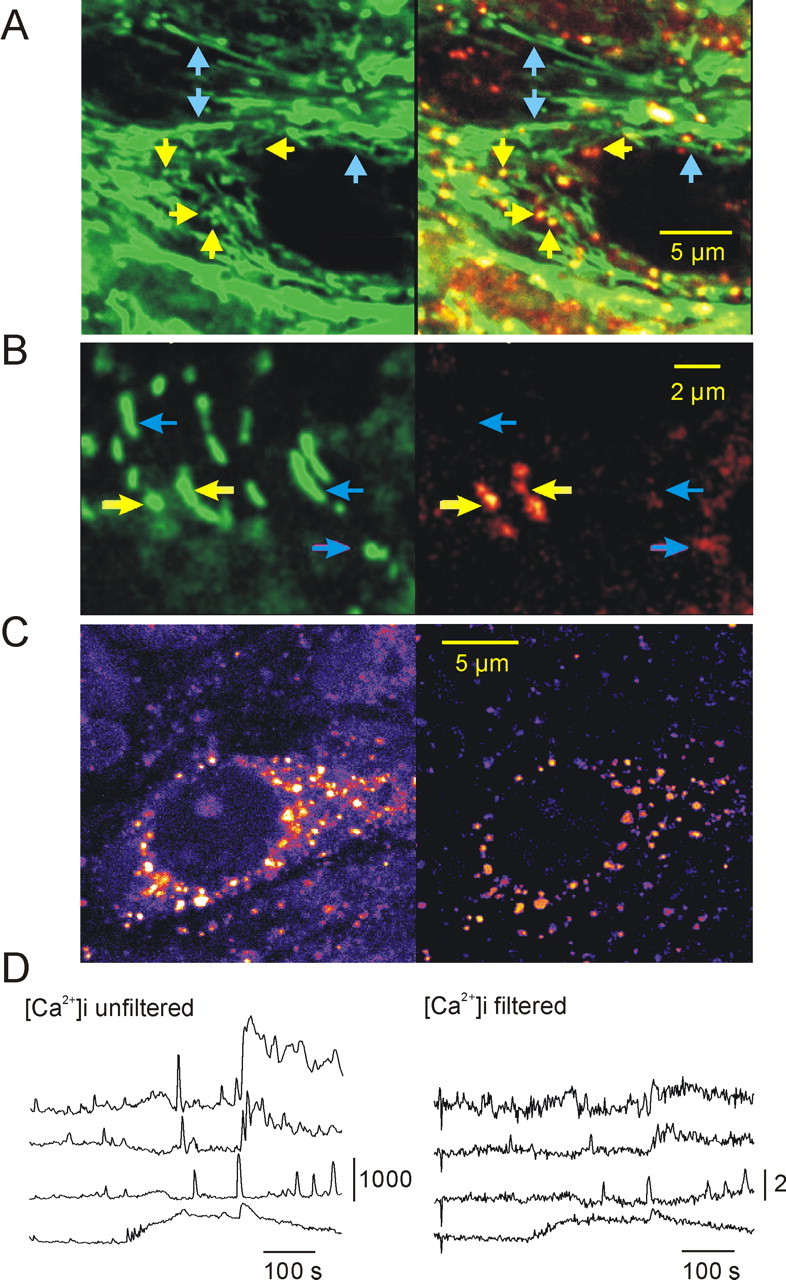
Differentiation between [Ca2+]m and [Ca2+]i in rhod-2-stained hippocampal slice cultures. A, Colocalization of the mitochondria-specific fluorescent marker MitoTracker Green (green fluorescence channel; A, B, left) and rhod-2 fluorescence (red fluorescence channel). On the superimposed picture (A, B, right), mitochondria with high resting [Ca2+]m are shown in yellow, and those with low [Ca2+]m are shown in green, whereas red represents the cytosolic Ca2+ ion level. A, B, Yellow and blue arrows point to mitochondria with high [Ca2+]m and low [Ca2+]m, respectively. B, MitoTracker Green-stained (green fluorescence channel) and rhod-2-stained (red fluorescence channel) mitochondria at a higher magnification. The high-[Ca2+]m spots within the elongated structures indicate that they may represent aggregates of several mitochondria. C, Spatial frequency-based image analysis (see Materials and Methods) for differentiation of mitochondrial and cytosolic fluorescence. The soma of a CA3 pyramidal cell (left) taken from the area CA3 of a rhod-2-stained slice culture is shown. The same cell is shown after bandpass filtering (right), displaying exclusively mitochondria. D, Effect of the bandpass filtering on the rhod-2 fluorescence transients. The traces on the left display [Ca2+]i transients as measured in the nucleus in four different cells during SLEs. The traces on the right represent [Ca2+]i changes measured in the same ROIs after filtering. To see the effect of bandpass filtering on the fluorescence intensity, the calibration bar of the traces represents absolute fluorescence intensity in arbitrary units (0-4096) instead of ΔF/F0 (supplemental time-lapse movies demonstrating the effect of filtering are available on-line at http://www.medichem.hu/mito/index.html).
Diffusion of the dye from the pipette resulted in a continuous increase in cytosolic Rh-123 fluorescence, which was corrected off-line (Materials and Methods). Although surrounding the neuropil, astrocytes and microglial cells were also stained during establishment of the seal, and their fluorescence vanished within a few minutes, thereby allowing selective measurements of ΔΨm in the patched neuron.
Mitochondria displayed two types of movement: Brownian motion and routed traffic within the soma and along the dendrites. Routed traffic either was continuous or consisted of a series of “rest-and-jump” sequences with an average speed of ∼0.35 ± 0.02 μm/s at 26°C (n = 34 mitochondria of seven slice cultures). Brownian motion of mitochondria resulted in small-amplitude fluctuations of the fluorescence baseline, regardless of the conditions (normal or low-[Mg2+] ACSF) or the mitochondrial dye used. Routed traffic resulted in fluorescence changes as mitochondria entered or left the focal plane. Thus, the analysis of ΔΨm and [Ca2+]m was restricted to mitochondria only displaying Brownian motion.
