

Abstract
Reductions in pain ratings when administered a placebo with expected analgesic properties have been described and hypothesized to be mediated by the pain-suppressive endogenous opioid system. Using molecular imaging techniques, we directly examined the activity of the endogenous opioid system on μ-opioid receptors in humans in sustained pain with and without the administration of a placebo. Significant placebo-induced activation of μ-opioid receptor-mediated neurotransmission was observed in both higher-order and sub-cortical brain regions, which included the pregenual and subgenual rostral anterior cingulate, the dorsolateral prefrontal cortex, the insular cortex, and the nucleus accumbens. Regional activations were paralleled by lower ratings of pain intensity, reductions in its sensory and affective qualities, and in the negative emotional state of the volunteers. These data demonstrate that cognitive factors (e.g., expectation of pain relief) are capable of modulating physical and emotional states through the site-specific activation of μ-opioid receptor signaling in the human brain.
Keywords: positron emission tomography, pain, stress, opioid receptors, placebo, human
Introduction
Placebo effects, the positive physiological or psychological changes associated with the administration of inert substances or procedures, can both enhance and obscure the effects of therapeutic interventions. However, they also represent an instance in which neural processes, through positive cognitive expectations, influence physical and neuropsychiatric states, a veritable example of “mind-body” interactions. It is therefore not surprising that interest is emerging in understanding their underlying mechanisms.
Substantial evidence implicates the endogenous opioid system in the mediation of placebo effects under conditions of expectation of analgesia. During both clinical and experimentally induced pain, placebo administration with expectation of analgesia has been associated with reductions in pain ratings that were reversed by either the open or hidden administration of naloxone (i.e., they were mediated by the activation of pain-suppressive endogenous opioid neurotransmission) (Gracely et al., 1983; Grevert et al., 1983; Levine and Gordon, 1984; Benedetti, 1996; Amanzio and Benedetti, 1999).
The endogenous opioid system, and specifically its activation of μ-opioid receptors, thought to primarily mediate the observed effects of placebo and naloxone, is implicated in a number of functions, from the regulation of stress responses and pain, particularly if sustained or threatening to the organism (Watkins and Mayer, 1982; Akil et al., 1984; Rubinstein et al., 1996; Sora et al., 1997; Zubieta et al., 2001), to reproductive and stress-related neuroendocrine functions (Smith et al., 1998; Drolet et al., 2001). Substantial evidence has also accumulated as to the involvement of this neurotransmitter system in the adaptation and response to novel and emotionally salient stimuli in both animal models and humans (Kalin et al., 1988; Nelson and Panksepp, 1998; Filliol et al., 2000; Zubieta et al., 2003b; Moles et al., 2004).
High placebo responders have shown more pronounced rostral anterior cingulate blood-flow responses to the systemic administration of a μ-opioid receptor agonist, remifentanil, suggesting the presence of variations in the responses of this receptor system as a function of placebo response (Petrovic et al., 2002). More recently, using functional magnetic resonance imaging (fMRI), the administration of a placebo with expectation of analgesia has also been associated with reductions in the activity of pain-responsive regions, namely the rostral anterior cingulate, the insular cortex, and the thalamus (Wager et al., 2004).
The present study directly examines whether the introduction of a placebo with expectation of analgesia indeed activates endogenous opioid neurotransmission, using positron emission tomography (PET) and molecular imaging with a μ-opioid receptor-selective radiotracer. Under these conditions, activation of this neurotransmitter system is evidenced by reductions in the in vivo availability of synaptic μ-opioid receptors to bind the radiolabeled tracer (Zubieta et al., 2001, 2002, 2003a; Bencherif et al., 2002).
From a methodological perspective, one of the difficulties inherent to the use of molecular imaging is the length of time required for the acquisition of quantitative measures of receptor availability (Zubieta et al., 2001, 2002), requiring that pain be maintained for a period of time. However, sustained pain also induces the activation of antinociceptive responses (Levine et al., 1978; Watkins and Mayer, 1982; Zubieta et al., 2001), which by substantially reducing pain ratings over time (Bencherif et al., 2002) may interfere with the subsequent formation of the placebo effect (Price et al., 1999). These issues were addressed by using an adaptive system that maintained pain over time by increasing the magnitude of the algesic stimulus as the rating of pain intensity reported by the volunteers declined over time (Zhang et al., 1993). The increases in algesic requirement then provided an objective psychophysical measure of antinociceptive activity, whereas the relationship between stimulus magnitude and pain ratings relate it to the subjective individual experiences of pain.
