

Abstract
In ataxia-telangiectasia (A-T), the loss of the ataxia-telangiectasia mutated (ATM) kinase leads to a failure of cell cycle checkpoints and DNA double-strand break detection resulting in cellular radiation sensitivity and a predisposition to cancer. There is also a significant loss of neurons, in particular cerebellar granule and Purkinje cells. Mice homozygous for null alleles of atm reproduce the radiation sensitivity and high-tumor incidence of the human disease but show no significant nerve cell loss. Using immunocytochemistry, we found the re-expression of cell cycle proteins in Purkinje cells and striatal neurons in both human and mouse A-T. In the mouse, we used fluorescent in situ hybridization (FISH) to document that DNA replication accompanies the reappearance of these proteins in at-risk neuronal cells. We also found the presence of significant cell cycle activity in the Purkinje cells of the atm+/- heterozygote mouse. The cell cycle events in mouse cerebellum occur primarily during the third postnatal week by both FISH and immunocytochemistry. Thus, the initiation of this ectopic cell division occurs just as the final stages of Purkinje cell development are being completed. These results suggest that loss of cell cycle control represents a common disease mechanism that underlies the defects in the affected tissues in both human and mouse diseases.
Keywords: ATM, cell cycle, cerebellum, neurodegenerative disorder, FISH, PCNA
Introduction
Ataxia-telangiectasia (A-T) is an autosomal recessive disorder characterized by cerebellar ataxia, retinal telangiectasia, immunodeficiency, radiosensitivity, infertility, predisposition to malignancies and progressive neuronal degeneration (Crawford, 1998; Kastan et al., 2000). Pathological examination reveals significant degeneration of cerebellar Purkinje and granule cells (Crawford, 1998) plus a later loss of neurons in striatum and substantia nigra (Ostetowska and Traczynska, 1964; Koepp et al., 1994). The disease is associated with mutations in ataxia-telangiectasia mutated (ATM), a gene encoding a 370 kDa serine/threonine kinase with sequence similarity to the catalytic subunit of phosphatidyl-inositol-3-kinase (Savitsky et al., 1995). Most of its substrates appear to be involved in cell cycle checkpoints and DNA double-strand break response (Abraham, 2001; Shiloh and Kastan, 2001).
Engineered mutations in the mouse ATM homolog (atm) accurately mimic the non-CNS features of human A-T (Barlow et al., 1996; Elson et al., 1996; Xu et al., 1996; Herzog et al., 1998), but the CNS phenotypes appear absent. There is no significant loss of atm-/- Purkinje cells (although many of these cells are ectopic and abnormally differentiated) (Barlow et al., 2000; Borghesani et al., 2000; Chiesa et al., 2000). There are reported reductions in the numbers of tyrosine hydroxylase-positive nigrostriatal neurons (Eilam et al., 1998, 2003), yet the levels of monoamine transmitters appear unchanged (Mount et al., 2004). Other CNS properties are also affected. The kinetics of adult neurogenesis is altered (Allen et al., 2001), and there is good evidence supporting a key role for ATM in the neuronal DNA damage response (Bakkenist and Kastan, 2003; Shiloh, 2003; McKinnon, 2004) as well as in p53 stabilization (Keramaris et al., 2003). Thus, there are hints that neuronal survival normally involves ATM. Nonetheless, a complete picture of ATM function in the nervous system (one that explains the pattern of cell loss in A-T) has yet to emerge.
Several laboratories have documented that when mature neurons attempt to reenter a cell cycle, the result is cell death rather than cell division (al-Ubaidi et al., 1992; Feddersen et al., 1992; Herrup and Busser, 1995; Kranenburg et al., 1996; Park et al., 1996). In addition, in a variety of disorders, cell cycle proteins reappear in neurons at risk for degeneration. These disorders include stroke (Love, 2003), amyotrophic lateral sclerosis (Ranganathan et al., 2001; Nguyen et al., 2003), Parkinson's disease (Jordan-Sciutto et al., 2003), and Alzheimer's disease (AD) (Arendt et al., 1996; Vincent et al., 1996; McShea et al., 1997; Nagy et al., 1997; Busser et al., 1998; Yang et al., 2003). In AD, fluorescence in situ hybridization (FISH) further reveals that DNA replication occurs in the at-risk neurons (Yang et al., 2001).
We report here that cell cycle proteins are ectopically reexpressed in the vulnerable neurons of both human and mouse ATM deficiency. In the atm-/- mouse, there is a narrow time window toward the end of Purkinje cell development in which most cell cycle events are initiated. Thus, loss of cell cycle control is a unifying hypothesis for the source of the cellular abnormalities in A-T.
Materials and Methods
Human subjects
Human autopsy tissue from six individuals diagnosed with A-T and six age-matched controls was obtained from Dr. Thomas O. Crawford (Johns Hopkins University, Baltimore, MD). Brain tissue was routinely formalin fixed, paraffin embedded, and sectioned at 10 μm. All A-T cases were confirmed both clinically and pathologically by Dr. Crawford. Three different regions of brain were sampled, including cerebellum, basal ganglia, and medial frontal gyrus. Cerebellum and basal ganglia are considered to undergo neurodegeneration by the late stage of A-T. As a control, we also obtained samples of medial frontal gyrus, which is not affected in A-T from these same subjects. The average age of the subjects was 22 years for the A-T patients and 22.5 years for non-A-T controls. The non-A-T controls were free of any known CNS neurological diseases at the time of death and nearly matched the age of the diagnosed A-T group.
atm-deficient mice
A breeding colony of mice with a targeted disruption of the ATM gene (Barlow et al., 1996) was established from founders obtained from The Jackson Laboratory (Bar Harbor, ME). Generation of mutants was achieved through the mating of heterozygous atm+/- males and atm+/- females. The mice were maintained on a 129/SvJ genetic background. Genotyping was performed on DNA extracted from tail biopsies using PCR techniques as described previously (Barlow et al., 1996). All animals were housed at Case Western Reserve University Medical School Animal Resource Center, a facility fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. All procedures for animals were approved by the Institutional Animal Care and Use Committee of the Case Western Reserve University.