Changes in ΔΨm associated with interictal activity
Under control conditions, changes in Rh-123 fluorescence, other than fluctuations attributable to Brownian motion, were observed neither in the soma nor in the dendrites. During interictal activity, Rh-123 fluorescence remained unchanged in the soma (n = 12 cells) (Fig. 3A,B), whereas in small dendrites and spines, Rh-123 fluorescence fluctuations occurred, indicating mitochondrial depolarization (n = 5 cells) (Fig. 3A-D). Because ROIs contained the mitochondrion and its immediate vicinity, dye release and subsequent unquenching of Rh-123 manifested in an increase in mean fluorescence intensity (Materials and Methods). Detailed analysis of Rh-123 fluorescence transients during interictal activity in dendrites was performed in two cells. Rh-123 fluctuations occurred simultaneously in mitochondria within a dendrite and occasionally in adjacent dendrites (Fig. 3B). However, some of the Rh-123 fluorescence fluctuations were restricted to a single mitochondrion, and even neighboring mitochondria could express independent depolarization (Fig. 3D). The Rh-123 fluorescence transients (61 ± 9%) lasted for 14 ± 1 s (n = 56 mitochondria of two cells), thereby comprising a period of interictal activity (∼900 ms) (Fig. 3B,D). Because interictal activity was measured at the soma, it represented the sum of synaptic input arriving at the entire dendritic arbor, whereas fluorescence measurements in a particular dendrite revealed ΔΨm fluctuations on localized synaptic events and conductance changes. Although reliable calibration of ΔΨm is hard to perform in slice preparations, the fact that mitochondria underwent several depolarization-repolarization cycles without any decrease in their fluorescence suggested that ΔΨm loss was only partial.
Figure 3.

ΔΨm fluctuations during interictal activity. A, A 3-D reconstruction of an Rh-123-filled CA3 pyramidal cell, with bright spots indicating mitochondria. Note that the sensitivity of the detection was increased in A and C to visualize cellular morphology. The patch pipette is marked with “P,” and the squares correspond to the higher-magnification images on the right. The fluorescence fluctuations of the numbered mitochondria in the dendrites (top) and in the soma (bottom) are shown in B. B, Rh-123 fluorescence fluctuations in the soma and dendrites during interictal activity. Short interictal discharges (top) were associated with fluctuations of Rh-123 fluorescence in the dendrites (middle) but not in the soma (bottom). Note that fluctuations coincide in adjacent dendrites (mitochondria labeled 2 and 3), whereas independent fluctuations may occur in remote dendrites (mitochondrion labeled 1). C, A 3-D reconstruction of an Rh-123-filled CA3 pyramidal cell, the square corresponds to the higher-magnification image on the right. The pseudocolor image on the right represents the part of the basal dendrites in which fluorescence monitoring was done. Fluorescence changes in the dendrites marked with 1 and 2 are presented in D (supplemental time-lapse movie is available on-line at http://www.medichem.hu/mito/index.html). D, Coincidence of the mitochondrial Rh-123 fluorescence fluctuations during interictal activity (top). Three adjacent mitochondria from two neighboring dendrites (1 and 2) were compared, and the dotted line indicates individual Rh-123 fluorescence transients. a, Rh-123 fluorescence transient occurring in dendrite 1, but not in dendrite 2; b, c, Rh-123 fluorescence transients, which remained localized around a single mitochondrion; d, Rh-123 fluorescence transients occurring simultaneously in both dendrites.
Changes in ΔΨm associated with SLEs
In the same dendrite, in which localized Rh-123 fluorescence fluctuations were observed during interictal activity, a homogeneous overall increase in cytosolic Rh-123 fluorescence occurred during an SLE (Fig. 4A), indicating dye release from mitochondria. Similarly, cytosolic Rh-123 fluorescence, as measured at the soma, increased slowly during the IBP, followed by a rapid increase, which reached its maximum of 69.7 ± 16% (n = 30 SLEs recorded in 12 cells) at the end of the TLP. Subsequently, the Rh-123 fluorescence slowly recovered during CLADPs (Fig. 4B, C). To give a relative estimation of the change in ΔΨm, we compared SLE-associated cytosolic Rh-123 elevation with the effect of protonophore application at the end of an SLE sequence. The protonophores FCCP and CCCP were shown to completely dissipate ΔΨm and to increase the intensity of cytosolic Rh-123 fluorescence (Duchen, 1992; Schuchmann et al., 2000; Kovács et al., 2001). Application of a protonophore resulted in an overall increase in the cytosolic Rh-123 fluorescence by 132 ± 9% (n = 3 cells), whereby fluorescence of single mitochondria disappeared (Fig. 5D). Because mitochondrial fluorescence did not vanish during recurrent SLEs, loss of ΔΨm should have been only partial.
Figure 4.

Changes in ΔΨm during seizure-like events. A, Rh-123 fluorescence changes in dendrites during transition from interictal activity to SLE from the same cell shown in Figure 3A. Independent Rh-123 fluorescence fluctuations (bottom) during interictal activity were followed by a simultaneous increase in the fluorescence. Note that the maximal depolarization of mitochondrial membranes occurred during the CLADP. B, An Rh-123-filled pyramidal cell indicating the position and shape of the cytosolic (Cy) and mitochondrial (1 and 2) ROIs, used for Rh-123 fluorescence monitoring in C and D. C, SLE-associated changes in Rh-123 fluorescence, as measured in the cytosol and in mitochondria. Rh-123 fluorescence in the cytosol increased to a plateau during the TLP, followed by a slow recovery during the CLADP (supplemental time-lapse movie is available on-line at http://www.medichem.hu/mito/index.html). Mitochondrial fluorescence followed the same pattern (2), although the relative amplitude was smaller as basal mitochondrial fluorescence intensity (F0) was higher. D, Changes in Rh-123 fluorescence in somatic mitochondria after five SLEs. In 1 of 17 cases, a few mitochondria underwent abrupt depolarization during an SLE, as revealed by the sudden large-amplitude elevation of Rh-123 fluorescence.
Figure 5.

