

Abstract
Mitochondrial uncoupling proteins dissociate ATP synthesis from oxygen consumption in mitochondria and suppress free-radical production. We show that genetic manipulation of uncoupling protein-2 (UCP2) directly affects substantia nigra dopamine cell function. Overexpression of UCP2 increases mitochondrial uncoupling, whereas deletion of UCP2 reduces uncoupling in the substantia nigra-ventral tegmental area. Overexpression of UCP2 decreased reactive oxygen species (ROS) production, which was measured using dihydroethidium because it is specifically oxidized to fluorescent ethidium by the superoxide anion, whereas mice lacking UCP2 exhibited increased ROS relative to wild-type controls. Unbiased electron microscopic analysis revealed that the elevation of in situ mitochondrial ROS production in UCP2 knock-out mice was inversely correlated with mitochondria number in dopamine neurons. Lack of UCP2 increased the sensitivity of dopamine neurons to 1-methyl-4-phenyl-1,2,5,6 tetrahydropyridine (MPTP), whereas UCP2 overexpression decreased MPTP-induced nigral dopamine cell loss. The present results expose the critical importance of UCP2 in normal nigral dopamine cell metabolism and offer a novel therapeutic target, UCP2, for the prevention/treatment of Parkinson's disease.
Keywords: uncoupling protein-2, mitochondria, reactive oxygen species, substantia nigra, MPTP, Parkinson's disease
Introduction
Mitochondria are critical for energy metabolism and storage and for cellular survival. The inner membrane potential of the mitochondrion determines ATP and free-radical production, calcium transport, and the integrity and stability of various proteins including cytochrome c, adenine nucleotide translocator, voltage-dependent anion channel, and Bcl-2 family members, all of which play roles in determining cellular fate (Kroemer and Reed, 2000; Nicholls and Ward, 2000). Mitochondrial membrane potential is dependent on a proton gradient that exists between the inter-membrane space and the matrix. Proton transport through the inner membrane is mainly by ATP synthase; however, nonspecific protonophores also exist that contribute to the stability and modulation of mitochondrial membrane potential. Mitochondrial uncoupling proteins (UCP) belong to this family of mitochondrial regulators. Since the original description of UCP1 in brown adipose tissue (Bouillaud et al., 1985), other UCPs have been discovered, including UCP2, which is expressed in the CNS (Fleury et al., 1997). We demonstrated previously that UCP2 is induced in acute brain injury and suppresses caspase 3 activation, which suggested a role for this protein in neuroprotection (Bechmann et al., 2002). Subsequently, it has been demonstrated that UCP2 can be neuroprotective in various neurodegenerative processes including experimental induction of epilepsy (Diano et al., 2003; Sullivan et al., 2003) and stroke (Mattiasson et al., 2003). These studies also indicated that UCP2 is neuroprotective, because it diminishes free radicals (Diano et al., 2003; Mattiasson et al., 2003), decreases calcium influx to the mitochondrion (Mattiasson et al., 2003), increases the number of mitochondria, and elevates cellular ATP levels (Diano et al., 2003).
Parkinson's disease (PD) is characterized by massive dopamine (DA) cell loss in the mesencephalic substantia nigra pars compacta (SNc), the underlying cause of which is unknown, although it appears to be related to mitochondrial metabolism. Indeed, oral administration of coenzyme Q10 (CoQ10), a critical cofactor in mitochondrial metabolism and UCP2 activity (Echtay et al., 2000), reduces dopamine cell loss in both mouse (Beal et al., 1998) and primate models of PD (Horvath et al., 2003b) and may also slow the progression of the disease in human PD patients (Shults et al., 2002). We found that an intriguing feature of CoQ10 therapy in an animal model of PD is that it induces mitochondrial uncoupling in the substantia nigra, most likely via activation of UCP2 (Horvath et al., 2003b), which preceded the CoQ10 prevention of cell death afflicted by 1-methyl-4-phenyl-1,2,5,6 tetrahydropyridine (MPTP) (Horvath et al., 2003b). Thus, we hypothesized that increased mitochondrial uncoupling by UCP2 may be an important step in prevention of dopamine cell loss during PD and models of PD.
To date, no known selective activators or inhibitors of UCP2 exist. Thus, to determine the role of UCP2 in physiological or pathological processes, we used animals in which the expression of UCP2 had been genetically modified. We studied UCP2 knock-out (KO) (Zhang et al., 2001) and UCP2-overexpressing (Bechmann et al., 2002; Diano et al., 2003; Horvath et al., 2003a; Mattiasson et al., 2003) transgenic (TG) mice and their wild-type (WT) littermates.
Materials and Methods
All male mice used in the following experiments were between 12 and 15 weeks of age at the time they were killed. All procedures were approved by the Institutional Animal Care and Use Committee of Yale University. All mice were maintained under standard laboratory conditions with water and food available ad libitum; lights were maintained on a 12 hr light/dark cycle.
hUCP-expressing transgenic animals. Human UCP2-expressing transgenic lines were generated as described previously (Horvath et al., 2003a). For the present study, line 32 animals were used. Founder transgenic mice (produced by Chrysalis DNX, Princeton, NJ) were back-crossed onto the C57BL/6J background to minimize phenotype variability attributable to mouse strain. Transmission of the transgene follows a simple Mendelian pattern. Presence or absence of the transgene was confirmed by PCR amplification of a conserved region of hUCP2 exon 4 in genomic DNA (Diano et al., 2003). For the experiments described in this paper, we used heterozygous mice of N7 or higher backcross generation. Thus, the phenotypes reported are from mice that are predominantly derived from the C57BL/6J strain. Nontransgenic littermates were used as controls for all experiments.
UCP2 knock-out animals. Breeding pairs of UCP2 KO animals were generously provided by Dr. Bradford Lowell (Department of Medicine, Beth Israel-Deaconess Medical Center, Harvard Medical School, Boston, MA), whose laboratory generated this knock-out line (Zhang et al., 2001). For the generation of these mice, c129/SVJ genomic library was used, and thus, it is anticipated that animals generated by the construct would retain genetic traces from the SV-129 strain regardless of repeated backcrossing on C57BL/6J strain (Zhang et al., 2001). Heterozygous wild-type littermates of UCP2 KO animals were used as controls for all experiments.
