

Abstract
Glutamatergic synapses are highly modifiable, suiting them for key roles in processes such as learning and memory. At crayfish glutamatergic neuromuscular junctions, hyperpolarization and cyclic nucleotide-activated (HCN) ion channels mediate hormonal modulation of glutamatergic synapses and a form activity-dependent long-term facilitation (LTF) of synaptic transmission. Here, we show that a new target for cAMP, exchange protein activated by cAMP (Epac) or cAMP-regulated guanine nucleotide exchange protein, is involved in the hormonal enhancement of synaptic transmission by serotonin. Induction of LTF “tags” synapses, rendering them responsive to cAMP in an HCN-independent manner. Epac also mediates the enhancement of tagged synapses. Thus, the cAMP-dependent enhancement of transmission is mediated by two separate pathways, neither of which involves protein kinase A.
Keywords: synaptic transmission, synaptic plasticity, neuromodulation, intracellular signaling, cAMP, Epac
Introduction
At cerebellar and cerebral mammalian synapses and Drosophila neuromuscular junctions, cAMP enhances transmitter release by directly modulating the secretory machinery (Zhong and Wu, 1991; Chavez-Noriega and Stevens, 1994; Chen and Regehr, 1997) by activating protein kinase A (PKA) (Capogna et al., 1995; Trudeau et al., 1996, 1998). At crayfish neuromuscular junctions, cAMP mediates an enhancement of glutamate release caused by serotonin, a native circulatory hormone (Glusman and Kravitz, 1982). Here, PKA is not a target of cAMP action. Instead, cAMP activates presynaptic hyperpolarization and cyclic nucleotide-activated (HCN) (also called Ih) channels, which couple to transmitter secretion by an actin-dependent process (Beaumont and Zucker, 2000).
HCN channels are also involved in a second form of synaptic plasticity at crayfish neuromuscular junctions. Extensive motor activity results in a cAMP-independent enhancement of transmission referred to as long-term facilitation (LTF). LTF is attributable to the accumulation of Na+ ions during electrical activity, resulting in a presynaptic hyperpolarization caused by electrogenic Na+ extrusion; this hyperpolarization activates HCN channels to induce an increase in glutamate release to action potentials (Beaumont et al., 2002; Zhong and Zucker, 2004). The long persistence of LTF requires the accumulation of presynaptic Ca2+ during electrical activity, activation of calcium-dependent phosphatases, presynaptic local protein synthesis, and an enzyme cascade including phosphoinositol 3-OH, MAP, and rapamycin-sensitive kinases (Beaumont et al., 2001).
Blocking HCN activation prevents both synaptic enhancement by cAMP and LTF. However, there are indications of an additional cAMP target in the serotonergic enhancement of transmission. Activation of adenylyl cyclase by forskolin is without effect in the presence of HCN channel blockers; however, if the blockers are added after exposure to forskolin, transmission remains elevated in the presence of those blockers until forskolin is withdrawn (Beaumont et al., 2002). This result implicates a second cAMP target in the maintenance of enhanced transmission, for which initiation requires HCN channel activation.
A second clue to the existence of an additional cAMP target lies in the abolition of the requirement for HCN activation by cAMP to enhance release after LTF induction. After LTF is induced, forskolin enhances release still further, but now this enhancement occurs in the presence of HCN channel inhibitors as well as PKA inhibitors (Beaumont et al., 2002). We use the term “temporal synaptic tagging” to describe this alteration in the state of the synapses after LTF induction. Apparently, cAMP acts on a separate target, distinct from HCN channels or PKA, to sensitize tagged synapses.
In this study, we identify exchange protein activated by cAMP (Epac) as the likely cAMP target responsible both for the maintenance of cAMP action after blocking HCN channels and for the response to cAMP at tagged synapses. We also introduce the use of brefeldin A (BFA) as an antagonist of Epac action and suggest that it might be a useful tool in searching for an involvement of the Epac pathway in other systems.
Materials and Methods
Preparation. Crayfish (Procambarus clarkia; 2-3 inches) were obtained from Niles Biological (Sacramento, CA). Preparation of the innervated dactyl opener muscle of the first walking leg has been described previously (Delaney et al., 1991). Autotomized legs were continuously superfused with Normal Van Harrevald's solution, containing the following (in mm): 195 NaCl, 13.5 CaCl2, 5.4 KCl, 2.6 MgCl2, and 10 Na-HEPES, pH 7.4, 14-17°C. All drugs were used at a dose giving near-maximal responses (>80%) rather than maximal responses to minimize nonspecific effects. 8-(4-chloro-phenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (8-CPT) was purchased from Axxora Biolog Biochemicals (San Diego, CA). Lamotrigine was a gift from GlaxoSmithKline (Research Triangle Park, NC). Brefeldin A and forskolin were obtained from EMD Biosciences (Pasadena, CA). The protein kinase A inhibitor KT 5720 was from A. G. Scientific (San Diego, CA). As a positive control for the lack of effect of KT 5720, we confirmed its efficacy against crayfish PKA, using the Promega (Madison, WI) Pep-Tag PKA Assay kit; a 1 μm concentration produced nearly complete inhibition of PKA. The HCN channel blocker ZD 7288 was obtained from Tocris Cookson (Ballwin, MO). In some cases (forskolin, lamotrigine), stock solutions of drugs were prepared in dimethylsulfoxide (DMSO) and dissolved before use in external media to a final concentration containing no more than 0.1% DMSO. Control experiments using 0.1% DMSO showed that synaptic transmission was not affected by addition of this solvent.
