

Abstract
Synaptic scaling is a form of homeostatic plasticity that scales synaptic strengths up or down to compensate for prolonged changes in activity. It has been controversial whether this plasticity is expressed presynaptically, postsynaptically, or both. Here we describe in detail the homeostatic changes that take place at excitatory synapses in visual cortical cultures after 1 or 2 d of activity blockade. After 7-10 d in vitro, activity blockade significantly increased postsynaptic accumulation of synaptic AMPA receptors via proportional increases in glutamate receptor 1 (GluR1) and GluR2. Time-lapse imaging of enhanced green fluorescent protein-tagged AMPA receptors revealed that receptor accumulation increased progressively over 2 d of activity blockade and affected the entire population of imaged synapses. The strength of synaptic connections between pyramidal neurons was more than doubled after activity blockade without affecting short-term depression or the coefficient of variation of the postsynaptic responses. Furthermore, uptake of the fluorescent styryl dye FM1-43 (N-(3-triethylammoniumpropyl)-4-[4-(dibutylamino)styryl] pyridinium dibromide) by presynaptic terminals was not different at control and activity-blocked synapses. In addition to the increased accumulation of postsynaptic AMPA receptors, boosting of dendritic AMPA currents by sodium channels was increased by activity blockade. These data indicate that, at young neocortical synapses, synaptic scaling has a predominantly postsynaptic locus and functions as a gain control mechanism to regulate neuronal activity without affecting the dynamics of synaptic transmission.
Keywords: homeostasis, short-term plasticity, synaptic plasticity, FM1-43, time-lapse imaging, AMPA receptors, sodium currents
Introduction
During development and learning synaptic connectivity is adjusted and refined via a number of activity-dependent processes. In addition to Hebbian plasticity, homeostatic forms of plasticity are needed to keep neuronal or network activity within a functional range (Miller, 1996; Marder and Prinz, 2002; Turrigiano and Nelson, 2004). Homeostatic synaptic plasticity has been identified in a variety of organisms and systems and appears to be a very general phenomenon (Davis and Bezprozvanny, 2001; Burrone and Murthy, 2003; Turrigiano and Nelson, 2004). Despite the growing amount of literature on homeostatic synaptic plasticity at central synapses, the expression mechanisms (presynaptic, postsynaptic, or both) are still not completely understood.
In principle, synaptic strength could be altered via changes in postsynaptic receptor accumulation, presynaptic release probability, the number of functional synaptic contacts between pairs of neurons, or all three mechanisms simultaneously. In central neurons, many studies have found that the amplitude (but not the frequency) of quantal glutamatergic currents is regulated bidirectionally by altered neuronal activity (O'Brien et al., 1998; Turrigiano et al., 1998; Ju et al., 2004). These changes are in the correct direction to stabilize firing, because lowering activity [using either glutamate receptor (GluR) blockers or TTX] increases quantal amplitude, whereas increasing activity (usually by blocking inhibition) reduces quantal amplitude. The changes in excitatory synaptic transmission are accompanied by changes in the postsynaptic sensitivity to glutamate (O'Brien et al., 1998; Turrigiano et al., 1998) and surface AMPA receptor (AMPAR) accumulation (Lissin et al., 1998; O'Brien et al., 1998), suggesting a major postsynaptic contribution to homeostatic synaptic plasticity. In contrast, other studies have found little or no change in quantal amplitude but have found an increase in miniature EPSC (mEPSC) frequency (Bacci et al., 2001; Thiagarajan et al., 2002). This increase in mEPSC frequency has been suggested to arise via increased presynaptic release probability, an interpretation supported by the observation that the uptake of the fluorescent styryl dyeN-(3-triethylammoniumpropyl)-4-[4-(dibutylamino)styryl] pyridinium dibromide (FM1-43) in hippocampal cultures increased after activity blockade (Murthy et al., 2001).
The locus of expression of homeostatic plasticity has important functional implications. Presynaptic changes in release probability will affect short-term synaptic plasticity profoundly (Zucker and Regehr, 2002), and these changes in the dynamics of synaptic transmission, in turn, are predicted to alter information flow in central circuits (Abbott et al., 1997; Markram et al., 1998). In contrast, postsynaptic changes in receptor accumulation can normalize postsynaptic activity without affecting the short-term dynamics of synaptic transmission. To date no studies on central synapses have examined both presynaptic and postsynaptic function in the same preparation or the effects of homeostatic plasticity on short-term synaptic dynamics.
Here we study in detail the effects of activity blockade on synaptic transmission at both sides of the synaptic cleft, using a preparation of cultured neocortical neurons after 7-10 d in vitro (DIV). We were unable to detect significant presynaptic changes after 2 d of TTX treatment. In contrast, 1 or 2 d of activity blockade significantly increased the synaptic accumulation of native AMPARs or of enhanced green fluorescent protein (EGFP)-tagged GluR2 in individual neurons. In addition, boosting of dendritic AMPA currents by TTX-sensitive sodium channels was increased by activity blockade. Our results suggest that, at young excitatory neocortical synapses, homeostatic plasticity occurs via coordinated changes in postsynaptic AMPAR and sodium channels and serves as a gain control mechanism that does not affect the short-term dynamics of synaptic transmission.
Materials and Methods
Neuronal cultures. Dissociated neuronal cultures were prepared from postnatal day 3 or 4 Long-Evans rat pups as described previously (Pratt et al., 2003). Cultures were used after 7-10 DIV. On each of the 2 d immediately before use, one-half of the dishes in a set of cultures was treated with 0.5 or 1 μm TTX, whereas the other half was left untreated.
Immunocytochemistry. Immunostaining of cultured neurons was performed as described previously (Rutherford et al., 1997). For the staining of surface GluR1 (Oncogene, Cambridge, MA) and GluR2 (Chemicon, Temecula, CA) receptors, live neurons were incubated with either anti-GluR1 or anti-GluR2 diluted with medium for 1 h in a 5% CO2 incubator at 37°C, followed by 1 h with 1.8 ml of medium to wash out unbound and nonspecifically bound antibodies. Then the cells were fixed as described above, and the secondary antibody Alexa Fluor 594 or 488 (Molecular Probes, Eugene, OR) was applied without permeabilization to label the surface receptors only. We verified that fixation alone did not permeabilize the membranes sufficiently to allow for detection of postsynaptic density-95 (PSD-95; which is localized intracellularly), indicating that this method allows us to detect surface receptors selectively (see supplemental figure, available at www.jneurosci.org as supplemental material). For double or triple labels with other synaptic markers, AMPARs were stained as above, and additional immunostaining was then performed as described previously (Rutherford et al., 1997). The following primary antibodies were used: rabbit or mouse anti-synapsin I (1:800; Chemicon), mouse anti-tau (1:1000; BD Transduction Laboratories, Lexington, KY), mouse anti-PSD-95 (1:100; Affinity BioReagents, Golden, CO), guinea pig anti-vesicular glutamate transporter 1 (anti-Vglut1) (1:500; Chemicon), rabbit anti-GluR1 (1:10; Oncogene), mouse anti-GluR2 (1: 20; Chemicon), and mouse anti-GFP (1:500; Molecular Probes). Secondary antisera were diluted 1:100 or 1:500.
Time-lapse microscopy. Transfection of plasmids for time-lapse imaging was performed by using the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's notes. The plasmid pBluescript, containing EGFP-tagged GluR2 DNA, was obtained from Dr. R. Malinow (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY) (Shi et al., 2001). The SmaI-XhaI fragment was subcloned into a pEF (elongation factor promoter) shooter vector (Invitrogen). PSD-95-Discosoma red (DsRed)T4 was made by inserting PSD-95 DNA (from Dr. D. Bredt, University of California, San Francisco, San Francisco, CA) (Craven et al., 1999) into a N1-DsRedT4 vector (from Dr. B. Glick, University of Chicago, Chicago, IL) (Bevis and Glick, 2002).
