

Abstract
Copper is an essential transition metal with a critical role in the CNS. This requirement is underscored by Menkes disease, a fatal neurodegenerative disorder of childhood resulting from the absence or dysfunction of a copper-transporting P-type ATPase. To elucidate the cell biological mechanisms of copper homeostasis in the CNS, a polyclonal antisera against Menkes ATPase was used in immunoblot and immunohistochemical studies, demonstrating abundant expression of this copper transporter in hippocampal neurons. Consistent with this observation, immunofluorescent analysis revealed Menkes ATPase in the late Golgi of hippocampal neurons in primary culture. Glutamate receptor activation was found to result in the rapid and reversible trafficking of Menkes ATPase to neuronal processes, independent of the intracellular copper concentration and specific for activation of the NMDA- but not AMPA/kainate-type glutamate receptors. Metabolic studies revealed that trafficking of Menkes ATPase after NMDA receptor activation is associated with rapid release of copper from hippocampal neurons. Menkes ATPase is directly required for this copper efflux, because similar studies in hippocampal neurons derived from mice lacking a functional Menkes ATPase demonstrated no copper release. Together, these data reveal a critical role for Menkes ATPase in the availability of an NMDA receptor-dependent, releasable pool of copper in hippocampal neurons and demonstrate a unique mechanism linking copper homeostasis and neuronal activation within the CNS.
Keywords: Menkes disease, neurodegeneration, copper, hippocampal neurons, P-type ATPase, NMDA receptor
Introduction
Copper is an essential transition metal with a critical role in the CNS. This requirement for copper is illustrated by Menkes disease, a fatal neurodegenerative disorder of childhood resulting from the loss of function of a copper-transporting P-type ATPase required for cellular copper homeostasis. Patients display gray matter degeneration, Purkinje cell abnormalities, and hippocampal neuronal loss (Okeda et al., 1991). Wilson disease, caused by loss of function of a homologous copper-transporting P-type ATPase, also results in neurological disease with basal ganglia neurodegeneration, Parkinsonian symptoms, and neuropsychiatric features (Lutsenko and Petris, 2003). Recently, dietary copper has been linked to neurodegeneration in experimental models of Alzheimer's disease (Bayer et al., 2003; Phinney et al., 2003; Sparks and Schreurs, 2003) and prion-mediated encephalopathy (Sigurdsson et al., 2003), findings that extend the role for copper and highlight the need for elucidating the cell biological mechanisms of copper homeostasis in the brain (Bush et al., 2003).
In mammals, copper serves as a cofactor for electron transfer by cuproenzymes required for processes critical to CNS function, including cellular respiration, catecholamine biosynthesis, anti-oxidant defense, and peptide amidation. Elucidation of the molecular genetics of Menkes and Wilson disease has revealed that copper homeostasis is predominantly determined by intracellular P-type ATPases that function to transport copper into the secretory pathway of the cell for incorporation into cuproenzymes and cellular excretion (Lutsenko and Petris, 2003). These ATPases reside in the late Golgi compartment but relocalize in response to increases in intracellular copper to a cytoplasmic vesicular compartment in which copper is accumulated for excretion, providing a sensitive posttranslational mechanism for maintaining intracellular copper homeostasis and avoiding toxicity. In non-neuronal cells, the trafficking of the ATPases is tied to their catalytic function. Copper also regulates noncatalytic phosphorylation of Menkes ATPase, a signal likely involved in trafficking (Petris et al., 1996; Hung et al., 1997).
Currently, the role of copper in CNS disease is poorly understood. The neurodegeneration in Menkes disease is not explained by the loss of function of cuproenzymes with known activity in the CNS. Furthermore, recent studies have demonstrated synaptosomal copper release, and application of copper has been shown to affect the excitability of cultured neurons, including directly the NMDA receptor, suggesting that copper may modulate neuronal transmission (Kardos et al., 1989; Trombley and Shepherd, 1996; Hopt et al., 2003). Moreover, because stimulation of the NMDA receptor controls permeability to Ca2+, modulation of receptor activity modulates activation of calcium-dependent cascades that contribute to synaptic modifications (Shen and Meyer, 1999; Liao et al., 2001; Lu et al., 2001).
Because redox reactive copper may additionally be a source of reactive oxygen species implicated in the pathogenesis of several neurodegenerative diseases, understanding normal brain copper homeostasis is critical to understanding these disorders. The availability of a specific antisera to Menkes ATPase as well as a murine model of this disease and methodology for primary hippocampal cultures provided the opportunity to directly examine the cellular mechanisms of copper homeostasis in the CNS.
Materials and Methods
Animals. Brindled (Mobr) C57BL/6 heterozygous female and C57BL/6 male mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and screened for the brindled mutation as described previously (Grimes et al., 1997). The brindled mouse contains an in-frame six nucleotide deletion in the coding region of the gene for Menkes ATPase, which results in impairment of copper-dependent trafficking of the Menkes ATPase with resultant marked diminution of copper homeostasis and the development of the Menkes phenotype in affected males. All mice were housed at the Washington University School of Medicine Vivarium under a 12 hr light/dark cycle. Food and water were provided ad libitum, and all care was given in compliance with National Institutes of Health guidelines on the use of laboratory and experimental animals.
Immunofluorescence microscopy. Anesthetized adult mice were perfused transcardially with 4% paraformaldehyde in ice-cold PBS, and 7-μm-thick coronal paraffin sections from brain were prepared and processed for immunohistochemistry. Sections were deparaffinized, hydrated, permeabilized with 0.1% saponin, and blocked with 3% normal goat serum in PBS. Menkes primary antibody was incubated with the sections overnight at 4°C, followed by detection with an Alexa fluorophore-conjugated secondary anti-rabbit antibody (Molecular Probes, Eugene, OR). Neurons were subsequently detected with NeuroTrace fluorescent Nissl stain (Molecular Probes). Sections were imaged by confocal laser scanning microscopy.