Electrophysiology. Sharp electrodes were used to impale proximal muscle fibers (electrode resistance, 10-20 MΩ) and primary or secondary branches of the excitor nerve axon (beveled electrode resistance, 20-40 MΩ). Nerve stimulation (2 Hz) used a suction electrode containing the excitor axon freed from the meropodite segment of the leg. Signals were amplified [excitatory junction potentials (EJPs): Neuroprobe 1600 Amplifier, A-M Systems, Carlsborg, WA; preterminal axonal recordings: Getting Microelectrode Amplifier, Getting, Iowa City, IA], filtered at 2 kHz, and digitized at 5 kHz, and averages of all EJPs in each minute were saved to a computer using pClamp7 software (Axon Instruments, Union City, CA). Responses were measured off-line (Clampfit 6.05; Axon Instruments). Because baseline EJP amplitudes were variable from fiber to fiber, results were expressed as percentage change from control EJP amplitude, taken as the average EJP amplitude over 10-20 min of continuous recording before experimental manipulations. Data are plotted as mean percentage ± SE of this control level. Statistical differences were tested using Student's two-tailed t tests. Only every fourth point is plotted, for the sake of clarity. Whenever comparisons were made between treatments, crayfish for both treatments were selected from the same shipment, and compared treatments were inter-leaved in successive experiments to minimize any trends in the responsiveness of batches of crayfish.
Results
Separate activation of Epac and HCN channels
In vertebrates, the type-2 isoform of Epac (Epac2), also called the type-2 isoform of cAMP-regulated guanine nucleotide exchange factor (cAMP-GEFII), is known to activate small GTPases such as rap1A (de Rooij et al., 1998; Kawasaki et al., 1998; Enserink et al., 2002) and to regulate hormonal secretion (Ozaki et al., 2000; Kashima et al., 2001). We used a recently developed agonist of cAMP activation of Epac2, 8-CPT (Christensen et al., 2003; Kang et al., 2003), to probe a role for a crayfish Epac homolog in regulating transmission at crayfish neuromuscular junctions.
8-CPT (50 μm) produced a modest increase (to 139 ± 6.0% of the initial amplitude) in EJPs. This is approximately one-fourth (p < 0.0001) the enhancement caused by the cyclase activator forskolin (to 200-250% of controls) (Beaumont and Zucker, 2000) (Fig. 1B). The effect of 8-CPT (Fig. 1A) remained in 30 μm of the HCN channel blocker ZD 7288, even when 1 μm of the PKA inhibitor KT 5720 was included (Fig. 1A) (average enhancement to 137 ± 7.5% in the presence of ZD 7288 and 141 ± 11% when KT 5720 was also present; both, p > 0.8 with respect to 8-CPT alone; because results are identical with and without KT 5720, they are lumped in Fig. 1A). As reported previously (Beaumont and Zucker, 2000), ZD 7288 alone had no effect on transmission. 8-CPT did not occlude responses to forskolin (to 216 ± 21% of amplitudes before forskolin) (Fig. 1B). However, forskolin responses were strongly reduced by ZD 7288 (to 134 ± 8.5% of controls; p < 0.03) (Fig. 1B), which again spared the enhancement by 8-CPT (open circles). This result is expected, because when 8-CPT is already activating Epac, forskolin acts only to activate HCN channels, so this enhancement should be blocked by ZD 7288. Forskolin depolarizes nerve terminals by ∼6 mV, resulting from activation of presynaptic HCN channels (Beaumont and Zucker, 2000). 8-CPT has no such effect (Fig. 2A). These results show an enhancement of synaptic transmission by 8-CPT independent of both HCN channel and PKA activation.
Figure 1.

The Epac agonist 8-CPT enhances synaptic transmission. A, 8-CPT (50 μm) induced a modest increase in EJP amplitude (filled circles; n = 12) that was not blocked by the HCN channel inhibitor ZD 7288 (30 μm; open circles; n = 8). In five of the latter experiments, the PKA inhibitor KT 5720 (1 μm) was also present. ZD 7288 alone had no effect on transmission (open triangles; n = 4). B, With continuous perfusion of 8-CPT, forskolin (30 μm) increased EJP amplitudes further, resulting from HCN activation (filled circles; n = 3), and ZD 7288 depressed this effect (open circles; n = 3). The dotted line marks baseline EJP amplitudes in this and all subsequent figures.
