

Abstract
Alzheimer's disease (AD) is characterized by the aggregation and deposition of the normally soluble amyloid-β (Aβ) peptide in the extracellular spaces of the brain as parenchymal plaques and in the walls of cerebral vessels as cerebral amyloid angiopathy (CAA). CAA is a common cause of brain hemorrhage and is found in most patients with AD. As in AD, the ϵ4 allele of the apolipoprotein E (apoE) gene (APOE) is a risk factor for CAA. To determine the effect of human apoE on CAA in vivo, we bred human APOE3 and APOE4 “knock-in” mice to a transgenic mouse model (Tg2576) that develops amyloid plaques as well as CAA. The expression of both human apoE isoforms resulted in a delay in Aβ deposition of several months relative to murine apoE. Tg2576 mice expressing the more fibrillogenic murine apoE develop parenchymal amyloid plaques and CAA by 9 months of age. At 15 months of age, the expression of human apoE4 led to substantial CAA with very few parenchymal plaques, whereas the expression of human apoE3 resulted in almost no CAA or parenchymal plaques. Additionally, young apoE4-expressing mice had an elevated ratio of Aβ 40:42 in brain extracellular pools and a lower 40:42 ratio in CSF, suggesting that apoE4 results in altered clearance and transport of Aβ species within different brain compartments. These findings demonstrate that, once Aβ fibrillogenesis occurs, apoE4 favors the formation of CAA over parenchymal plaques and suggest that molecules or treatments that increase the ratio of Aβ 40:42 may favor the formation of CAA versus parenchymal plaques.
Keywords: Alzheimer's disease, apolipoprotein E, cerebral amyloid angiopathy, amyloid-β, ratio, transgenic models
Introduction
Alzheimer's disease (AD) is characterized by the extracellular aggregation and deposition of the normally soluble amyloid-β (Aβ) peptide in the form of diffuse and fibrillar plaques. Aβ can also deposit in the walls of cerebral vessels, which is known as cerebral amyloid angiopathy (CAA). The ϵ4 allele of the apolipoprotein E (apoE) gene (APOE) is a risk factor for developing both AD and CAA (Schmechel et al., 1993; Greenberg et al., 1995; Nicoll et al., 1997). Conversely, although the ϵ2 allele is protective for AD, it is associated with an increased risk factor for developing hemorrhage when CAA is present (Nicoll et al., 1996, 1997; Greenberg et al., 1998). The exact molecular mechanism by which apoE affects AD or CAA is not completely clear, although increasing evidence suggests that this is likely to be related in large part to the ability of apoE to act as an Aβ chaperone (Wisniewski et al., 1997). apoE is a 299 aa protein secreted at high levels in the liver and the brain (Mahley, 1988). In the brain, it is secreted in unique high-density lipoprotein-like particles predominantly by glial cells (Pitas et al., 1987). apoE has been shown to bind directly to the Aβ peptide and influence its fibrillogenesis and clearance in vitro (Strittmatter et al., 1993; Wisniewski et al., 1993; LaDu et al., 1994; Munson et al., 2000) and in vivo (Naslund et al., 1995; Wisniewski et al., 1995; Ghersi-Egea et al., 1996; Permanne et al., 1997; Russo et al., 1998; Shibata et al., 2000; Ji et al., 2001; DeMattos et al., 2004). apoE has also been shown to be critically important for the formation of fibrillar Aβ (Congo red or thioflavine positive) in brain parenchyma and CAA in vivo (Bales et al., 1997; Holtzman et al., 2000b; Fryer et al., 2003).
The Aβ peptide is 38-43 aa in length and is generated from proteolytic processing of the amyloid-β precursor protein (APP), and the most common species is Aβ40 and Aβ42. The Tg2576 [Swedish mutation in the amyloid precursor protein (APPsw)] transgenic mouse model of AD develops parenchymal plaque pathology and prominent CAA beginning at 9 months of age (Hsiao et al., 1996). In the absence of endogenous murine apoE, APPsw mice still develop Aβ deposits, but the vast majority of the plaques are nonfibrillar, and the formation of CAA and CAA-associated hemorrhage is completely absent even at very old ages (Holtzman et al., 2000b; Fryer et al., 2003). Although murine apoE is homologous to human apoE, it differs by ∼25% at the amino acid level. To further our understanding of the effects of human apoE isoforms on Aβ metabolism and CAA in vivo, we used APPsw mice expressing human apoE3 or apoE4 under the control of endogenous apoE regulatory elements (“knock-in” mice). These studies demonstrate early effects of apoE4 on increasing the Aβ 40:42 ratio and demonstrate that, once Aβ fibrillogenesis occurs, apoE4 results in a shift of Aβ deposition from brain parenchyma to the vasculature.