Dependence of ΔΨm changes on mitochondrial Ca2+ ion cycling and opening of the mitochondrial permeability transition pore. A, Representative trace of Rh-123 fluorescence changes during an SLE in the presence of Ru360 (180 μm) in the pipette solution. Inhibition of the mitochondrial Ca2+ ion uniporter prevented elevation of cytosolic Rh-123 fluorescence. B, Representative trace of Rh-123 fluorescence changes during an SLE in the presence of the mitochondrial Na+/Ca2+ ion exchanger inhibitor CGP-37157 (15 μm) in the pipette solution. CGP-37157 decreased SLE-associated Rh-123 elevation. C, Representative trace of Rh-123 fluorescence elevation in the presence of the mitochondrial permeability transition pore inhibitor cyclosporin A in the perfusion. Note the lengthening of the CLADP. D, Comparison of the effects of mitochondrial Ca2+ ion cycling and mitochondrial permeability transition pore inhibitors as well as protonophores on the mitochondrial depolarization during SLEs. Application of a protonophore (a; n = 3 cells), SLE-associated elevations (b; n = 30 SLEs from 12 cells), SLE-associated changes in the presence of Ru360 (c; n = 19 SLEs from 9 cells), application of a protonophore in the presence of Ru360 (d; n = 6 cells), SLE-associated changes in the presence of CGP-37157 (e; n = 12 SLEs from 5 cells), and SLE-associated elevations in the presence of cyclosporin A (f; n = 10 SLEs from 5 cells). Asterisks indicate significant differences. Error bars represent SEM.
In most cases, the time course and amplitude of cytosolic and mitochondrial fluorescence elevations were rather similar (Fig. 4B, C). However, in 1 of 17 cells, few mitochondria displayed an abrupt increase in Rh-123 fluorescence (Fig. 4B,D), indicating stronger depolarization.
Involvement of Ca2+ ion uptake and extrusion in changes of ΔΨm
It was shown that changes in ΔΨm evoked by a prolonged (15 s) depolarization step above the threshold of -40 mV were dependent on Ca2+ ion influx through voltage-gated calcium channels in pyramidal cells (Schuchmann et al., 2000). Therefore, we hypothesized that the loss of ΔΨm during SLE was dependent on an increase in [Ca 2+]i. There may be at least three mechanisms by which a [Ca 2+]i rise can lead to mitochondrial depolarization, i.e., the mitochondrial Ca 2+ ion uptake via the Ca2+ ion uniporter, which represents the major pathway of Ca2+ ion uptake into mitochondria, the Ca2+ ion extrusion via the electrogenic mitochondrial Na+/Ca2+ ion exchanger, and the Ca2+ ion-induced opening of the mitochondrial permeability transition pore (Zoratti and Szabó, 1995; Gunter et al., 2004).
To test for the first possibility, Ru360 (180 μm), a selective inhibitor of the mitochondrial Ca2+ ion uniporter (Zhou and Bers, 2002), was added to the patch solution, allowing diffusion of Ru360 to mitochondria before induction of epileptiform activity. Ru360 completely inhibited the SLE-associated Rh-123 fluorescence elevations (-0.7 ± 3 vs 69.7 ± 16%; p < 0.01; n = 19 SLEs in 9 Ru360-treated cells vs n = 30 SLEs in 12 control cells) (Fig. 5A,D). In some cases, even a deflection of the fluorescence baseline occurred during SLEs, indicating plasma-membrane depolarization-dependent Rh-123 release from the cytosol and/or dye dilution attributable to swelling of the cell. The Ca2+ ion uniporter inhibitors, such as ruthenium red and Ru360, can quench the fluorescence of Rh-123. Although Ru360 is less colored than ruthenium red, we tested whether unspecific fluorescence quenching masked the mitochondrial depolarization. Bath application of a protonophore (10 μm) for 1 min after an SLE sequence elevated cytosolic Rh-123 fluorescence by 83 ± 25% (n = 6 cells), with simultaneous disappearance of mitochondrial fluorescence, clearly indicating that fluorescence quenching did not prevent the detection of ΔΨm changes. Thus, the inhibitory effect of Ru360 on the SLE-associated Rh-123 fluorescence elevations was attributable to the blockade of the mitochondrial Ca2+ ion uniporter.
The mitochondrial Na+/Ca2+ ion exchange, the main pathway for Ca2+ ion extrusion, mediates electrogenic transport of Ca2+ ions (Cox et al., 1993; Wang and Thayer, 2002). The mitochondrial Na+/Ca2+ ion exchanger is different from the Na+/Ca2+ ion exchanger of the plasma membrane and can be selectively blocked by CGP-37157 (White and Reynolds, 1995; Scanlon et al., 2000). Application of CGP-37157 (15 μm) through the patch pipette resulted in a significant reduction of Rh-123 fluorescence elevation during SLEs, occasionally followed by a slow increase in the baseline, likely because of mitochondrial Ca2+ ion accumulation-dependent depolarization (9.9 ± 3 vs 69.7 ± 16%, p = 0.01; n = 12 SLEs in 5 CGP-37157-treated cells vs n = 30 SLEs in 12 control cells) (Fig. 5B,D).
To test whether mitochondrial depolarization during epileptiform activity was attributable to Ca2+ ion-dependent opening of the mitochondrial permeability transition pore, we added cyclosporin A (10 μm), an inhibitor of pore opening (Zoratti and Szabó, 1995; White and Reynolds, 1996), continuously with the perfusion with low-[Mg2+] ACSF. Cyclosporin A did not significantly alter the Rh-123 fluorescence elevations during SLEs (74.1 ± 23 vs 69.7 ± 16% p = 0.7; n = 10 SLEs in 5 cyclosporin A-treated cells vs n = 30 SLEs in 12 control cells) (Fig. 5C,D), suggesting that opening of the mitochondrial permeability pore did not contribute to SLE-associated mitochondrial depolarization. The abrupt elevation of Rh-123 fluorescence in a few mitochondria, observed in 1 of 17 cases under low-[Mg2+] perfusion, did not occur in the presence of cyclosporin A. It is noteworthy that the CLADP was lengthened, and the transition to late recurrent discharges occurred after 1-2 SLEs under this condition.