Figure 2.

LTG activates presynaptic HCN channels, but 8-CPT does not. A, Membrane potential (RMPaxon) recorded from a preterminal axon branch is unaffected by 8-CPT (50 μm; n = 3). B, LTG (50 μm) reversibly depolarized nerve terminals (n = 8). Superfusing ZD 7288 (30 μm) produced a 4-5 mV hyperpolarization resulting from block of the resting HCN activation (Beaumont and Zucker, 2000). ZD 7288 also blocked subsequent actions of LTG. C, HCN channels activated by hyperpolarizing current (500 msec) displayed a slowly relaxing afterdepolarization enhanced by LTG and blocked by ZD 7288. Sample recordings (C1) are shown for 40 nA currents: ZD 7288 reduced the afterdepolarization from 14 to 2.5 mV in the absence of LTG and from 22 to 6.9 mV in its presence. Dashed lines show the initial membrane potential (-74 mV); responses during the pulse were truncated. Plots (C2) indicate afterdepolarization amplitudes versus current magnitude in different solutions (n = 8 for each solution; the four sets of data are significantly different at p < 0.05 by ANOVA).
We used the drug lamotrigine (LTG) to directly activate HCN channels without elevating cAMP. LTG is a use-dependent blocker of some sodium channels (Kuo and Lu, 1997), but this drug activates HCN channels in pyramidal neurons without affecting sodium channels (Poolos et al., 2002). As in pyramidal neurons, 50 μm LTG reversibly depolarized motor neuron terminals by 4 mV (3.9 ± 0.32 mV), without affecting action potentials, and the depolarization was antagonized by ZD 7288 (Fig. 2B). This is similar to the presynaptic effect of forskolin, which depolarizes terminals by 6.3 ± 0.6 mV (Beaumont and Zucker, 2000). HCN channels that are activated by a hyperpolarizing current pulse evoke a slowly relaxing depolarizing afterpotential that is blocked by ZD 7288 (Beaumont and Zucker, 2000). LTG enhanced this afterdepolarization, and this effect also was opposed by ZD 7288 (Fig. 2C). We conclude that LTG can be used to activate HCN channels in this preparation, as in pyramidal neurons. The somewhat smaller activation of HCN channels by LTG (∼4 mV) than by forskolin (∼6 mV) suggests that LTG is a weaker HCN activator than cAMP.
LTG, like 8-CPT, induced a modest enhancement (to 133 ± 0.75%) in synaptic transmission. The enhancement was not fully reversed by a 2 hr rinse, which is surprising, because its effect on HCN channels did seem to reverse fully in this time. Unlike the case of 8-CPT, this enhancement was antagonized by ZD 7288 (99.9 ± 1.1% of control EJP amplitude; p < 0.0001) (Fig. 3A). After exposure to LTG (activating only HCN channels), forskolin still produced a large enhancement of transmission (to 248 ± 19% of the initial level or 199 ± 19% of the slightly increased responses in LTG just before adding forskolin) (Fig. 3B). We expected forskolin responses to include activation of HCN channels plus a recruitment of Epac activation; their reduction by ZD 7288 (p < 0.04) to a level (133 ± 13% of controls) resembling activation of Epac alone by 8-CPT (to 138 ± 6.0%) (Fig. 1 A) was consistent with this expectation. Therefore, LTG activates only a ZD 7288-sensitive HCN-related enhancement of transmission (Fig. 3A), without involving Epac, whereas 8-CPT activates only an Epac-dependent enhancement, without involving HCN channels, and thus is insensitive to ZD 7288 (Fig. 1A).
Figure 3.

The HCN channel agonist LTG activates the HNC-dependent portion of cAMP-dependent enhancement of synaptic transmission and LTG plus 8-CPT mimic forskolin action. A, LTG (50 μm) increased EJP amplitude modestly (filled circles; n = 5); this effect was blocked by ZD 7288 (open circles; n = 3). B, After LTG, forskolin enhanced EJP amplitudes by additional activation of HCN channels and by recruiting Epac activation (filled circles; n = 8); ZD 7288 reduced these responses to a level resembling Epac responses (open circles; n = 5). C, Concurrent HCN channel activation with LTG and Epac with 8-CPT produces a large EJP enhancement (filled circles; n = 8); ZD 7288 resulted in a response enhancement similar to the effect of Epac activation alone (open circles; n = 5). D, Forskolin occludes the enhancement by LTG and 8-CPT (n = 4). E, Sequential activation of HCN channels with LTG and Epac with 8-CPT (filled circles; n = 3) induced a smaller enhancement; ZD 7288 (open circles; n = 3) had no effect on the subsequent Epac responses.