Mitochondrial isolation and uncoupling activity. Mitochondria were pooled from the substantia nigra-ventral tegmental area (SN-VTA) (four animals = 1; total n = 4) and were isolated by differential centrifugation. Because of the size of the tissue dissected, it was impossible to isolate solely the SNc, and as such, we report our results as SN-VTA. Briefly, the SN-VTA was dissected rapidly and was homogenized in isolation buffer (215 mm mannitol, 75 mm sucrose, 0.1% fatty acid-free BSA, 20 mm HEPES, 1 mm EGTA, pH adjusted to 7.2 with KOH). The homogenate was spun at 1300 × g for 3 min, the supernatant was removed, and the pellet was resuspended with isolation buffer and spun again at 1300 × g for 3 min. The two sets of supernatant from each sample were topped of with isolation buffer and spun at 13,000 × g for 10 min. The supernatant was discarded, and the step was repeated. After this second spin at 13,000 × g, the supernatant was discarded, and the pellets were resuspended with isolation buffer without EGTA and spun at 10,000 × g for 10 min. The final mitochondrial pellet was resuspended with 50 μl of isolation buffer without EGTA. Protein concentrations were determined with a BCA protein assay kit (Pierce, Rockford, IL).
Mitochondrial respirations were assessed using a Clark-type oxygen electrode (Hansatech Instruments, Norfolk, UK) at 37°C with pyruvate and malate (5 and 2.5 mm) as oxidative substrates in respiration buffer (215 mm mannitol, 75 mm sucrose, 0.1% fatty acid-free BSA, 20 mm HEPES, 2 mm MgCl, 2.5 mm KH2PO4, pH adjusted to 7.2 with KOH). After the addition of ADP and oligomycin, UCP-mediated proton conductance was measured as increased fatty acid-induced respiration (Echtay et al., 2002), which was then compared with state 4 respiration induced by oligomycin, an inhibitor of H+-transporting ATP synthase.
UCP expression levels in the substantia nigra. Frozen substantia nigra tissue was thawed into Trizol. RNA and cDNA were prepared as described previously (Diano et al., 2003). Real-time PCR was performed as described previously (Horvath et al. 2003b) using primers for UCP2 (forward, 5′-CTACAAGACCATTGCACGAGAGG-3′; reverse, 5′-AGCTGCTCATAGGTGACAAACAT-3′), UCP4 (forward, 5′-GTGAAGGTCCAGATGCAAATG-3′; reverse, 5′-CATTCTCAGCCACGAGGG-3′), and brain mitochondrial carrier protein 1 (BMCP1) (forward, 5′-TGGGGTAGTGTCAGGAGTGATTTC-3′; reverse, 5′-AATGATGTTCCAGGGTCCAAGTC-3′). Specificity of amplification was confirmed by sequencing bands from test reactions. Amplification threshold values were measured, and endpoint reaction samples were run on 1% agarose gels in ethidium bromide to confirm the size and intensity of bands detected.
In situ detection of reactive oxygen species. Dihydroethidium (Molecular Probes, Eugene, OR) was used to investigate the local in situ production of reactive oxygen species (ROS), because it is specifically oxidized by superoxide ( ) to red fluorescent ethidium (Bindokas et al., 1996). An intravenous injection of 200 μl of dihydroedithium (DHE; stock solution, 100 mg/ml in DMSO, diluted 1:100 with sterile saline before injection) was administered through the femoral vein to anesthetized mice. Three hours after DHE injection, mice were overdosed with dimethyl ether and transcardially perfused with fixative [4% paraformaldehyde, 0.1% glutaraldehyde, and 15% picric acid in phosphate buffer (PB)]. Brains were removed and were postfixed overnight in fixative without glutaraldehyde. Serial 50 μm coronal sections were cut through the SN using a vibratome and were collected in microtiter plates wrapped in foil. To observe
) to red fluorescent ethidium (Bindokas et al., 1996). An intravenous injection of 200 μl of dihydroedithium (DHE; stock solution, 100 mg/ml in DMSO, diluted 1:100 with sterile saline before injection) was administered through the femoral vein to anesthetized mice. Three hours after DHE injection, mice were overdosed with dimethyl ether and transcardially perfused with fixative [4% paraformaldehyde, 0.1% glutaraldehyde, and 15% picric acid in phosphate buffer (PB)]. Brains were removed and were postfixed overnight in fixative without glutaraldehyde. Serial 50 μm coronal sections were cut through the SN using a vibratome and were collected in microtiter plates wrapped in foil. To observe 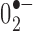 production in SNc dopaminergic cells, sections were incubated overnight with a monoclonal tyrosine hydroxylase (TH) antibody (1:1000). After a series of washes, sections were incubated with the fluorescent secondary antibody, donkey anti-mouse IgG 488 Alexafluore (1:200; Molecular Probes), to visualize TH-immunoreactive cells. After appropriate washing, sections were then incubated with Hoechst 33258 (100 ng/ml; Sigma, St. Louis, MO) as a nuclear stain in PB, washed, and mounted with vectashield. The localization and expression pattern of ethidium, TH, and Hoechst 33258 fluorescence was observed at high magnification (100×) with a fluorescent microscope, and images were captured using AxioVision 2.2 image software (Carl Zeiss, Thornwood, NY). Because the major source of
production in SNc dopaminergic cells, sections were incubated overnight with a monoclonal tyrosine hydroxylase (TH) antibody (1:1000). After a series of washes, sections were incubated with the fluorescent secondary antibody, donkey anti-mouse IgG 488 Alexafluore (1:200; Molecular Probes), to visualize TH-immunoreactive cells. After appropriate washing, sections were then incubated with Hoechst 33258 (100 ng/ml; Sigma, St. Louis, MO) as a nuclear stain in PB, washed, and mounted with vectashield. The localization and expression pattern of ethidium, TH, and Hoechst 33258 fluorescence was observed at high magnification (100×) with a fluorescent microscope, and images were captured using AxioVision 2.2 image software (Carl Zeiss, Thornwood, NY). Because the major source of 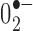 is located within the mitochondria (Bindokas et al., 1996), it was possible to directly count red fluorescent mitochondria after the oxidation of DHE to ethidium. Fluorescent mitochondria were counted blindly in neurons with visible nuclei in a minimum of five neurons per section in three sections per animal, and the mean number of fluorescent mitochondria per cell was calculated for each animal.
is located within the mitochondria (Bindokas et al., 1996), it was possible to directly count red fluorescent mitochondria after the oxidation of DHE to ethidium. Fluorescent mitochondria were counted blindly in neurons with visible nuclei in a minimum of five neurons per section in three sections per animal, and the mean number of fluorescent mitochondria per cell was calculated for each animal.