Temporal and spatial pattern of [Ca2+]m fluctuations during epileptiform activity
The observation that interictal activity as well as SLEs were associated with Ca2+ ion-dependent mitochondrial depolarization suggested that mitochondria might accumulate Ca2+ ions during epileptiform activity. We therefore monitored [Ca2+]m by using the mitochondria-specific Ca2+ ion-sensitive fluorescent dye rhod-2 AM in bulk-stained slice cultures (Kovács et al., 2001; Kann et al., 2003a,b).
The pattern of rhod-2 fluorescence was more complex than that of Rh-123. Although similar mitochondria-like organelles appeared in both the soma and dendrites, a considerable part of the dye remained in the cytosol. Colocalization with MitoTracker Green identified the bright spots as rounded mitochondria. However, several MitoTracker Green-labeled mitochondria were not stained by rhod-2, suggesting differences either in resting [Ca2+]m or in the dye-loading capability of cells (Fig. 6A). Occasionally, bright rhod-2 spots were localized within filaments as visualized by MitoTracker Green. At a higher magnification, these structures appeared as aggregates of several mitochondria (Fig. 6B).
Because of the cytosolic fraction of rhod-2, we applied spatial frequency-based image analysis (Gerencsér and Ádám-Vizi, 2001) to clearly differentiate between mitochondrial and cytosolic fluorescence changes (Fig. 6C,D). Neuronal [Ca2+]i transients did not occur in normal ACSF. In low-[Mg+] ACSF, both dendritic [Ca2+]i and [Ca2+]m transients appeared together with the development of epileptiform activity. During interictal activity, only a few mitochondria expressed [Ca2+]m transients (80 ± 12%; 14 ± 0.7 s; n = 36 mitochondria; seven slice cultures), and the signal averaged from several mitochondria showed only small-amplitude fluctuations (Fig. 7A,B). At the onset of an SLE, [Ca2+]i increased almost instantaneously in neighboring cells, and the transients followed the temporal pattern of the SLE, particularly during the CLADP (Fig. 7A,B). Also, [Ca2+]m transients, similar to those observed during interictal activity (104 ± 12%; 13 ± 0.8 s; n = 25 mitochondria; seven slice cultures), appeared in most of the mitochondria (Fig. 7C). Bath application of Ru360 (10 μm) together with low-[Mg2+] ACSF did not affect the SLE-associated [Ca2+]i transients, but [Ca2+]m transients were absent (n = 38 mitochondria; two slice cultures) (Fig. 7D). Although low-amplitude fluctuations of mitochondrial rhod-2 fluorescence signal could be still observed, they were not associated with epileptic discharges and likely reflected Brownian motion. Because the affinity of rhod-2 is relatively high (Kd = 570 nm), it might underestimate large [Ca2+]m transients (Billups and Forsythe, 2002). Therefore, its low-affinity derivative, rhod-ff (Kd = 19 μm) was applied in another set of experiments (n = 4 slice cultures; data not shown). Rhod-ff gave the same staining and colocalization pattern as rhod-2, allowing the use of spatial frequency-based image analysis. Rhod-ff fluorescence transients in individual mitochondria were similar to those observed using rhod-2 during interictal activity as well as SLEs, suggesting that Ca2+ ion buffering by the fluorescent probe did not significantly alter the time course of [Ca2+]m transients.
Figure 7.
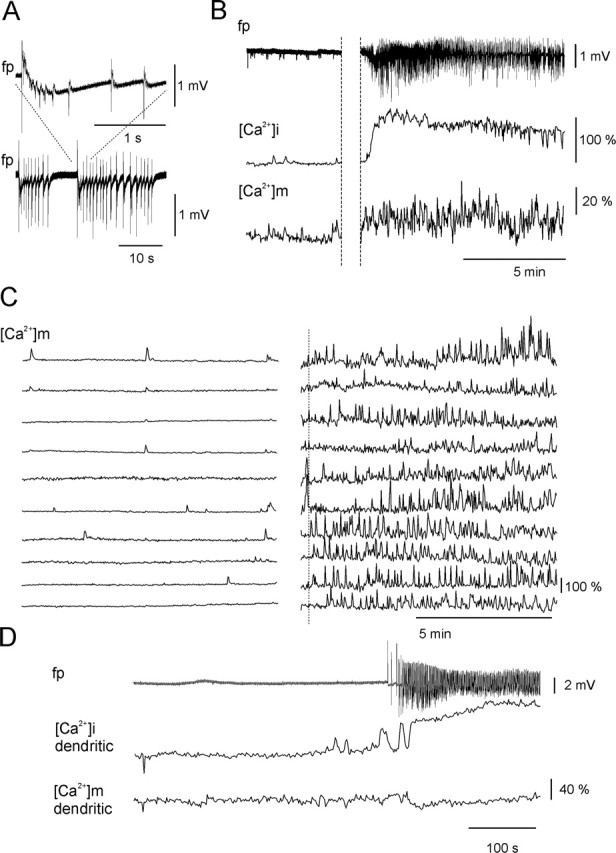
Changes in rhod-2 fluorescence during SLEs. A, Field potential (fp) recording during SLEs revealed recurrent synchronous population discharges. B, Time course of field potential, [Ca2+]i, and [Ca2+]m transients during interictal activity (left) and during an SLE (right). The [Ca2+]i accumulation was associated with an increase in the amplitude of the [Ca2+]m fluctuation (averaged from the 10 mitochondria shown in C) during SLE. C, [Ca2+]m fluctuations as measured in individual mitochondria in the soma and dendrites during interictal activity (traces on the left) and an SLE (traces on the right). The onset of the SLE is marked with a dotted line (supplemental time-lapse movies are available on-line at http://www.medichem.hu/mito/index.html). D, Block of [Ca2+]m transients by Ru360. Blockade of the mitochondrial Ca2+ ion uniporter by Ru360 left the dendritic [Ca2+]i transients unaltered (middle trace), whereas SLE-associated [Ca2+]m transients were absent in the mitochondria from the same dendrite (bottom trace). The remaining fluctuations were solely attributable to mitochondrial Brownian motion. Ru360 was continuously present in the perfusion.