Combined activation of Epac and HCN channels
Our hypothesis is that cAMP elevated by forskolin activates both HCN channels and Epac to evoke increases in transmission to 200-250% of control levels. Consistent with this hypothesis, simultaneous application of LTG and 8-CPT enhanced release to 198 ± 1.3% (Fig. 3C), mimicking the effect of forskolin (200-250% in different data sets) (see Fig. 5C,E). Moreover, forskolin completely occluded effects of 8-CPT and LTG when these were added to the bath 30 min after including forskolin (which remained present) (Fig. 3D), which is also consistent with forskolin acting on the same targets (Epac and HCN channels) as 8-CPT and LTG. ZD 7288 reduced (p < 0.0001) responses in 8-CPT plus LTG to only 137 ± 7.4% of controls, almost identical to the effect of 8-CPT activating Epac alone (138 ± 6.0%) (Fig. 1A). The sequential activation of HCN channels and Epac enhanced transmission to 168 ± 8.9% (Fig. 3E), close to the sum (171%) of the individual enhancements over baseline (100%) of 8-CPT (33%) and LTG (38%). As expected, ZD 7288 added later, just before the 8-CPT, did not alter the response to 8-CPT after exposure to LTG alone (Fig. 3E). The enhancement to sequential agonist addition (168 ± 8.9%) (Fig. 3E) is somewhat less (p < 0.05) than the enhancement to forskolin (200-250%) or to simultaneous application of 8-CPT and LTG (198 ± 1.3%) (Fig. 3C), raising the possibility of nonlinear or cooperative interactions between HCN and Epac pathways.
Figure 5.

BFA antagonizes Epac enhancement of transmission. A, The small G-protein antagonist BFA (100 μm) has no effect on synaptic transmission (n = 3). B, The enhancement of transmission by 50 μm 8-CPT (filled circles; from Fig. 1 A) was blocked by BFA (open circles; n = 5). C, The enhancement of transmission by 30 μm forskolin (filled circles; n = 9) was reduced to approximately one-half in BFA (open circles; n = 4). D, The enhancement by 50 μm LTG (filled circles; from Fig. 1C) was not blocked by BFA (open circles; n = 3). E, Concurrent application of BFA and 30 μm ZD 7288 (ZD) (open circles; n = 4) blocked fully the forskolin-induced enhancement of EJP amplitude (filled circles; n = 5).
We suspected Epac to be responsible for maintaining the enhancement of transmission when HCN channels are inhibited after exposure to forskolin (Beaumont et al., 2002) (see Fig. 6B). Confirming this hypothesis, replacing forskolin with 8-CPT sustained enhanced EJPs (to 202 ± 23% of baseline) until 8-CPT was washed out. Moreover, the maintained enhancement was not affected by blocking HCN channels (Fig. 4B).
Figure 6.
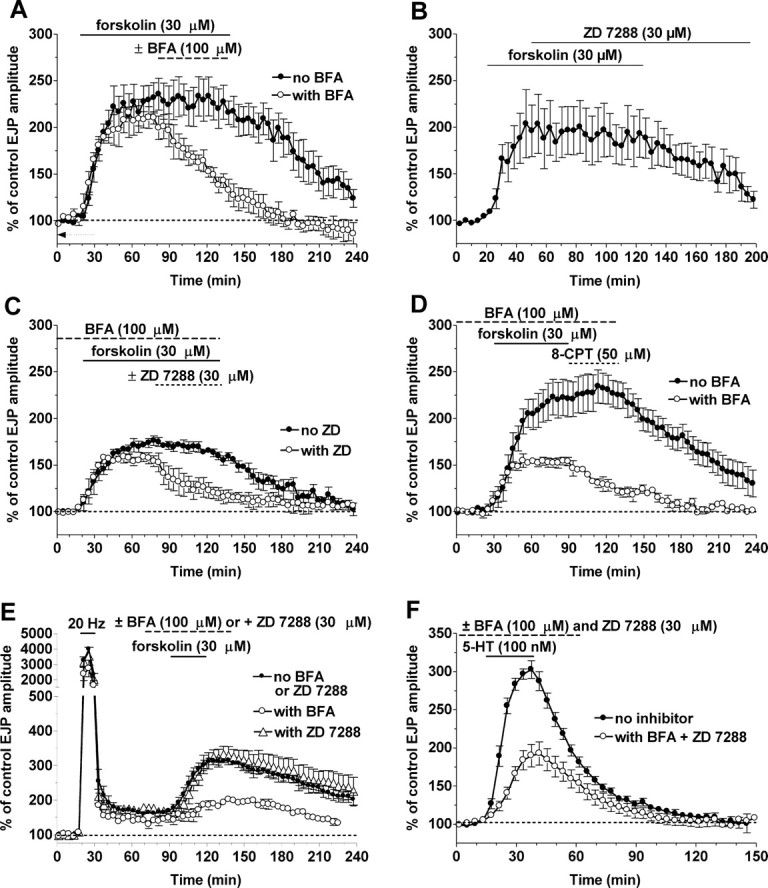
The role of Epac in the maintenance of enhanced transmission by cAMP and in synaptic tagging. A, The delayed addition of BFA antagonized the maintenance of synaptic enhancement by forskolin (n = 3). B, The delayed addition of ZD 7288 does not affect the maintained enhancement of transmission by forskolin, which only subsides when forskolin is removed (n = 4) (Beaumont et al., 2002). C, When forskolin enhancement is confined to HCN activation by BFA, the maintenance of this enhancement, which normally lasts as long as forskolin is present (filled circles; n = 4), is now sensitive to the delayed addition of ZD 7288 (ZD) (open circles; n = 3). D, The maintenance of forskolin enhancement by 8-CPT (filled circles) (from Fig. 3 A) was blocked by BFA (open circles; n = 3). E, The enhancement of EJP amplitude by forskolin at tagged synapses after LTF induction (filled circles; n = 3) was unaffected by ZD 7288 (open triangles; n = 5) but was inhibited by BFA (open circles; n = 3). F, The enhancement of transmission by 1 μm serotonin (5-HT; filled circles; n = 8) was reduced to approximately one-half by the combined block of HCN channel activation with ZD 7288 and Epac-dependent enhancement by BFA (open circles; n = 6).