Histology
After animals were deeply anesthetized with Avertin (0.02 cc/mg body weight), they were transcardially perfused with PBS, followed by 4% paraformaldehyde in 0.1 m sodium phosphate buffer (PB). The brain was dissected, immediately removed from the cranium, and transferred to fresh 4% paraformaldehyde at 4°C overnight. The brains were then cryoprotected by sinking in 30% sucrose/PB at 4°C overnight. After bisecting along the midline, the brains were embedded in OCT, and 10 μm cryostat sections were cut and allowed to air dry on SuperPlus glass slides overnight.
Single and double immunocytochemistry
Antibodies.The proliferating cell nuclear antigen (PCNA) mouse monoclonal antibody recognizing the PCNA p36 protein (Dako, High Wycombe, UK) was diluted 1:250 in 10% goat serum/PBS blocking buffer before use. The rabbit polyclonal cyclin A antibody (ab 7956; working dilution, 1:200; Abcam, Cambridge, UK) was raised against the C-terminal domain of cyclin A2. The cyclin B1 mouse monoclonal IgG2b (Upstate Biotechnology, Lake Placid, NY) was raised against the human protein and applied at a dilution of 1:100. The rabbit polyclonal calbindin antibody (dilution, 1: 500; Chemicon, Temecula, CA) and the mouse monoclonal calbindin antibody (dilution, 1:1000; Sigma, St. Louis, MO) were used as Purkinje cell markers.
All paraffin-embedded human sections were deparaffinized in xylene and then rehydrated through graded ethanol to water. For staining using HRP-conjugated secondary antibodies, the sections were pretreated in 0.3% hydrogen peroxide in methanol for 30 min to remove endogenous peroxidase activity, rinsed in Tris-buffered saline (TBS), and then treated with 0.1 m citrate buffer in a microwave at sufficient power to keep the solution at 100°C for 10 min. Sections were cooled in the same buffer at room temperature (RT) for 30 min and rinsed in TBS. Slides were incubated in 10% goat serum in PBS blocking solution for 1 h at RT, after which primary antibody was applied to the sections that were then incubated at 4°C overnight. The sections were washed three times in TBS before applying the secondary antibody (Vector Laboratories, Burlingame, CA), which was diluted in blocking solution at 1:300. Secondary antibody was applied for 1 h at RT. Afterward, sections were rinsed three times in TBS. Rinsed sections were then incubated in Vectastain ABC Elite reagent for 1 h and developed using diaminobenzidine, according to the protocol of the manufacturer (Vector Laboratories). The sections were counterstained with hematoxylin, and after dehydration, all sections were mounted in Permount under a glass coverslip. Control sections were subjected to the identical staining procedure, except for the omission of the primary antibody.
Double fluorescence immunocytochemistry was used solely on the mouse cryosections. Sections were first rinsed in PBS, followed by pretreatment in 0.1 m citrate buffer for 6-8 min at 100°C. After the slides had cooled in buffer for 30 min at room temperature, slides were rinsed in PBS. Sections were incubated in 10% goat serum in PBS to block nonspecific binding for 1 h at room temperature. All primary antibodies were diluted in PBS containing 0.4% Triton X-100 and 10% goat serum and then were applied to sections and incubated overnight at 4°C. After rinsing in PBS, they were incubated for 2 h with secondary antibody, which was conjugated with various fluorescent Alexa dyes (dilution, 1:500; Molecular Probes, Eugene, OR). The sections were then rinsed in PBS and reincubated in 10% goat serum blocking solution for 1 h, followed by addition of the second primary antibody (raised in a different species from the first primary antibody) for a second overnight incubation at 4°C. Sections were then rinsed in PBS, and both secondary antibodies conjugated with different fluorescence dyes were applied to the sections for 2 h at room temperature. After rinsing, all sections were mounted in PBS/glycerol under a glass coverslip.
FISH
FISH probes. Three mouse-specific DNA probes were generated from bacterial artificial chromosomes (BACs), which carried individual specific mouse genomic DNA sequences. One of the DNA probes (480C6) was made from the region that encodes the β-amyloid precursor protein located on mouse chromosome 16. The other two probes were generated from overlapping BACs (170L21 and 566) containing the structural gene for mouse aldolase C on mouse chromosome 11. Each of the three probes covers 100-300 kb of unique genomic sequence. They were labeled by standard nick translation protocols using digoxygenin-labeled dUTP. After labeling, probes were concentrated with mouse Cot-1 DNA (Invitrogen, San Diego, CA) to block hybridization to repetitive sequences.
Before hybridization, all sections were rinsed in PBS and then pretreated with 30% pretreatment powder (Oncor, Gaithersburg, MD) for 15 min at 45°C followed by treatment with protease (0.25 mg/ml; Oncor) for 25 min at 45°C. After rinsing in 2× SSC, the slides were dehydrated through graded alcohols and allowed to air dry. Labeled probe was applied to the individual sections, which were then covered with a glass coverslip and sealed with rubber cement. To denature DNA, slides were heated at 90-92°C on a heated block for 12 min and hybridized with probe overnight at 37°C. After rinsing in 50% formamide/2× SSC at 37°C for 15 min, the slides were transferred to 0.1× SSC buffer for 30 min at 37°C and then rinsed in 0.5 m phosphate buffer with 0.5% NP-40 (PN buffer) at room temperature. To block nonspecific antibody binding, 10% goat serum in PBS was applied to the sections, followed by incubation in mouse anti-digoxygenin primary antibody (dilution, 1:200; Boehringer Mannheim, Indianapolis, IN) for 30 min at 37°C. After rinsing, secondary anti-mouse antibody conjugated with Alexa 488 (dilution, 1:250) was applied to the sections for 30 min at 37°C. Slides were rinsed three times in PN buffer, and the sections were counterstained with either 4′,6′-diamidino-2-phenylindole (DAPI) or propidium iodide and covered with a coverslip. The number of spots of hybridization in each nucleus was determined at 1000× under fluorescent illumination. Images were captured on a Leitz (Wetzlar, Germany) research microscope equipped with a digital camera (Prog C14). Purkinje cells were identified on the basis of the size of their nucleus and their position in the Purkinje cell layer.