TH light and electron microscopic immunohistochemistry. Total dopamine cell number in the SNc of mice treated with MPTP or vehicle was assessed by light microscopic TH immunocytochemistry and unbiased stereology. All animals were heavily anesthetized and killed, and their brains were removed and postfixed in 4% paraformaldehyde and 15% picric acid in PB. Coronal sections (50 μm) were cut through the entire SNc using a vibratome and were washed thoroughly. Before incubation of primary antibody (TH; 1:5000 in 0.1% Triton PB at 4°C for 48 hr), sections were blocked with 1% H2O2, washed again, and then incubated with goat anti-mouse IgG to avoid cross reaction between the secondary antibody and endogenous mouse IgG. After primary antibody incubation, sections were washed thoroughly and incubated in the secondary antibody (biotinylated horse anti-mouse IgG; 1:250 in PB; Vector Laboratories, Burlingame, CA). Sections were then incubated for 2 hr at room temperature with avidinbiotin complex. The immunoreaction was visualized using 3,3′-diaminobenzidine (DAB) as the chromagen. Specificity of TH immunostaining was confirmed by omitting primary antibody incubation. TH was detected using a mouse monoclonal antibody (DiaSorin, Stillwater, MN) diluted at 1:10,000 in PBS containing 0.2% Triton X-100 (PBS-TX) and 0.1% sodium azide.
Endogenous peroxidase activity was quenched by incubation for 10 min with 3% hydrogen peroxide in PBS. After washing with PBS, sections were incubated for 24 hr at room temperature with TH antibody. After washing with PBS, sections were incubated for 1 hr in biotin-conjugated goat anti-mouse IgG diluted 1:500 in PBS-TX (Vector Laboratories). The sections were washed again and were incubated for 1 hr in a streptavidin-peroxidase complex (Sigma) diluted 1:2000 in PBS-TX. Peroxidase activity was visualized with 0.05% DAB (Sigma) and 0.002% H2O2, and staining was intensified with 0.8% nickel ammonium sulfate. The sections were mounted on gelatin-coated slides, air dried, dehydrated in xylene, and coverslipped with a mixture of distyrene, tricresyl phosphate, and xylene mounting medium.
For electron microscopic analysis of mitochondria, TH immunohistochemistry was performed as described above except for the addition of a freeze-thaw step to disrupt cellular membranes before the incubation with the primary antibody (antibody diluent minus Triton X-100). Mitochondria were counted blindly from randomly selected sections, and Scion (Frederick, MD) Image was used to normalize cytoplasmic area so that mitochondrial number per cell are expressed as square micrometers.
Stereology. The central feature of unbiased stereology is the use of a systematic random sampling method, which meets the statistical requirements necessary to ensure an unbiased estimate of the feature of interest. In selecting a sample series, the first section to be analyzed was selected randomly from an initial interval of sections, which then defines the spacing of the remaining sections to be examined. Thus, when we chose to sample every 10th section to measure TH cell number, the starting point of the series for each animal was randomly selected from the first 10 sections, and then every 10th section was taken from this starting point through the remainder of the series. A grid area was chosen from within each section in a similar systematic random manner. These areas (typically 100 μm2) were then used to count neurons. Within each area, an optical section was established between the surfaces of the tissue section, thus creating a three-dimensional sampling area of known dimensions. All cells with nuclei entering the focal plane within this area were counted, provided they did not cross three of the borders of the sampling boxes considered as exclusionary borders. However, cells crossing the other three inclusion borders were included in the counts. The counts obtained from these sampling boxes were determined and represent the number of cells per unit volume of the structure of interest.
MPTP treatment. MPTP in PBS was administered using an acute dosing regimen of 15 mg/kg intraperitoneally every 2 hr for four doses. Control animals were treated with a volume of PBS equal to the injection volume in the MPTP-treated animals. All animals were killed 7 d after the MPTP or PBS treatment. Experimental groups included MPTP-treated hUCP2-expressing transgenic mice (n = 6), their wild-type littermates (n = 6), UCP2 KO animals (n = 6), and their wild-type littermates (n = 6). Control animals included PBS-treated hUCP2-expressing transgenic mice (n = 6), their wild-type littermates (n = 6), UCP2 KO animals (n = 6), and their wild-type littermates (n = 6). All animals were males.
Striatal dopamine levels. At the time the mice were killed (7 d after MPTP treatment), both striata were rapidly dissected on a chilled glass plate and were frozen at -70°C. The samples were subsequently thawed in 0.4 ml of chilled 0.1 m perchloric acid and sonicated. Aliquots were taken for protein quantification using a spectrophotometric assay. Other aliquots were centrifuged, and dopamine levels were measured in supernatants by HPLC with electrochemical detection. Concentrations of dopamine and metabolites were expressed as nanograms per milligram of protein (mean ± SEM).
Striatal 1-methyl-4-phenylpyridinium levels. Assessment of 1-methyl-4-phenylpyridinium (MPP+) levels in the various genotypes was done according to previously published protocols (Klivenyi et al., 2004). MPTP (20 mg/kg) was administered intraperitoneally to male UCP2 knock-out mice (n = 3), UCP2-overexpressing mice (n = 3), and their respective wild-type littermates (n = 3 each). Mice were killed 90 min after the injection. MPP+ levels were quantified by HPLC with UV detection at 295 nm. Samples were sonicated in 0.1 m perchloric acid, and an aliquot of supernatant was injected on to a Brownlee aquapore X 03-224 cation exchange column (Rainin, Woburn, MA). Samples were eluted isocratically with 90% 0.1 mm acetic acid and 75 mm triethylamine HCl, pH 2.3, adjusted with formic acid and 10% acetonitrile. Protein assay (Bio-Rad Laboratories, Hercules, CA) measurements were conducted at 595 nm on a spectrophotometric microtiter plate reader (Molecular Devices, Menlo Park, CA).