Imaging started 3 d after transfection. Only pyramidal neurons with moderate EGFP-GluR2 expression were selected for our experiments. Imaging solution contained the following (in mm): 117 NaCl, 5.3 KCl, 1.8 CaCl2, 0.814 MgSO4, 1 NaH2PO4, 20 HEPES, 50 dextrose, and 100 mg BSA, with a final osmolarity between 315 and 320 mOsm, pH 7.3. Between imaging sessions, the culture dishes were returned to the incubator. To minimize bleaching and photo damage, we used a 3% neutral density filter. The average peak intensity of puncta was well correlated with dendritic intensity in the same neuron (data not shown), indicating that neurons that expressed higher amounts of the EGFP-GluR2 protein showed higher peak and dendritic intensity and vice versa.
Photography and image analysis. Cultures were viewed on an inverted Olympus Optical (Tokyo, Japan) IX70 microscope with a 60× oil immersion objective with a numerical aperture of 1.25 (except for FM1-43 experiments; see below). Digital photographs were taken with a SenSys CCD camera (PhotoMetrics, Huntington Beach, CA) (see Fig. 6) or an Orca ER CCD camera (Hamamatsu Photonics, Hamamatsu, Japan) (see Figs. 1, 2, 3, 5). For Figures 1, 2, 3, seven or eight z-stack images were taken for each region by using a z-section focus drive. The exposure time and image bit range for each label were fixed within an experiment to allow for comparison between conditions. Digital images were quantified with Openlab software (Improvision, Lexington, MA).
Figure 6.

Number of putative synaptic contacts between pairs of pyramidal neurons. A, Pyramidal neurons were filled with biocytin through patch pipettes and then fixed and stained. Arrowheads indicate the course of the axon of the presynaptic neuron (pre). Action potentials in the top pyramidal neuron evoked a postsynaptic (post) response in the bottom pyramidal neuron. Scale bar, 30 μm. B, Magnification of an axon-dendrite crossing. Both presynaptic and postsynaptic structures are filled with biocytin (blue), but only the axon contains tau protein (red). Synapsin stain is shown in green. Scale bar, 2 μm. C, Summary of number of putative synaptic contacts between pairs of pyramidal neurons in control cultures (n = 11) and TTX-treated cultures (n = 8; p = 0.76). D, Dendrites of pyramidal neurons were visualized with a biocytin fill through a patch pipette (blue). Synapses were marked with antibodies against the presynaptic protein synapsin (green) and the postsynaptic protein PSD-95 (red), a marker for glutamatergic synapses. Scale bar, 1 μm. E, Number of synapsin puncta per micrometer of dendrite of pyramidal neurons for control (n = 6) and TTX-treated (n = 8) cultures. F, Percentages of synapsin puncta that were associated with PSD-95 puncta.
Figure 1.
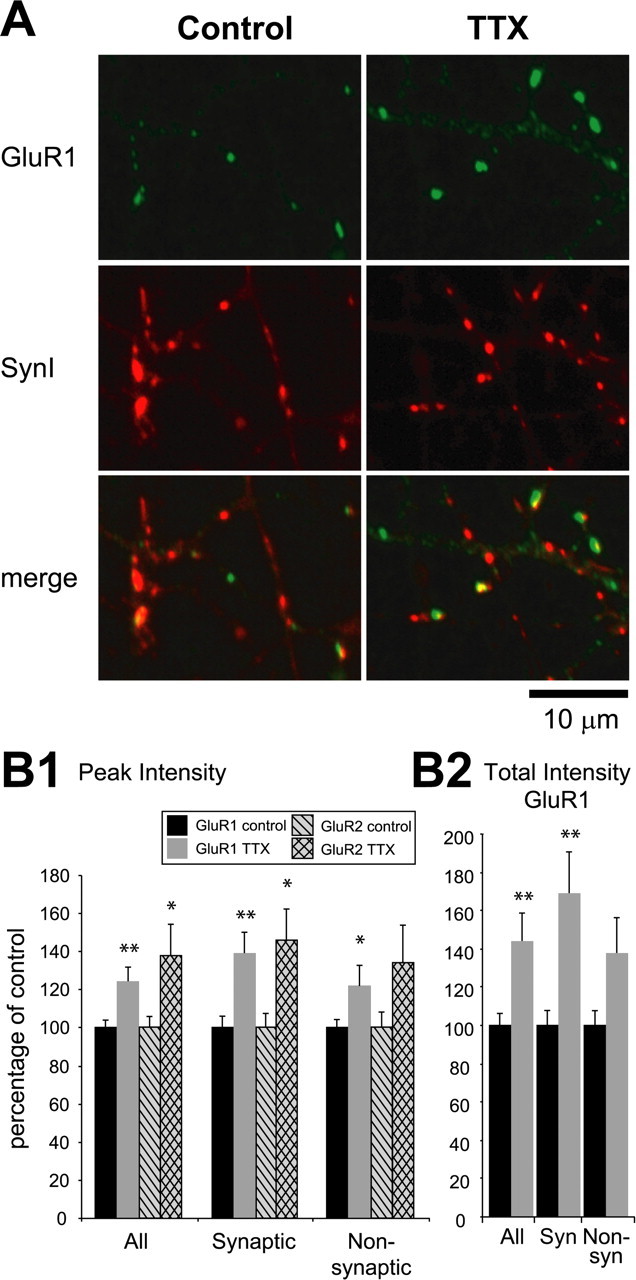
Activity blockade increases surface AMPA receptor accumulation. A, Representative immunofluorescent images of staining for control and TTX-treated cortical neurons. Cell-surface GluR1 receptors were immunolabeled (green) on live neurons, after which the neurons were fixed and permeabilized for synapsin I (SynI) staining (red). Green puncta associated with red puncta were considered synaptic GluR1 (61 ± 5% of anti-GluR1 puncta). B1, Quantification of GluR1 and GluR2 staining. Peak intensities of GluR1 or GluR2 puncta were measured, and cell averages were computed for control neurons or neurons treated with TTX for 2 d; values are expressed as a percentage of control values. TTX treatment significantly increased both surface GluR1 and GluR2. Synaptic GluR1 and GluR2 increased more than nonsynaptic GluR1 and GluR2. B2, Measuring total (instead of peak) intensity of GluR1 puncta gave similar results. Syn, Synaptic; Non-syn, nonsynaptic. n represents 20 control and 19 TTX-treated neurons (GluR1) and 7 control and 7 TTX-treated neurons (GluR2). Here and in the following figures, error bars represent SEM. Asterisks indicate significant differences (*p < 0.05; **p < 0.01.)
Figure 2.

Effect of TTX treatment on GluR1 and GluR2 colocalization. A, Colocalization of synaptic GluR1 and GluR2. Cell-surface GluR1 (red) and GluR2 (green) were immunolabeled on live neurons, after which the neurons were fixed and permeabilized for Vglut1 staining. B, TTX treatment increased GluR1/GluR2 colocalization at puncta that colocalized with the presynaptic marker Vglut1 [n = 10 neurons for control (Cont) and 13 for TTX]. Bars are normalized to the total number of synaptic GluR1 (top 2 bars) and GluR2 (bottom 2 bars) punctain each condition. C, Correlation of GluR1 and GluR2 total intensities from a representative control (left) and TTX-treated (right) neuron. Dots represent individual puncta. For these neurons, Pearson's correlation coefficients and p values from Spearman's rank test were as follows: control, r = 0.91 and p = 0.0002; TTX, r = 0.91 and p = 0.0005. a.u., Arbitrary units. D, Average correlations and slopes were not different between control and TTX-treated neurons. Correlations were calculated only in neurons that had at least five puncta with intensities above the group average (control, n = 6; TTX, n = 11).
Figure 3.