Cell counts
For each of the three genotypes (atm+/+, atm+/-, and atm-/-), we began at a randomly selected location in the Purkinje cell layer within a given section and continued counting until a total of 200 calbindin-positive cells had been scored for the presence or absence of the cell cycle marker. Only cells with a discernable portion of their nucleus in the section were scored. Two parasagittal sections (a total of 400 cells) were scored for each animal.
Results
Human A-T disease
Cell cycle proteins in the cerebellum of human A-T patients
Skin fibroblasts, myeloid cells, and other cells cultured from A-T patients (or atm-/- mice) have significant deficits in cell cycle control. There is growing evidence that a variety of neurodegenerative conditions are associated with the reinitiation of an abortive neuronal cell division. Thus, we reasoned that the neurological phenotype in A-T disease might also be attributable to an ATM-dependent failure of cell cycle control. To test this, human autopsy materials from three different brain regions (cerebellum, basal ganglia, and medial frontal gyrus) of A-T patients and age-matched controls were examined. As expected, the density of Purkinje cells was significantly reduced in the A-T cerebellum, and some of the remaining Purkinje cells were misplaced in the molecular layer. We immunostained this material with an antibody to PCNA. PCNA is a subunit of the DNA polymerase holoenzyme that is elevated primarily during S-phase. As shown in Figure 1A, we observed PCNA expressed ectopically in the nuclei of Purkinje cells in all of the A-T subjects. There was little or no background staining in the non-A-T controls (Fig. 1B).
Figure 1.

Expression of cell cycle proteins in the cerebellar Purkinje cells of human A-T disease. Immunostaining shows that cell cycle proteins PCNA (A, arrows), cyclin B (C, arrows), and cyclin A (E, arrows) are reexpressed in the postmitotic Purkinje cells of human A-T specimens. B, D, and F show that representative fields from age-matched controls lack significant expression of PCNA, cyclin B, and cyclin A, respectively. Scale bar, 10 μm.
To validate these findings, we stained our materials with an antibody to a second cell cycle protein, cyclin B. Cyclin B is the regulatory subunit of the Cdc2 kinase, usually found in the G2-phase of the cycle. This marker also appears in A-T Purkinje cells at levels of staining comparable with PCNA (Fig. 1C). As with PCNA, cyclin B staining is not found in the nuclei of Purkinje cells in the control group (Fig. 1D). A third cell cycle protein, cyclin A (an S-G2-phase marker) is also upregulated in the cytoplasm of the A-T Purkinje cells (Fig. 1E) but not in controls. In addition to the Purkinje cells, we found that some granule cells were immunopositive for the cell cycle protein markers in the cerebellum of A-T patients (data not shown); no staining was found in the granule cells of the control group.
Cell cycle proteins in basal ganglia and medial frontal gyrus
Neuronal cell death is observed in other brain regions at late stages of A-T. These include the striatum and basal ganglia. Immunohistochemistry of this region using antibodies against PCNA (Fig. 2 A), cyclin B (Fig. 2 B), and cyclin A (Fig. 2C) distinctly labeled a number of nerve cells. The fraction of striatal neurons that were immunopositive was less than the fraction of immunopositive Purkinje cells. Nonetheless, the number of labeled cells contrasted markedly with the control group, in which no cell cycle-positive cells were found (Fig. 2 D-F). The medial frontal gyrus is considered to be unaffected in A-T. Consistent with our hypothesis, we observed no cell cycle events in the nerve cells of this region in either A-T (Figure 2G-I) or control groups (data not shown).
Figure 2.

Cell cycle proteins reexpressed in noncerebellar brain regions of A-T disease. PCNA (A, arrow), cyclin B (B, arrow), and cyclin A (C, arrows) reappeared in the neuronal cells of striatum, which is considered an affected region in the late stage of A-T. D-F show that no cell cycle-immunopositive neuronal cells were found in the striatum of non-A-T controls. The medial frontal gyrus, which is considered to be an unaffected region in A-T, is shown. G-I show that there is no cell cycle staining found in this region of the A-T brain. Scale bar, 10 μm.
The atm mouse model
Cell cycle events in adult atm-/- mice
We established a mouse breeding colony of which members carry the targeted disruption of the atm gene generated by Barlow et al. (1996). As in humans, the life expectancy of mutant mice is reduced; in our colony, death resulting from tumor growth usually occurs when the animals are 3-4 months of age. Despite the lack of overt Purkinje cell degeneration in these animals, we sought to determine whether cell cycle events were initiated. Littermates of all three genotypes (atm-/-, atm+/-, and atm+/+) were examined by double-label immunocytochemistry. We found that a significant number of the Purkinje cells express PCNA in the atm-/- mutant (Fig. 3A) as well as the atm+/- heterozygous (Fig. 3D) animals; no PCNA immunostaining was found in wild-type mice (Fig. 3G). Figure 3, B, E, and H, shows calbindin Purkinje cell staining. Cyclin A immunoreactivity was also found in the Purkinje cells of the mutant (Fig. 3J) and heterozygous mice (Fig. 3M). The staining intensity was not significantly different between these two genotypes. Only very low background staining was found in the wild-type mice (Fig. 3P). Calbindin staining is shown in Figure 3, K, N, and Q.
Figure 3.