Figure 4.

Epac activation substitutes for cAMP production in maintaining enhanced transmission, and Epac is the cAMP target at tagged synapses. A, 8-CPT (50 μm) maintained the enhancement of transmission produced by 30 μm forskolin (open circles; n = 6). B, The 8-CPT maintenance of enhancement was not blocked by delayed addition of 30 μm ZD 7288 (open circles; n = 6). The filled circles in A and B show responses to forskolin alone (n = 6). C, LTF, measured as an enhancement in EJP amplitude at least 20 min after the end of the tetanus, is induced by presynaptic stimulation at 20 Hz for 10 min in the absence of any drugs (filled green triangles; n = 9). LTF induced in the presence of Epac activation by 8-CPT was similar (filled red circles; n = 3). LTF in the presence of 8-CPT remained dependent on HCN activation and was blocked by ZD 7288 (open blue circles; n = 3). D, After LTF induction, synapses respond to forskolin (FSK) even when HCN channels are blocked, a process we call temporal synaptic tagging. 8-CPT strongly enhanced tagged synapses (filled red circles; n = 4), and this enhancement was insensitive to ZD 7288 (open blue circles; n = 4), just like responses to forskolin (open green triangles; n = 5).
The role of Epac in temporal synaptic tagging
HCN channels also play a critical role in the induction of LTF after sustained motor neuron activity (Beaumont et al., 2002). A curious property of LTF is that after its induction, forskolin produces an additional enhancement of synaptic transmission that is independent of HCN activation (Fig. 6E) [Beaumont et al. (2002), their Fig. 6B]. It is as if LTF induction leaves the synapses marked or temporally “tagged,” such that subsequent enhancement by cAMP involves only a second target different from the activation of HCN channels. We suspected Epac to be this target. We tested this hypothesis by recording responses to 8-CPT after LTF induction.
We first checked that 8-CPT did not occlude LTF induction by tetanic motor neuron stimulation (increasing EJPs to 180 ± 24% of controls in the presence of 8-CPT, compared with 169 ± 9.3% in the absence of 8-CPT; p > 0.6) (Fig. 4C). 8-CPT also left untouched the inhibition of LTF induction seen when HCN channels were blocked by ZD 7288 throughout the experiment (to 106.8 ± 1.8% of controls, compared with 111 ± 11% when ZD 7288 is present alone) (Beaumont et al., 2002) (data not shown). We then confirmed our prediction that after tagging of synapses by LTF induction, 8-CPT enhanced transmission (to 194 ± 28%) as much as forskolin (to 197 ± 8.7%; p > 0.9) (Fig. 4D). Because 8-CPT enhancement is attributable to Epac activation, it was unaffected by HCN channel block (increase to 191 ± 28% of controls; p > 0.9).
A new Epac antagonist
The evidence for a role for Epac in regulating synaptic transmission would be strengthened by the use of an Epac antagonist, but none has been reported thus far. A candidate is BFA, which inhibits vesicular traffic from the Golgi apparatus by slowing GDP-GTP exchange on ADP-ribosylation factors (ARF); the latter are targets of GEFs for ARF that are structurally related to Epac, although they are not activated by cAMP (Morinaga et al., 1996, 1999; Chardin and McCormick, 1999). We found that BFA also antagonizes 8-CPT action on synaptic transmission, presumably by similarly blocking a downstream action of Epac. First, we showed that BFA alone (100 μm) had no effect on synaptic transmission (Fig. 5A). Figure 5B shows that the enhancement of transmission (to 138 ± 6.0%) by 8-CPT was eliminated (107.3 ± 6.1% of controls; p < 0.03) by BFA. Lower doses of BFA had a smaller effect, with a half-maximal block at ∼10 μm (data not shown). Moreover, the enhancement of transmission by forskolin (to 220 ± 13%) was reduced to an increase to 165 ± 15% in BFA (p < 0.03) (Fig. 5C). We observed no effect of BFA against LTG responses (enhancement by LTG to 133 ± 0.75% of controls vs 134 ± 8.8% in BFA; p > 0.8) (Fig. 5D). This rules out any action of this drug on HCN channel-dependent enhancement of transmission and supports its specificity for Epac-mediated effects. Therefore, the remaining enhancement by forskolin in the presence of BFA should be attributable to HCN activation. Confirming this supposition, the combination of BFA and ZD 7288 nearly abolished (111 ± 12% of controls; p < 0.0003) the usual enhancement (to 225 ± 21%) in transmission produced by forskolin (Fig. 5E).