Time-lapse imaging of EGFP-GluR2. A, Cortical neurons transfected with EGFP-GluR2 showed punctate EGFP signals (green), which could be stained by anti-GFP antibody (red) in the nonpermeabilized condition, indicating surface expression of the EGFP-GluR2 receptors. A small fraction of dim EGFP-GluR2 puncta (20%) was not colocalized with surface antibodies and likely represents internal receptor clusters (arrow). B, For EGFP-GluR2 clusters that were detectable with surface staining, there was a strong correlation between the intensity of EGFP and Alexa Fluor 594 signals (anti-GFP antibody) from the same puncta. From this example, r = 0.89 and p < 0.0001. TTX treatment did not change the slope of the relationship (control, 261 puncta; TTX, 253 puncta; 7 neurons in each condition). a.u., Arbitrary units; Contr, control. C, PSD-95-DsRed was used as a postsynaptic marker to identify synaptic EGFP-GluR2 puncta. D, Example of time-lapse analysis in an EGFP-GluR2-expressing neuron: effect of 1 d of TTX treatment on EGFP intensity. E, TTX-treated neurons showed increased EGFP-GluR2 intensity. Data are from 10 control neurons (7 for day 2) and 12 TTX-treated neurons (10 for day 2). Dendrite intensity slightly decreased in both control and TTX-treated neurons. Intensity values were normalized to values at day 0. F, Ratio between peak intensity of EGFP-GluR2 puncta and dendritic intensity. The synaptic/dendritic intensity ratio increased significantly for TTX neurons after 1 d and to a lesser extent during day 2 of TTX treatment. For control neurons, this ratio did not change (p > 0.2). G, Cumulative distribution of synaptic/dendritic intensity ratio for all EGFP-GluR2 puncta in TTX-treated neurons. Day 0, 245 puncta; day 1, 306 puncta; day 2, 286 puncta. The control distribution did not change (day 0, 193 puncta; day 1, 244 puncta; day 2, 200 puncta) (data not shown). Asterisks in E and F indicate significance levels from paired Student's t tests (*p < 0.05; **p < 0.01; ***p < 0.001). Scale bars: A, C, 5 μm; D, 10 μm.
Figure 5.

Synaptic scaling does not affect vesicle recycling dramatically. A1, Distal dendrites of pyramidal neuron, labeled with Alexa Fluor 594. A2, Presynaptic terminals were labeled with electrical stimulation (30 s at 20 Hz) in the presence of 15 μm FM1-43. A3, After destaining (1500 stimuli at 10 Hz), FM1-43 puncta have disappeared. Only FM1-43 puncta that touched the dendrites of the filled neuron were included in the analysis. Scale bars, 5 μm. All images were taken from the same field of view. B, Cumulative distribution of intensity values of FM1-43 puncta labeled by weak electrical stimulation (20 stimuli at 0.5 Hz). Data are from 111 control and 113 puncta; there are nine neurons in each condition. Intensity values are shown in arbitrary units (a.u.). C, Cumulative distribution of intensity values of FM1-43 puncta labeled by high K+ protocol. Data are from 1404 control and 1191 TTX puncta; there are 14 neurons in each condition. D, Destaining time course for individual puncta in control neurons. Thick black dots are average values. E, Average destaining time course for control (black) and TTX puncta (gray). Error bars represent SD for 160 control and 103 TTX puncta (6 neurons in each condition). Puncta were destained maximally with 1500 stimuli at 10 Hz, because additional stimulation did not result in more destaining.
AMPAR intensity values from immunocytochemical or time-lapse images were analyzed as follows. Dendritic intensity was measured with respect to background intensity. Puncta were selected based on a fixed intensity difference from local dendrite intensity. For all selected puncta, the peak intensity (brightest pixel; 1 pixel = 0.11 × 0.11 μm) and total intensity (summed intensity of all pixels above threshold) were measured. Both intensity measures gave qualitatively similar results. Local dendritic intensity was subtracted from all puncta intensities. For the time-lapse analysis, only EGFP-GluR2 puncta that were associated with PSD-95-DsRed were selected to enhance the selection of synaptic puncta. All analysis was done blind to experimental condition.
Paired recordings. For electrophysiological recordings, the cultures were placed on the stage of a Nikon (Tokyo, Japan) Diaphot inverted microscope and perfused with artificial CSF (ACSF; brought to 320 mOsm with dextrose and bubbled with 5% CO2/95% O2) (Rutherford et al., 1997; Turrigiano et al., 1998; Watt et al., 2000). Internal solution contained the following (in mm): 100 K-gluconate, 20 KCl, 10 K-HEPES, 0.5 EGTA, 4 Mg-ATP, 3 Na-GTP, 10 Na2 phosphocreatine, and 0.1% biocytin, pH ∼7.4, with KOH (290 mOsm with sucrose). Junction potentials (∼15 mV) were left uncompensated. Whole-cell voltage-clamp recordings were obtained with Axopatch 200B amplifiers (Axon Instruments, Foster City, CA). Recordings were accepted only if the resting membrane potential (Vm) was below -50 mV and series resistance (Rs) was <30 MΩ and if these parameters changed <20% in the course of the recording. Average cell parameters included the following: Vm = -66 ± 3 mV, input resistance = 529 ± 91 MΩ, Rs = 19.3 ± 1.5 MΩ, and whole-cell capacitance = 21 ± 2 pF; they were not different between conditions. For the amplitude and coefficient of variation (CV) measurements, we restricted our data set to neurons with Rs <20 MΩ. This did not change any of the average cell parameters significantly.
Whole-cell recordings were made at room temperature from two to four pyramidal neurons simultaneously. Monosynaptic connections were identified by a short latency (<4 ms) and small temporal jitter (<1 ms). Short-term plasticity of monosynaptic excitatory connections (reversal potential, ∼0 mV) was measured by evoking trains of 10 action potentials at 20 Hz in the presynaptic neuron. The ACSF contained 50 μm APV to prevent the induction of any long-term changes in synaptic transmission. In some of the recordings, 2-5 μm DNQX was added to the ACSF to reduce spontaneously occurring synaptic events that interfered with our recordings. At this low concentration, DNQX reduced the EPSC amplitude by no more than 10%, and we verified that neither APV nor DNQX affected short-term depression. So that we could probe the recovery from short-term depression, an additional action potential followed the 20 Hz train by a variable interval (100, 200, 300, 500, or 700 ms). Trains of presynaptic action potentials were separated by 25-30 s to allow for full recovery of short-term plasticity. Traces were sampled at 10 kHz.
The amplitude of the postsynaptic current to each stimulus of the train was determined by assuming linear addition of the synaptic responses. For this purpose, we fit the decay of the preceding responses with an exponential function, using in-house software written with IgorPro (WaveMetrics, Lake Oswego, OR). We used the mean amplitude of the postsynaptic response (9-50 repetitions) to each action potential in the train (weighed by their SE) to fit an exponential function, yielding an estimate of the time constant of depression in each pair. The depression in all pairs was well described by a single exponential. Recovery from depression also was well described by an exponential function with a single time constant.
Fluorescent styryl dye experiments. Pyramidal neurons were transfected with DsRed [using a gene gun (Kilman et al., 2002) or Lipofectamine 2000] or filled through a patch pipette with the red dye Alexa Fluor 594 before FM1-43 labeling. Presynaptic terminals were labeled by evoking presynaptic activity with an extracellular electrode (two silver wires separated by ∼5 mm, at ∼2-3 mm distance from the imaged neuron) in the presence of the fluorescent styryl dye FM1-43. We verified that the stimulus reliably evoked action potentials in neurons in the vicinity of the imaged neuron. Labeling stimuli were applied in a HEPES-buffered solution containing the following (in mm): 119 NaCl, 2.5 KCl, 2 MgSO4,2 CaCl2, 25 HEPES, 30 glucose, pH 7.35 (∼320 mOsm), plus (in μm) 10 DNQX, 50 APV, 20 bicuculline, and 15 FM1-43. Images of distal dendrites of labeled pyramidal neurons were taken after a period of 15 min of wash with ACSF containing synaptic blockers. Images were taken with an Orca ER CCD camera (Hamamatsu Photonics) with a 40× oil immersion objective (1.3 numerical aperture) with 500-550 nm (green) and 590-650 nm (red) filters. Then the terminals were destained with 1500 pulses at 10 Hz, after which images were taken again. Fluorescence values of FM1-43-positive puncta were taken as the difference of fluorescence in these two images. In experiments in which the destaining time course was measured, images were taken every 10 s during the destaining stimulation. A second destaining stimulation was applied to ensure that destaining was maximal. Puncta intensities were normalized to their starting value (average of three to five images before the destaining protocol), and end values were subtracted (average of three images after the second destaining protocol). Bleaching was estimated by a linear fit through the last three intensity values of each punctum. After intensity values were corrected for bleaching (∼1% per image), the destaining of individual FM1-43 puncta was fit with a monoexponential function. Only puncta in which the intensity before destaining did not vary >20% were included.