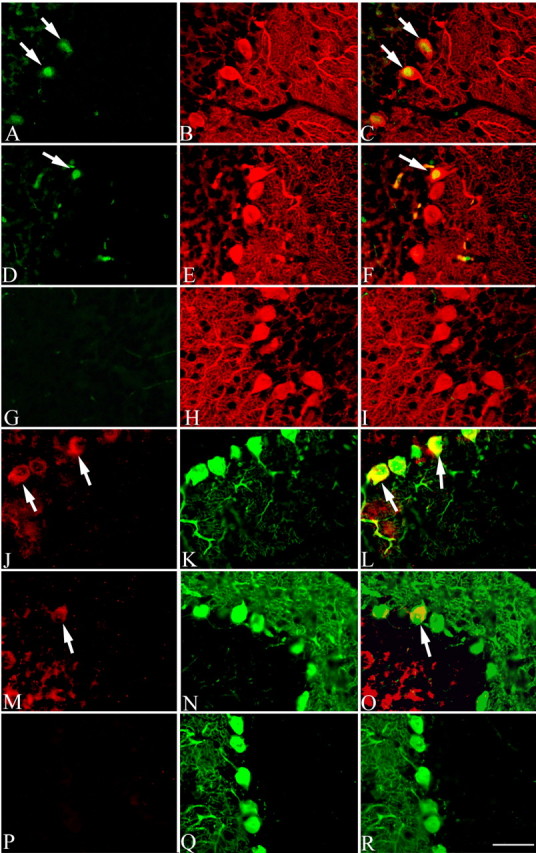
Reappearance of cell cycle proteins in the cerebellar Purkinje cells of adult atm-/- mouse. PCNA (green) was reexpressed in the postmitotic Purkinje cells (calbindin; red) of atm-/- (A-C, arrows) and atm+/- (D-F, arrow). No immunostaining was found in wild-type atm animals (G-I). Similar to PCNA, the cell cycle protein cyclin A (red) reappears in the Purkinje cells (calbindin; green) of atm-/- (J-L, arrows) and atm+/- (M-O, arrow). No staining was found in the adult atm+/+ mouse (P-R). Scale bar, 10 μm.
Because the literature suggests that neuronal cell death is also found in the atm-/- striatum, we examined this structure as well. We observed cells double stained with cyclin A and the neuronal marker neuronal-specific nuclear protein (NeuN) in atm-/- (Fig. 4A) and atm+/- (Fig. 4B). No staining was found in wild-type atm+/+ controls (Fig. 4C). Figure 4, D and E, shows PCNA immunostaining and DAPI counterstaining in the striatum of atm-/- and atm+/- mice. No labeling is found in the wild-type atm+/+ (Fig. 4F).
Figure 4.

Cell cycle proteins revealed in the neuronal cells of the striatum of 3-month-old atm-/- mice. Cyclin A (red) coexpressed with the neuronal marker (NeuN; green) in the striatum of atm-/- (A, arrows) and atm+/- (B, arrows) mice. No cyclin A immunostaining was found in atm+/+ mice (C). D and E show PCNA (green) in the striatal neuronal cells of atm-/- and atm+/- mice, but not in the atm+/+ mice (F). Cell nuclei stained with DAPI (blue) are shown. Scale bar, 10 μm.
DNA replication in the atm mouse model
To confirm our immunocytochemistry results, we used the previously developed FISH technique (Yang et al., 2001) on the interphase nuclei of the mouse cerebellar neurons. We used mouse BAC probes, each of which recognizes unique genetic sequences on one of two different mouse chromosomes, 16 and 11. In atm-/- (Fig. 5A) and heterozygous atm+/- (Fig. 5B) mice, hybridization with the chromosome 16 probe revealed Purkinje cells with three or four hybridization signals (three to four spots), indicating that DNA replication has occurred in these neurons. Two or fewer hybridization signals were found in every Purkinje cell examined in wild types (Fig. 5C). To determine whether this apparent DNA replication was unique to mouse chromosome 16, a pair of probes (BACs 556 and 170L21) was used to repeat these experiments. These two BACs are partially overlapping DNA sequences found on mouse chromosome 11. Both probes revealed numerous Purkinje cells with three and four spots of FISH hybridization in mutant (Fig. 5D) and heterozygous (Fig. 5E) animals. No more than two spots were found in any wild-type nucleus (Fig. 5F). In both mutant atm-/- and heterozygous atm+/- mice, we also found hyperploid granule cells in the granule cell layer of cortical cerebellum (data not shown). No such cells were detected in wild-type animals.
Figure 5.

FISH analysis of the cerebellar Purkinje cells of atm-/- mice. In A (atm-/-) and B (atm+/-), three and four bright hybridization signals of mouse chromosome 16 (480C6) were found (arrows). Insets in A and B show two cells contained three and four hybridization signals. Another BAC probe (170L21) for sequences on chromosome 11 showed that nuclei of Purkinje cells of atm-/- (D) and atm+/- (E) can be found, which contain more than two spots of hybridization, indicating that DNA replication has occurred in these cells. Only two hybridized signals were found in the nuclei of Purkinje cells of atm-/- (C, F). Cells were counterstained with propidium iodide. Scale bar, 10 μm.