If the maintenance of synaptic enhancement by forskolin was dependent on Epac activation, we would expect it to be antagonized by BFA. Figure 6A confirms this prediction. Thus, the late addition of BFA (Fig. 6A), unlike the late addition of ZD 7288 (Fig. 6B), interfered with the maintenance of enhanced synaptic transmission. Normally, forskolin responses are maintained when ZD 7288 is added late (Fig. 6B) because of continued action of cAMP on Epac. Consistent with this expectation, responses to forskolin in the presence of BFA were no longer maintained after addition of ZD 7288 (Fig. 6C). Finally, the maintenance of enhancement by 8-CPT was blocked by BFA (Fig. 6D), just like the forskolin enhancement itself (Fig. 6A).
After the induction of LTF, synapses are temporally tagged such that they now respond to forskolin with an additional enhancement of EJP amplitude that is resistant to block by HCN channel inhibitors. We propose that these tagged responses to forskolin are attributable to the action of cAMP on Epac after HCN channels have already been activated (by LTF), much like the sequential activation of HCN channels by LTG followed by Epac activation by 8-CPT (Fig. 3E). If this hypothesis is correct, BFA should inhibit this HCN channel-independent tagged response to forskolin. Figure 6E shows this to be the case, where an enhancement to 189 ± 4.0% of the immediately preceding EJPs by forskolin after LTF induction was unaffected by the presence of ZD 7288 with the forskolin (196 ± 7.7% of controls) but was reduced (p < 0.03) to an increase to only 139 ± 14% when BFA was present with the forskolin, which is not significantly different from the enhancement (to 111 ± 12%) in BFA plus ZD 7288 (p ≈ 0.2). Together, our results implicate Epac as an important second target for cAMP in modulating transmission at these synapses.
The original impetus for these studies was the observation that a portion (approximately one-half) (Beaumont and Zucker, 2000) of the enhancement of transmitter release by serotonin is mediated by cAMP. If the cAMP-dependent portion of serotonin action is attributable to its combined activation of HCN channels and Epac, antagonists of these two pathways together should reduce serotonin responses by approximately one-half. This prediction was tested in the experiments of Figure 6 F, which confirmed that enhancement of transmission by 0.1 μm serotonin to 303 ± 11% was reduced to 192 ± 16% (p < 0.0001) by the combination of BFA and ZD 7288.
Discussion
Our results implicate Epac as a target, along with HCN channels, in modulating transmitter release at a fast glutamatergic synapse. Activation of these targets by cAMP mediates part of the enhancement of transmission by serotonin, and Epac is the target of cAMP in the activation of tagged synapses after LTF induction. The effects of HCN and Epac activation do not appear to be strictly additive (Fig. 3C, compare with Figs. 1A and 3A), suggesting the possibility of cooperative or otherwise nonlinear interactions between the two parallel pathways (Fig. 7A).
Figure 7.

Enhancement of synaptic transmission by cAMP. A, Schematic of pathways enhancing transmission to cAMP and actions of agonists and antagonists. B, Effects of agonists activating Epac and HCN separately and together: green, 8-CPT activating Epac; blue, LTG activating HCN channels; orange, 8-CPT and LTG applied together; red, forskolin (from Figs. 1 A, 3A, C, and 5E). C, Effects of antagonists of Epac and HCN separately and together on forskolin enhancement: green, ZD 7288 (ZD) blocking HCN channels and leaving Epac; blue, BFA blocking Epac and leaving HCN channels; black, ZD 7288 and BFA applied together; red, forskolin alone (from Fig. 5C, E) (Beaumont and Zucker, 2000).
These conclusions rely on pharmacological activators and inhibitors of Epac and HCN channels. The drugs used are not perfect; 8-CPT is a strong activator of Epac, even stronger than cAMP (Enserink et al., 2002; Christensen et al., 2003), but it also weakly activates PKA, albeit with much lower affinity. Because PKA has already been ruled out as a target for cAMP in modulating transmission at the synapses under study (Beaumont and Zucker, 2000), the imperfect specificity of 8-CPT is not an issue here. We have demonstrated that 8-CPT has no effect on HCN channels or their effect on transmission. We used LTG as an agonist of HCN channels, causing a presynaptic depolarization resembling that in forskolin, and when combined with 8-CPT, it enhances transmission nearly as much as forskolin. ZD 7288 is an antagonist of HCN activation and its effects, without action on Epac-dependent enhancement. Finally, we introduced BFA as a downstream inhibitor of Epac. We have shown it to be without effect on HCN-dependent enhancement, but it is likely to have effects on other guanine exchange factors besides Epac.