Materials and Methods
Subjects. Volunteers were 20- to 30-year-old, right-handed males, who were nonsmokers and had no personal history of medical, psychiatric illness, substance abuse or dependence, and no family history of inheritable illnesses. This study was restricted to males within a narrow age range, because both age and sex effects have been described for μ-opioid receptor concentrations and the capacity to activate this neurotransmitter system (Gabilondo et al., 1995; Zubieta et al., 1999, 2002). Volunteers were not taking psychotropic medications or hormone treatments and did not exercise in excess of 1 h three times a week. Subjects were instructed not to drink alcohol for at least 24 h nor to exercise or eat for at least 3 h before the study. Written informed consent was obtained in all cases. All the procedures used were approved by the University of Michigan Investigational Review Board and the Subcommittee for Human Use of Radioisotopes.
Experimental design. Each volunteer was scanned three times with PET and [11C]carfentanil. An initial study was performed without any intervention (baseline), and the second and third studies included either a sustained pain challenge or sustained pain with placebo with implied analgesic properties. Radiotracer administrations were separated by at least 2 h to allow for radiotracer decay and eliminate any possible residual effects of the preceding challenge.
Pain and pain plus placebo conditions were introduced 40 min after radiotracer administration in a blind, randomized, and counterbalanced design (one-half of the volunteers receiving pain first and one-half receiving pain plus placebo). Deep sustained muscle pain was maintained from 40 to 60 min after radiotracer administration by the infusion of medication-grade 5% hypertonic saline into the relaxed masseter (jaw) muscle via a computer-controlled pump (total volume, 2.91 ± 0.96 ml), as described previously (Zhang et al., 1993; Stohler and Kowalski, 1999). Briefly, after the standardized bolus injection of 0.15 ml of hypertonic saline, infused over 15 s, subjects were required to report the present pain intensity every 15 s on an electronic version of 100 mm visual analog scale (VAS), with the lower and upper bound of the scale marked with numbers 0 and 100, representing the range from “no pain” to “the most pain intensity imaginable.” Based on pain intensity scores provided by the subject every 15 s for the reminder of the experiment, individual infusion requirements were continuously modeled and updated to keep the present pain intensity scores in the preset range for the full duration of the experiment. Infusion volumes, required to maintain the preset pain intensity, were recorded every 15 s, and the cumulative infusion volume required over time was used as an indicator of subjects' pain sensitivity. Using this model, pain disappears 5-10 min after completion of the algesic infusion. To avoid swelling and possible tissue damage, the maximum infusion rate was limited to 250 μl/min.
Immediately after completion of the trials, the subjective pain experience was evaluated using the comprehensive version of the McGill Pain Questionnaire (MPQ) (Melzack and Torgerson, 1971) and overall VAS scores (rated from 0 to 100) of pain intensity and pain unpleasantness. The internal affective state of the volunteers was rated with the Positive and Negative Affectivity Scale (PANAS) (Watson et al., 1988) and the Profile of Mood States (POMS). The composite Total Mood Disturbance score (TMD) was used to evaluate the transient negative mood elicited by the pain stimulus (POMS-TMD) (McNair et al., 1992). These scores were related to the algesic input (subjective scores per vol of hypertonic saline, in milliliters) to provide with assessments of the pain experience as a function of the pain stimulus administered to the subject in each of the experimental conditions.
Subjects were given the following clinical trial-type instructions before administration of the placebo, so that the conditions of the study would be similar to those encountered in typical placebo-controlled drug trials: “We are studying the effect of a medication that may or may not relieve pain. This medication is thought to have analgesic effects through the activation of brain systems that suppress pain.” In the written consent form, it was further explained that they could receive an active drug or a substance with no intrinsic pain relief properties. Subjects were also informed that they may not be able to ascertain whether the drug was actually working to relieve the pain or not but that the investigators would be able to ascertain its analgesic properties through their monitoring equipment.