Alternatively, synaptic activity was evoked by superfusion for 2 min with a high K+ HEPES-buffered solution (same as above, but with 81.5 mm NaCl and 40 mm KCl) with 15 μm FM1-43. During washout, the synaptic activity was blocked in ACSF containing 0.2 mm Ca2+, 5 mm Mg2+, and 0.1 μm TTX (15 min). Puncta were destained by a 5 min application of the high K+ solution in the absence of FM1-43.
Images were quantified with custom-made software written in IgorPro (WaveMetrics). Fluorescence values presented in Figure 5 were obtained by averaging an area of 2 × 4 of the brightest pixels of each visually selected punctum (1 pixel = 0.16 × 0.16 μm). Results were qualitatively indistinguishable if we used a computer algorithm [based on Nägler et al. (2001)] to select only puncta that had fluorescence values at least 3 SDs from the mean background or if intensity measures were taken over a 5 × 5 pixel area.
Glutamate responses. Whole-cell recordings were made with an internal solution based on KMeSO4 (Watt et al., 2000). Glutamate (5 mm in ACSF) was applied by a glass pipette (∼2 μm in diameter) for 5-7 ms at a pressure of 15-25 psi and located at the apical dendrite at distances of 100-250 μm from the soma. All recordings were done in the presence of 50 μm APV to isolate AMPA responses. After a steady baseline response was established in ACSF, 1 μm TTX was washed in. In approximately one-half of the recordings, we started in TTX and washed in ACSF to avoid any bias. During the wash-in period, the responses were monitored in voltage clamp, and we always checked whether the wash-in was complete before the responses were measured. Recordings were accepted only if input resistance did not change by >30% and membrane potential did not change by >16 mV. The mean amplitude and kinetics of the glutamate responses in ACSF were similar in control and TTX-treated neurons (data not shown). In some TTX-treated neurons, the glutamate application in ACSF induced action potentials in current-clamp mode. For these neurons, the glutamate responses were measured at more hyperpolarized potentials (-80 to -100 mV), resulting in a small under-estimation of the TTX effect in these cells.
Statistics. Statistical analyses were performed with unpaired Student's t tests, except for comparing different time points in time-lapse experiments and for glutamate responses in different perfusion media, for which a paired Student's t test was used. Correlations were described with Pearson's correlation coefficient, and their significance was tested with Spearman's ranked test.
Results
Experiments were performed on postnatal cultures of visual cortical neurons after 7-10 DIV. All recordings and cell fills were obtained from visually identified pyramidal neurons as described previously (Rutherford et al., 1998; Pratt et al., 2003). All TTX treatments were for 2 d, unless otherwise indicated.
Effect of activity blockade on cell-surface GluR1 and GluR2
Blocking activity in visual cortical cultures with TTX for 2 d results in a homeostatic increase in mEPSC amplitude and in evoked excitatory transmission (Rutherford et al., 1998; Turrigiano et al., 1998; Watt et al., 2000). In this study, we ask whether this increase is expressed presynaptically or postsynaptically. To examine whether synaptic accumulation of AMPAR is influenced by activity blockade, we began by immunolabeling cell-surface GluR1 and GluR2 in cortical pyramidal neurons. Surface staining that used antibodies directed against the extracellular N-terminal regions of these AMPAR subunits showed a punctate staining pattern (Fig. 1A). To discriminate between synaptic and nonsynaptic GluR puncta, we counterstained neurons for the presynaptic markers synapsin I or Vglut1 after fixing and permeabilization (Fig. 1A). The peak intensity of surface GluR1 puncta was increased significantly in neurons that were treated with TTX for 2 d (Fig. 1B1), with synaptic GluR1 puncta showing a more pronounced enhancement than nonsynaptic puncta. The density of synaptic GluR1 puncta was not altered significantly by TTX treatment (control, 0.10 ± 0.01 puncta/μm; 1122 puncta from 20 cells; TTX, 0.09 ± 0.01 puncta/μm; 773 puncta from 19 cells). The intensity of GluR2 puncta also increased in TTX-treated neurons (Fig. 1B1). Measuring total intensity (summed intensity of all pixels above threshold within a punctum) (Fig. 1B1) or peak intensity (Fig. 1B2) gave qualitatively similar results. These data indicate that blocking neuronal activity for 2 d increased both synaptic and nonsynaptic surface AMPARs, although the increase was more pronounced at synaptic sites.
To investigate the effect of inactivity on the composition of GluR1 and GluR2 within synapses, we performed anti-GluR1 and anti-GluR2 double immunolabeling in pyramidal neurons (Fig. 2A). TTX treatment produced a small increase in the percentage of synaptic puncta with colocalized GluR1 and GluR2 (Fig. 2B). Single-labeled puncta might represent synapses containing only one type of AMPA subunit or synapses in which one subunit is below our detection threshold. The apparent increase in colocalization therefore could result from the increased intensity of GluR staining after TTX treatment. This interpretation is supported by the observation that single-labeled puncta had significantly lower intensity values than double-labeled puncta (single-label intensity was 69 ± 9% of double-label intensity; p < 0.05). The intensities of the GluR1 and GluR2 signals in individual puncta showed a significant positive correlation in both control and TTX-treated neurons, and the mean slopes were similar (Fig. 2C,D). These results indicate that TTX treatment increased GluR1 and GluR2 synaptic receptor accumulation proportionally.
Time-lapse analysis of EGFP-GluR2 puncta
Using antibodies to detect changes in native receptor accumulation requires comparisons across groups of neurons grown under different conditions. To follow synaptic receptor accumulation in individual neurons over time, we performed time-lapse imaging of neurons that expressed an EGFP-tagged GluR2. Transfected neurons showed bright puncta, which were well colocalized with surface anti-GFP immunolabeling (Fig. 3A) and surface anti-GluR2 antibody staining (data not shown), and the intensities of the EGFP and antibody signals were well correlated (Fig. 3B). Importantly, this relationship was not altered by TTX treatment (Fig. 3B) (Pearson's r = 0.72 ± 0.07 for control and 0.75 ± 0.06 for TTX-treated neurons), indicating that the intensity of individual EGFP-GluR2 puncta could be used as a reliable indicator of the amount of surface receptors.
Approximately 20% of the EGFP-GluR2 puncta were not colocalized with surface anti-GluR2 or anti-EGFP staining (Fig. 3A, arrow) and likely represent intracellular EGFP-GluR2 receptor clusters. To follow EGFP-GluR2 surface accumulation at synaptic sites, we needed a way to select synaptic pools of EGFP-GluR2 receptors preferentially. We therefore cotransfected the neurons with the postsynaptic marker PSD-95-DsRed (Fig. 3C). Most (69 ± 7%) of EGFP-GluR2 puncta that colocalized with PSD-95-DsRed also were colocalized with synapsin, and the majority of synapsin puncta reflect functional synapses that can be labeled by FM1-43 (Pratt et al., 2003). PSD-95-DsRed cotransfection did not affect targeting of EGFP-GluR2 to synapses, because the percentage of EGFP-GluR2 puncta that were associated with synapsin was similar in neurons expressing only EGFP-GluR2 and in neurons expressing both EGFP-GluR2 and PSD-95-DsRed (cotransfection with PSD-95 was 112.8 ± 11.5% of GluR2 alone).