Discussion
The present findings demonstrate a strong correlation between epileptiform activity and changes in ΔΨm and [Ca2+]m. During interictal activity, localized ΔΨm fluctuations in dendritic mitochondria occurred, which developed into a widespread synchronous mitochondrial depolarization during SLEs both in dendrites and in the soma. Mitochondrial depolarization was critically dependent on Ca2+ ion cycling across mitochondrial membranes, but not on opening of the permeability transition pore. This was also supported by the finding that [Ca2+]m transients occurred in a few mitochondria during interictal activity, whereas a large part of the mitochondrial compartment expressed [Ca2+]m transients during SLEs. Because synchronized loss of ΔΨm will reduce the formation of ATP, and because low ΔΨm together with increased [Ca2+]m favors mitochondrial free radical formation, these events might significantly contribute to the pathophysiology of epilepsy.
Morphological and functional heterogeneity of the mitochondrial compartment
Despite their omnipresence in presynaptic and postsynaptic structures and their important role in cell physiology (Shepherd and Harris, 1998; Duchen, 2000; Nicholls and Budd, 2000), investigation of mitochondria is hampered because of their relative inaccessibility for experimental procedures (Rizzuto et al., 1992; Colegrove et al., 2000a,b), particularly in brain-slice preparations (Schuchmann et al., 2000). To overcome these difficulties, we combined confocal laser-scanning microscopy and patch-clamp or field potential recordings to study the effects of epileptiform activity on mitochondrial function in hippocampal slice cultures.
Monitoring ΔΨm and [Ca2+]m as well as visualization with MitoTracker Green showed that the mitochondrial compartment of CA3 neurons is heterogeneous. High-[Ca2+]m spots mostly corresponded to rounded mitochondrial structures, whereas mitochondrial filaments had rather low [Ca2+]m and expressed weak Rh-123 fluorescence. Interestingly, [Ca2+]m spots were also found within or at the end of a filament, indicating that the latter might be composed of several mitochondria. We cannot distinguish at present whether these structures represent individual mitochondria or chains and aggregates of several mitochondria (Collins et al., 2002; Müller et al., 2005). Regardless of their morphology, however, the differences in ΔΨm and [Ca2+]m may indicate functional differences. This has been shown previously for motoneurons in crayfish, in which ΔΨm differed between mitochondria in axons of tonic and phasic motoneurons, depending on their workload (Nguyen et al., 1997). Alternatively, the differences might represent a Ca2+ ion-dependent “thread-to-grain” transition of mitochondria, as involved in aging and apoptosis (Rintoul et al., 2003; Skulachev et al., 2004).
The presence of independent ΔΨm and [Ca2+]m fluctuations in neighboring mitochondria strongly suggested that mitochondria in hippocampal CA3 cells are electrically and spatially discontinuous, which is in line with previous observations in different cell types (Collins et al., 2002; Collins and Bootman, 2003). As a functional consequence, mitochondrial Ca2+ ion uptake and ATP synthesis may adapt to local requirements, which is of importance in structures with high ion-channel density and ion-transport activity (i.e., dendrites and spines) (Billups and Forsythe, 2002; Zenisek and Matthews, 2000).
[Ca2+]i, ΔΨm, and [Ca2+]m during interictal activity
In contrast to the synaptic activity during control condition, interictal activity was associated with [Ca2+]i, [Ca2+]m, and ΔΨm transients, which were often restricted to a single dendrite or an individual branch. Dendritic [Ca2+]i transients can originate from synaptic events (Kovalchuk et al., 2000), back-propagating action potentials (Magee and Johnston, 1997), or release from endoplasmatic reticulum stores (Nakamura et al., 2000). Mitochondria are involved in shaping Ca2+ ion signaling either by Ca2+ ion buffering or by ATP supply to Ca2+ ion pumps (Zenisek and Matthews, 2000; Billups and Forsythe, 2002), which might in turn influence ΔΨm (Duchen, 2000). In the present experiments, ΔΨm fluctuations to physiological [Ca2+]i transients were never observed under control condition. However, small [Ca2+]i transients could have been partially suppressed by the presence of the Ca2+ ion buffer (EGTA) in the patch solution (Kovalchuk et al., 2000). During interictal activity, Ca2+ ion conductance (de Curtis et al., 1999; Empson and Jefferys, 2001) and synaptic Ca2+ ion flux are enhanced (Mody et al., 1987; Gloveli et al., 1999), which allow the build up of high-[Ca2+]i domains in narrow dendrites and spines. The subsequent Ca2+ ion uptake of mitochondria may be responsible for the ΔΨm fluctuations in the dendrites. In contrast, ΔΨm transients of somatic mitochondria were not observed during interictal activity. This can be explained by their greater distance to the plasma membrane and the high-Ca2+ ion buffer capacity of the cytosol. Alternatively, detection of mitochondrial depolarization in the soma could have been prevented by immediate dilution of released Rh-123 and/or reuptake by neighboring mitochondria. These mechanisms may be more pronounced compared with dendrites with a lower density of mitochondria and smaller cytosolic compartment.
Although ΔΨm fluctuations seemed to depend on interictal activity, there was not a strict correlation between locally measured ΔΨm transients and short membrane potential changes measured at the soma, suggesting that synaptic input during interictal activity was less synchronous.
[Ca2+]i, ΔΨm, and [Ca2+]m during SLEs
In contrast to interictal activity, SLEs were associated with an almost simultaneous mitochondrial depolarization in the soma and primary dendrites, indicating a common trigger mechanism. During the TLP and single discharges of the CLADP, plasma membrane depolarized for several tens of seconds to values near -30 mV, thereby allowing activation of voltage-gated calcium channels. Consequently, an increase in [Ca2+]i may be causal to widespread synchronized depolarization of mitochondria. Concomitantly, large-amplitude [Ca2+]m transients appeared and the amplitude of averaged [Ca2+]m signal increased, suggesting coupling of mitochondria during epileptic discharges. In contrast to [Ca2+]m, widespread mitochondrial depolarization did not display fluctuations and outlasted the entire SLE, indicating that mechanisms underlying mitochondrial depolarization are continuously active during the SLE.