Materials and Methods
Animals and tissue preparation. The production, genotyping, and background strain (C57BL/6 × SJL) of APPsw murine Apoe+/+ and APPsw murine Apoe-/- mice used in this study have been described previously (Hsiao et al., 1996; Holtzman et al., 2000b). To determine the role of human apoE3 and apoE4 on CAA, we used mice in which the endogenous murine Apoe gene is replaced with the human APOE3 or APOE4 gene (knock-in mice). The production and genotyping of human APOE3 and APOE4 knock-in mice has been described previously (Sullivan et al., 1997). We bred APOE3 and APOE4 knock-in mice onto a C57BL/6 × SJL background so that all of the mice used in this study were on an identical genetic background. APPsw mice overexpress a human APP695 transgene containing the familial Swedish AD mutations at positions 670/671 under the control of the prion promoter and were a generous gift from Dr. K. Hsaio-Ashe (University of Minnesota, Minneapolis, MN). Animals were anesthetized with pentobarbital (150 mg/kg, i.p.) and perfused transcardially with 0.1 m PBS containing heparin (3 U/ml), pH 7.4, for 10 min. All of the mice used in this study were well perfused, as determined in the remaining hemibrain during histological assessment. For 15-month-old animals, one hemibrain was immersion-fixed overnight at 4°C in PBS containing 4% paraformaldehyde. After fixation, the brain was cryoprotected in PBS containing 30% sucrose at 4°C. The cerebellum of the remaining hemibrain was removed, and cerebral vessels were isolated from the forebrain, as described previously (Fryer et al., 2003). All experimental protocols used were approved by the Animal Studies Committee at Washington University.
Histological and biochemical analysis. Coronal sections (50 μm) were cut on a freezing-sliding microtome, mounted on Superfrost Plus slides (Fisher Scientific, Houston, TX), and permeabilized for 30 min at room temperature with PBS containing 0.25% Triton X-100. To visualize fibrillar Aβ plaques and CAA, sections were stained with thioflavine S or the Congo red derivative X-34 dye in 40% ethanol in PBS (a generous gift from Dr. W. Klunk, University of Pittsburgh, Pittsburgh, PA). Sections were quickly rinsed twice in 40% ethanol in PBS and twice in PBS before being mounted with Vectashield (Vector Laboratories, Burlingame, CA). X-34 has the advantage of a greater signal-to-noise ratio, as well as a narrow emission spectra, compared with thioflavine S (Styren et al., 2000). For PBS-soluble extracts, tissue was briefly Dounce homogenized in ice-cold PBS containing protease inhibitors and immediately spun at 20,000 × g for 5 min. Aβ1-40 and Aβ1-42 were quantified by ELISA with specific monoclonal antibodies, as described previously (Cirrito et al., 2003). Total tissue cholesterol was determined using the Amplex Red cholesterol kit (Sigma, St. Louis, MO).
apoE ELISA. Plates (96 well) were coated overnight with the monoclonal antibody WU-E4 (Krul et al., 1988) at a concentration of 10 μg/ml in 35 mm NaHCO3, 16 mm NaCO3, 0.02% NaN3, pH 9.6. Plates were washed and blocked for 1 h at 37°C with 1% dry milk-PBS. Plates were washed again, and samples and standards were loaded in ELISA buffer (0.5% BSA-PBS and 0.025% Tween 20) and incubated overnight at 4°C. Human apoE isolated from plasma β-very-low-density lipoprotein was used as standard (BIODESIGN International, Saco, ME). Plates were washed again and incubated for 90 min at 37°C with goat anti-apoE antisera at a 1:500 dilution (Calbiochem, La Jolla, CA) in ELISA buffer. Plates were washed again and incubated with HRP-conjugated horse anti-goat antibody (Vector Laboratories) at a 1:5000 dilution in ELISA buffer. Plates were washed and developed with tetramethylbenzidine substrate (Sigma) and monitored for absorbance at 650 nm. The Student's t test was used to compare apoE levels in human apoE3- and apoE4-expressing mice.
Quantitation of CAA and Aβ load. The percentage of cross-sectional area covered by CAA vessels (percentage of CAA load), as defined by X-34-positive vessels, was quantified using unbiased stereological principles, as described previously (Fryer et al., 2003). Surface and penetrating CAA load, as well as amyloid plaque load, was determined in the cortex and overlying leptomeningeal vessels immediately dorsal to the striatum in four sections, each separated by 300 μm. Stereo Investigator image-analysis software (MicroBrightField, Williston, VT) was used to quantify percentage of CAA load, as well as amyloid plaque load, using Cavalieri's point-counting method. One-way ANOVA, followed by a post hoc Tukey's t test, was used to compare APPsw mice expressing murine apoE, human apoE3, or human apoE4. Because the data were not normally distributed, the Mann-Whitney t test was used to compare deposits in human apoE3- or apoE4-expressing mice.
Isolation of cerebral vessels, parenchymal tissue, and CSF. The isolation of cerebral vessels and parenchymal tissue was performed exactly as described previously (Fryer et al., 2003). Murine CSF was isolated as described previously (DeMattos et al., 2002).
Results
APPsw mice develop prominent amyloid plaques and CAA: human apoE delays Aβ deposition
In this study, we used human APOE knock-in mice to study the effects of human apoE3 and apoE4 as well as murine apoE on Aβ deposition in the APPsw mouse model of AD. In this model, Aβ is deposited predominantly as fibrillar Aβ plaques (i.e., thioflavine S- or X-34-positive amyloid). In addition to parenchymal amyloid plaques, this model also develops substantial CAA. An ex vivo amyloid stain demonstrates the extensive CAA in the leptomeningeal vessels on the surface of the brain (Fig. 1A) in this model, which is perhaps underappreciated when simply histologically examining individual brain tissue sections. In the APPsw model, Aβ begins to deposit as parenchymal plaques and CAA at ∼9 months of age and is present in all of the APPsw mice by 12 months of age (Fig. 2A,D) (Hsiao et al., 1996). We found previously that the expression of human apoE isoforms in another APP transgenic mouse model (PDAPP) with an APPV717F mutation (Holtzman et al., 2000a; Fagan et al., 2002) resulted in a delay in deposition of several months. In this APPsw model, we also found that the expression of human apoE under the control of endogenous regulatory elements results in a delay of several months or longer in Aβ deposition compared with animals expressing endogenous murine apoE (Fig. 2B,C,E,F).