The mottled mouse (Mobr-J) fibroblast cell line 802-1Mnk (Mo-/Y), which carries a mutation in Menkes ATPase that results in <2% protein, and the corresponding wild-type cell line 802-5 (Mo+/Y) were cultured as described previously (Kelly and Palmiter, 1996). Rat hippocampal cultures were prepared from 18-d-old rat embryos as described previously (Goslin et al., 1998). Hippocampi were dissected and were dissociated using trypsin and trituration through a Pasteur pipette. Neurons were plated on coverslips coated with poly-l-lysine in MEM with 10% horse serum, allowed to attach for 4 hr, transferred to glial monolayers, and maintained for up to 3 weeks in serum-free MEM. Cultures were plated at low density (4800 cells/cm2) for immunofluorescence studies or high density (14,400 cells/cm2) for analysis of copper secretion. All analysis was performed at 2 weeks in vitro, except for a study of the localization of Menkes ATPase during neuronal development (2 d in vitro) (see Fig. 1C) and the study of the localization of Menkes ATPase within axons and dendrites (1 week in vitro) (see Fig. 3), which was performed on more immature neurons to visualize isolated processes. Mouse hippocampal cultures were similarly prepared from postnatal day 0 (P0) male offspring of Mobr heterozygous female, wild-type male matings. Neurons were stimulated in extracellular solution [ECS; containing (in mm): 150 NaCl, 4 KCl, 10 HEPES, 10 d-glucose, 2 CaCl2] containing 5 μm glycine and 50 μm glutamate, or 100 μm glycine alone, and rinsed in ECS with 2 mm MgCl2. In other experiments, cells were incubated in 200 μm bathocuproine disulfonic acid (BCS), a cell-impermeant copper specific chelator, for 24 hr or 100 μm CuCl2 for 3 hr in media before fixation. Neurons were fixed with 4% paraformaldehyde/4% sucrose in PBS for 15 min at 37°C and permeabilized for 10 min with 0.25% Triton X-100 in PBS. Neurons were incubated with primary antibodies overnight at 4°C, followed by detection with appropriate Alexa fluorophore-conjugated secondary antibodies (Molecular Probes).
Figure 1.
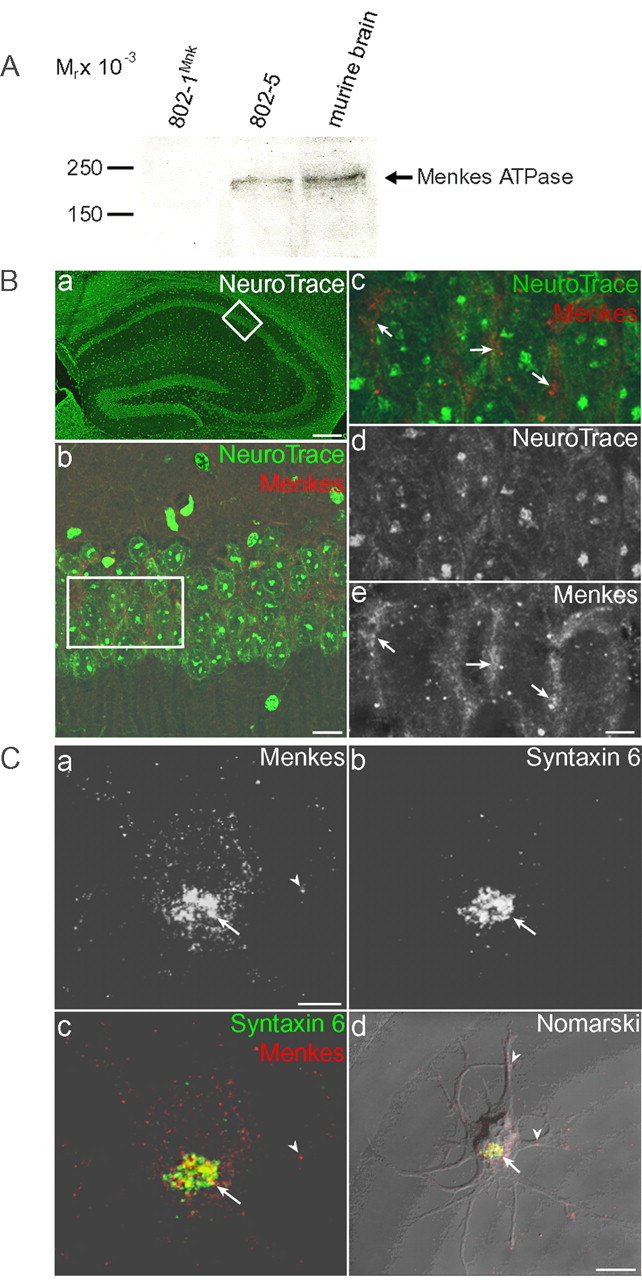
Immunolocalization of Menkes ATPase. A, Immunoblot analysis of Menkes ATPase in immortalized mouse embryonic fibroblasts and whole murine brain. Lysate (100 μg) was separated by 7.5% SDS-PAGE, transferred to nitrocellulose, and analyzed with Menkes antisera followed by chemiluminescent detection. The specificity of this murine Menkes ATPase antisera was confirmed by the lack of this signal in Menkes-deficient 802-1Mnk immortalized mouse embryonic fibroblast cells, compared with the strong signal present in the corresponding wild-type cell line 802-5 and in lysate from whole murine brain. B, Immunolocalization of Menkes ATPase in murine brain. Double-label indirect immunofluorescence in coronal sections of the murine hippocampus stained for neurons (NeuroTrace; green) and Menkes (red). Ba, NeuroTrace staining revealing overall architecture of the mouse hippocampus, with boxed area enlarged in b. Bb-Be, Merged image of double-label fluorescence using Menkes antibody (b, c, e) and NeuroTrace (b-d) for analysis by confocal microscopy. Boxed area is enlarged in c-e. Arrows depict localization of Menkes ATPase to the perinuclear region of CA1 neurons. Scale bars: a, 150 μm; b, 20 μm; (in e) c-e, 5 μm. C, Menkes ATPase immunolocalization in developing rat neurons. Hippocampal neurons were cultured for 2 d at low density, fixed, and processed for double-label immunofluorescence using Menkes (a) and syntaxin 6 (b) antibodies for analysis with confocal microscopy. Cc, Images of Menkes ATPase and syntaxin 6 are merged to depict overlapping regions. Cd, Merged images of Menkes ATPase and syntaxin 6 overlaid on a Nomarski differential interference contrast image of the developing neuron. Arrows depict localization of Menkes ATPase in late Golgi; arrowheads indicate Menkes ATPase in processes. Scale bars: (in a) a-c, 10 μm; d, 25 μm.
Figure 3.

Trafficking of Menkes ATPase to axons and dendrites. Neurons 1 week in culture at low density were treated with 100 μm CuCl2 for 3 hr, fixed, triple immunolabeled for Menkes (a, d), the dendrite marker MAP2 (b, e), and the axon marker dephospho-tau (Tau 1) (c, f), and analyzed by confocal microscopy. Arrowheads indicate overlap in dendrites (a-c) and axons (d-f). Scale bar: (in a) a-f,10 μm.