To follow receptor accumulation in individual neurons, we imaged distal dendrites at 1 d time intervals (Fig. 3D). In control neurons, the peak intensity of EGFP-GluR2 puncta was stable over a 2 d imaging period, whereas in neurons that were treated with 1 μm TTX, the intensity of EGFP-GluR2 puncta significantly increased (Fig. 3E). The magnitude of the increase in peak intensity of the EGFP-GluR2 signal was comparable with that detected by using surface staining of native receptors (compare Figs. 1B, 3E). These data show for the first time that activity blockade increases the accumulation of synaptic receptors in individual neurons over time.
In both control and TTX-treated neurons dendritic fluorescence slightly decreased over the 1 d period (Fig. 3E), which could indicate a small decline in the expression level of the EGFP-GluR2 receptors. Bleaching was estimated to reduce fluorescence by no more than 2-4%. The average ratio of the peak synaptic intensity to the dendritic intensity (synaptic/dendritic ratio) at day 0 was similar in control and TTX neurons, indicating that there was no initial bias in overall expression levels between the two groups (Fig. 3F).
To control for differences in receptor expression over time, we reexpressed our time-lapse data as the synaptic/dendritic ratio. This ratio increased significantly and progressively in neurons that were treated with TTX for 2 d, whereas the ratio for control neurons remained quite constant (Fig. 3F). TTX treatment affected the entire population of EGFP-GluR2 puncta, which is clear from the shift to the right in the cumulative intensity distribution (Fig. 3G).
Overexpression of PSD-95 has been reported to affect AMPAR localization (El-Husseini et al., 2000; Stein et al., 2003; Ehrlich and Malinow, 2004). In our neurons, mild overexpression did not affect any of the synaptic parameters that we measured. The proportion of AMPAR puncta associated with presynaptic markers was not different (see above). The ratio of dendritic to synaptic EGFP-GluR2 intensities was similar in neurons transfected with both PSD-95-DsRed and EGFP-GluR2 or with EGFP-GluR2 alone (5.4 ± 0.3 and n = 18 neurons vs 6.4 ± 0.7 and n = 15 neurons; p = 0.21), suggesting that overexpression of PSD-95 did not by itself affect synaptic GluR2 accumulation. The lack of effect of PSD-95 coexpression on AMPAR synaptic localization and accumulation, as well as the similar magnitude increase in receptor intensity for native receptors and for EGFP-GluR2 coexpressed with PSD-95, suggests that coexpression of PSD-95 does not affect the behavior of AMPARs significantly in this assay.
Short-term plasticity
The data presented above suggest that changes in postsynaptic AMPAR accumulation can account for the increased quantal amplitude induced by activity blockade. However, whereas 2 d of activity blockade increase quantal amplitude by 1.5- to 2-fold (Turrigiano et al., 1998), evoked unitary EPSCs increase by twofold to threefold (Fig. 4A,B) (Turrigiano et al., 1998; Watt et al., 2000). This suggests that changes in quantal amplitude cannot account fully for the increase in evoked transmission. In the previous sections, evidence was given for a postsynaptic change induced by prolonged TTX treatment. In hippocampal neurons, activity blockade can induce presynaptic changes in FM1-43 recycling (Murthy et al., 2001). A presynaptic increase in release probability will increase EPSC amplitude, but it also is predicted to increase short-term depression (Zucker and Regehr, 2002). To determine whether short-term plasticity is altered by activity blockade, we recorded from pairs of pyramidal neurons in control and “sister” TTX-treated neocortical cultures. We evoked trains of 10 precisely timed action potentials at 20 Hz in the presynaptic neuron (held in current clamp) and recorded synaptic currents in the postsynaptic neuron held in voltage clamp at -70 mV (Fig. 4A). Two days of TTX treatment increased the amplitude of the evoked EPSCs twofold to threefold (Fig. 4A,B), but despite this large increase in amplitude, the short-term plasticity was not different in control and TTX-treated pairs. Monosynaptic connections showed similar depression in both conditions (Fig. 4A,C). Short-term depression of EPSC amplitudes could be fit by a single exponential, with a time constant of 86 ± 24 ms in control and 81 ± 9 ms in TTX-treated pairs. The EPSC amplitudes reached similar steady-state levels under the two conditions (22 ± 4% of initial amplitude for control and 22 ± 6% for TTX-treated pairs). Recovery from depression was probed by evoking a single action potential in the presynaptic neuron at various intervals after the train. There was no difference in the time constant of recovery between control and TTX-treated pairs (Fig. 4D). Mean latency (2.6 ± 0.2 ms) and decay time constant (13.4 ± 2.2 ms) of the evoked EPSCs were not different in control and TTX-treated pairs. In addition, there were no significant differences in resting membrane potential, input resistance, and other whole-cell parameters (see Materials and Methods).
Figure 4.

Synaptic scaling does not alter short-term synaptic plasticity. A, Representative examples of the postsynaptic response (bottom traces) to a 20 Hz train of 10 action potentials (top trace) in pairs of pyramidal neurons from control (CTL) and TTX-treated (TTX) cortical cultures. B, EPSC amplitude (first of the train) for control and TTX-treated pairs. Asterisk indicates a significant difference (p < 0.02). C, Normalized EPSC amplitudes averaged over all neurons as a function of the time of the presynaptic action potentials. Exponential curves were constructed from the mean time constant of depression for all neurons (control, τ = 86 ms; TTX, τ = 81 ms; 7 pairs in both conditions). D, Time constants of single exponential functions describing the recovery from depression (control, n = 7; TTX, n = 6; p = 0.41). E, CV of EPSCs from control and TTX-treated pairs.
A simple binomial model of a synaptic connection between two neurons predicts that the CV of the EPSC amplitude is strongly dependent on the presynaptic parameters N and p:
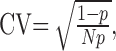 |
1 |
where N is the number of independent release sites and p is the average release probability (Faber and Korn, 1991; Korn and Faber, 1991). An unchanged CV while synaptic efficacy is altered reflects a postsynaptic change (Faber and Korn, 1991; Korn and Faber, 1991), whereas a presynaptic increase in p or N is expected to reduce the CV. We measured the CV of the evoked EPSCs from our pyramidal neuron pairs and found that the CV values for control and TTX-treated pairs were not different (control, 0.34 ± 0.03; TTX, 0.34 ± 0.04; p = 0.92) (Fig. 4E) and were very close to values we have reported previously for control pairs (Pratt et al., 2003). Together with the lack of effect on short-term depression, this suggests that a change in p did not contribute significantly to the increase in synaptic transmission induced by activity blockade.
FM1-43 experiments
We used the styryl dye FM1-43 to probe further the possible changes in the presynaptic terminals induced by activity blockade. An increase in the number of functional release sites or an increased release and recycling of vesicles is expected to result in an increased uptake of FM1-43 by the presynaptic terminals during synaptic activity. Synaptic activity was evoked by extracellular electrical stimulation or perfusion with a high K+ solution. We determined the intensity of FM1-43-labeled puncta that were touching the distal dendrites of pyramidal neurons (Fig. 5A), which were labeled with a red dye through a patch pipette or by DsRed transfection. From our immunostaining results (see below), we estimate that at least 70% of these puncta represent excitatory synapses. In some experiments, FM4-64 (N-(3-triethylammoniumpropyl)-4-(6-(4-(diethylamino)phenyl)hexatrienyl)pyridinium dibromide) was used, and pyramidal neurons were labeled with EGFP; similar results were obtained with both combinations of labels.