Statistical analysis. All data are presented as mean ± SEM. Statistical comparisons were made using one-way ANOVA followed by Newman-Keuls post hoc tests. p < 0.05 was considered significant.
Results
UCP2 increases mitochondrial uncoupling activity
UCP2 is a highly active H+ transporter in the presence of CoQ10 and free fatty acids (Echtay et al., 2000, 2001). Whereas CoQ10, the electron acceptor for complexes I and II of the electron transfer chain, is constitutively expressed in mitochondria, endogenous free fatty acids are sequestered by BSA (Echtay et al., 2002). Thus, isolated mitochondria were incubated with palmitic acid (300 μm), and the increase in respiration was expressed relative to state 4 respiration induced by the ATP synthase inhibitor oligomycin to give an index of uncoupling activity. UCP2 TG mice showed a significant 462 ± 21% increase in uncoupling activity in the SN-VTA with respect to UCP2 WT controls (282 ± 19%; p < 0.001) (Fig. 1). Conversely, UCP2 KO mice exhibited a significantly suppressed level of uncoupling activity when compared with WT controls (169 ± 9 vs 244 ± 11%; p = 0.0016) (Fig. 1).
Figure 1.

Fatty acid-induced uncoupling in UCP2 WT/TG and WT/KO mice and UCP expressions. A, Exposure to the free fatty acid palmitate induces increased mitochondrial uncoupling in UCP2 TG mice when compared with WT controls. Asterisk indicates p < 0.001; n = 4. UCP2 KO mice show reduced mitochondrial uncoupling compared with WT controls. Asterisk indicates p = 0.002; n = 4. Data are expressed as the percentage of increase above oligomycin-induced state 4 respiration. Error bars represent SEM. B, Real-time PCR analysis of the SN revealed expressions of both UCP4 and BMCP1 mRNAs without significant differences between their expression levels in UCP2 KO, hUCP2-expressing mice an their respective wild-type littermates.
UCP expression levels in the substantia nigra
To determine whether genetic downregulation or upregulation of the UCP2 gene affects the expression of other brain-associated mitochondrial uncoupling proteins, we assessed UCP4 and BMCP1 transcript levels in the substantia nigra of UCP2 knockout, UCP2-overexpressing transgenic animals, and in their respective wild-type littermates. As expected, knock-out mice had no detectable UCP2 message. Transgenic overexpressing mice showed on average one cycle less to reach threshold, suggesting approximately twofold more UCP2 mRNA. Real-time PCR analysis of samples revealed the presence of both UCP4 and BMCP1 transcripts in the substantia nigra (Fig. 1B); UCP4 mRNA was more robustly expressed than BMCP1. The levels of these transcripts did not differ between the various strains and genotypes (Fig. 1B)
UCP2 decreases ROS production in vivo
In situ ROS production in SNc TH cells was measured using DHE, because it is specifically oxidized to ethidium by superoxide. Microscopic analysis revealed small red fluorescent staining strictly located to the cytosol of each cell. The fine punctate red fluorescent staining was presumed mitochondria specific, because production of superoxide is a byproduct of mitochondrial respiration (Bindokas et al., 1996) and therefore allowed quantitative analysis in animals studied.
Three hours after DHE injection, the number of ethidium-fluorescent mitochondria in TH cells was significantly (p < 0.05) lower in UCP2 TG mice compared with their WT controls (6.22 ± 1.27 vs 13.93 ± 2.51 ethidium-fluorescent mitochondria per TH cell) (Fig. 2). In UCP2 KO mice, there was a significant increase (p < 0.05) in the number of ethidium-fluorescent mitochondria in TH cells compared with WT controls (16.47 ± 1.55 vs 10.08 ± 0.79 ethidium-fluorescent mitochondria per TH cell) (Fig. 3).
Figure 2.

Free-radical production in TH neurons of the SNc in UCP2 WT/TG mice. A, B, Representative photomicrographs of in situ ROS production assessed by the oxidation of DHE to ethidium by 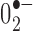 . Left photomicrographs show a representation of double exposure (A) for ethidium (red) and Hoechst nuclear staining (blue) or triple exposure (B) for ethidium, Hoechst nuclear staining, and TH immunofluorescence (green) in UCP2 WT mice. C, D, Representative findings of ethidium and Hoechst nuclear staining (C) with TH immunofluorescence (D) in UCP2 TG mice. Scale bar: (in C) A-D, 10 μm. E, Quantification of the number of red fluorescent mitochondria within TH cells revealed a significant decrease in UCP2-overexpressing animals compared with their WT controls (n = 4, n = 3, respectively). *Significant with respect to WT controls (p < 0.05). Error bars represent SEM.
. Left photomicrographs show a representation of double exposure (A) for ethidium (red) and Hoechst nuclear staining (blue) or triple exposure (B) for ethidium, Hoechst nuclear staining, and TH immunofluorescence (green) in UCP2 WT mice. C, D, Representative findings of ethidium and Hoechst nuclear staining (C) with TH immunofluorescence (D) in UCP2 TG mice. Scale bar: (in C) A-D, 10 μm. E, Quantification of the number of red fluorescent mitochondria within TH cells revealed a significant decrease in UCP2-overexpressing animals compared with their WT controls (n = 4, n = 3, respectively). *Significant with respect to WT controls (p < 0.05). Error bars represent SEM.
Figure 3.

Free-radical production in TH neurons of the SNc in UCP2 WT/KO mice. A, B, Representative photomicrographs of in situ ROS production assessed by the oxidation of DHE to ethidium by 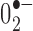 . Left photomicrographs show a representation of double exposure (A) for ethidium (red) and Hoechst nuclear staining (blue) or triple exposure (B) for ethidium, Hoechst nuclear staining, and TH immunofluorescence (green) in UCP2 WT mice. C, D, In UCP2 KO mice (right photomicrographs), representative findings for ethidium and Hoechst nuclear staining (C) with TH immunofluorescence (D). Scale bar: (in C) A-D, 10 μm. E, Quantification of the number of red fluorescent mitochondria within TH cells revealed a significant increase in UCP2 KO mice compared with their WT controls (n = 6, n = 5, respectively). *Significant with respect to WT controls (p < 0.05). Error bars represent SEM.