Figure 1.
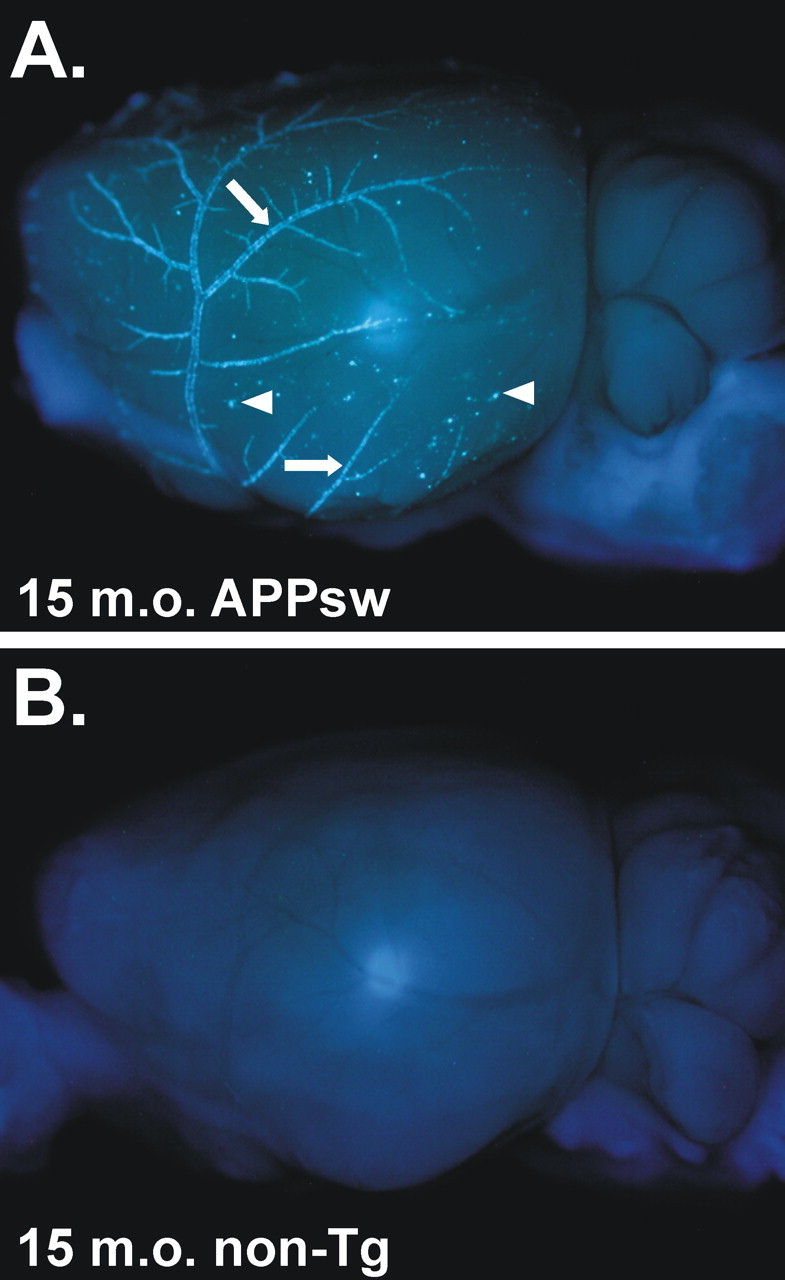
Ex vivo image of an APPsw brain demonstrating extensive CAA. A, A 15-month-old (15 m.o.) APPsw mouse brain stained with X-34 and imaged with UV epifluorescence to denote fibrillar amyloid. Extensive CAA in leptomeningeal vessels can be seen on the cortical surface (arrows) as well as on plaques near the cortical surface (arrowheads). B, A 15-month-old non-transgenic (non-Tg) mouse brain stained with X-34, demonstrating the specificity of the stain.
Figure 2.

Expression of human apoE3 or apoE4 delays deposition compared with murine apoE in the APPsw model at 12 months of age. A, D, Anti-Aβ immunostaining with monoclonal antibody m3D6 (A) or X-34 staining (D) to denote fibrillar amyloid of APPsw mice expressing endogenous murine apoE demonstrating CAA (arrows) and parenchymal plaques (arrowheads). However, 12-month-old (12 m.o.) APPsw mice expressing human apoE3 (B, E) or apoE4 (C, F) had little or no detectable plaques.