These findings are in accordance with our previous observations on SLE-associated large fluctuating Ca2+ ion influx in hippocampal slice cultures and concomitant mitochondrial depolarization (Kovács et al., 2000, 2001). Mutual information analysis of field potential, extracellular [Ca2+], and [Ca2+]i transients revealed that these parameters became associated in the course of recurrent epileptiform activity (Kovács et al., 2000). The present finding substantiates a role of mitochondrial Ca2+ ion cycling in shaping intracellular Ca2+ ion signaling during SLEs, as proposed in our model of recurrent Ca2+ ion dynamics (Szilágyi et al., 2000).
Ca2+ ion cycling-dependent loss of ΔΨm
Although the coincidence of [Ca2+]m fluctuations and mitochondrial depolarization suggests that the electrogenic Ca2+ ion cycling might be responsible for mitochondrial depolarization, other mechanisms, i.e., an increase in the ADP/ATP and the NAD+/NADH ratio, may also influence metabolic activity and thereby ΔΨm (Cortassa et al., 2003). Selective inhibition of mitochondrial Ca2+ ion uptake (Ru360) and extrusion (CGP-37157) clearly indicated that both processes determined mitochondrial depolarization during SLEs and suggested that the activation of both processes was necessary to maintain [Ca2+]m transients. This is in line with our observation that [Ca2+]m is characterized by fluctuating dynamics rather than by a lasting elevation during SLEs.
The opening of the mitochondrial permeability transition pore plays a substantial role in NMDA-induced mitochondrial depolarization (White and Reynolds, 1996). Nevertheless, our experiments with cyclosporin A suggested that pore opening did not contribute to mitochondrial depolarization during SLEs. In contrast, the sudden loss of ΔΨm in individual mitochondria, which was observed in one case after a sequence of five recurrent SLEs, but not in the presence of cyclosporin A, might have been mediated by opening of the permeability transition pore. In that case, the opening would have been promoted by enhanced formation of reactive oxygen species, which has been reported for laser illumination of high Rh-123 concentrations in mitochondria (Buckman and Reynolds, 2001; Collins et al., 2002).
We also observed a prolongation of the CLADP in the presence of cyclosporin A, which is in line with the reported epileptogenic effect of low doses of cyclosporin A (Gorji et al., 2002), particularly under low-[Mg2+] conditions (Wong and Yamada, 2000). However, this is likely attributable to its inhibitory effects on calcineurin and subsequent modulation of GABAA currents (Jones and Westbrook, 1997) rather than on the blockade of permeability transition pore.
Consequences of synchronized mitochondrial depolarization
Although mild decreases in ΔΨm during interictal activity could be counterbalanced by an increase in ΔpH to maintain proton motive force, large ΔΨm loss during SLEs most probably disturbs this homeostasis. As a consequence, ATP synthesis cannot keep pace with the increased ATP demand of the ion pumps, which increase seizure susceptibility and compromise termination of a seizure (McNamara, 1994; Vaillend et al., 2002). Although mild depolarization decreases the mitochondrial superoxide radical formation at complex III (Papa and Skulachev, 1997; Nicholls and Budd, 2000), ΔΨm-independent free radical formation at complex I (Votyakova and Reynolds, 2001) can be enhanced by high [Ca2+]m (Gyulkhandanyan and Pennefather, 2004). Therefore, enhanced mitochondrial Ca2+ ion cycling, low ΔΨm values, and subsequent oxidative stress during seizures might lead to chronic mitochondrial and neuronal damage (Kovács et al., 2001, 2002; Vergun et al., 2003).
Footnotes
This work was supported by “Training and Excellence” Grant ICA1-CT-2002-70007, Center of Excellence on Biomolecular Chemistry Grant QLK2-CT-2002-90436, MU00025/2002, Hungarian Scientific Research Fund (OTKA)-F043589, OTKA-T035225, Economic Competitiveness Operational Program 3.1.1.-2004-05-0068/3.0, 1/A/005/2004 National Office for Research and Technology MediChem2, and Deutsche Forschungsgemeinschaft Grants Sonderforschungsbereiche (SFB) 507 and SFB TR3. We thank Dr. Sebastian Schuchmann for technical advice and Dr. Siegrun Gabriel for critical reading of this manuscript.