. Left photomicrographs show a representation of double exposure (A) for ethidium (red) and Hoechst nuclear staining (blue) or triple exposure (B) for ethidium, Hoechst nuclear staining, and TH immunofluorescence (green) in UCP2 WT mice. C, D, In UCP2 KO mice (right photomicrographs), representative findings for ethidium and Hoechst nuclear staining (C) with TH immunofluorescence (D). Scale bar: (in C) A-D, 10 μm. E, Quantification of the number of red fluorescent mitochondria within TH cells revealed a significant increase in UCP2 KO mice compared with their WT controls (n = 6, n = 5, respectively). *Significant with respect to WT controls (p < 0.05). Error bars represent SEM.
Moreover, it was apparent at high magnification that the intensity of ethidium fluorescence was decreased in UCP2 TG and increased in UCP2 KO animals compared with respective WT controls (Figs. 2, 3).
UCP2 KO mice have reduced mitochondria number
Unbiased electron microscopic analysis of SNc dopaminergic neurons revealed that UCP2 KO mice (n = 14) have a significant reduction mitochondrial number per square micrometer compared with WT controls (n = 27; 0.348 ± 0.039 vs 0.457 ± 0.042; p < 0.05). There was no difference in mitochondrial number between UCP2 TG mice (n = 18) and their wild-type controls (n = 15; 0.448 ± 0.052 vs 0.440 ± 0.029) (Fig. 4).
Figure 4.

UCP2 KO mice have reduced mitochondria in SNc TH cells. A, Representative electron micrograph shows numerous mitochondria in UCP2 WT mice in the cytoplasm of immunoreactive TH cells examined with electron microscopy. B, Representative electron micrographs of UCP2 KO mice illustrate reduced mitochondrial number in the cytoplasm of immunoreactive TH cells examined with electron microscopy. Asterisks in A and B indicate mitochondria. Scale bar: (in B) A, B, 1 μm. C, Quantification of the number of mitochondria in UCP2 WT/TG and UCP2 WT/KO mice. Data are normalized to cytoplasmic area and expressed as mitochondrial number per square micrometer. UCP2 KO mice (n = 14) had reduced mitochondria within TH neurons compared with WT controls (n = 27; 0.460 ± 0.042 vs 0.318 ± 0.028; p < 0.05). There was no difference between UCP2 TG mice (n = 18) and their WT controls (n = 15; 0.448 ± 0.052 vs 0.440 ± 0.029, respectively). *Significant differences in mitochondria counts between UCP2 KO and wild-type animals. Error bars represent SEM.
UCP2 restricts MPTP-induced cell loss and striatal dopamine levels
There was no difference between the volume (data not shown) or TH cell counts in the SNc of UCP2 TG mice and their WT littermates before MPTP treatment (8875.64 ± 850.25 vs 8733.13 ± 837.86 TH cells per SNc) (Fig. 5). In contrast, MPTP treatment resulted in significantly (p < 0.05) less dopamine cell loss (∼36%) in UCP2 TG mice (5595.11 ± 511.44 vs 8875.64 ± 850.25 TH cells per SNc) than in their WT littermates (∼62%; 3342.22 ± 132.88 vs 8733.13 ± 837.86 TH cells per SNc) (Fig. 5). On the other hand, MPTP-induced loss of striatal dopamine levels was equal between transgenic and their wild-type animals (Fig. 5). Vehicle-treated hUCP2-expressing transgenic animals had 94.4 ± 5.54 ng DA/mg protein in the striatum, which was reduced by ∼74% to 24.9 ± 1.58 ng DA/mg protein in MPTP-treated hUCP2-expressing animals. In vehicle-treated wild-type animals, striatal DA levels were 96.2 ± 1.8 ng DA/mg protein, which was reduced by MPTP treatment to 25.5 ± 3.07 ng DA/mg protein, representing an approximate 73% loss of dopamine levels. Neither vehicle-treated nor the MPTP-treated DA values differed between transgenic and wild-type animals (p > 0.05)
Figure 5.

MPTP-induced TH cell loss in UCP2 TG and WT mice. A-D, Dopamine cell number assessed by tyrosine hydroxylase immunocytochemistry showed decreased number of cells in MPTP-treated WT and UCP2 TG mice (n = 5, 6). Scale bar: (in B) A-D, 50 μm. E, Stereological quantification of dopamine cell number revealed that UCP2 TG mice had significantly (p < 0.05) more surviving neurons than their WT littermates after MPTP treatment. *Significant with respect to wild-type controls. F, Analysis of striatal dopamine levels showed equally diminished dopamine levels after MPTP injections in transgenic and wild-type animals. Error bars represent SEM.
In UCP2 KO mice and their WT littermates, similar to the TG mice, there was no difference between the volume (data not shown) or TH cell number of the SNc before MPTP treatment (9847.53 ± 403.74 vs 9664.45 ± 347.92 TH cells per SNc) (Fig. 6). In UCP2 KO mice, ∼62% of TH cells were lost after MPTP treatment in the SNc (3830.32 ± 578.15 vs 9847.53 ± 403.74 TH cells per SNc) (Fig. 6). This cell loss was significantly higher (p < 0.05), approximately double, than that observed in WT littermates (∼32%; 6509.83 ± 690.43 vs 9664.45 ± 347.92 TH cells per SNc) (Fig. 6). In this case, MPTP-induced loss of striatal dopamine levels was significantly different between KO and wild-type animals (Fig. 6). Vehicle-treated UCP2 KO animals had 118.2 ± 3.46 ng DA/mg protein in the striatum, which was reduced by ∼73% to 31.3 ± 6.17 ng DA/mg protein in MPTP-treated UCP2 KO animals. In vehicle-treated wild-type animals, striatal DA levels were 110.2 ± 4.56 ng DA/mg protein, which was reduced by MPTP treatment to 57.5 ± 11.84 ng DA/mg protein, representing an approximate 48% loss of dopamine levels. Whereas vehicle-treated DA values did not differ statistically between UCP2KO and wild-type animals, the DA values after MPTP treatment were significantly lower in UCP2 KO animals compared with those of wild-type littermates (31.3 ± 6.17 vs 57.5 ± 11.84 ng DA/mg protein; p < 0.05).
Figure 6.