apoE4 shifts amyloid deposition from parenchyma to vessels
APPsw mice expressing human apoE4 did begin to deposit Aβ between 12 and 15 months of age. Seven of 11 APPsw mice expressing human apoE4 had Aβ deposition at 15 months of age, whereas 2 of 12 APPsw mice expressing human apoE3 had Aβ deposition. Strikingly, the Aβ deposited in the presence of apoE4 was almost exclusively in cerebral vessels and was all fibrillar, with very few parenchymal plaques, at 15 months of age (Fig. 3C,F). This is in contrast to the mixed pathology of parenchymal plaques and CAA in APPsw mice expressing endogenous murine apoE (Fig. 3A,D). APPsw mice expressing human apoE3 developed very few plaques at this age (Fig. 3B,E), with no histologically detectable deposition of Aβ in the majority of these animals. To more thoroughly analyze the deposition of Aβ in brain parenchyma versus CAA, we performed a stereological analysis to quantify area covered by CAA, as well as fibrillar parenchymal plaques. The expression of apoE4 resulted in a marked and significant increase in CAA, as well as parenchymal amyloid plaques, compared with mice expressing apoE3 (Fig. 4). Although murine apoE resulted in the greatest amount of amyloid deposition compared with either of the human apoE isoforms, the ratio of amyloid deposition in vessels versus parenchyma was much lower in the presence of murine apoE (3:1 ratio) versus apoE4 (21:1 ratio). Strikingly, virtually all Aβ deposits in the apoE4-expressing mice at 15 months of age were in the form of CAA (Fig. 4). The expression of apoE4 resulting in mostly CAA at 15 months of age is not merely attributable to an overall delay in parenchymal Aβ deposition, because 12-month-old APPsw mice expressing murine apoE develop comparable levels of parenchymal plaques and CAA (Fig. 2A,D). Interestingly, although APPsw mice expressing apoE4 had substantial levels of CAA, the expression of both apoE3 and apoE4 in APPsw mice resulted in a dramatic reduction in the amount of fibrillar parenchymal plaques in the cortex compared with APPsw mice expressing murine apoE (Fig. 4D).
Figure 3.

Expression of apoE determines the level of CAA and parenchymal plaque pathology in an isoform-dependent manner in the 15-month-old (15 m.o.) APPsw model. A-F, Anti-Aβ immunostaining with m3D6 antibody (A-C) or X-34 staining (D-F) to visualize fibrillar Aβ deposits demonstrates CAA (arrows) and parenchymal plaques (arrowheads). A, D, APP swmice expressing endogenous murine apoE had both CAA and parenchymal plaques. B, C, E, F, APPsw mice expressing human apoE3 (B, E) had only occasional Aβ deposits, whereas APPsw mice expressing human apoE4 (C, F) had extensive CAA with infrequent parenchymal plaques.
Figure 4.

Stereological quantitation of CAA and fibrillar plaque load. A-C, In 15-month-old APPsw mice, expression of human apoE4 (hApoE) and murine apoE (mApoE) resulted in significantly greater CAA load in leptomeningeal vessels (A), penetrating cortical vessels (B), and total cortical vessels (C) and parenchymal plaque load compared with mice expressing human apoE3. B-D, Expression of murine apoE also resulted in significantly higher levels of penetrating (B), total CAA (C), and parenchymal plaque load (D) compared with expression of human apoE4. D, Expression of both human apoE3 and apoE4 resulted in a dramatic reduction in parenchymal amyloid plaque load compared with mice expressing murine apoE. Error bars represent SEM.
To further examine the amount and type of Aβ deposited in APPsw mice expressing human apoE3 or apoE4 at 15 months of age, we isolated cerebral vessels and brain parenchymal tissue to measure the absolute level of Aβ by ELISA. As would be expected from the histological quantitation and from previous biochemical studies of CAA (Joachim et al., 1988; Suzuki et al., 1994; Fryer et al., 2003), cerebral vessels isolated from 15-month-old APPsw mice expressing human apoE4 had substantially more Aβ1-40 and Aβ1-42 than human apoE3-expressing mice (Fig. 5A,B). Also reflective of the histological quantitation, brain parenchymal levels of Aβ in 15-month-old APPsw mice expressing human apoE4 were considerably higher than in human apoE3-expressing mice (Fig. 5C,D).
Figure 5.

Fifteen-month-old APPsw mice expressing human apoE4 (hApoE4) had dramatically more deposition of Aβ incerebral vessels than mice expressing human apoE3. A, B, Human apoE3-expressing mice deposited 21.26 ± 5.097 ng/mg Aβ1-40 and 25.58 ± 9.282 ng/mg Aβ1-42 in cerebral vessels, whereas human apoE4-expressing mice deposited 21,040 ± 13,530 ng/mg Aβ1-40 and 4535 ± 2094 ng/mg Aβ1-42 in cerebral vessels. C, D, Additionally, human apoE3-expressing mice deposited 8.493 ± 1.918 ng/mg Aβ1-40 and 5.827 ± 1.421 ng/mg Aβ1-42 in brain parenchymal tissue, whereas human apoE4-expressing mice deposited 60.89 ± 20.53 ng/mg Aβ1-40 and 57.38 ± 17.48 ng/mg Aβ1-42 in brain parenchymal tissue. Error bars represent SEM.