Cell cycle events in developing atm mice
We have shown that cell cycle events are associated with the degenerative process of Purkinje cell death in human and mouse A-T cerebellum. One important value of the mouse model is that it enables longitudinal studies to be performed on animals of known genotype. This enables us to address the important question of when the cell cycle process begins in the at-risk cells. We examined atm-/-, atm+/-, and atm+/+ mice at several postnatal ages, including postnatal day 10 (P10), P20, and P30. In P10 mice, we found no positive immunostaining for either PCNA (Fig. 6A-C) or cyclin A (Fig. 7A-C) in Purkinje cells of any of the three genotypes. As expected, the granule cells in the external granule cell layer expressed PCNA (Fig. 6A-C) at this age (an internal positive control). At P20 and P30, however, we found abundant PCNA (Fig. 6D,E,G,H) and cyclin A (Fig. 7D,E,G,H) immunopositive Purkinje cells in both mutant and heterozygous mice. Very low levels of background staining were seen in the wild-type mouse (Figs. 6C,F,I, 7C,F,I). We counted the percentages of immunopositive Purkinje cells in the three different genotypes at all three ages. This analysis revealed that >1 in 10 Purkinje cells in both mutant and heterozygous animals was immunopositive for either PCNA (13%) or cyclin A (12%), compared with 1% in wild-type atm+/+ animals (Table 1). Surprisingly, we observed that heterozygous atm+/- mice had levels of immunopositive Purkinje cells that were indistinguishable from those found in atm-/- mutants (11% for PCNA and 10.5% for cyclin A).
Figure 6.
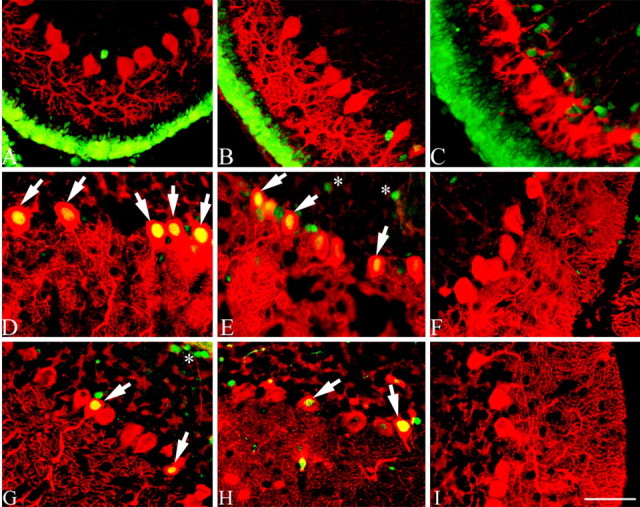
Cell cycle protein expression in developing atm mice. In P10 atm mice, no PCNA immunostaining was found in the Purkinje cells of any of the three genotypes (A-C). As expected, cells in the external granule cell layer expressed PCNA at P10. For P20 (D, E, arrows) and P30 (G, H, arrows) mice, PCNA is unexpectedly reexpressed in the Purkinje cells of mutant and heterozygous animals, suggesting that cell cycle initiation had begun during a narrow time window as the late stages of Purkinje cell development are completed. Asterisks in E and G showed PCNA-positive immunostaining in internal granule cells. F and I show no PCNA-immunopositive staining in the wild-type mice at ages P20 and P30. PCNA, Green; calbindin, red. Scale bar, 10 μm.
Figure 7.

Cyclin A is also reexpressed in the Purkinje cells of young atm-/- mice. Similar to PCNA expression, cyclin A is reexpressed in the nuclei of Purkinje cells of atm-/- and heterozygous atm+/- animals at P20 (D, E, arrows) and P30 (G, H, arrows), but no staining is found in any of the three genotypes at P10 (A-C) or in wild-type animals at P20 (F) or P30 (I). Arrows indicate cyclin A colocalized with calbindin in Purkinje cells. Scale bar, 10 μm.
Table 1.
Developmental appearance of cell cycle markers in ATM-deficient mice
|
|
Percentage of immunopositive neurons |
||
|---|---|---|---|
|
|
PCNA |
Cyclin A |
|
| P10 | |||
| +/+ | 0 | 0 | |
| +/− | 0 | 0 | |
| −/− | 0 | 0 | |
| P20 | |||
| +/+ | 1.5 | 1.5 | |
| +/− | 11.5 | 10.0 | |
| −/− | 12.0 | 13.5 | |
| P30 | |||
| +/+ | 1.0 | 1.0 | |
| +/− | 11.0 | 10.5 | |
| −/− |
13.0 |
12.0 |
|
Cells were immunostained for calbindin and either PCNA or cyclin A. Beginning at random within a given section, 200 sequential calbindin-positive cells in the Purkinje cell layer were scored for the presence or absence of the cell cycle marker. Two parasagittal sections (a total of 400 cells) were scored for each animal.
We confirmed this finding using the patterns of FISH labeling as a metric. At either P20 or P30, three or four hybridization spots (hyperploid content) were consistently found in the nuclei of Purkinje cells of atm-/- (Fig. 8D,G) and atm+/- (Fig. 8E,H) mice. Three or four spots were also found in a few granule cells in the internal granular cell layer at P20 and P30 in both mutants (data not shown). Only two or fewer hybridization signals were found in the Purkinje cells of P10 mice of all three genotypes (Fig. 8A-C) as well as in wild-type animals at P20 (Fig. 8F) and P30 (Fig. 8I). Taken as a group, these findings demonstrate that significant DNA replication has occurred in these “postmitotic” cells, suggesting a cell cycle process that has proceeded through most or all of the S-phase.
Figure 8.

FISH evidence confirms that cell cycle events in the Purkinje cells of developing atm mice lead to DNA replication. Three or four hybridization signals were found in the Purkinje cells of mutant and heterozygous atm mice at P20 (D, E) and P30 (G, H) using mouse BAC DNA probes (480C6 or 556) localized on chromosomes 16 or 11. Only two copies of these chromosomes were found in wild-type animals (F, I) and in all three genotypes of mice at P10 (A-C). Arrows indicate the hybridization signals in the nuclei of Purkinje cells. Scale bar, 10 μm.