MPTP-induced dopamine cell loss in UCP2 KO and WT mice. A-D, Dopamine cell number assessed by tyrosine hydroxylase immunocytochemistry showed decreased number of cells in MPTP-treated WT and UCP2 KO mice (n = 5, 6). Scale bar: (in B) A-D, 50 μm. E, Stereological quantification revealed that UCP2 KO mice had significantly (p < 0.05) less surviving dopamine neurons than their WT littermates after MPTP treatment. *Significant with respect to WT controls. F, Analysis of striatal dopamine levels showed diminished dopamine levels after MPTP injections in both wild-type and UCP2 KO animals. *Significant differences in striatal dopamine levels between UCP2 KO and wild-type animals. However, in MPTP-treated UCP2 KO animals, there was significantly (p < 0.05) lower levels of dopamine remaining in the striatum compared with the values of wild-type animals. Error bars represent SEM.
Striatal MPP+ levels
There was no difference in striatal MPP+ levels between the hUCP2-expressing transgenic and wild-type animals after peripheral MPTP administrations (5.9 ± 1.4 vs 5.56 ± 0.85 ng MPP+/mg wet tissue; p > 0.05). Similarly, there was no difference in striatal MPP+ levels between the UCP2 KO and wild-type animals after peripheral MPTP administrations (3.02 ± 0.38 vs 2.97 ± 0.41 ng MPP+/mg wet tissue; p > 0.05). It is of significance to note that when the values from the strains of transgenic and KO animals are compared, the MPP+ values are significantly higher in the hUCP2 transgenic strain compared with the KO strain (5.69 ± 0.65 vs 2.99 ± 0.25 ng MPP+/mg wet tissue; p < 0.05).
Discussion
Mitochondrial uncoupling in the substantia nigra
The premise of the present results is that mitochondrial UCP2 protects SNc dopaminergic neurons from neurotoxic insults. Although initial reports questioned the ability of UCP2 to induce functional uncoupling (Jezek, 2002), the fact that uncoupling activity in UCP2 TG and KO mice deviates from the their respective controls in opposite directions indicates that UCP2 plays a functional and physiologically important role in mitochondrial uncoupling activity in the SN-VTA. Indeed, this is supported by recent evidence showing functional UCP2-induced uncoupling activity (Echtay et al., 2001; Diano et al., 2003). WT mice from both mice strains also show increased uncoupling activity compared with oligomycin-induced state 4 respiration, because they possess genetically “normal” levels of UCP2 expression. Interestingly, UCP2 KO mice also display some uncoupling activity. This uncoupling activity may be attributed to BMCP1 or UCP4, which are predominantly expressed in the CNS and display uncoupling activity (Sanchis et al., 1998; Mao et al., 1999). Indeed, we detected transcripts for both of these putative mitochondrial uncouplers in the SN of both the transgenic and knock-out strains. Their levels, however, were not affected either by UCP2 down (UCP2 KO animals) or upregulation (hUCP2-expressing animals). Nevertheless, it maybe suggested that if BMCP1 and UCP4 are functional uncouplers in the SN, they may also contribute to neuroprotective processes.
UCP2 and ROS production in nigral dopamine cells
The effect of UCP2 on in situ ROS production in SNc dopaminergic neurons was inversely correlated to UCP2 levels, demonstrating that the lack of UCP2 increases, whereas UCP2 overexpression decreases, in situ ROS production. Increased ROS production was observed in macrophages of UCP KO mice exposed to infection (Arsenijevic et al., 2000). Similarly, UCP2 deficiency promotes ROS production and delays liver regeneration in mice (Horimoto et al., 2004). Interestingly, the ability of UCP2 to regulate ROS production in different cell types and tissues, as evident above, upholds the hypothesis that the function of UCP2 is associated more with ROS and oxidative stress reduction than with thermogenesis (Echtay et al., 2002).
The altered generation of ROS in SNc dopaminergic neurons of UCP2 KO or UCP2 TG mice is probably related to the mitochondrial membrane potential. This assumption is based first on the ability of UCP2 to directly regulate mitochondrial membrane potential through H+ transportation (Echtay et al., 2001) and second on the fact that mitochondrial ROS production is intimately associated with the mitochondrial membrane potential such that an increase in mitochondrial membrane potential promotes ROS production (Liu et al., 2002; Starkov and Fiskum, 2003). Previous studies have shown that increased mitochondrial uncoupling via UCP2 decreases ROS or oxidative stress and is neuroprotective in response to pharmacological and physical insults (Diano et al., 2003; Horvath et al., 2003b; Mattiasson et al., 2003; Sullivan et al., 2003). Although these observations help to underscore the importance of UCP2 in neuroprotection after injury/insult, our results are novel and intriguing because they imply that UCP2 has the intrinsic ability to buffer in vivo ROS production, at least in SNc dopaminergic neurons, in the absence of cellular stress.
UCP2 and mitochondria number in nigral dopamine cells
It was important to determine that increased or decreased in vivo ROS production in SNc dopaminergic neurons in UCP2 KO and TG mice, respectively, was not simply a product of increased or decreased absolute mitochondrial number. Quantification of mitochondrial number in the SNc revealed that UCP2 KO mice had significantly lower number of mitochondria compared with WT controls. Given that increased ROS initiates apoptosis (Pedersen, 1999), it seems likely that the decrease in mitochondrial number is a product of increased ROS in UCP2 KO mice. UCP2 TG mice, however, showed no significant difference relative to WT controls. We have shown previously that UCP2 TG mice have increased mitochondria in the hippocampus (Diano et al., 2003). The lack of difference between UCP2 TG and WT controls may reflect regional differences in mitochondria biogenesis. Thus, it appears that although increased UCP2 in TG mice does not immediately heighten normal cell function, it has the intrinsic ability to buffer cell metabolism after toxic insults, such as MPTP treatment, as shown in the present study. Conversely, there appears to be a threshold level of UCP2, below which normal cell metabolism is compromised.
Nigral UCP2 and MPTP-induced dopamine loss
MPTP-induced toxicity produced a reduction in SNc TH neuronal number in mice lacking or overexpressing UCP2 and their respective WT controls. Nevertheless, because the direction and extent of TH cell loss in UCP2 KO and UCP2 TG animals deviated from their respective WT littermates in the opposite direction, it is reasonable to conclude that the lack of UCP2 in the SNc decreases, whereas the increased availability of UCP2 increases, the survivability of TH neurons during MPTP exposure. Strain differences between the UCP2 KO (Zhang et al., 2001) and UCP2 TG mice (Horvath et al., 2003a) do not allow us to directly compare the results collected from the UCP2 KO and TG mice. Additionally, the different level of cell loss observed in wild-type mice of the two strains further confirms the existence of strain differences in susceptibility to MPTP in mice (Dauer and Przedborski, 2003).