Drugs used were 100 μm 2-amino-5-phosphonovalerate (APV) (Research Biochemicals, Natick, MA), 10 μm dizocilpine (MK-801) (Research Biochemicals), 20 μm nifedipine (EMD Biosciences, San Diego, CA), 20 μm glycine, BAPTA-AM (Molecular Probes), 10 μm 6-cyano-7-nitroquinozaline-2,3-dione (CNQX) (Sigma, St. Louis, MO), 1 μm tetrodotoxin (TTX) (Sigma), 200 μm CdCl2 (Sigma), 200 μm BCS (Sigma), 10 μm neocuproine (Sigma), and 100 μm CuCl2 (Sigma). Antibodies against dephospho-tau (tau-1; Chemicon International, Temecula, CA), MAP2 (Chemicon International), syntaxin 6 (BD Biosciences, San Jose, CA), gephyrin (Alexis Biochemicals, Carlsbad, CA), Ca2+-calmodulin-dependent protein kinase II (CaMKIIα) (clone 6G9; Affinity Bioreagents, Golden, CO), and postsynaptic density-95 (PSD-95) (clone 6G6-1C9; Affinity Bioreagents) were used according to manufacturers' specifications. Primary antibodies were visualized with Alexa Fluor 488 goat anti-mouse IgG1, Alexa Fluor 568 goat anti-rabbit IgG, and Alexa Fluor 633 goat anti-mouse IgG2a (Molecular Probes).
Immunoblotting. Cell and tissue lysates were prepared in 50 mm HEPES, pH 7.4/0.1% Nonidet P-40/150 mm NaCl supplemented with protease inhibitor mixture (EMD Biosciences) on ice for 15 min, followed by centrifugation for 10 min at 6000 × g at 4°C. Protein concentration for all samples was determined by Bradford's method (Bio-Rad, Hercules, CA). Lysates were incubated for 10 min in the presence of SDS sample buffer containing 2-mercaptoethanol and centrifuged for 5 min at 16,000 × g at 4°C before loading the supernatant onto SDS-PAGE. Proteins were separated, transferred to nitrocellulose, and detected by the SuperSignal West Pico Chemiluminescence kit (Pierce, Rockford, IL) using goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (Pierce).
Image analysis. Sets of neurons that were compared for quantitation were stained simultaneously. Cells were analyzed using a laser-scanning microscope (BX61WI FV500; Olympus, Melville, NY), equipped with argon 488, krypton 568, and HeNe 633 lasers and fitted with a PlanApo 60× objective with 1.4 numerical aperture. Imaged neurons were chosen from random areas of each coverslip based on phase contrast showing healthy morphology and imaged using identical confocal settings. For multichannel imaging, fluorophores were imaged sequentially to eliminate cross talk between the channels, and serial Z stacks of images were acquired using a pinhole aperture of 150 μm. The quantities of Menkes ATPase within the neuronal soma and the late Golgi were determined by double-blinded quantitation of maximal projection images of neurons immunolabeled for syntaxin 6 and Menkes using Photoshop Version 7.0 (Adobe Systems, San Jose, CA).
Values in Figure 6 represent the integrated Menkes ATPase immunofluorescence intensity in the late golgi as a percentage of the integrated intensity for the entire neuronal soma. Menkes ATPase was classified as somatic if the signal fell inside a manually traced region of interest corresponding to the neuronal soma. Menkes ATPase was classified as within the late golgi if the signal overlapped with syntaxin 6-immunoreactive areas. The syntaxin 6 images were subjected to a user-defined intensity threshold to define the area of the late golgi. Signal overlap was determined by first generating a binary mask from the thresholded syntaxin 6 image. Any pixel in the Menkes ATPase images that overlapped with a pixel from the binarized syntaxin 6 mask was considered to be within the late golgi. Values obtained were imported into Excel (Microsoft, Redmond, WA), and each data point was calculated as the percentage ratio of the integrated somatic Menkes signal outside the of syntaxin 6 mask to Menkes signal from the entire soma. Control ratios were subtracted from the experimental ratios to yield final values expressed as mean percentage increase ± SEM (n = 3 independent experiments of ≥15 cells per condition). Statistical analyses for p values were performed by using a one-way ANOVA with a Student-Newman-Keuls multiple-comparison test using SigmaStat 2 software (SPSS, Chicago, IL).
Figure 6.

Quantitation of Menkes ATPase trafficking. Neurons were treated with a 200 μm concentration of the copper chelator BCS for 24 hr and then exposed to extracellular solution with 2 mm Mg2+ (control) or pretreated with the indicated drugs and then exposed to 50 μm glutamate (glu), 5 μm glycine (gly), and the indicated drugs in Mg2+-free extracellular solution, fixed, immunolabeled for Menkes and syntaxin 6, and analyzed by quantitative confocal microscopy. The percentage increase in Menkes ATPase outside the late Golgi was calculated relative to control (see Materials and Methods). *p < 0.001. Error bars represent SD.
64Cu and 59Fe release studies. 64Cu was obtained from the Washington University cyclotron, and 59Fe was obtained from PerkinElmer Life Sciences (Emeryville, CA). Neurons were plated at high density and used at 13 d in culture. Neurons were incubated for 6 hr at 37°C with 100 μCi/ml 64Cu or 10 μCi/ml 59Fe, at which point the coverslips on which the neurons were plated were removed from the glial feeder layer and washed extensively in ECS with equimolar Ca2+ and Mg2+. Stimulation of the neurons with combined glutamate and glycine in Mg2+-free solution was then performed for 2 min, during which time samples from the bath were taken. Buffer was collected at multiple time points for 64Cu or 59Fe analysis in a Packard (Meridian, CT) gamma counter. In all cases, after the final wash, measured 64Cu or 59Fe activity in the bath was comparable with background levels. An aliquot of the initial 64Cu or 59Fe added at the start of incubation was collected, and the cpm was determined for each culture dish. After metabolic labeling of the cells, extensive washing, and the treatments indicated above, the cpm in the bath were determined, and the relative increase in 64Cu or 59Fe in the bath was calculated by dividing the output cpm from the experimental time point by the input cpm from the initial incubation time point. Statistical analyses for p values were performed by using a one-way ANOVA with a Student-Newman-Keuls multiple-comparison test using SigmaStat 2 software (SPSS).