apoE4 alters the Aβ 40:42 ratio in brain tissue and CSF in young APPsw mice
Given the dramatic effect of human apoE isoforms and species on Aβ-related pathology at 15 months of age, we examined young mice at 3 months of age, an age well before the development of Aβ deposition, for changes that may explain the shift from mixed CAA/parenchymal pathology to predominantly CAA by apoE4 expression. We examined the levels of apoE and Aβ in both cortical PBS extracts (as a reflection of the extracellular parenchymal pool) as well as in CSF. We found that APPsw mice expressing human apoE4 had significantly higher levels of Aβ1-40 in PBS extracts compared with mice expressing murine apoE (Fig. 6A). Aβ1-42 levels were also slightly lower in these PBS extracts of APPsw mice expressing human apoE4, but this did not reach statistical significance (Fig. 6A). Aβ1-40 levels in CSF were slightly lower in APPsw mice expressing human apoE4, but this also did not reach statistical significance (Fig. 6B). However, there was a significantly higher ratio of Aβ 40:42 in PBS extracts of cortical tissue of mice expressing apoE4 compared with mice expressing endogenous murine apoE (Fig. 6C). Interestingly, this ratio was reversed in CSF, and human apoE4-expressing mice had a significantly lower Aβ 40:42 ratio than the other genotypes, suggesting that apoE4 results in a less-efficient transport of Aβ between brain parenchymal tissues to other CNS compartments such as CSF (Fig. 6D). These results also highlight the importance of examining the Aβ 40:42 ratio, in addition to absolute levels of different Aβ peptides, and suggest that a higher 40:42 ratio in the brain predisposes it to CAA. The level of Aβ40, Aβ42, or the ratio of Aβ 40:42 did not differ between any of the genotypes in the plasma of these mice (data not shown). This highlights the observation that different apoE-dependent mechanisms seem to be involved in Aβ transport within the CNS versus peripheral circulation. The levels of apoE in apoE3- and apoE4-expressing mice were also not significantly different in either cortex (Fig. 7A) or CSF (Fig. 7B), indicating that differences in the level of apoE do not account for the pathological differences observed between APOE3 and APOE4 knock-in mice. It was not possible to directly compare the levels of murine to human apoE in an ELISA format, because the murine and human apoE standards are not identical, and the detection antibody does not recognize murine and human apoE equally. Additionally, we found no differences in brain cholesterol in mice expressing murine apoE or human apoE3 or apoE4, suggesting that apoE isoform and species did not influence brain cholesterol to somehow influence Aβ (Fig. 7C).
Figure 6.

Expression of human apoE4 (hApoE4) alters the ratio of Aβ 40:42 in extracellular pools of the CNS. A, The level of Aβ1-40 in PBS-soluble brain extracts was significantly higher in APPsw mice expressing human apoE4 compared with mice expressing murine apoE (mApoE). Aβ1-42 levels were slightly, but not significantly, lower in APPsw mice expressing human apoE4 compared with mice expressing murine apoE. B, The level of Aβ1-40 in the CSF was slightly, but not significantly, lower in APPsw mice expressing human apoE4 compared with mice expressing murine apoE. Also, Aβ1-42 levels were slightly, but not significantly, higher in APPsw mice expressing human apoE4 compared with mice expressing murine apoE. C, The Aβ 40:42 ratio in PBS-soluble extracts was significantly higher in APPsw mice expressing human apoE4 compared with murine apoE. D, However, the Aβ 40:42 ratio in the CSF was significantly lower in APPsw mice expressing human apoE4 compared with mice expressing human apoE3 or murine apoE. Error bars represent SEM.
Figure 7.

Expression of murine apoE (mApoE) or human apoE3 (hApoE3) or apoE4 (hApoE4) does not alter apoE levels or total brain cholesterol. A, B, Levels of human apoE were not significantly different in PBS-soluble brain extracts of cortical tissue (A) or in CSF (B). C, Additionally, the level of hippocampal cholesterol did not significantly differ among mice expressing murine apoE, human apoE3, or human apoE4. Error bars represent SEM.
Discussion
Although much is known about the factors that lead to the deposition of Aβ in brain parenchyma, much less is known about the mechanisms of Aβ deposition in cerebral vessels. We used the APPsw mouse model of AD and human APOE knock-in mice to explore the effects of human apoE expression on Aβ deposition in brain parenchyma and cerebral vessels. As we saw previously in another mouse model of AD (Holtzman et al., 1999, 2000a), expression of human apoE isoforms resulted in a marked delay in deposition of Aβ until ∼15 months of age. Strikingly, the expression of human apoE4 in APPsw mice at 15 months of age led to a shift in Aβ deposits (relative to the expression of endogenous murine apoE), resulting in substantial CAA with very little parenchymal plaque deposition. Expression of human apoE3 resulted in almost no Aβ deposition at this age. Reflective of these histological observations, biochemical analysis of Aβ levels in cerebral vessels of aged mice showed a dramatic increase in the level of Aβ1-40 and Aβ1-42 in APPsw mice expressing human apoE4 compared with human apoE3-expressing mice. Biochemical analysis of young mice well before Aβ deposition showed no differences in the level of human apoE in the brain or CSF. There were also no differences in brain cholesterol in mice expressing human apoE3, human apoE4, or endogenous murine apoE. Interestingly, APPsw mice expressing human apoE4 had a significant increase in the Aβ 40:42 ratio in extracellular brain pools, whereas this ratio was reversed in CSF. Aβ levels or 40:42 ratios were not different between any of the genotypes in the plasma of these young mice, further indicating that apoE-dependent Aβ clearance seems to occur by different mechanisms within the CNS versus peripheral circulation. Thus, there are two main effects of human apoE expression on Aβ deposition in APPsw mice: (1) a delay in Aβ deposition by both human apoE3 and apoE4 (likely attributable to the fact that murine apoE is more efficient in facilitating aggregation of Aβ into fibrillar forms) and (2) a striking redistribution of Aβ deposition to the cerebral vasculature once Aβ fibrillogenesis begins in the presence of apoE4 (likely facilitated by an increased Aβ 40:42 ratio present at young ages). Together, these data suggest that apoE isoforms differentially alter the clearance and transport of Aβ species within brain extracellular space and that, once Aβ fibrillogenesis occurs, an increased 40:42 ratio results in a shift of Aβ deposition from the brain parenchyma to vessels.