We first used a modest loading protocol [20 stimuli at 0.5 Hz (Murthy et al., 2001); destaining with 1500 stimuli at 10 Hz] to label presynaptic terminals. This relatively weak loading protocol presumably releases only a subset of available vesicles at each terminal and will fail to label terminals with very low p. An increase in p therefore should result in an increased number or intensity of labeled terminals. TTX treatment did not affect FM1-43 uptake with this labeling protocol (mean puncta intensity for TTX-treated neurons was 94.3 ± 30.5% of control) (Fig. 5B). The density of FM1-43 puncta (puncta/unit length of dendrite) also was unaffected (TTX density was 97.8 ± 18.0% of control).
We next used a protocol designed to label all vesicles available for release. Cultures were perfused for 2 min with a high K+ solution to depolarize strongly all presynaptic terminals. A change in the number of release sites or the number of vesicles that are available for release after TTX treatment is expected to result in altered labeling with this protocol. There was only a small increase in the mean fluorescence value of FM1-43 puncta on pyramidal dendrites (mean puncta intensity of TTX neurons was 107.5 ± 13.8% of control) (Fig. 5C). There was no effect on the density of labeled terminals (TTX was 93.4 ± 11.0% of control density). Extending the period of TTX treatment to 4 d also did not affect FM1-43 uptake by synaptic activity (data not shown), suggesting that the absence of an effect on FM1-43 uptake could not be explained by a slow time course to develop presynaptic changes.
We then examined the rate at which vesicles are released and recycled. Presynaptic terminals were labeled with a strong electrical stimulation (30 s stimulation at 20 Hz) (Chi et al., 2003), after which we imaged the destaining of the terminals during the application of 1500 stimuli at 10 Hz (Fig. 5D,E). As before, there was no difference between the density and intensity of FM1-43 puncta in control and TTX-treated neurons after the staining protocol (data not shown). The density of labeled terminals (0.14 ± 0.07 μm-1) was comparable with the density of synapsin puncta (see below), indicating that the majority of presynaptic terminals were labeled. Destaining of each FM1-43 punctum was described with a single exponential function. The destaining of FM1-43 puncta was slightly but not significantly slower in TTX-treated neurons (mean time constant was 33.0 ± 3.6 s for control and 38.0 ± 2.3 s for TTX-treated neurons; p = 0.26). Together, these results indicate that 2 d of activity blockade produced, at most, subtle changes in vesicle release and recycling in the presynaptic terminal.
Number of excitatory synapses
Another mechanism for increasing synaptic strength is to increase the number of synaptic contacts between presynaptic and postsynaptic neurons. To determine whether activity blockade affects the number of contacts, we filled pyramidal neurons with biocytin and fixed and stained them for the presynaptic protein synapsin I and the axonal protein tau. Using these fluorescent markers, we easily could follow the presynaptic axon branching over the dendritic tree of the postsynaptic pyramidal neuron, and we counted axon-dendrite crossings that were associated with synapsin (Fig. 6A,B) as described previously (Pratt et al., 2003). Pairs of pyramidal neurons made, on average, 7.8 ± 0.9 putative synaptic contacts in control cultures and 7.0 ± 1.0 contacts in TTX-treated cultures, indicating that TTX treatment did not affect the number of contacts between pairs (Fig. 6C). There was no correlation between the number of synaptic contacts between two neurons and the distance between their somata for the range of distances in our data set (25-400 μm; p = 0.65), as found previously for EPSC amplitude (Pratt et al., 2003).
TTX treatment also did not alter overall synaptic density, because the number of synapsin-positive puncta per unit length of dendrite did not change (Fig. 6D,E). This is consistent with the results from the FM1-43 experiments, which showed no change in the density of FM1-43-labeled presynaptic puncta. In addition, ∼70% of all synapsin puncta on the distal dendrites of pyramidal neurons were associated with PSD-95 (a selective marker of excitatory synapses) for both control and TTX-treated neurons (Fig. 6F). These data show that TTX treatment did not affect the number of synaptic contacts between pyramidal neurons or the overall density of excitatory synapses.
Amplification of postsynaptic response by sodium channels
The experiments described above suggest that changes in release probability and the number of release sites are unlikely to contribute significantly to the increase in evoked transmission induced by activity blockade. This suggests that the larger effect of activity blockade on evoked transmission than on quantal currents is attributable to an additional postsynaptic mechanism. Evoked synaptic currents are much larger than quantal events, and the local depolarization they generate might be strong enough to activate dendritic voltage-gated sodium channels, which are known to boost synaptic currents (Magee and Johnston, 1995; Schwindt and Crill, 1995; Stuart and Sakmann, 1995; Lipowsky et al., 1996). We have shown previously that 2 d of activity blockade increases sodium current density (Desai et al., 1999), suggesting that this boosting might be enhanced. We therefore examined the contribution of voltage-gated sodium channels to the postsynaptic response to glutamate in control neurons and neurons treated for 2 d with TTX.
To test the contribution of TTX-sensitive currents, we found it necessary to measure the amplitude of evoked AMPA responses in the same neuron before and after wash-in of TTX. Because TTX blocks synaptic transmission, we could not measure synaptically evoked responses, so instead we used direct glutamate application to the dendrites to evoke AMPA-mediated responses, as described previously (Turrigiano et al., 1998; Watt et al., 2000). Glutamate (5 mm) was applied through a patch pipette close to the dendrite to evoke postsynaptic responses with amplitudes in the same range as synaptically evoked EPSCs (50-300 pA). Responses were measured in both voltage clamp and current clamp, always from a membrane potential of -70 mV, before and after wash-in of 1 μm TTX. In current clamp, TTX reduced glutamate responses only slightly in control neurons, whereas in TTX-treated neurons, the responses were reduced in amplitude and in area by 20-30% (Fig. 7A,B). This indicates that in TTX-treated neurons, but not in control neurons, dendritic glutamate responses are amplified significantly by TTX-sensitive sodium channels. This was not attributable to a difference in initial amplitude of the glutamate-evoked responses, because we adjusted our glutamate application so that TTX-treated and control neurons had similar initial amplitudes (control, 19.1 ± 3.7 mV; TTX-treated, 20.4 ± 4.6 mV). In voltage clamp, the effect of TTX was slightly smaller but in the same direction (Fig. 7C). We noticed a clear dependence of the degree of effect of TTX wash-in on the distance of the glutamate application from the soma (Pearson's r = -0.56). TTX wash-in had the strongest effect when the glutamate application was >150 μm from the soma (Fig. 7C). Average dendritic distances between glutamate application and soma were similar in control and TTX-treated neurons (control, 149 ± 15 μm; TTX, 151 ± 9 μm). This suggests that significant amplification of postsynaptic responses occurs locally in distal dendrites of TTX-treated neurons in which the local membrane potential escapes the somatic voltage clamp and that this amplification is stronger at more distal locations.
Figure 7.

Amplification of glutamate responses by sodium channels. A, Current-clamp recordings of responses to dendritic glutamate application for a control and a TTX-treated neuron. Black traces are responses in ACSF; gray traces are responses after wash-in of 1 μm TTX. Holding membrane potential was -70 mV. Calibration: 10 mV, 100 ms. B, Black bars represent control neurons (n = 6); gray bars represent TTX-treated neurons (n = 10). Asterisk indicates a significant difference (p < 0.05) between responses before and after TTX wash-in (paired Student's t test). C, Mean amplitude of the glutamate responses recorded in voltage clamp (at -70 mV) after wash-in of TTX, expressed as a percentage of values in ACSF alone. Left bars represent data from all dendritic locations, and right bars represent data from dendritic locations >150 μm from the soma. Data are from 8 control and 12 TTX-treated neurons.
To estimate the location of synapses between pyramidal neurons, we calculated the dendritic length between putative synaptic contacts and the postsynaptic soma from the data set from Figure 6, A and B. These contacts were distributed in an apparently random manner over the dendritic tree at distances ranging from 10 to 270 μm from the soma (average distance was 112 ± 8 μm). In both control and TTX-treated pairs, ∼40% of the putative synaptic contacts were located at distances >150 μm from the soma. Many of the contacts were located on thin dendrites, which removes them even further from the influence of the somatic voltage clamp. This suggests that a significant fraction of synapses between pyramidal neurons is at remote locations where the evoked synaptic depolarization can be amplified by sodium channels.