Results
Immunoblot analysis of tissue lysates from normal mice demonstrated expression of Menkes ATPase in the brain. The specificity of this murine Menkes ATPase antisera was confirmed by a significant decrease in this signal in Menkes-deficient 802-1 cells (Fig. 1A). Immunohistochemistry revealed that this expression was confined to the perinuclear region of neurons in the hippocampus (Fig. 1B) and other brain regions as well as epithelial cells of the choroid plexus (data not shown). Consistent with these observations, immunofluorescent analysis of primary rat hippocampal neurons cultured for 2 d revealed that Menkes ATPase is present during early development in a perinuclear distribution overlapping with late Golgi markers including syntaxin 6 (Fig. 1C), adaptor protein complex (AP-1), Vti-1a, and trans-Golgi network (TGN) 38 (data not shown). As observed previously in nonpolarized cell lines (Petris et al., 1996; Hamza et al., 2003), localization of Menkes ATPase in mature cultured hippocampal neurons (2 weeks in culture) was dramatically altered by changes in the intracellular copper concentration (Fig. 2). Analysis of neurons grown in 200 μm BCS for 24 hr to reduce the intracellular copper content demonstrated predominant localization of Menkes ATPase to the late Golgi as judged by overlap with syntaxin 6, whereas incubation for 3 hr in 100 μm CuCl2 revealed trafficking of Menkes ATPase to a cytoplasmic vesicular compartment that extended down processes (Fig. 2). In mature cultured neurons, imaging of endogenous MAP2 and dephospho-tau cannot unequivocally distinguish dendrites from axons, because the processes run in tandem. These same experiments were repeated on neurons aged 1 week in culture, a point in cultured neuron development at which dendritic and axonal process identities are established, but synapses have just begun to form and isolated processes are still apparent. When neurons were treated with 100 μm CuCl2 for 3 hr and examined using antisera for MAP2 and dephospho-tau, markers of mature dendrites and axons, respectively, Menkes ATPase was readily detected in both dendrites and axons in primary hippocampal neurons (Fig. 3).
Figure 2.

Copper-mediated trafficking of Menkes ATPase in hippocampal neurons. Neurons were cultured for 2 weeks at low density, treated with a 200 μm concentration of the copper chelator BCS overnight (a-c) or 100 μm CuCl2 for 3 hr (d-f), fixed, double immunolabeled for Menkes (a, d; red in c, f) and syntaxin 6 (b, e; green in c, f), and analyzed by confocal microscopy. Arrowheads indicate Menkes ATPase outside the late Golgi. Scale bar: (in a) a-f, 10 μm.
Because trafficking of Menkes ATPase is the key cellular event determining copper homeostasis, and the primary function of neurons is to transmit and receive information via changes in membrane potential, we next examined the effect of neuronal activity on the trafficking of Menkes ATPase. Primary hippocampal neurons were incubated for 24 hr with 200 μm BCS and then treated in the presence of this chelator with 50 mm KCl in Mg2+-free ECS for 2 min to induce depolarization, followed by a recovery period of 10 min. Under these conditions, depolarization was found to induce the identical trafficking of Menkes ATPase to the cell processes as observed with CuCl2 (Fig. 4). This finding suggested that neuronal activation may mediate rapid changes in neuronal copper homeostasis independent of the intracellular copper concentration. To directly examine this possibility and to determine the nature of this activation, hippocampal neurons were incubated for 24 hr with 200 μm BCS and then stimulated with glutamate-glycine (in Mg2+-free ECS) for 1 min in the presence of this chelator, followed by a recovery period of 10 min. As can be seen in Figure 4, this treatment was sufficient to cause trafficking of Menkes ATPase from the late Golgi in a manner similar to that observed with elevated copper. Experiments using process-specific markers revealed that Menkes ATPase trafficked to both axons and dendrites after treatment with glutamate-glycine (data not shown). This effect of glutamate-glycine was not dependent on action potential-mediated synaptic activity in the cultures, because blocking voltage-gated sodium channels with TTX before and during treatment did not inhibit Menkes ATPase trafficking (see Fig. 6).
Figure 4.

Glutamate-glycine-dependent trafficking of Menkes ATPase. Neurons were treated with a 200 μm concentration of the copper chelator BCS for 24 hr and then exposed to extracellular solution with 2 mm Mg2+ (a-c), 50 mm KCl (d-f), or 50 μm glutamate (Glu) and 5 μm glycine (Gly) (g-i), fixed, and immunolabeled for Menkes (a, d, g; red in c, f, i) and syntaxin 6 (b, e, h; green in c, f, i) and analyzed by confocal microscopy. Arrowheads (d, f, i) indicate Menkes ATPase outside late Golgi and in processes. Scale bar: (in a) a-i, 10 μm. Glu, Glutamate; Gly, glycine.
Because the above experiments were performed in the presence of the copper-specific chelator BCS, a treatment previously shown to inhibit copper-mediated trafficking of Menkes ATPase (Hamza et al., 2003), these data reveal evidence of Menkes ATPase trafficking independent of the intracellular copper concentration. In support of this concept, treatment of hippocampal neurons with the cell-permeant copper chelator neocuproine for 4 hr before and during glutamate-glycine stimulation also failed to abrogate Menkes ATPase trafficking (data not shown). The effect of glutamate-glycine was specific to neurons, because no such trafficking of either Menkes or Wilson ATPase was observed after similar treatment in multiple cell lines known to express these copper transporters (data not shown). To determine whether ionotropic glutamate receptor activity was required for glutamate-glycine-induced translocation of Menkes ATPase, these experiments were repeated using receptor-specific inhibitors. Whereas blockade of the AMPA/kainate (KA) receptors with CNQX was without effect on Menkes ATPase trafficking, antagonism of the NMDA receptor with APV abrogated dispersal of Menkes ATPase from the late Golgi during stimulation (Fig. 5). Quantitative confocal microscopy revealed an average 17% ± 1.1 relative increase in cytoplasmic Menkes ATPase after glutamate-glycine stimulation compared with control-treated cells (Fig. 6). Treatment with either CNQX or TTX in combination with NMDA receptor stimulation did not abrogate the trafficking of Menkes ATPase (average, 21% ± 1.1 and 20% ± 1.2, respectively). Treatment with glycine alone (in Mg2+-free ECS), a stimulus that relies on synaptic release of glutamate to activate synaptic but not extrasynaptic NMDA receptors (Liao et al., 2001; Lu et al., 2001), resulted in Menkes ATPase trafficking equal to that of coapplication of glutamate-glycine (average, 21% ± 1.2). In contrast, activation of nonsynaptic NMDA receptors through treatment with glycine and the open NMDA channel blocker MK-801, followed by washout and subsequent glutamate-glycine stimulation, did not induce Menkes ATPase translocation (average, 2.5% ± 1.3). As noted above, antagonism of the NMDA receptor with APV abrogated Menkes ATPase movement in response to glutamate-glycine (average, 0.1% ± 0.9) (Fig. 6). Like-wise, treatment with APV abrogated the trafficking of Menkes ATPase after neuronal depolarization with 50 mm KCl in Mg2+-free ECS. The APV treatment was a specific inhibitor of the NMDA receptor activity-induced Menkes ATPase trafficking, because trafficking caused by the independent stimulus CuCl2 was not abrogated by coapplication with APV (data not shown).