Discussion
Abortive cell cycle entry leads to nerve cell death rather than cell division in most CNS neurons. This has been shown both in vivo and in vitro. In vivo, cell cycle initiation has been forced by ectopically driving T-antigen with a neuronal-specific promoter. In these circumstances, the result is death of the T-antigen-positive neuron rather than its division (al-Ubaidi et al., 1992; Feddersen et al., 1992). In addition, in several examples of target-related cell death, the dying neurons initiate a cell cycle that proceeds through DNA synthesis in the hours before their pyknotic demise (Herrup and Busser, 1995; Wu et al., 2000). Increasing evidence has also documented the appearance of unscheduled cell cycle events in postmitotic neurons in several different neurodegenerative disorders. These include amyotrophic lateral sclerosis (Ranganathan et al., 2001; Ranganathan and Bowser, 2003), stroke (Love, 2003), and both early and late stages of Alzheimer's disease (Nagy et al., 1997; Busser et al., 1998; Vincent et al., 1998; Yang et al., 2003).
The data reported here are the first evidence that unscheduled cell cycle events are also involved in another neurodegenerative disease, ataxia-telangiectasia. In A-T patients, the extent of the Purkinje cell loss is correlated with the severity of the clinical phenotype. We found that cell cycle proteins PCNA, cyclin A, and cyclin B reappeared in postmitotic Purkinje cells and granule cells of the human A-T cerebellum. This evidence points to a situation in which mature Purkinje cells have reinitiated a lethal cell cycle. Thus, the loss of Purkinje and granule cells in A-T may share a common pathogenic mechanism with other neurodegenerative disorders such as Alzheimer's disease. We found that all regions of the A-T brain that have been identified with neuronal cell loss have evidence of ectopic reexpression of cell cycle proteins in neuronal cell bodies, and those regions that are spared have little to no expression of these proteins. This correlation argues that the immunolabeled proteins are proximal to the neurodegeneration itself and mark, in a cell autonomous manner, a neuron under stress and at high risk for death.
This linkage of the neurodegeneration with events of the cell cycle has important implications in understanding the etiology of A-T. Inactivation of ATM and the subsequent deficiencies in DNA damage detection and cell cycle checkpoints are appreciated as playing an important role in the tumorigenesis and ionizing radiation sensitivity that are found in A-T cells (Vorechovsky et al., 1997; Bullrich et al., 1999; Stankovic et al., 1999). Our findings suggest that failure of cell cycle regulation is a unifying feature that extends this concept to the death of CNS neurons as well. These observations are consistent with the accumulating evidence of impaired DNA damage response as a factor in the death of adult neurons. Mutations in NBS1 and hypomorphic mutations of the MRE11 gene, for example, are associated with human neurological defects, Nijimegen breakage syndrome (NBS) and ataxia-telangiectasia-like disorder (Shiloh, 1997; Stewart et al., 1999; Delia et al., 2004). Nbs1 and Mre11 are two proteins that, together with Rad50, form the MRZ11/RAD50/NBS1 complex. The complex is a target of ATM kinase, and it plays a role in double-strand break detection and cell cycle checkpoint signaling. Unfortunately, our data offer little by way of explanation of the differential sensitivity of neurons in different brain regions or what the stimulus is that triggers the initial attempt of the cell at division.
Our data can also be viewed in light of the reports that chromosomal aneuploidies are found in 5-7% of normal cortical neuronal nuclei and that neurons that are genetically atm-/- are more likely to carry such imbalances (McConnell et al., 2004). The events reported by these workers are unlikely to be the equivalent of the cell cycle events reported here. First, we found virtually no evidence of hyperploidy or reexpression of cell cycle proteins in the wild-type animals. Second, the differences we found between the mutant and the wild type is ∼10-fold, not double as reported previously. The discrepancies have several possible origins. The methods of analysis used by McConnell et al. (2004) are different from our own. For example, they isolated their nuclei from brain homogenates before DNA hybridization. A second difference is that their analysis was restricted to cells of the cerebral cortex. Finally, the anomalies they reported were attributable to predominantly chromosome loss, a situation we are unlikely to detect with confidence in 10 μm cryostat sections.
Our analyses of the atm mouse model show that Purkinje cells in heterozygous atm mice have levels of cell cycle reinitiation similar to those found in null atm mice (Table 1). This observation is intriguing given the consistent reports of increased cancer risks in carrier individuals, both mice and humans. There is no direct evidence for increased tumorigenesis in mouse carriers of truncation atm mutations (the mutation we studied), but heterozygotes of this type of mutation are more sensitive to ionizing radiation if tumorigenesis is used as an outcome measure (Barlow et al., 1999). Also noteworthy in this regard is the recurring observation that human A-T carriers are prone to develop malignant tumors (Vorechovsky et al., 1997; Bullrich et al., 1999; Spring et al., 2002). Unlike these non-CNS phenotypes, however, the Purkinje cell phenotypes behave as fully dominant traits (the levels of cell cycle anomalies are equivalent in heterozygotes and wild types). Clearly, the prohibition against entering a cell cycle in a Purkinje cell or striatal neuron is quite sensitive to the absence of even half of the normal quantities of ATM protein. Equally clear, however, is the fact that the death of the neuron is not an immediate and unavoidable consequence of the cell cycle processes we describe. Our finding that mouse atm-/- and atm+/- neurons can survive for months with a replicated genome suggests that the cell cycle events themselves are permissive, but the final death of the neuron likely requires a second “hit” (an event requiring months in the human and never occurring in the relatively short life span of the mouse).
Our finding that the initiation of the cell cycle events in cerebellum occurs during a narrow time window between P10 and P20 is unexpected. This period marks the final stages of Purkinje cell development. Cell migration and the development of a single-cell Purkinje cell layer are complete, but expansion and refinement of the dendritic arbor and synaptogenesis will continue for several weeks. It would appear that the cell cycle initiation is not a slow and steady accretion of at-risk cells; rather, the triggering event occurs during a relatively brief period of time during the final stage of Purkinje cell development. Indeed, the timing of events suggests that one normal function of the ATM protein is to intervene during a vulnerable period in the life history of the Purkinje cell and block any attempt at cell division.