In humans, APOE4 is associated with larger amounts of CAA in patients with AD, and some APOE4 homozygous individuals examined at older ages have been shown to have substantial CAA (Schmechel et al., 1993; Vidal et al., 2000). In a study of 125 AD cases, the severity of CAA was strongly associated with the number of APOE4 alleles, whereas parenchymal Aβ was independent of APOE genotype (Chalmers et al., 2003). Interestingly, cases with severe CAA had a significantly lower parenchymal Aβ load (Chalmers et al., 2003). Although APOE2 seems to protect against developing AD, it is a known risk factor for CNS hemorrhage if CAA is present (Nicoll et al., 1996, 1997; Greenberg et al., 1998). Additional studies in AD mouse models expressing human apoE2 will be useful in determining the mechanism of this CAA-associated hemorrhage, and we are in the process of generating these mice. Despite these genetic associations discovered over a decade ago, little is known about the molecular mechanisms by which apoE influences the development of CAA. Previously, we demonstrated a critical role of murine apoE in CAA. APPsw mice lacking endogenous murine apoE do not develop CAA even up to 24 months of age, despite significant parenchymal plaque deposition, some of which is fibrillar (Fryer et al., 2003). Together with the results presented herein, these data suggest that apoE may play as prominent a role in the deposition of Aβ in cerebral vessels as it does in brain parenchyma.
Cerebral vessels in both human cases of CAA as well as mouse models with extensive CAA have been shown to have high levels of Aβ1-40 deposited compared with Aβ1-42 (Joachim et al., 1988; Prelli et al., 1988; Suzuki et al., 1994; Alonzo et al., 1998; McCarron et al., 2000; Fryer et al., 2003; Ingelsson et al., 2004). Additionally, as with the APPsw mice used in this study, familial AD patients with the Swedish APP 670/671 mutation have substantial CAA (Lannfelt et al., 1994). Recent evidence has also suggested that the Aβ 40:42 ratio may be a key pathologic factor favoring deposition of Aβ in cerebral vessels versus brain parenchyma. APP transgenic mice expressing the APP mutation that results in hereditary cerebral hemorrhage with amyloidoses [i.e., E22Q (Dutch type)] have a high Aβ 40:42 ratio and develop extensive CAA with few parenchymal plaques (Herzig et al., 2004). However, when this model was bred to a presenilin transgenic mouse model (PS45) harboring a mutation favoring generation of Aβ1-42 over Aβ1-40, young mice had a low Aβ 40:42 ratio, and the pathology in aged mice was shifted from CAA to parenchymal plaque deposition (Herzig et al., 2004). Because the expression of apoE4 versus apoE3 and murine apoE in APPsw mice results in a high 40:42 ratio in brain parenchymal tissue and the later development of a significantly elevated ratio of CAA to parenchymal plaques, this supports the hypothesis that a high Aβ 40:42 ratio favors the formation of CAA over parenchymal plaque pathology. The interesting aspect of this finding in relation to apoE4 is that a preponderance of current evidence suggests that the effect of apoE on Aβ metabolism, including the 40:42 ratio, is via apoE acting as an Aβ chaperone and not by affecting Aβ production from APP.
We showed previously that, although apoE clearly affects the clearance of Aβ in vivo, additional studies will be necessary to determine the molecular mechanisms underlying this effect. Several in vitro studies suggest that apoE influences cellular Aβ clearance, and our recent in vivo study suggests that apoE in some way directly influences the half-life of soluble Aβ in the interstitial fluid of the brain (DeMattos et al., 2004). The finding that apoE4-expressing mice have a significantly higher Aβ 40:42 ratio in the PBS-soluble brain fraction and a significantly lower 40:42 ratio in the CSF, compared with mice expressing apoE3 and murine apoE, also strongly supports the idea that apoE can directly influence soluble Aβ metabolism by altering its clearance and transport between brain compartments. Additionally, the fact that APP transgenic mice that are Apoe-/- develop Aβ deposits in a different anatomical distribution (Bales et al., 1997; Holtzman et al., 1999; Irizarry et al., 2000) also suggests that apoE is involved in extracellular Aβ transport. The clearance of Aβ from brain parenchyma has been proposed to occur along periarterial interstitial fluid drainage pathways (Weller et al., 1998). Interestingly, in recent in vivo imaging experiments with APPsw mice, we find that all CAA is present only in arterioles and not venules (D. M. Holtzman, unpublished observations). The local environment along these pathways has abundant molecules known to bind apoE, such as heparan sulfate proteoglycans and low-density lipoprotein receptor family members (Shibata et al., 2000; Strittmatter and Bova Hill, 2002). It is possible that apoE-Aβ complexes are transported from brain parenchymal tissue to these interstitial fluid drainage pathways in which they bind to apoE-binding molecules, and the local environment promotes the fibrillogenesis of Aβ. Other secreted molecules present in the brain extracellular space, such as TGF-β, also likely contribute to the pathogenesis of CAA (Wyss-Coray et al., 1997). For example, overexpression of TGF-β increases CAA in APP transgenic mice, and this may be attributable to the upregulation of extracellular matrix proteins (Wyss-Coray et al., 1997, 2000). Finally, the results presented here suggest that caution should be taken in AD therapies that may subtly alter the Aβ 40:42 ratio in brain tissue, either intentionally or unintentionally, because they may be effective in reducing parenchymal plaque deposition at the expense of developing CAA (Davis et al., 2004; Deane et al., 2004).