Correspondence should be addressed to Dr. Richard Kovács, Department of Neurochemistry, Institute of Biomolecular Chemistry, Chemical Research Center, Hungarian Academy of Sciences, Pusztaszeri út 59-67, 1025 Budapest, Hungary. E-mail: mozsafa@gmx.net.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254260-10$15.00/0
References
- Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B (1997) Mitochondrial participation in the intracellular Ca2+ network. J Cell Biol 136: 833-844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar S, Fayuk D, Somjen GG, Aitken PG, Turner DA (2000) Mitochondrial and intrinsic optical signals imaged during hypoxia and spreading depression in rat hippocampal slices. J Neurophysiol 84: 311-324. [DOI] [PubMed] [Google Scholar]
- Billups B, Forsythe ID (2002) Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci 22: 5840-5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindokas VP, Lee CC, Colmers WF, Miller RJ (1998) Changes in mitochondrial function resulting from synaptic activity in the rat hippocampal slice. J Neurosci 18: 4570-4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckman JF, Reynolds IJ (2001) Spontaneous changes in mitochondrial membrane potential in cultured neurons. J Neurosci 21: 5054-5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegrove SL, Albrecht MA, Friel DD (2000a) Dissection of mitochondrial Ca2+ uptake and release fluxes in situ after depolarization-evoked [Ca2+]i elevations in sympathetic neurons. J Gen Physiol 115: 351-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegrove SL, Albrecht MA, Friel DD (2000b) Quantitative analysis of mitochondrial Ca2+ uptake and release pathways in sympathetic neurons. Reconstruction of the recovery after depolarization-evoked [Ca2+]i elevations. J Gen Physiol 115: 371-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins TJ, Bootman MD (2003) Mitochondria are morphologically heterogeneous within cells. J Exp Biol 206: 1993-2000. [DOI] [PubMed] [Google Scholar]
- Collins TJ, Berridge MJ, Lipp P, Bootman MD (2002) Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J 21: 1616-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortassa S, Aon MA, Marban E, Winslow RL, O'Rourke B (2003) An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J 84: 2734-2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DA, Conforti L, Sperelakis N, Matlib MA (1993) Selectivity of inhibition of Na+-Ca2+ exchange of heart mitochondria by benzothiazepine CGP-37157. J Cardiovasc Pharmacol 21: 595-599. [DOI] [PubMed] [Google Scholar]
- de Curtis M, Radici C, Forti M (1999) Cellular mechanisms underlying spontaneous interictal spikes in an acute model of focal cortical epileptogenesis. Neuroscience 88: 107-117. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Heinemann U (1991) Regional and time dependent variations of low Mg2+ induced epileptiform activity in rat temporal cortex slices. Exp Brain Res 87: 581-596. [DOI] [PubMed] [Google Scholar]
- Duchen MR (1992) Ca2+-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem J 283: 41-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR (2000) Mitochondria and calcium: from cell signalling to cell death. J Physiol (Lond) 529: 57-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Empson RM, Jefferys JG (2001) Ca2+ entry through L-type Ca2+ channels helps terminate epileptiform activity by activation of a Ca2+ dependent afterhyperpolarisation in hippocampal CA3. Neuroscience 102: 297-306. [DOI] [PubMed] [Google Scholar]
- Gerencsér AA, Ádám-Vizi V (2001) Selective, high-resolution fluorescence imaging of mitochondrial Ca2+ concentration. Cell Calcium 30: 311-321. [DOI] [PubMed] [Google Scholar]
- Gloveli T, Egorov AV, Schmitz D, Heinemann U, Müller W (1999) Carbachol-induced changes in excitability and [Ca2+]i signalling in projection cells of medial entorhinal cortex layers II and III. Eur J Neurosci 11: 3626-3636. [DOI] [PubMed] [Google Scholar]
- Gorji A, Scheld HH, Speckmann EJ (2002) Epileptogenic effect of cyclosporine in guinea-pig hippocampal slices. Neuroscience 115: 993-997. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Yule DI, Gunter KK, Eliseev RA, Salter JD (2004) Calcium and mitochondria. FEBS Lett 567: 96-102. [DOI] [PubMed] [Google Scholar]
- Gyulkhandanyan AV, Pennefather PS (2004) Shift in the localization of sites of hydrogen peroxide production in brain mitochondria by mitochondrial stress. J Neurochem 90: 405-421. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Konnerth A, Pumain R, Wadman WJ (1986) Extracellular calcium and potassium concentration changes in chronic epileptic brain tissue. Adv Neurol 44: 641-661. [PubMed] [Google Scholar]
- Jones MV, Westbrook GL (1997) Shaping of IPSCs by endogenous calcineurin activity. J Neurosci 17: 7626-7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R (1999) Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci USA 96: 13807-13812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann O, Schuchmann S, Buchheim K, Heinemann U (2003a) Coupling of neuronal activity and mitochondrial metabolism as revealed by NAD(P)H fluorescence signals in organotypic hippocampal slice cultures of the rat. Neuroscience 119: 87-100. [DOI] [PubMed] [Google Scholar]
- Kann O, Kovács R, Heinemann U (2003b) Metabotropic receptor-mediated Ca2+ signaling elevates mitochondrial Ca2+ and stimulates oxidative metabolism in hippocampal slice cultures. J Neurophysiol 90: 613-621. [DOI] [PubMed] [Google Scholar]
- Kovács R, Gutierrez R, Kivi A, Schuchmann S, Gabriel S, Heinemann U (1999) Acute cell damage after low-Mg2+-induced epileptiform activity in organotypic hippocampal slice cultures. NeuroReport 10: 207-213. [DOI] [PubMed] [Google Scholar]
- Kovács R, Szilágyi N, Barabás P, Heinemann U, Kardos J (2000) Low-[Mg2+]-induced Ca2+ fluctuations in organotypic hippocampal slice cultures. NeuroReport 11: 2107-2111. [DOI] [PubMed] [Google Scholar]
- Kovács R, Schuchmann S, Gabriel S, Kardos J, Heinemann U (2001) Ca2+ signalling and changes of mitochondrial function during low-Mg2+-induced epileptiform activity in organotypic hippocampal slice cultures. Eur J Neurosci 13: 1311-1319. [DOI] [PubMed] [Google Scholar]
- Kovács R, Schuchmann S, Gabriel S, Kann O, Kardos J, Heinemann U (2002) Free radical-mediated cell damage after experimental status epilepticus in hippocampal slice cultures. J Neurophysiol 88: 2909-2918. [DOI] [PubMed] [Google Scholar]
- Kovalchuk Y, Eilers J, Lisman J, Konnerth A (2000) NMDA receptor-mediated subthreshold Ca2+ signals in spines of hippocampal neurons. J Neurosci 20: 1791-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger C, Duchen MR (2002) Mitochondria, Ca2+ and neurodegenerative disease. Eur J Pharmacol 447: 177-188. [DOI] [PubMed] [Google Scholar]