The increased vulnerability of SNc dopamine cells to MPTP in wild-type mice of the transgenic strain compared with the wild-type mice the KO strain may be explained by the significantly lower level of MPP+ accumulation in the striatum of the KO strain animals after peripheral MPTP administration. There was an approximately twofold increase of MPP+ in the striatum of the transgenic strain relative to the knock-out strain. This may also underlie no observable difference in striatal dopamine levels in hUCP2-expressing transgenic mice and wild-type littermates after MPTP treatment, although there was significantly lower striatal dopamine levels measured in MPTP-treated UCP2 KO animals compared with their wild-type littermates. At low exposure levels to MPP+, the elimination of UCP2 reduces striatal dopamine levels (KO strain); however, double exposure of MPP+ is not sufficiently compensated by the elevated uncoupling (in hUCP2 transgenic mice), leading to equally compromised axonal function in the wild-type and transgenic mice. Nevertheless, attributable likely to the effect of hUCP2 on ROS production in the SN, the retrograde degeneration induced by injured striatal dopamine axons resulted in lower loss of dopamine cells in hUCP2-expressing transgenic mice compared with that observed in wild-type littermates.
The notion that UCP2 provides protection against SNc dopaminergic neuro-degeneration is supported by anatomical evidence illustrating UCP2/TH coexpression in the substantia nigra (Horvath et al., 2003b). Moreover, CoQ10, an obligatory cofactor required for the activation of UCPs (Echtay et al., 2000, 2001), induces mitochondrial uncoupling and results in decreased MPTP-induced toxicity in non-human primates (Horvath et al., 2003b), mice (Beal et al., 1998), and may even slow the progression of human PD (Shults et al., 2002). CoQ10 also reduced ROS production in cells undergoing biochemically induced apoptosis (Alleva et al., 2001), thus providing a plausible mechanistic link between CoQ10-slowing human PD and ROS-induced cell death in dopamine neurons, such as that seen after MPTP intoxication (Cleeter et al., 1992).
MPTP arrests electron flow through complex 1 leading to cell death, and MPTP stimulates the production of ROS, especially superoxide (Cleeter et al., 1992). Although the nature of ROS was not identified in our studies, superoxide is the primary reactant in the formation of all ROS and reactive nitrogen species, which are also implicated in the etiology of PD (Kuhn et al., 2004). The importance of MPTP-induced ROS production on dopamine neurotoxicity in vivo is underscored in transgenic mice overexpressing copper/zinc superoxide dismutase, a key ROS scavenging enzyme (Przedborski et al., 1992).
X-chromosome-linked inhibitor of caspases, including caspase 3, prevents SNc dopamine cell death after the administration of MPTP (Eberhardt et al., 2000). UCP2 inhibited caspase-3 activation in acute brain injury (Bechmann et al., 2002) and during oxygen and glucose deprivation (Mattiasson et al., 2003). Because caspase-3 activation is downstream of ROS production in apoptotic cell death, the increased neuroprotection observed in UCP2 TG mice exposed to MPTP may be a direct result of decreased ROS. Conversely, UCP2 KO mice are more susceptible to MPTP neurotoxicity than their WT littermates because of increase ROS production, which leads caspase-3 activation and decreased mitochondrial number ultimately leading to ATP depletion and increased cell death (Pedersen, 1999).
In summary, our data reveal that UCP2 contributes to the regulation of cellular metabolism in SNc dopamine neurons, including mitochondrial uncoupling activity, in situ ROS production, and mitochondrial number, implying that it is an essential homeostatic protein regulating cell survival and vulnerability to harmful toxins. This assertion is strengthened by evidence that UCP2 protects against dopamine cell loss caused by the mitochondrial complex 1 toxin MPTP. Given that degeneration of SNc dopaminergic neurons in human PD is associated with mitochondrial complex 1 dysfunction and free-radical toxicity (Dauer and Przedborski, 2003), we provide evidence that the pharmacological targeting of UCP2 activity is a novel therapeutic avenue for the prevention and treatment of PD in humans.
Footnotes
This work was supported by National Institutes of Health Grant R01 NS-41725. Breeding pairs of UCP2 knock-out animals were generously provided by Dr. Bradford Lowell.