Figure 5.

NMDA receptor-dependent trafficking of Menkes ATPase. Neurons were treated for 24 hr with a 200 μm concentration of the copper chelator BCS and then exposed to extracellular solution with 2 mm Mg2+ (a), exposed to 50 μm glutamate (Glu) and 5 μm glycine (Gly) in Mg2+-free extracellular solution (b), or pretreated with the indicated drugs and then exposed to 50 μm glutamate, 5 μm glycine, and the indicated drugs in Mg2+-free extracellular solution (c-f). Neurons were treated with the AMPA receptor antagonist CNQX (c), the NMDA receptor antagonist APV (d), the cell-permeable Ca2+ chelator BAPTA-AM (e), or the voltage-gated calcium channel antagonist CdCl2 (f). Cells were fixed and immunolabeled for Menkes for analysis by confocal microscopy. Arrowheads (b, c, f) indicate Menkes ATPase outside late Golgi and in processes. Scale bar: (in a) a-f, 10 μm.
Because Ca2+ entry through the NMDA receptor underlies the ability of the receptor to activate calcium-dependent biochemical cascades, we sought to determine whether Ca2+ flux was necessary for the induction of movement of Menkes ATPase after glutamate-glycine stimulation. Neurons were incubated for 24 hr with 200 μm BCS and were then treated for 70 min with a 20 μm concentration of the cell-permeant calcium chelator BAPTA-AM and analyzed for Menkes ATPase trafficking after glutamate-glycine stimulation. Under these conditions, chelation of intracellular Ca2+ was found to abrogate the effect of receptor stimulation on Menkes ATPase trafficking (Fig. 5e). This signal is likely to be mediated by Ca2+ flow from the extracellular milieu through the NMDA receptor, because general antagonism of voltage-gated calcium channels through 15 min pretreatment with CdCl2 or a 15 min pretreatment with nifedipine to specifically block L-type calcium channels, which are the predominant species of voltage-gated calcium channels in the neurons in this culture system, did not diminish trafficking of Menkes ATPase in response to glutamate-glycine stimulation coapplied with the inhibitors (Fig. 5f).
Previous studies have shown that activation of the NMDA receptor results in translocation of CaMKIIα to postsynaptic densities where it colocalizes with PSD-95, a postsynaptic density scaffolding protein (Shen and Meyer, 1999; Gleason et al., 2003). To determine more precisely the subcellular localization of Menkes ATPase after NMDA receptor activation, hippocampal neurons were treated with glutamate-glycine as indicated above, and the localization of CaMKIIα, PSD-95, and Menkes ATPase was determined. In control-treated cells, CaMKIIα was distributed relatively homogeneously along individual processes and concentrated in a few spines (Fig. 7a), changing after NMDA receptor activation to a more punctate distribution that overlapped extensively with PSD-95 (Fig. 7c). NMDA receptor activation resulted in trafficking of Menkes ATPase to processes, but only a minor portion was found to colocalize with CaMKIIα and PSD-95 (Fig. 7d, arrows). Similar experiments using antibodies to gephyrin, a marker for GABAergic synapses in hippocampal neurons (Levi et al., 2004), failed to reveal any colocalization with Menkes ATPase in processes after glutamate-glycine treatment (data not shown). Thus, whereas CaMKIIα translocates to glutamatergic synapses, Menkes ATPase translocates primarily to extrasynaptic sites. In all cases, these observed changes in CaMKIIα and Menkes ATPase trafficking were inhibited by the NMDA receptor antagonist APV (Fig. 7e,f).
Figure 7.

Characterization of NMDA receptor activation-mediated Menkes ATPase trafficking. Neurons were treated for 24 hr with a 200 μm concentration of the copper chelator BCS, exposed to extracellular solution with 2 mm Mg2+ (Control) (a, b), 50 μm glutamate (Glu) and 5 μm glycine (Gly) (c, d), or 50 μm glutamate and 5 μm glycine and the NMDA receptor antagonist APV (e, f), and then fixed, triple immunolabeled for CaMKIIα, PSD-95, and Menkes, and analyzed by confocal microscopy (see Materials and Methods). Menkes ATPase associated (arrows) and unassociated (arrowheads) with glutamatergic synapses is shown. Scale bar: (in a) a-f, 10 μm.
Together, these data indicate a direct effect of NMDA receptor activation on the localization of Menkes ATPase. Because copper-induced trafficking of the copper transporting ATPases from the late Golgi is a critical component of copper efflux and homeostasis in other cell types, we next sought to directly examine the effect of NMDA receptor stimulation on copper homeostasis in hippocampal neurons. As can be seen in Figure 8, A and B, when copper efflux from these neurons was examined after metabolic labeling with 64Cu, cells treated with glutamate-glycine showed an average eightfold increase in 64Cu levels in the bath solution when compared with mock-treated cells. These findings were highly specific, because the 64Cu cpm from control-treated cells was equivalent to that from coverslips prepared without neurons, incubated with 64Cu, and treated with glutamate-glycine (data not shown). Application of TTX to cultures treated with glutamate-glycine did not have an effect on copper release, indicating that this process is not dependent on action potential-mediated activity (Fig. 8B). As observed for the effect of glutamate-glycine on Menkes ATPase trafficking, these findings were the direct result of NMDA receptor activation, because application of APV before and during treatment abrogated the efflux of 64Cu from hippocampal neurons (Fig. 8B). The removal of protein from the efflux solution by acetone or trichloroacteic acid precipitation did not alter the amount of detectable 64Cu in the media after glutamate-glycine treatment (data not shown), suggesting that the released copper was either free in solution or bound to small molecules and not incorporated into specific cuproproteins before efflux. Furthermore, these findings were specific for copper, because no such efflux was observed when these same experiments were repeated in hippocampal neurons metabolically labeled with 59Fe (Fig. 8C). To determine whether Menkes ATPase plays a direct role in copper efflux after NMDA receptor activation, these experiments were repeated using hippocampal neurons derived from newborn (P0) male mice hemizygous for wild-type (Mobr +/Y) or brindled mutant Menkes ATPase (Mobr-/Y) (see Materials and Methods). As can be seen in Figure 8D, whereas glutamate-glycine stimulation of neurons derived from mice with a fully functional Menkes ATPase resulted in copper efflux, this same treatment in cells derived from Mobr-/Y failed to show any increase in 64Cu in the media when compared with control-treated neurons. These findings were not the result of genetic differences in 64Cu uptake during the labeling period, because hippocampal neurons from Mobr +/Y and Mobr-/Y animals showed equivalent 64Cu uptake (data not shown), consistent with previous observations on copper uptake in Mobr cells (La Fontaine et al., 1999).