- Lux HD, Heinemann U, Dietzel I (1986) Ionic changes and alterations in the size of the extracellular space during epileptic activity. Adv Neurol 44: 619-639. [PubMed] [Google Scholar]
- Magee JC, Johnston D (1997) A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science 275: 209-213. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Denton RM (1993) Mitochondrial Ca2+ transport and the role of intramitochondrial Ca2+ in the regulation of energy metabolism. Dev Neurosci 15: 165-173. [DOI] [PubMed] [Google Scholar]
- McNamara JO (1994) Cellular and molecular basis of epilepsy. J Neurosci 14: 3413-3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P (1966) Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Bodmin, UK: Glynn Research. [DOI] [PubMed]
- Mody I, Lambert JD, Heinemann U (1987) Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J Neurophysiol 57: 869-888. [DOI] [PubMed] [Google Scholar]
- Müller M, Mironov SL, Ivannikov MV, Schmidt J, Richter DW (2005) Mitochondrial organization and motility probed by two-photon microscopy in cultured mouse brainstem neurons. Exp Cell Res 303: 114-127. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Nakamura K, Lasser-Ross N, Barbara JG, Sandler VM, Ross WN (2000) Inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release evoked by metabotropic agonists and backpropagating action potentials in hippocampal CA1 pyramidal neurons. J Neurosci 20: 8365-8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PV, Marin L, Atwood HL (1997) Synaptic physiology and mitochondrial function in crayfish tonic and phasic motor neurons. J Neurophysiol 78: 281-294. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL (2000) Mitochondria and neuronal survival. Physiol Rev 80: 315-360. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Ward MW (2000) Mitochondrial membrane potential and neuronal excitotoxicity: mortality and millivolts. Trends Neurosci 23: 166-174. [DOI] [PubMed] [Google Scholar]
- Papa S, Skulachev VP (1997) Reactive oxygen species, mitochondria, apoptosis and aging. Mol Cell Biochem 174: 305-319. [PubMed] [Google Scholar]
- Pitter JG, Maechler P, Wollheim CB, Spät A (2002) Mitochondria respond to Ca2+ already in the submicromolar range: correlation with redox state. Cell Calcium 31: 97-104. [DOI] [PubMed] [Google Scholar]
- Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ (2003) Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci 23: 7881-7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Simpson AW, Brini M, Pozzan T (1992) Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358: 325-327. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Brini M, Chiesa A, Filippin L, Pozzan T (1999) Mitochondria as biosensors of calcium microdomains. Cell Calcium 26: 193-199. [DOI] [PubMed] [Google Scholar]
- Robb-Gaspers LD, Rutter GA, Burnett P, Hajnoczky G, Denton RM, Thomas AP (1998) Coupling between cytosolic and mitochondrial calcium oscillations: role in the regulation of hepatic metabolism. Biochim Biophys Acta 1366: 17-32. [DOI] [PubMed] [Google Scholar]
- Scanlon JM, Brocard JB, Stout AK, Reynolds IJ (2000) Pharmacological investigation of mitochondrial Ca2+ transport in central neurons: studies with CGP-37157, an inhibitor of the mitochondrial Na+-Ca2+ exchanger. Cell Calcium 28: 317-327. [DOI] [PubMed] [Google Scholar]
- Schuchmann S, Luckermann M, Kulik A, Heinemann U, Ballanyi K (2000) Ca2+- and metabolism-related changes of mitochondrial potential in voltage-clamped CA1 pyramidal neurons in situ. J Neurophysiol 83: 1710-1721. [DOI] [PubMed] [Google Scholar]
- Shepherd GM, Harris KM (1998) Three-dimensional structure and composition of CA3→CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J Neurosci 18: 8300-8310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skulachev VP, Bakeeva LE, Chernyak BV, Domnina LV, Minin AA, Pletjushkina OY, Saprunova VB, Skulachev IV, Tsyplenkova VG, Vasiliev JM, Yaguzhinsky LS, Zorov DB (2004) Thread-grain transition of mitochondrial reticulum as a step of mitoptosis and apoptosis. Mol Cell Biochem 256-257: 341-358. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Müller D (1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37: 173-182. [DOI] [PubMed] [Google Scholar]
- Szalai G, Csordás G, Hantash BM, Thomas AP, Hajnóczky G (2000) Calcium signal transmission between ryanodine receptors and mitochondria. J Biol Chem 275: 15305-15313. [DOI] [PubMed] [Google Scholar]
- Szilágyi N, Kovács R, Kardos J (2000) Coupled intra- and extracellular Ca2+ dynamics in recurrent seizure-like events. Eur J Neurosci 12: 3893-3899. [DOI] [PubMed] [Google Scholar]
- Vaillend C, Mason SE, Cuttle MF, Alger BE (2002) Mechanisms of neuronal hyperexcitability caused by partial inhibition of Na+-K+-ATPases in the rat CA1 hippocampal region. J Neurophysiol 88: 2963-2978. [DOI] [PubMed] [Google Scholar]
- Vergun O, Votyakova TV, Reynolds IJ (2003) Spontaneous changes in mitochondrial membrane potential in single isolated brain mitochondria. Biophys J 85: 3358-3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votyakova TV, Reynolds IJ (2001) DeltaPsi(m)-dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem 79: 266-277. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Thayer SA (2002) NMDA-induced calcium loads recycle across the mitochondrial inner membrane of hippocampal neurons in culture. J Neurophysiol 87: 740-749. [DOI] [PubMed] [Google Scholar]
- Ward MW, Rego AC, Frenguelli BG, Nicholls DG (2000) Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci 20: 7208-7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ (1995) Mitochondria and Na+/Ca2+ exchange buffer glutamate-induced calcium loads in cultured cortical neurons. J Neurosci 15: 1318-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ (1996) Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci 16: 5688-5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M, Yamada KA (2000) Cyclosporine induces epileptiform activity in an in vitro seizure model. Epilepsia 41: 271-276. [DOI] [PubMed] [Google Scholar]
- Zenisek D, Matthews G (2000) The role of mitochondria in presynaptic calcium handling at a ribbon synapse. Neuron 25: 229-237. [DOI] [PubMed] [Google Scholar]
- Zhang CL, Dreier JP, Heinemann U (1995) Paroxysmal epileptiform discharges in temporal lobe slices after prolonged exposure to low magnesium are resistant to clinically used anticonvulsants. Epilepsy Res 20: 105-111. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Bers DM (2002) Time course of action of antagonists of mitochondrial Ca2+ uptake in intact ventricular myocytes. Pflügers Arch 445: 132-138. [DOI] [PubMed] [Google Scholar]
- Zoratti M, Szabó I (1995) The mitochondrial permeability transition. Biochim Biophys Acta 1241: 139-176. [DOI] [PubMed] [Google Scholar]