The finding of cell cycle events in the at-risk populations of neurons in both human A-T patients and mouse models of the disease offers several important insights. The most important among these is that it appears that a failure of cell cycle regulation is a unifying feature of the disease, a defining characteristic of the cells in all affected tissues. Because reentrance into a cell cycle is a lethal event for a mature neuron, loss of cell cycle control results in nerve cell death, not tumorigenesis. Second, the relatively narrow time window during which the cell cycle events appear suggests a linkage with processes at work during the normal developmental program of the Purkinje cell. If these could be identified, therapeutic interventions might be sensibly targeted to specific times in the early childhood of affected individuals. Last, the finding that heterozygotes are affected by the cell cycle changes at levels equivalent to their homozygous littermates encourages the exploration of whether carriers (of either truncation or missense mutations) might be at higher risk for other forms of neurodegenerative disease.
Footnotes
This work was supported by grants from the Ataxia-Telangiectasia Children's Project to Y.Y. and the National Institutes of Health (NS20591 and AG08012) to K.H. We thank Dr. Thomas O. Crawford for providing the human tissue used in this study. We declare that we have no competing financial interests.
Correspondence should be addressed to Yan Yang, Department of Neurology, Alzheimer Research Laboratory (E504), Case School of Medicine, 10900 Euclid Avenue, Cleveland, OH 44106. E-mail: yan.yang@case.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252522-08$15.00/0
References
- Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15: 2177-2196. [DOI] [PubMed] [Google Scholar]
- Allen DM, van Praag H, Ray J, Weaver Z, Winrow CJ, Carter TA, Braquet R, Harrington E, Ried T, Brown KD, Gage FH, Barlow C (2001) Ataxia telangiectasia mutated is essential during adult neurogenesis. Genes Dev 15: 554-566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- al-Ubaidi MR, Hollyfield JG, Overbeek PA, Baehr W (1992) Photoreceptor degeneration induced by the expression of simian virus 40 large tumor antigen in the retina of transgenic mice. Proc Natl Acad Sci USA 89: 1194-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Rodel L, Gartner U, Holzer M (1996) Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer's disease. NeuroReport 7: 3047-3049. [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421: 499-506. [DOI] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A (1996) Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86: 159-171. [DOI] [PubMed] [Google Scholar]
- Barlow C, Eckhaus MA, Schaffer AA, Wynshaw-Boris A (1999) Atm haplo-insufficiency results in increased sensitivity to sublethal doses of ionizing radiation in mice. Nat Genet 21: 359-360. [DOI] [PubMed] [Google Scholar]
- Barlow C, Ribaut-Barassin C, Zwingman TA, Pope AJ, Brown KD, Owens JW, Larson D, Harrington EA, Haeberle AM, Mariani J, Eckhaus M, Herrup K, Bailly Y, Wynshaw-Boris A (2000) ATM is a cytoplasmic protein in mouse brain required to prevent lysosomal accumulation. Proc Natl Acad Sci USA 97: 871-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghesani PR, Alt FW, Bottaro A, Davidson L, Aksoy S, Rathbun GA, Roberts TM, Swat W, Segal RA, Gu Y (2000) Abnormal development of Purkinje cells and lymphocytes in Atm mutant mice. Proc Natl Acad Sci USA 97: 3336-3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullrich F, Rasio D, Kitada S, Starostik P, Kipps T, Keating M, Albitar M, Reed JC, Croce CM (1999) ATM mutations in B-cell chronic lymphocytic leukemia. Cancer Res 59: 24-27. [PubMed] [Google Scholar]
- Busser J, Geldmacher DS, Herrup K (1998) Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci 18: 2801-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa N, Barlow C, Wynshaw-Boris A, Strata P, Tempia F (2000) Atm-deficient mice Purkinje cells show age-dependent defects in calcium spike bursts and calcium currents. Neuroscience 96: 575-583. [DOI] [PubMed] [Google Scholar]
- Crawford TO (1998) Ataxia telangiectasia. Semin Pediatr Neurol 5: 287-294. [DOI] [PubMed] [Google Scholar]
- Delia D, Piane M, Buscemi G, Savio C, Palmeri S, Lulli P, Carlessi L, Fontanella E, Chessa L (2004) MRE11 mutations and impaired ATM-dependent responses in an Italian family with ataxia-telangiectasia-like disorder. Hum Mol Genet 13: 2155-2163. [DOI] [PubMed] [Google Scholar]
- Eilam R, Peter Y, Elson A, Rotman G, Shiloh Y, Groner Y, Segal M (1998) Selective loss of dopaminergic nigro-striatal neurons in brains of Atm-deficient mice. Proc Natl Acad Sci USA 95: 12653-12656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilam R, Peter Y, Groner Y, Segal M (2003) Late degeneration of nigrostriatal neurons in ATM-/- mice. Neuroscience 121: 83-98. [DOI] [PubMed] [Google Scholar]
- Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-Torres J, Leder P (1996) Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci USA 93: 13084-13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feddersen RM, Ehlenfeldt R, Yunis WS, Clark HB, Orr HT (1992) Disrupted cerebellar cortical development and progressive degeneration of Purkinje cells in SV40 T antigen transgenic mice. Neuron 9: 955-966. [DOI] [PubMed] [Google Scholar]
- Herrup K, Busser JC (1995) The induction of multiple cell cycle events precedes target-related neuronal death. Development 121: 2385-2395. [DOI] [PubMed] [Google Scholar]
- Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ (1998) Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science 280: 1089-1091. [DOI] [PubMed] [Google Scholar]