Figure 8.

Copper efflux from hippocampal neurons. A, Neurons were incubated with 300 μCi of 64Cu for 6 hr, washed, and exposed to ECS with 2 mm Mg2+ (Control) or 50 μm glutamate (Glu) and 5 μm glycine (Gly), and the bath solution was analyzed for 64Cu for the indicated times. Data points are from a single representative experiment. B, Neurons were incubated with 300 μCi of 64Cu for 6 hr, washed, and exposed to ECS with 2 mm Mg2+ (Control), 50 μm glutamate and 5 μm glycine or glutamate-glycine in combination with the NMDA receptor antagonist APV (Glu/Gly + APV), the bath solution was analyzed for 64Cu for the first minute of efflux, and the relative cpm were calculated as described in Materials and Methods. Data points represent a minimum of three separate experiments using separate culture preparations. C, Neurons were incubated with 30 μCi of 59Fe for 6 hr, washed, and exposed to ECS with 2 mm Mg2+ (Control), or 50 μm glutamate and 5 μm glycine, the bath solution was analyzed for 59Fe for the first minute of efflux, and the relative cpm were calculated as described in Materials and Methods. Data points were collected in triplicate from one experiment. D, Mouse hippocampal neurons were isolated and cultured at P0 from male offspring of Mobr heterozygous female, wild-type male matings. Individual neuronal cultures were prepared from each pup, followed by metabolic labeling with 64Cu, treatment with ECS with 2 mm Mg2+ (Control) or 50 μm glutamate and 5 μm glycine, measurement of 64Cu for the first minute of efflux, and calculation of the relative cpm, as described above and in Materials and Methods. After the analysis, the genotype of the cultures at the Menkes locus was determined by PCR as either wild type (Mobr +/Y) or functionally null (Mobr-/Y). Data points represent a minimum of three separate experiments using separate culture preparations. All means and SDs were calculated and differences compared using SigmaStat. *p < 0.001.
Discussion
The data in this study reveal that activation of synaptic NMDA receptors in hippocampal neurons results in trafficking of Menkes ATPase and an associated efflux of copper dependent on Menkes ATPase function. Although previous studies have demonstrated that copper stimulates the exocytic movement of Wilson and Menkes copper-transporting P-type ATPases from the late Golgi and that mutations affecting this trafficking process directly cause disease as a result of a resultant marked diminution of copper homeostasis (Petris et al., 1996; Hung et al., 1997; Lutsenko and Petris, 2003), this is the first evidence for a mechanism activating trafficking of one of the copper-transporting ATPases independent of copper concentration. This process is not attributable to an influx of extracellular copper, because all studies were performed in the presence of excess copper chelators. Although it is possible that NMDA receptor activation results in the release of copper from an intracellular storage pool, such a possibility is unlikely given the paucity of available intracellular copper in the cytoplasm of mammalian cells (Rae et al., 1999) and the inability of cell-permeant copper chelators to abrogate the glutamate-glycine-stimulated trafficking of Menkes ATPase. Although Menkes ATPase trafficking is coupled to copper transport, the finding that Menkes ATPase traffics in hippocampal neurons independently of copper suggests that Menkes ATPase trafficking in these neurons may be independent of catalytic activity (Lutsenko and Petris, 2003). Importantly, these findings demonstrate a direct link between ionotropic receptor activity in hippocampal neurons, a process critical to neuronal function, and copper homeostasis in these same cells. Our observation that Menkes ATPase is expressed in several brain regions is consistent with previous studies of Menkes mRNA expression in the mouse brain (Iwase et al., 1996; Murata et al., 1997) and suggests that similar mechanisms linking neuronal activation and copper homeostasis may exist in other neuronal cell types in the CNS.
Quantitative analysis of Menkes ATPase movement after NMDA receptor activation revealed that 5-10 min after glutamate-glycine stimulation, ∼20% of this transporter had trafficked from the Golgi, an amount similar to what was observed in this time period after copper treatment of mouse fibroblasts (Hamza et al., 2003). These kinetics, as well as the fact that a similar pattern of Menkes trafficking is observed in hippocampal neurons after increased copper (Fig. 2), suggest that the underlying biochemical mechanisms for this trafficking may be similar in all cells. Analysis of this vesicular compartment revealed no overlap with known markers of the late endosomal or lysosomal pathways (data not shown), similar to what has been observed previously in cell lines after copper stimulation (Tao and Gitlin, 2003). Moreover, in the present study, Menkes ATPase was not found to localize to the cell surface. This may be because only a small amount endogenously cycles to the surface or, as is the case for Wilson ATPase, it may be that the ATPase itself does not reach the surface and that the final exit of copper is via a vesicular pathway once the ATPase has cycled back to the TGN. Consistent with this possibility, cell surface biotinylation studies did not detect Menkes ATPase on the plasma membrane of hippocampal neurons under any of the experimental conditions (data not shown). Studies of the homologous Wilson ATPase have revealed that although it is critical for excretion of copper into the bile from hepatocytes, before copper exocytosis, the ATPase is recycled back to the Golgi, precluding use of this protein as a maker for following the release process. A similar occurrence in hippocampal neurons may explain why only a portion of Menkes ATPase was found to colocalize with PSD-95 and CaMKIIα after glutamate-glycine stimulation, underscoring the importance of further characterization of markers for this compartment to critically assess the mechanism and sites of copper release.
Copper efflux after NMDA receptor activation was observed to occur very rapidly, suggesting that Menkes ATPase may accumulate copper in a vesicular compartment that is readily available for exocytosis after Ca2+ influx through the NMDA receptor. In this regard, activation of the NMDA receptor may additionally play a critical role in neuronal copper homeostasis by stimulating movement of Menkes ATPase to processes to maintain availability of a readily releasable copper pool. Stimulation of AMPA/KA receptors in the absence of NMDA receptor activation was not sufficient to induce rapid copper efflux, and treatment with TTX was not sufficient to block the glutamate-glycine-induced rapid copper efflux (Fig. 8B), indicating that the major copper-carrying exocytotic organelles are not classic synaptic vesicles. Rather, the organelles that mediate copper efflux in an NMDA receptor-dependent manner may be dendritically localized. Furthermore, the selective induction of Menkes ATPase trafficking by activation of synaptic or total cellular but not extrasynaptic NMDA receptors further reveals a specific pathway, likely triggered by a specific route of calcium entry. Activation of synaptic NMDA receptors has been shown to induce membrane insertion of new AMPA receptors through an exocytotic process (Liao et al., 2001; Lu et al., 2001), raising the possibility that synaptic NMDA receptors may be inducing the excretion of copper through a similar process. Activation of synaptic but not extrasynaptic NMDA receptors induces phosphoactivation of the transcription factor cAMP response element-binding protein (CREB) (Hardingham and Bading, 2003), suggesting that Menkes ATPase trafficking and copper excretion may use enzymes previously found to link synaptic NMDA receptor activation to CREB phosphorylation or to induction of long-term potentiation, such as CaMKIV or MAPK (mitogen-activated protein kinase) family members (Zhu et al., 2002; Hardingham and Bading, 2003; Man et al., 2003).