Despite this limitation, it is difficult to imagine any interpretation of our results that does not implicate Epac in the cAMP-dependent regulation of these synapses. Activators of Epac and HCN channels each modestly enhance transmission, and together they mimic the action of forskolin (Fig. 7B). Antagonists of Epac and HCN channels each reduce cAMP-dependent enhancement of transmission, and together they almost abolish the enhancement (Fig. 7C). Our introduction of BFA as an antagonist of Epac action may provide a useful tool for delineating other cellular functions for this molecule.
The exact mechanism of Epac action in our preparation is unclear, but other studies suggest two possibilities: Epac is an activator of the Ras-like GTPase Rap1 (de Rooij et al., 1998; Kawasaki et al., 1998; Enserink et al., 2002), but no role for Rap1 in secretion has been reported. The cAMP-dependent potentiation of insulin secretion in pancreatic β-cells, and of growth hormone in PC12 cells transfected with growth hormone and Epac2, is mediated by interactions of Epac2 with the Rab3-binding partner Rab3 interacting molecule 2 (Rim2) (Ozaki et al., 2000; Kashima et al., 2001). Rab3 is a small GTPase concentrated in synaptic vesicles, whereas Rim2 is closely related to Rim1, a plasma membrane protein at nerve terminals that can also bind to Epac (Ozaki et al., 2000); Rab3 and Rim1 have been implicated in vesicle trafficking and the late priming of vesicles, regulating their ability to release by an increase in [Ca2+]i (Geppert and Südhof, 1998; Koushika et al., 2001). The enhancement in transmitter release caused by serotonin (Wang and Zucker, 1998) and forskolin (V. Beaumont and R. S. Zucker, unpublished results) has also been shown to result from an increase in the size of the pool of releasable vesicles. Thus, we speculate that at crayfish nerve terminals, Epac exerts its influence on transmitter release by interacting with an isoform of Rim to regulate the availability of docked vesicles for release.
Epac probably regulates transmission at other synapses as well. cAMP modulates long-term potentiation (LTP) at hippocampal CA1 synapses by acting on a target other than PKA (Otmakhov and Lisman, 2002). Recent work (Morozov et al., 2003) showing a role for Rap1 in coupling cAMP signaling to a mitogen-activated protein kinase that regulates LTP raises the possibility that Epac may be the cAMP target in that pathway. Rim1 influences several forms of short-term plasticity (Castillo et al., 2002). Epac has recently been implicated in the regulation by γ-amino butyric acid of vesicle priming during short-term depression (Sakaba and Neher, 2003), and this effect might also involve Rim1. Rab3, Rim1, and cAMP have all been implicated in the presynaptic expression of LTP at mossy fiber synapses (Weisskopf et al., 1994; Castillo et al., 1997; Castillo et al., 2002), and Epac may be a cAMP target in that process as well.
Footnotes
This work was supported by grants from the National Science Foundation and the National Institutes of Health. We thank Dr. J. L. Bos for prepublication data on 8-CPT and a sample of the drug and Dr. Kevin Staras for valuable suggestions.
Correspondence should be addressed to Prof. Robert S. Zucker, Molecular and Cell Biology Department, University of California, 111 Life Sciences Addition, Berkeley, CA 94720-3200. E-mail: zucker@socrates.berkeley.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250208-07$15.00/0
References
- Beaumont V, Zucker RS (2000) Enhancement of synaptic transmission by cyclic AMP modulation of presynaptic Ih channels. Nat Neurosci 3: 133-141. [DOI] [PubMed] [Google Scholar]
- Beaumont V, Zhong N, Fletcher R, Froemke RC, Zucker RS (2001) Phosphorylation and local presynaptic protein synthesis in calcium- and calcineurin-dependent induction of crayfish long-term facilitation. Neuron 32: 489-501. [DOI] [PubMed] [Google Scholar]
- Beaumont V, Zhong N, Froemke RC, Ball RW, Zucker RS (2002) Temporal synaptic tagging by Ih activation and actin: involvement in long-term facilitation and cAMP-induced synaptic enhancement. Neuron 33: 601-613. [DOI] [PubMed] [Google Scholar]
- Capogna M, Gahwiler BH, Thompson SM (1995) Presynaptic enhancement of inhibitory synaptic transmission by protein kinases A and C in the rat hippocampus in vitro J Neurosci 15: 1249-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Janz R, Südhof TC, Tzounopoulos T, Malenka RC, Nicoll RA (1997) Rab3A is essential for mossy fibre long-term potentiation in the hippocampus. Nature 388: 590-593. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Südhof TC, Malenka RC (2002) RIM1α is required for presynaptic long-term potentiation. Nature 415: 327-330. [DOI] [PubMed] [Google Scholar]