Correspondence should be addressed to Tamas L. Horvath, Department of Obstetrics, Gynecology, and Reproductive Sciences, Yale University School of Medicine, 333 Cedar Street FMB 339, New Haven, CT 06520. E-mail: tamas.horvath@yale.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250184-08$15.00/0
References
- Alleva R, Tomasetti M, Andera L, Gellert N, Borghi B, Weber C, Murphy MP, Neuzil J (2001) Coenzyme Q blocks biochemical but not receptor-mediated apoptosis by increasing mitochondrial antioxidant protection. FEBS Lett 503: 46-50. [DOI] [PubMed] [Google Scholar]
- Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, Bouillaud F, Richard D, Collins S, Ricquier D (2000) Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet 26: 435-439. [DOI] [PubMed] [Google Scholar]
- Beal MF, Matthews RT, Tieleman A, Shults CW (1998) Coenzyme Q10 attenuates the 1-methyl-4-phenyl-1,2,3, tetrahydropyridine (MPTP) induced loss of striatal dopamine and dopaminergic axons in aged mice. Brain Res 783: 109-114. [DOI] [PubMed] [Google Scholar]
- Bechmann I, Diano S, Warden CH, Bartfai T, Nitsch R, Horvath TL (2002) Brain mitochondrial uncoupling protein 2 (UCP2): a protective stress signal in neuronal injury. Biochem Pharmacol 64: 363-367. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Jordan J, Lee CC, Miller RJ (1996) Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci 16: 1324-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillaud F, Ricquier D, Thibault J, Weissenbach J (1985) Molecular approach to thermogenesis in brown adipose tissue: cDNA cloning of the mitochondrial uncoupling protein. Proc Natl Acad Sci USA 82: 445-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleeter MW, Cooper JM, Schapira AH (1992) Irreversible inhibition of mitochondrial complex I by 1-methyl-4-phenylpyridinium: evidence for free radical involvement. J Neurochem 58: 786-789. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S (2003) Parkinson's disease: mechanisms and models. Neuron 39: 889-909. [DOI] [PubMed] [Google Scholar]
- Diano S, Matthews RT, Patrylo P, Yang L, Beal MF, Barnstable CJ, Horvath TL (2003) Uncoupling protein 2 prevents neuronal death including that occurring during seizures: a mechanism for preconditioning. Endocrinology 144: 5014-5021. [DOI] [PubMed] [Google Scholar]
- Eberhardt O, Coelln RV, Kugler S, Lindenau J, Rathke-Hartlieb S, Gerhardt E, Haid S, Isenmann S, Gravel C, Srinivasan A, Bahr M, Weller M, Dichgans J, Schulz JB (2000) Protection by synergistic effects of adenovirus-mediated X-chromosome-linked inhibitor of apoptosis and glial cell line-derived neurotrophic factor gene transfer in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J Neurosci 20: 9126-9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echtay KS, Winkler E, Klingenberg M (2000) Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature 408: 609-613. [DOI] [PubMed] [Google Scholar]
- Echtay KS, Winkler E, Frischmuth K, Klingenberg M (2001) Uncoupling proteins 2 and 3 are highly active H(+) transporters and highly nucleotide sensitive when activated by coenzyme Q (ubiquinone). Proc Natl Acad Sci USA 98: 1416-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand MD (2002) Superoxide activates mitochondrial uncoupling proteins. Nature 415: 96-99. [DOI] [PubMed] [Google Scholar]
- Fleury C, Neverova M, Collins S, Raimbault S, Champigny O, Levi-Meyrueis C, Bouillaud F, Seldin MF, Surwit RS, Ricquier D, Warden CH (1997) Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat Genet 15: 269-272. [DOI] [PubMed] [Google Scholar]
- Horimoto M, Fulop P, Derdak Z, Wands JR, Baffy G (2004) Uncoupling protein-2 deficiency promotes oxidant stress and delays liver regeneration in mice. Hepatology 39: 386-392. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Diano S, Miyamoto S, Barry S, Gatti S, Alberati D, Livak F, Lombardi A, Moreno M, Goglia F, Mor G, Hamilton J, Kachinskas D, Horwitz B, Warden CH (2003a) Uncoupling proteins-2 and 3 influence obesity and inflammation in transgenic mice. Int J Obes Relat Metab Disord 27: 433-442. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Diano S, Leranth C, Garcia-Segura LM, Cowley MA, Shanabrough M, Elsworth JD, Sotonyi P, Roth RH, Dietrich EH, Matthews RT, Barnstable CJ, Redmond Jr DE (2003b) Coenzyme Q induces nigral mitochondrial uncoupling and prevents dopamine cell loss in a primate model of Parkinson's disease. Endocrinology 144: 2757-2760. [DOI] [PubMed] [Google Scholar]
- Jezek P (2002) Possible physiological roles of mitochondrial uncoupling proteins-UCPn. Int J Biochem Cell Biol 34: 1190-1206. [DOI] [PubMed] [Google Scholar]
- Klivenyi P, Calingasan NY, Starkov A, Stavrovskaya IG, Kristal BS, Yang L, Wieringa B, Beal MF (2004) Neuroprotective mechanisms of creatine occur in the absence of mitochondrial creatine kinase. Neurobiol Dis 15: 610-617. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Reed JC (2000) Mitochondrial control of cell death. Nat Med 6: 513-519. [DOI] [PubMed] [Google Scholar]
- Kuhn DM, Sakowski SA, Sadidi M, Geddes TJ (2004) Nitrotyrosine as a marker for peroxynitrite-induced neurotoxicity: the beginning or the end of the end of dopamine neurons? J Neurochem 89: 529-536. [DOI] [PubMed] [Google Scholar]
- Liu Y, Fiskum G, Schubert D (2002) Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem 80: 780-787. [DOI] [PubMed] [Google Scholar]
- Mao W, Yu XX, Zhong A, Li W, Brush J, Sherwood SW, Adams SH, Pan G (1999) UCP4, a novel brain-specific mitochondrial protein that reduces membrane potential in mammalian cells. FEBS Lett 443: 326-330. [DOI] [PubMed] [Google Scholar]
- Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, Warden CH, Castilho RF, Melcher T, Gonzalez-Zulueta M, Nikolich K, Wieloch T (2003) Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med 9: 1062-1068. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Ward MW (2000) Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci 23: 166-174. [DOI] [PubMed] [Google Scholar]
- Pedersen PL (1999) Mitochondrial events in the life and death of animal cells: a brief overview. J Bioenerg Biomembr 31: 291-304. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Kostic V, Jackson-Lewis V, Naini AB, Simonetti S, Fahn S, Carlson E, Epstein CJ, Cadet JL (1992) Transgenic mice with increased Cu/Zn-superoxide dismutase activity are resistant to N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. J Neurosci 12: 1658-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchis D, Fleury C, Chomiki N, Goubern M, Huang Q, Neverova M, Gregoire F, Easlick J, Raimbault S, Levi-Meyrueis C, Miroux B, Collins S, Seldin M, Richard D, Warden C, Bouillaud F, Ricquier D (1998) BMCP1, a novel mitochondrial carrier with high expression in the central nervous system of humans and rodents, and respiration uncoupling activity in recombinant yeast. J Biol Chem 273: 34611-34615. [DOI] [PubMed] [Google Scholar]
- Shults CW, Oakes D, Kieburtz K, Beal MF, Haas R, Plumb S, Juncos JL, Nutt J, Shoulson I, Carter J, Kompoliti K, Perlmutter JS, Reich S, Stern M, Watts RL, Kurlan R, Molho E, Harrison M, Lew M (2002) Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol 59: 1541-1550. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Fiskum G (2003) Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem 86: 1101-1107. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Dube C, Dorenbos K, Steward O, Baram TZ (2003) Mitochondrial uncoupling protein-2 protects the immature brain from excitotoxic neuronal death. Ann Neurol 53: 711-717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, Hagen T, Vidal-Puig AJ, Boss O, Kim YB, Zheng XX, Wheeler MB, Shulman GI, Chan CB, Lowell BB (2001) Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 105: 745-755. [DOI] [PubMed] [Google Scholar]