The finding that NMDA receptor activation results in Menkes ATPase-dependent copper efflux suggests the possibility that this copper release may play a role in neuronal function. Previous studies in neuronal cultures have demonstrated that micromolar copper can inhibit amino-acid receptor-mediated currents, including NMDA receptor-mediated currents, in rat olfactory bulb neurons (Trombley and Shepherd, 1996) and in cultured mouse and rat hippocampal and cortical neurons (Trombley and Shepherd, 1996; Vlachova et al., 1996; Weiser and Wienrich, 1996) and modulate synaptic plasticity in hippocampal slices (Doreulee et al., 1997). Copper has also been shown to reduce the rate of repetitive firing of action potentials through action on transient and delayed rectifier-type potassium channels and to inhibit voltage-gated calcium channels, thereby exerting an inhibitory effect on neurotransmitter release (Horning and Trombley, 2001; Castelli et al., 2003). Because catalytic amounts of copper can function as electron acceptors promoting the reaction of nitric oxide with thiols, release of this metal could function as a molecular switch to control extracellular S-nitrosylaton of the NMDA receptor, a posttranslational mechanism shown to be critical for modulating receptor function (Lipton et al., 1998; Choi et al., 2000).
Although the absolute amount of copper released in our experiments cannot be determined using 64Cu, previous analysis of extracellular copper concentrations after brief depolarization of cortical synaptosomes demonstrated the release of attogram amounts of copper that result in a local micromolar concentration when the volume of the synaptic cleft is taken into account (Kardos et al., 1989). This is consistent with the concentrations observed to modulate neuronal function in the studies noted above, and therefore our data suggest a physiological role for copper independent of incorporation into specific proteins. Interestingly, a similar neuromodulatory role has been proposed for zinc, a divalent transition metal released from excitatory synapses in the hippocampus, where it inhibits NMDA receptors and influences signaling pathways (Peters et al., 1987). Inappropriate zinc release is proposed to result in neuronal cell death under conditions such as ischemia, and this may also be true for copper efflux under conditions of prolonged or inappropriate NMDA receptor activation (Choi and Koh, 1998). In this case, activation-dependent copper efflux may provide a substrate for reactive oxygen species implicated in the pathogenesis of Alzheimer's and other neurodegenerative diseases (Bush et al., 2003).
Together, the data in this study reveal a unique model of copper homeostasis within the CNS and raise the hypothesis that copper may function as an activity-dependent modulator of hippocampal synaptic transmission, suggesting a potential role for Menkes ATPase in memory and learning. The data also lend new insight into the role of Menkes ATPase in the pathophysiology of Menkes disease. Although this disorder results in very characteristic neuronal pathology, this is not easily explained by evoking the loss of function of any of the known cuproenzymes expressed in the brain. Our data suggest that impairment of NMDA receptor-mediated copper efflux in affected patients could directly alter receptor function contributing both to seizures and rapid neuronal degeneration. As such, these findings point toward novel therapeutic approaches aimed directly at pharmacological antagonism of NMDA receptor activity in affected patients.
Footnotes
This work was supported by a National Science Foundation Graduate Research Fellowship (M.L.S.) and by National Institutes of Health Grants NS33184 (A.M.C.) and DK44464 (J.D.G.). We thank Gary Banker for insightful discussion of the data, Huaiyang Wu and Fernanda Laezza for help with neuronal cultures and advice, Maureen Boyle and Penny Jensen for help with brain tissue preparations, and Louis Muglia, David Holtzman, and Guojun Bu for critical review of this manuscript.
Correspondence should be addressed to Dr. Jonathan D. Gitlin, Washington University School of Medicine, McDonnell Pediatric Research Building, 660 South Euclid Avenue, St. Louis, MO 63110. E-mail: gitlin@kids.wustl.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250239-08$15.00/0
References
- Bayer TA, Schèafer S, Simons A, Kemmling A, Kamer T, Tepest R, Eckert A, Schèussel K, Eikenberg O, Sturchler-Pierrat C, Abramowski D, Staufenbiel M, Multhaup G (2003) Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Abeta production in APP23 transgenic mice. Proc Natl Acad Sci USA 100: 14187-14192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush AI, Masters CL, Tanzi RE (2003) Copper, beta-amyloid, and Alzheimer's disease: tapping a sensitive connection. Proc Natl Acad Sci USA 100: 11193-11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli L, Tanzi F, Taglietti V, Magistretti J (2003) Cu2+, Co2+, and Mn2+ modify the gating kinetics of high-voltage-activated Ca2+ channels in rat palaeocortical neurons. J Membr Biol 195: 121-136. [DOI] [PubMed] [Google Scholar]
- Choi DW, Koh JY (1998) Zinc and brain injury. Annu Rev Neurosci 21: 347-375. [DOI] [PubMed] [Google Scholar]
- Choi YB, Tenneti L, Le DA, Ortiz J, Bai G, Chen HS, Lipton SA (2000) Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat Neurosci 3: 15-21. [DOI] [PubMed] [Google Scholar]
- Doreulee N, Yanovsky Y, Hass HL (1997) Suppression of long-term potentiation in hippocampal slices by copper. Hippocampus 7: 666-669. [DOI] [PubMed] [Google Scholar]
- Gleason MR, Higashijima S, Dallman J, Liu K, Mandel G, Fetcho JR (2003) Translocation of CaM kinase II to synaptic sites in vivo. Nat Neurosci 6: 217-218. [DOI] [PubMed] [Google Scholar]
- Goslin K, Asmussen H, Banker G (1998) Rat hippocampal neurons in lowdensity culture. In: Culturing nerve cells (Banker GK, ed), pp 339-370. Cambridge, MA: MIT.