Discussion
Synaptic scaling is a form of homeostatic plasticity that regulates synaptic strength in response to prolonged changes in activity. To understand the functional consequences of synaptic scaling, we believe it is important to understand its expression mechanism or mechanisms. Here we describe in detail the homeostatic changes that take place at excitatory synapses in visual cortical cultures after spiking activity in the network is blocked for 1-2 d. Our results indicate that synaptic scaling at young neocortical synapses is expressed mostly postsynaptically. Activity blockade more than doubled the strength of synaptic connections between pyramidal neurons via a combination of increased accumulation of AMPARs in the postsynaptic membrane and increased boosting of AMPA currents by postsynaptic sodium channels. The numbers of excitatory synapses as well as vesicle release and recycling in presynaptic terminals were not significantly affected. Most importantly from a functional point of view, short-term dynamics of synaptic transmission and variability of EPSC amplitude were unaltered. Lowering visual cortical activity through visual deprivation also adjusts excitatory quantal amplitude without much effect on synaptic dynamics (Desai et al., 2002; Maffei et al., 2004), suggesting that this feature of homeostatic plasticity is conserved in vitro and in vivo. These results suggest that homeostatic synaptic scaling in young neocortical neurons functions as a gain control mechanism that regulates neuronal activity without affecting synaptic dynamics.
We used several methods to demonstrate postsynaptic changes in receptor accumulation. Consistent with previous results in spinal cultures (O'Brien et al., 1998), 2 d of TTX treatment significantly increased the intensity of native AMPAR surface staining via proportional increases in GluR1 and GluR2 subunits. This accords well with careful measurements of mEPSC kinetics, which have shown no change in decay kinetics after blockade of activity that used either TTX or glutamate receptor blockers at these neocortical synapses (Turrigiano et al., 1998), as expected if subunit composition of AMPAR does not change (Geiger et al., 1995; Thiagarajan et al., 2002; Grosskreutz et al., 2003). In contrast, recent reports have suggested that in hippocampal cultures, activity blockade leads to preferential increases in GluR1 synthesis and/or synaptic localization without significant changes in GluR2 and alters decay kinetics and pharmacological sensitivity of mEPSCs in a manner consistent with a greater GluR1 content (Thiagarajan et al., 2002; Ju et al., 2004). The reported increase in density of GluR1 puncta after activity blockade is likely to reflect increased GluR1 insertion at existing synapses, because mEPSC amplitude increased but frequency was not altered (Ju et al., 2004). These subunit-specific differences in receptor accumulation between neocortical and spinal neurons on the one hand, and hippocampal neurons on the other, suggest that there are fundamental differences in activity-dependent receptor trafficking at these different types of central synapses. The mechanism by which the increased AMPA receptor accumulation at postsynaptic sites occurs remains to be unraveled. Several possibilities exist: posttranscriptional regulation, posttranslational regulation, or a change in protein turnover perhaps because of interactions with other (synaptic) proteins.
As a second method of quantifying altered receptor accumulation, we used time-lapse imaging of EGFP-tagged AMPAR. With this method, we could follow for the first time changes in receptor accumulation in individual neurons during the period of TTX treatment. Following the same population of synapses over time in individual neurons demonstrated that the entire distribution of synaptic receptor intensities increased during activity blockade, similar to the increase in the mEPSC amplitude distribution reported previously (Turrigiano et al., 1998). These data suggest that synaptic AMPAR accumulation increases 50-100% during 2 d of activity blockade, a magnitude of change that matches closely the change in quantal amplitude (Turrigiano et al., 1998; Watt et al., 2000).Together, these data indicate that increased receptor accumulation can account for the increase in quantal amplitude and contributes significantly to the enhancement of evoked excitatory transmission.
In contrast to the clear effects on receptor accumulation, activity blockade induced no detectable changes in presynaptic parameters of transmission. A thorough analysis of presynaptic function, including short-term plasticity, variability of synaptic transmission, and FM1-43 dye uptake and release, revealed no significant changes in any of these parameters. We did not find any difference in FM1-43 uptake in control and TTX-treated neurons. The destaining of FM1-43 puncta was somewhat slower in TTX-treated neurons, possibly indicating a small decrease in release probability or a subtle difference in presynaptic vesicle recycling, but these effects are in the wrong direction to contribute to the increase in EPSC amplitude induced by activity blockade. The electrophysiological measures are less sensitive to changes in N than in p. However, an increase in N is predicted to increase the number of FM1-43-labeled puncta and/or the intensity of FM1-43 labeling, but no such changes were detected. In addition, our immunostaining experiments did not show any difference in the number of putative synapses between pyramidal neurons or in overall excitatory synaptic density. Together, these data suggest that there are, at best, only subtle changes in presynaptic function at these neocortical synapses.
Synaptic currents in TTX-treated neurons were more than doubled in amplitude compared with control EPSCs (Fig. 4A,B), whereas quantal amplitude (and AMPAR accumulation) increased by only 50-100% (Turrigiano et al., 1998; Watt et al., 2000). Our data suggest that the additional increase in evoked transmission is attributable in part to an enhanced amplification of synaptic currents by sodium channels. Sodium currents increase in amplitude by ∼30% after 2 d of TTX treatment (Desai et al., 1999), and a recent report suggested that dendritic sodium channels are upregulated after a 2 d of TTX treatment (Aptowicz et al., 2004). Dendritically localized sodium channels can boost the amplitude of synaptic currents (Magee and Johnston, 1995; Schwindt and Crill, 1995; Stuart and Sakmann, 1995; Lipowsky et al., 1996). We found little decrement in glutamate-evoked currents when sodium channels were blocked in control neurons but found a 20-30% decrease in TTX-treated neurons. A 30% increase in amplification in addition to the increase in quantal amplitude would result in a 2- to 2.5-fold total increase in EPSC amplitude, as observed. Other voltage-dependent conductances also change after TTX treatment (Desai et al., 1999), and it is possible that additional dendritic conductances contribute to the differential boosting of synaptic currents in control and TTX-treated neurons.
In hippocampal cultures, prolonged inactivity can increase synapse size and the number of vesicles available for release (Murthy et al., 2001). This has led to the suggestion that homeostatic plasticity occurs via an increase in synapse size, which concomitantly expands the presynaptic and postsynaptic area and generates coupled increases in release probability and in postsynaptic receptor number. One difficulty with this model is that synaptic growth is slow [only a 30% increase in synaptic area after 2 d (Murthy et al., 2001)], whereas changes in mEPSC amplitude are larger and faster [a doubling in amplitude after 2 d (Turrigiano et al., 1998)]. We now show that increased receptor accumulation can occur independently of any measurable changes in presynaptic function. This argues strongly against the “synapse growth” model of homeostatic plasticity in which changes in presynaptic and postsynaptic function are coupled inextricably. Instead, our data support the idea that presynaptic and postsynaptic changes reflect separable and independent homeostatic mechanisms, with the potential for differential expression at different synapses, at different developmental stages, or in response to different experimental manipulations (Turrigiano et al., 1998; Paradis et al., 2001; Burrone et al., 2002; Thiagarajan et al., 2002; Ju et al., 2004).
Synapses transmit information from one neuron to the next. An important determinant of how information gets transferred is the dynamic behavior of the synapse. Facilitating synapses can serve to emphasize bursts of action potentials (Lisman, 1997; Kepecs and Lisman, 2003), whereas synaptic depression can serve as a dynamic gain control mechanism that emphasizes changes in presynaptic firing rates independent of absolute rates (Abbott et al., 1997; Markram et al., 1998). Plasticity mechanisms that alter short-term dynamics thus will affect the information that a synapse is able to transmit. Here we show that homeostatic synaptic scaling does not affect the short-term plasticity or variability of synaptic transmission. Interestingly, another important aspect of excitatory synaptic transmission, the ratio of current through AMPA and NMDA receptors, also is maintained after activity blockade (Watt et al., 2000). These results suggest that synaptic scaling at young neocortical synapses is expressed postsynaptically and adjusts the gain without fundamentally altering the information content of excitatory synaptic transmission.