- Jordan-Sciutto KL, Dorsey R, Chalovich EM, Hammond RR, Achim CL (2003) Expression patterns of retinoblastoma protein in Parkinson disease. J Neuropathol Exp Neurol 62: 68-74. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Lim DS, Kim ST, Xu B, Canman C (2000) Multiple signaling pathways involving ATM. Cold Spring Harb Symp Quant Biol 65: 521-526. [DOI] [PubMed] [Google Scholar]
- Keramaris E, Hirao A, Slack RS, Mak TW, Park DS (2003) Ataxia telangiectasia-mutated protein can regulate p53 and neuronal death independent of Chk2 in response to DNA damage. J Biol Chem 278: 37782-37789. [DOI] [PubMed] [Google Scholar]
- Koepp M, Schelosky L, Cordes I, Cordes M, Poewe W (1994) Dystonia in ataxia telangiectasia: report of a case with putaminal lesions and decreased striatal [123I]iodobenzamide binding. Mov Disord 9: 455-459. [DOI] [PubMed] [Google Scholar]
- Kranenburg O, van der Eb AJ, Zantema A (1996) Cyclin D1 is an essential mediator of apoptotic neuronal cell death. EMBO J 15: 46-54. [PMC free article] [PubMed] [Google Scholar]
- Love S (2003) Neuronal expression of cell cycle-related proteins after brain ischaemia in man. Neurosci Lett 353: 29-32. [DOI] [PubMed] [Google Scholar]
- McConnell MJ, Kaushal D, Yang AH, Kingsbury MA, Rehen SK, Treuner K, Helton R, Annas EG, Chun J, Barlow C (2004) Failed clearance of aneuploid embryonic neural progenitor cells leads to excess aneuploidy in the Atm-deficient but not the Trp53-deficient adult cerebral cortex. J Neurosci 24: 8090-8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ (2004) ATM and ataxia telangiectasia. EMBO Rep 5: 772-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShea A, Harris PL, Webster KR, Wahl AF, Smith MA (1997) Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol 150: 1933-1939. [PMC free article] [PubMed] [Google Scholar]
- Mount HT, Martel JC, Fluit P, Wu Y, Gallo-Hendrikx E, Cosi C, Marien MR (2004) Progressive sensorimotor impairment is not associated with reduced dopamine and high energy phosphate donors in a model of ataxia-telangiectasia. J Neurochem 88: 1449-1454. [DOI] [PubMed] [Google Scholar]
- Nagy Z, Esiri MM, Cato AM, Smith AD (1997) Cell cycle markers in the hippocampus in Alzheimer's disease. Acta Neuropathol (Berl) 94: 6-15. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Boudreau M, Kriz J, Couillard-Despres S, Kaplan DR, Julien JP (2003) Cell cycle regulators in the neuronal death pathway of amyotrophic lateral sclerosis caused by mutant superoxide dismutase 1. J Neurosci 23: 2131-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostetowska E, Traczynska H (1964) On ataxia with telangiectasia, an anatomical-clinical observation. Acta Neuropathol (Berl) 27: 319-325. [DOI] [PubMed] [Google Scholar]
- Park DS, Farinelli SE, Greene LA (1996) Inhibitors of cyclin-dependent kinases promote survival of post-mitotic neuronally differentiated PC12 cells and sympathetic neurons. J Biol Chem 271: 8161-8169. [DOI] [PubMed] [Google Scholar]
- Ranganathan S, Bowser R (2003) Alterations in G(1) to S phase cell-cycle regulators during amyotrophic lateral sclerosis. Am J Pathol 162: 823-835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan S, Scudiere S, Bowser R (2001) Hyperphosphorylation of the retinoblastoma gene product and altered subcellular distribution of E2F-1 during Alzheimer's disease and amyotrophic lateral sclerosis. J Alzheimers Dis 3: 377-385. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S (1995) A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268: 1749-1753. [DOI] [PubMed] [Google Scholar]
- Shiloh Y (1997) Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu Rev Genet 31: 635-662. [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3: 155-168. [DOI] [PubMed] [Google Scholar]
- Shiloh Y, Kastan MB (2001) ATM: genome stability, neuronal development, and cancer cross paths. Adv Cancer Res 83: 209-254. [DOI] [PubMed] [Google Scholar]
- Spring K, Ahangari F, Scott SP, Waring P, Purdie DM, Chen PC, Hourigan K, Ramsay J, McKinnon PJ, Swift M, Lavin MF (2002) Mice heterozygous for mutation in Atm, the gene involved in ataxia-telangiectasia, have heightened susceptibility to cancer. Nat Genet 32: 185-190. [DOI] [PubMed] [Google Scholar]
- Stankovic T, Weber P, Stewart G, Bedenham T, Murray J, Byrd PJ, Moss PA, Taylor AM (1999) Inactivation of ataxia telangiectasia mutated gene in B-cell chronic lymphocytic leukaemia. Lancet 353: 26-29. [DOI] [PubMed] [Google Scholar]
- Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM (1999) The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99: 577-587. [DOI] [PubMed] [Google Scholar]
- Vincent I, Rosado M, Davies P (1996) Mitotic mechanisms in Alzheimer's disease? J Cell Biol 132: 413-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent I, Zheng JH, Dickson DW, Kress Y, Davies P (1998) Mitotic phosphoepitopes precede paired helical filaments in Alzheimer's disease. Neurobiol Aging 19: 287-296. [DOI] [PubMed] [Google Scholar]
- Vorechovsky I, Luo L, Dyer MJ, Catovsky D, Amlot PL, Yaxley JC, Foroni L, Hammarstrom L, Webster AD, Yuille MA (1997) Clustering of missense mutations in the ataxia-telangiectasia gene in a sporadic T-cell leukaemia. Nat Genet 17: 96-99. [DOI] [PubMed] [Google Scholar]
- Wu Q, Combs C, Cannady SB, Geldmacher DS, Herrup K (2000) β-Amyloid activated microglia induce cell cycling and cell death in cultured cortical neurons. Neurobiol Aging 21: 797-806. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D (1996) Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev 10: 2411-2422. [DOI] [PubMed] [Google Scholar]
- Yang Y, Geldmacher DS, Herrup K (2001) DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci 21: 2661-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Mufson EJ, Herrup K (2003) Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J Neurosci 23: 2557-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]