- Grimes A, Hearn C, Lockhart P, Newgreen D, Mercer JF (1997) Molecular basis of the brindled mouse mutant (Mo(br)): a murine model of Menkes disease. Hum Mol Genet 6: 1037-1042. [DOI] [PubMed] [Google Scholar]
- Hamza I, Prohaska J, Gitlin J (2003) Essential role for Atox1 in the copper-mediated intracellular trafficking of the Menkes ATPase. Proc Natl Acad Sci USA 100: 1215-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham G, Bading H (2003) The Yin and Yang of NMDA receptor signalling. Trends Neurosci 26: 81-89. [DOI] [PubMed] [Google Scholar]
- Hopt A, Korte S, Fink H, Panne U, Niessner R, Jahn R, Kretzschmar H, Herms J (2003) Methods for studying synaptosomal copper release. J Neurosci Methods 128: 159-172. [DOI] [PubMed] [Google Scholar]
- Horning MS, Trombley PQ (2001) Zinc and copper influence excitability of rat olfactory bulb neurons by multiple mechanisms. J Neurophysiol 86: 1652-1660. [DOI] [PubMed] [Google Scholar]
- Hung I, Suzuki M, Yamaguchi Y, Yuan D, Klausner R, Gitlin J (1997) Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae J Biol Chem 272: 21461-21466. [DOI] [PubMed] [Google Scholar]
- Iwase T, Nishimura M, Sugimura H, Igarashi H, Ozawa F, Shinmura K, Suzuki M, Tanaka M, Kino I (1996) Localization of Menkes gene expression in the mouse brain; its association with neurological manifestations in Menkes model mice. Acta Neuropathol (Berl) 91: 482-488. [DOI] [PubMed] [Google Scholar]
- Kardos J, Kovacs I, Hajos F, Kalman M, Simonyi M (1989) Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci Lett 103: 139-144. [DOI] [PubMed] [Google Scholar]
- Kelly EJ, Palmiter RD (1996) A murine model of Menkes disease reveals a physiological function of metallothionein. Nat Genet 13: 219-222. [DOI] [PubMed] [Google Scholar]
- La Fontaine S, Firth SD, Lockhart PJ, Brooks H, Camakaris J, Mercer JF (1999) Intracellular localization and loss of copper responsiveness of Mnk, the murine homologue of the Menkes protein, in cells from blotchy (Mo blo) and brindled (Mo br) mouse mutants. Hum Mol Genet 8: 1069-1075. [DOI] [PubMed] [Google Scholar]
- Levi S, Logan SM, Tovar KR, Craig AM (2004) Gephyrin is critical for glycine receptor clustering but not for the formation of functional GABAergic synapses in hippocampal neurons. J Neurosci 24: 207-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Scannevin RH, Huganir R (2001) Activation of silent synapses by rapid activity-dependent synaptic recruitment of AMPA receptors. J Neurosci 21: 6008-6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Rayudu PV, Choi YB, Sucher NJ, Chen HS (1998) Redox modulation of the NMDA receptor by NO-related species. Prog Brain Res 118: 73-82. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble W, MacDonald J, Wang YT (2001) Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29: 243-254. [DOI] [PubMed] [Google Scholar]
- Lutsenko S, Petris MJ (2003) Function and regulation of the mammalian copper-transporting ATPases: insights from biochemical and cell biological approaches. J Membr Biol 191: 1-12. [DOI] [PubMed] [Google Scholar]
- Man HY, Wang Q, Lu WY, Ju W, Ahmadian G, Liu L, D'Souza S, Wong TP, Taghibiglou C, Lu J, Becker LE, Pei L, Liu F, Wymann MP, MacDonald JF, Wang YT (2003) Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron 38: 611-624. [DOI] [PubMed] [Google Scholar]
- Murata Y, Kodama H, Abe T, Ishida N, Nishimura M, Levinson B, Gitschier J, Packman S (1997) Mutation analysis and expression of the mottled gene in the macular mouse model of Menkes disease. Pediatr Res 42: 436-442. [DOI] [PubMed] [Google Scholar]
- Okeda R, Gei S, Chen I, Okaniwa M, Shinomiya M, Matsubara O (1991) Menkes' kinky hair disease: morphological and immunohistochemical comparison of two autopsied patients. Acta Neuropathol (Berl) 81: 450-457. [DOI] [PubMed] [Google Scholar]
- Peters S, Koh J, Choi DW (1987) Zinc selectively blocks the action of N-methyl-d-aspartate on cortical neurons. Science 236: 589-593. [DOI] [PubMed] [Google Scholar]
- Petris MJ, Mercer JF, Culvenor JG, Lockhart P, Gleeson PA, Camakaris J (1996) Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. EMBO J 15: 6084-6095. [PMC free article] [PubMed] [Google Scholar]
- Phinney AL, Drisaldi B, Schmidt SD, Lugowski S, Coronado V, Liang Y, Horne P, Yang J, Sekoulidis J, Coomaraswamy J, Chishti MA, Cox DW, Mathews PM, Nixon RA, Carlson GA, St George-Hyslop P, Westaway D (2003) In vivo reduction of amyloid-beta by a mutant copper transporter. Proc Natl Acad Sci USA 100: 14193-14198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae T, Schmidt P, Pufahl R, Culotta V, O'Halloran T (1999) Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science 284: 805-808. [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T (1999) Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science 284: 162-166. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM, Brown DR, Alim MA, Scholtzova H, Carp R, Meeker HC, Prelli F, Frangione B, Wisniewski T (2003) Copper chelation delays the onset of prion disease. J Biol Chem 278: 46199-46202. [DOI] [PubMed] [Google Scholar]
- Sparks DL, Schreurs BG (2003) Trace amounts of copper in water induce beta-amyloid plaques and learning deficits in a rabbit model of Alzheimer's disease. Proc Natl Acad Sci USA 100: 11065-11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao TY, Gitlin JD (2003) Hepatic copper metabolism: insights from genetic disease. Hepatology 37: 1241-1247. [DOI] [PubMed] [Google Scholar]
- Trombley P, Shepherd G (1996) Differential modulation by zinc and copper of amino acid receptors from rat olfactory bulb neurons. J Neurophysiol 76: 2536-2546. [DOI] [PubMed] [Google Scholar]
- Vlachova V, Zemkova H, Vyklicky LJ (1996) Copper modulation of NMDA responses in mouse and rat cultured hippocampal neurons. Eur J Neurosci 8: 2257-2264. [DOI] [PubMed] [Google Scholar]
- Weiser T, Wienrich M (1996) The effects of copper ions on glutamate receptors in cultured rat cortical neurons. Brain Res 742: 211-218. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R (2002) Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 110: 443-455. [DOI] [PubMed] [Google Scholar]