Footnotes
This work was supported by the Netherlands Organization for Scientific Research (C.J.W.), the Sloan-Schwartz Foundation (C.J.W.), the Uehara Memorial Foundation (K.I.), and National Institutes of Health Grant NS 36853. We thank M. Herman and M. Walsh for technical assistance and R. Malinow (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY), D. Bredt (University of California, San Francisco, San Francisco, CA), and B. Glick (University of Chicago, Chicago, IL) for kindly providing DNA constructs.
Correspondence should be addressed to Gina G. Turrigiano at the above address. E-mail: turrigiano@brandeis.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252895-11$15.00/0
C.J.W. and K.I. contributed equally to this work.
References
- Abbott LF, Varela JA, Sen K, Nelson SB (1997) Synaptic depression and cortical gain control. Science 275: 220-224. [DOI] [PubMed] [Google Scholar]
- Aptowicz CO, Kunkler PE, Kraig RP (2004) Homeostatic plasticity in slice cultures involves changes in voltage-gated Na+ channel expression. Brain Res 998: 155-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacci A, Coco S, Pravettoni E, Schenk U, Armano S, Frassoni C, Verderio C, De Camilli P, Matteoli M (2001) Chronic blockade of glutamate receptors enhances presynaptic release and downregulates the interaction between synaptophysin-synaptobrevin-vesicle-associated membrane protein 2. J Neurosci 21: 6588-6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevis BJ, Glick BS (2002) Rapidly maturing variants of the Discosoma red fluorescent protein (DsRed). Nat Biotechnol 20: 83-87. [DOI] [PubMed] [Google Scholar]
- Burrone J, Murthy VN (2003) Synaptic gain control and homeostasis. Curr Opin Neurobiol 13: 560-567. [DOI] [PubMed] [Google Scholar]
- Burrone J, O'Byrne M, Murthy VN (2002) Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 420: 414-418. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA (2003) Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron 38: 69-78. [DOI] [PubMed] [Google Scholar]
- Craven SE, El-Husseini AE, Bredt DS (1999) Synaptic targeting of the postsynaptic density protein PSD-95 mediated by lipid and protein motifs. Neuron 22: 497-509. [DOI] [PubMed] [Google Scholar]
- Davis GW, Bezprozvanny I (2001) Maintaining the stability of neural function: a homeostatic hypothesis. Annu Rev Physiol 63: 847-869. [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG (1999) Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat Neurosci 2: 515-520. [DOI] [PubMed] [Google Scholar]
- Desai NS, Cudmore RH, Nelson SB, Turrigiano GG (2002) Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci 5: 783-789. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R (2004) Postsynaptic density-95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci 24: 916-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS (2000) PSD-95 involvement in maturation of excitatory synapses. Science 290: 1364-1368. [PubMed] [Google Scholar]
- Faber DS, Korn H (1991) Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J 60: 1288-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JRP, Melcher T, Koh D-S, Sakmann B, Seeburg PH, Jonas P, Monyer H (1995) Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron 15: 193-204. [DOI] [PubMed] [Google Scholar]
- Grosskreutz J, Zoerner A, Schlesinger F, Krampfl K, Dengler R, Bufler J (2003) Kinetic properties of human AMPA-type glutamate receptors expressed in HEK293 cells. Eur J Neurosci 17: 1173-1178. [DOI] [PubMed] [Google Scholar]
- Ju W, Morishita W, Tsui J, Gaietta G, Deerinck TJ, Adams SR, Garner CC, Tsien RY, Ellisman MH, Malenka RC (2004) Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci 7: 244-253. [DOI] [PubMed] [Google Scholar]
- Kepecs A, Lisman JE (2003) Information encoding and computation with spikes and bursts. Network: Comput Neural Syst 14: 103-118. [PubMed] [Google Scholar]
- Kilman V, van Rossum MCW, Turrigiano GG (2002) Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABAA receptors clustered at neocortical synapses. J Neurosci 22: 1328-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn H, Faber DS (1991) Quantal analysis and synaptic efficacy in the CNS. Trends Neurosci 14: 439-445. [DOI] [PubMed] [Google Scholar]
- Lipowsky R, Gillessen T, Alzheimer C (1996) Dendritic Na+ channels amplify EPSPs in hippocampal CA1 pyramidal cells. J Neurophysiol 76: 2181-2191. [DOI] [PubMed] [Google Scholar]
- Lisman JE (1997) Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci 20: 38-43. [DOI] [PubMed] [Google Scholar]
- Lissin DV, Gomperts SN, Carroll RC, Christine CW, Kalman D, Kitamura M, Hardy S, Nicoll RA, Malenka RC, von Zastrow M (1998) Activity differentially regulates the surface expression of synaptic AMPA and NMDA glutamate receptors. Proc Natl Acad Sci USA 95: 7097-7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, Nelson SB, Turrigiano GG (2004) Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nat Neurosci 7: 1353-1359. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D (1995) Synaptic activation of voltage-gated channels in the dendrites of hippocampal pyramidal neurons. Science 268: 301-304. [DOI] [PubMed] [Google Scholar]
- Marder E, Prinz AA (2002) Modeling stability in neuron and network function: the role of activity in homeostasis. BioEssays 24: 1145-1154. [DOI] [PubMed] [Google Scholar]
- Markram H, Wang Y, Tsodyks M (1998) Differential signaling via the same axon of neocortical pyramidal neurons. Proc Natl Acad Sci USA 95: 5323-5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KD (1996) Synaptic economics: competition and cooperation in synaptic plasticity. Neuron 17: 371-374. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Schikorski T, Stevens CF, Zhu Y (2001) Inactivity produces increases in neurotransmitter release and synaptic size. Neuron 32: 673-682. [DOI] [PubMed] [Google Scholar]
- Nägler K, Mauch DH, Pfrieger FW (2001) Glia-derived signals induce synapse formation in neurones of the rat central nervous system. J Physiol (Lond) 533: 665-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL (1998) Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 21: 1067-1078. [DOI] [PubMed] [Google Scholar]
- Paradis S, Sweeney ST, Davis GW (2001) Homeostatic control of presynaptic release is triggered by postsynaptic membrane depolarization. Neuron 30: 737-749. [DOI] [PubMed] [Google Scholar]
- Pratt KG, Watt AJ, Griffith LC, Nelson SB, Turrigiano GG (2003) Activity-dependent remodeling of presynaptic inputs by postsynaptic expression of activated CaMKII. Neuron 39: 269-281. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, DeWan A, Lauer HM, Turrigiano GG (1997) Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. J Neurosci 17: 4527-4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, Turrigiano GG (1998) BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron 21: 521-530. [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Crill WE (1995) Amplification of synaptic current by persistent sodium conductance in apical dendrite of neocortical neurons. J Neurophysiol 74: 2220-2224. [DOI] [PubMed] [Google Scholar]
- Shi S-H, Hayashi Y, Esteban JA, Malinow R (2001) Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105: 331-343. [DOI] [PubMed] [Google Scholar]
- Stein V, House DRC, Bredt DS, Nicoll RA (2003) Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci 23: 5503-5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart GJ, Sakmann B (1995) Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron 15: 1065-1076. [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Piedras-Renteria ES, Tsien RW (2002) α- and βCaMKII: inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron 36: 1103-1114. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB (2004) Homeostatic synaptic plasticity in the developing nervous system. Nat Rev Neurosci 5: 97-107. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB (1998) Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391: 892-896. [DOI] [PubMed] [Google Scholar]
- Watt AJ, van Rossum MCW, MacLeod KM, Nelson SB, Turrigiano GG (2000) Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron 26: 659-670. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355-405. [DOI] [PubMed] [Google Scholar]