- Chardin P, McCormick F (1999) Brefeldin A: the advantage of being un-competitive. Cell 97: 153-155. [DOI] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Stevens CF (1994) Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci 14: 310-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Regehr WG (1997) The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci 17: 8687-8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen AE, Selheim F, de Rooij J, Dremier S, Schwede F, Dao KK, Martinez A, Maenhaut C, Bos JL, Genieser HG, Doskeland SO (2003) cAMP analog mapping of Epac1 and cAMP-kinase. Discriminating analogs demonstrate that Epac and cAMP-kinase act synergistically to promote PC-12 cell neurite extension. J Biol Chem 278: 35394-35402. [DOI] [PubMed] [Google Scholar]
- Delaney K, Tank DW, Zucker RS (1991) Presynaptic calcium and serotonin-mediated enhancement of transmitter release at crayfish neuromuscular junction. J Neurosci 11: 2631-2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396: 474-477. [DOI] [PubMed] [Google Scholar]
- Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Doskeland SO, Blank JL, Bos JL (2002) A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol 4: 901-906. [DOI] [PubMed] [Google Scholar]
- Geppert M, Südhof TC (1998) Rab3 and synaptotagmin: the yin and yang of synaptic membrane fusion. Annu Rev Neurosci 21: 75-95. [DOI] [PubMed] [Google Scholar]
- Glusman S, Kravitz EA (1982) The action of serotonin on excitatory nerve terminals in lobster nerve-muscle preparations. J Physiol (Lond) 325: 223-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang G, Joseph JW, Chepurny OG, Monaco M, Wheeler MB, Bos JL, Schwede F, Genieser HG, Holz GG (2003) Epac-selective cAMP analog 8-pCPT-2′-O-Me-cAMP as a stimulus for Ca2+-induced Ca2+ release and exocytosis in pancreatic beta-cells. J Biol Chem 278: 8279-8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima Y, Miki T, Shibasaki T, Ozaki N, Miyazaki M, Yano H, Seino S (2001) Critical role of cAMP-GEFII-Rim2 complex in incretin-potentiated insulin secretion. J Biol Chem 276: 46046-46053. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM (1998) A family of cAMP-binding proteins that directly activate Rap1. Science 282: 2275-2279. [DOI] [PubMed] [Google Scholar]
- Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML (2001) A post-docking role for active zone protein Rim. Nat Neurosci 4: 997-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CC, Lu L (1997) Characterization of lamotrigine inhibition of Na+ channels in rat hippocampal neurones. Br J Pharmacol 121: 1231-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinaga N, Tsai SC, Moss J, Vaughan M (1996) Isolation of a brefeldin A-inhibited guanine nucleotide-exchange protein for ADP ribosylation factor (ARF) 1 and ARF3 that contains a Sec7-like domain. Proc Natl Acad Sci USA 93: 12856-12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinaga N, Adamik R, Moss J, Vaughan M (1999) Brefeldin A inhibited activity of the sec7 domain of p200, a mammalian guanine nucleotide-exchange protein for ADP-ribosylation factors. J Biol Chem 274: 17417-17423. [DOI] [PubMed] [Google Scholar]
- Morozov A, Muzzio IA, Bourtchouladze R, Van-Strien N, Lapidus K, Yin D, Winder DG, Adams JP, Sweatt JD, Kandel ER (2003) Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron 39: 309-325. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Lisman JE (2002) Postsynaptic application of a cAMP analogue reverses long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurophysiol 87: 3018-3032. [DOI] [PubMed] [Google Scholar]
- Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, Takai Y, Seino S (2000) cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol 2: 805-811. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Migliore M, Johnston D (2002) Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci 5: 767-774. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E (2003) Direct modulation of synaptic vesicle priming by GABAB receptor activation at a glutamatergic synapse. Nature 424: 775-778. [DOI] [PubMed] [Google Scholar]
- Trudeau LE, Emery DG, Haydon PG (1996) Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron 17: 789-797. [DOI] [PubMed] [Google Scholar]
- Trudeau LE, Fang Y, Haydon PG (1998) Modulation of an early step in the secretory machinery in hippocampal nerve terminals. Proc Natl Acad Sci USA 95: 7163-7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Zucker RS (1998) Regulation of synaptic vesicle recycling by calcium and serotonin. Neuron 21: 155-167. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA (1994) Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science 265: 1878-1882. [DOI] [PubMed] [Google Scholar]
- Zhong N, Zucker RS (2004) Roles of Ca2+, hyperpolarization and cyclic nucleotide-activated channel activation, and actin in temporal synaptic tagging. J Neurosci 24: 4205-4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Wu CF (1991) Altered synaptic plasticity in Drosophila memory mutants with a defective cyclic AMP cascade. Science 251: 198-201. [DOI] [PubMed] [Google Scholar]