Footnotes
This work was supported by National Institutes of Health Grants AG13956, AG05681, AG11355, and NS034467. We thank Guojun Bu, Laura Dugan, Alison Goate, Gene Johnson, Joel Price, and Ron Demattos for helpful comments and discussion of this manuscript.
Correspondence should be addressed to Dr. David M. Holtzman, Washington University School of Medicine, Department of Neurology, 660 South Euclid Avenue, Box 8111, St. Louis, MO 63110. E-mail: holtzman@neuro.wustl.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252803-08$15.00/0
References
- Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM (1998) Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol 57: 353-359. [DOI] [PubMed] [Google Scholar]
- Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B, Paul SM (1997) Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet 17: 263-264. [DOI] [PubMed] [Google Scholar]
- Chalmers K, Wilcock GK, Love S (2003) APOE epsilon 4 influences the pathological phenotype of Alzheimer's disease by favouring cerebrovascular over parenchymal accumulation of A beta protein. Neuropathol Appl Neurobiol 29: 231-238. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM (2003) In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-β metabolism and half-life. J Neurosci 23: 8844-8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Van Nostrand WE (2004) Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem 279: 20296-20306. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV (2004) LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43: 333-344. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Parsadanian M, O'Dell MA, Foss EM, Paul SM, Holtzman DM (2002) Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer's disease. J Neurochem 81: 229-236. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Cirrito JR, Parsadanian M, May PC, O'Dell MA, Taylor JW, Harmony JA, Aronow BJ, Bales KR, Paul SM, Holtzman DM (2004) ApoE and clusterin cooperatively suppress Abeta levels and deposition. Evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron 41: 193-202. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM (2002) Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer's disease. Neurobiol Dis 9: 305-318. [DOI] [PubMed] [Google Scholar]
- Fryer JD, Taylor JW, DeMattos RB, Bales KR, Paul SM, Parsadanian M, Holtzman DM (2003) Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J Neurosci 23: 7889-7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghersi-Egea JF, Gorevic PD, Ghiso J, Frangione B, Patlak CS, Fenstermacher JD (1996) Fate of cerebrospinal fluid-borne amyloid beta-peptide: rapid clearance into blood and appreciable accumulation by cerebral arteries. J Neurochem 67: 880-883. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Rebeck GW, Vonsattel JP, Gomez-Isla T, Hyman BT (1995) Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol 38: 254-259. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Vonsattel JP, Segal AZ, Chiu RI, Clatworthy AE, Liao A, Hyman BT, Rebeck GW (1998) Association of apolipoprotein E epsilon2 and vasculopathy in cerebral amyloid angiopathy. Neurology 50: 961-965. [DOI] [PubMed] [Google Scholar]
- Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Sturchler-Pierrat C, Burki K, van Duinen SG, Maat-Schieman ML, Staufenbiel M, Mathews PM, Jucker M (2004) Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci 7: 954-960. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, Chang LK, Sun Y, Paul SM (1999) Expression of human apolipoprotein E reduces amyloid-beta deposition in a mouse model of Alzheimer's disease. J Clin Invest 103: R15-R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM (2000a) Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 97: 2892-2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Fagan AM, Mackey B, Tenkova T, Sartorius L, Paul SM, Bales K, Ashe KH, Irizarry MC, Hyman BT (2000b) Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer's disease model. Ann Neurol 47: 739-747. [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: 99-102. [DOI] [PubMed] [Google Scholar]
- Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC (2004) Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 62: 925-931. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Cheung BS, Rebeck GW, Paul SM, Bales KR, Hyman BT (2000) Apolipoprotein E affects the amount, form, and anatomical distribution of amyloid beta-peptide deposition in homozygous APP(V717F) transgenic mice. Acta Neuropathol (Berl) 100: 451-458. [DOI] [PubMed] [Google Scholar]
- Ji Y, Permanne B, Sigurdsson EM, Holtzman DM, Wisniewski T (2001) Amyloid beta40/42 clearance across the blood-brain barrier following intra-ventricular injections in wild-type, apoE knock-out and human apoE3 or E4 expressing transgenic mice. J Alzheimers Dis 3: 23-30. [DOI] [PubMed] [Google Scholar]
- Joachim CL, Duffy LK, Morris JH, Selkoe DJ (1988) Protein chemical and immunocytochemical studies of meningovascular beta-amyloid protein in Alzheimer's disease and normal aging. Brain Res 474: 100-111. [DOI] [PubMed] [Google Scholar]
- Krul ES, Tikkanen MJ, Schonfeld G (1988) Heterogeneity of apolipoprotein E epitope expression on human lipoproteins: importance for apolipoprotein E function. J Lipid Res 29: 1309-1325. [PubMed] [Google Scholar]
- LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE (1994) Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem 269: 23403-23406. [PubMed] [Google Scholar]
- Lannfelt L, Bogdanovic N, Appelgren H, Axelman K, Lilius L, Hansson G, Schenk D, Hardy J, Winblad B (1994) Amyloid precursor protein mutation causes Alzheimer's disease in a Swedish family. Neurosci Lett 168: 254-256. [DOI] [PubMed] [Google Scholar]
- Mahley RW (1988) Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240: 622-630. [DOI] [PubMed] [Google Scholar]
- McCarron MO, Nicoll JA, Stewart J, Cole GM, Yang F, Ironside JW, Mann DM, Love S, Graham DI (2000) Amyloid beta-protein length and cerebral amyloid angiopathy-related haemorrhage. NeuroReport 11: 937-940. [DOI] [PubMed] [Google Scholar]
- Munson GW, Roher AE, Kuo YM, Gilligan SM, Reardon CA, Getz GS, LaDu MJ (2000) SDS-stable complex formation between native apolipoprotein E3 and beta-amyloid peptides. Biochemistry 39: 16119-16124. [DOI] [PubMed] [Google Scholar]
- Naslund J, Thyberg J, Tjernberg LO, Wernstedt C, Karlstrom AR, Bogdanovic N, Gandy SE, Lannfelt L, Terenius L, Nordstedt C (1995) Characterization of stable complexes involving apolipoprotein E and the amyloid beta peptide in Alzheimer's disease brain. Neuron 15: 219-228. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Burnett C, Love S, Graham DI, Ironside JW, Vinters HV (1996) High frequency of apolipoprotein E epsilon 2 in patients with cerebral hemorrhage due to cerebral amyloid angiopathy. Ann Neurol 39: 682-683. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Burnett C, Love S, Graham DI, Dewar D, Ironside JW, Stewart J, Vinters HV (1997) High frequency of apolipoprotein E epsilon 2 allele in hemorrhage due to cerebral amyloid angiopathy. Ann Neurol 41: 716-721. [DOI] [PubMed] [Google Scholar]
- Permanne B, Perez C, Soto C, Frangione B, Wisniewski T (1997) Detection of apolipoprotein E/dimeric soluble amyloid beta complexes in Alzheimer's disease brain supernatants. Biochem Biophys Res Commun 240: 715-720. [DOI] [PubMed] [Google Scholar]
- Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW (1987) Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta 917: 148-161. [DOI] [PubMed] [Google Scholar]
- Prelli F, Castano E, Glenner GG, Frangione B (1988) Differences between vascular and plaque core amyloid in Alzheimer's disease. J Neurochem 51: 648-651. [DOI] [PubMed] [Google Scholar]
- Russo C, Angelini G, Dapino D, Piccini A, Piombo G, Schettini G, Chen S, Teller JK, Zaccheo D, Gambetti P, Tabaton M (1998) Opposite roles of apolipoprotein E in normal brains and in Alzheimer's disease. Proc Natl Acad Sci USA 95: 15598-15602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD (1993) Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA 90: 9649-9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV (2000) Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 106: 1489-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Bova Hill C (2002) Molecular biology of apolipoprotein E. Curr Opin Lipidol 13: 119-123. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 90: 1977-1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styren SD, Hamilton RL, Styren GC, Klunk WE (2000) X-34, a fluorescent derivative of Congo red: a novel histochemical stain for Alzheimer's disease pathology. J Histochem Cytochem 48: 1223-1232. [DOI] [PubMed] [Google Scholar]
- Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, Quarfordt SH, Maeda N (1997) Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem 272: 17972-17980. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Iwatsubo T, Odaka A, Ishibashi Y, Kitada C, Ihara Y (1994) High tissue content of soluble beta 1-40 is linked to cerebral amyloid angiopathy. Am J Pathol 145: 452-460. [PMC free article] [PubMed] [Google Scholar]
- Vidal R, Calero M, Piccardo P, Farlow MR, Unverzagt FW, Mendez E, Jimenez-Huete A, Beavis R, Gallo G, Gomez-Tortosa E, Ghiso J, Hyman BT, Frangione B, Ghetti B (2000) Senile dementia associated with amyloid beta protein angiopathy and tau perivascular pathology but not neuritic plaques in patients homozygous for the APOE-epsilon4 allele. Acta Neuropathol (Berl) 100: 1-12. [DOI] [PubMed] [Google Scholar]
- Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE (1998) Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. Am J Pathol 153: 725-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Golabek A, Matsubara E, Ghiso J, Frangione B (1993) Apolipoprotein E: binding to soluble Alzheimer's beta-amyloid. Biochem Biophys Res Commun 192: 359-365. [DOI] [PubMed] [Google Scholar]
- Wisniewski T, Lalowski M, Golabek A, Vogel T, Frangione B (1995) Is Alzheimer's disease an apolipoprotein E amyloidosis? Lancet 345: 956-958. [DOI] [PubMed] [Google Scholar]
- Wisniewski T, Ghiso J, Frangione B (1997) Biology of A beta amyloid in Alzheimer's disease. Neurobiol Dis 4: 313-328. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L (1997) Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer's disease. Nature 389: 603-606. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Sanan DA, Mucke L, Masliah E (2000) Chronic over-production of transforming growth factor-beta1 by astrocytes promotes Alzheimer's disease-like microvascular degeneration in transgenic mice. Am J Pathol 156: 139-150. [DOI] [PMC free article] [PubMed] [Google Scholar]