The placebo condition consisted of the introduction of 1 ml of 0.9% physiological saline into one of the intravenous ports, every 4 min, starting 4 min after the initiation of the pain challenge and lasting for 15 s each time. Subjects were informed that the study drug was to be administered by means of a warning that was followed by a second-by-second count of the infusion timing (15 s), using a computer-generated human voice recording. Subjects were also asked to estimate the expected analgesia before the introduction of the placebo and afterward, estimating the analgesic properties using a VAS scale from 0 (no analgesic effect) to 100 (maximum analgesia).
Neuroimaging methods. MRI scans were acquired in all subjects on a 1.5 tesla scanner (Signa; General Electric, Milwaukee, WI). Acquisition sequences were axial spoiled gradient-recalled echo (SPGR) inverse recovery-Prep MR [echo time (TE), 5.5; repetition time (TR), 14; inversion time, 300; flip angle, 20°; number of excitations (NEX), 1; 124 contiguous images; 1.5 mm thickness], followed by axial T2 and proton density images (TR, 4000; TE, 20 and 100, respectively; NEX, 1; 62 contiguous images; 3 mm thick).
PET scans were acquired with a Siemens AG (Erlangen, Germany) HR+ scanner in three-dimensional mode [reconstructed full width at half maximum (FWHM) resolution, ∼ 5.5 mm in-plane and 5.0 mm axially], with septa retracted and scatter correction. Participants were positioned in the PET scanner gantry, and two intravenous (antecubital) lines were placed. A light forehead restraint was used to eliminate intrascan head movement. [11C]carfentanil was synthesized at high specific activity (>2000 Ci/mmol) by the reaction of 11C-methyliodide and a nonmethyl precursor as described previously (Dannals et al., 1985), with minor modifications to improve its synthetic yield (Jewett, 2001); 10-15 mCi (370-555 MBq) were administered to each subject for each of the three PET scans. The baseline study was always performed first, typically 24-48 h before the challenge studies. Pain and pain plus placebo studies were separated by at least 2 h to allow for tracer decay. The maximum mass of carfentanil injected was 0.03 μg/kg per scan, ensuring that the compound was administered in tracer quantities (i.e., subpharmacological doses). Receptor occupancy by carfentanil was calculated to be between 0.2 and 0.6% for brain regions with low, intermediate, and high μ-opioid receptor concentrations, based on the mass of carfentanil administered and the known concentration of μ-opioid receptors in the postmortem human brain (Gross-Isseroff et al., 1990; Gabilondo et al., 1995). Fifty percent of the [11C]carfentanil dose was administered as a bolus, and the remainder as a continuous infusion using a computer-controlled pump to achieve steady-state tracer levels at ∼35-40 min after tracer administration. Twenty-eight frames were acquired over 90 min with an increasing duration (30 s to 5 min).
Images were reconstructed using iterative algorithms (brain mode; FORE/OSEM, 4 iterations; 16 subsets; no smoothing) into a 128 × 128 pixel matrix in a 28.8-cm-diameter field of view. Attenuation correction was performed through a 6 min transmission scan (68Ge source) obtained before the PET study, also with iterative reconstruction of the blank/transmission data followed by segmentation of the attenuation image. Small head motions during emission scans were corrected by an automated computer algorithm for each subject before analysis, and the images coregistered to each other with the same software (Minoshima et al., 1993). Time points were then decay-corrected during reconstruction of the PET data. Image data were then transformed on a voxel-by-voxel basis into two sets of parametric maps: (1) a tracer transport measure (K1 ratio) and (2) a receptor-related measure at equilibrium (DVReq), the latter using data obtained from 40-90 min after tracer administration. The tracer transport and binding measures were calculated using a modified Logan graphical analysis (Logan et al., 1996), using the occipital cortex (an area devoid of μ-opioid receptors) as the reference region. With the bolus-continuous infusion tracer administration protocol used, all regions achieved steady state by between 35 and 40 after tracer administration (e.g., they are not susceptible to biases introduced by blood flow and therefore tracer transport changes). The slope of the Logan plot was used for the estimation of the distribution volume ratio (DVReq), a measure equal to the (Bmax/Kd) + 1 for this receptor site and radiotracer. Bmax/Kd (or DVReq - 1) was the receptor-related measure (μ-opioid receptor availability or binding potential). K1 and DVReq images for each experimental period and MR images were coregistered to each other and to the International Consortium for Brain Mapping (ICBM) stereotactic atlas orientation (Meyer et al., 1997).
Statistical parametric maps of differences between conditions (control-pain, pain plus placebo-pain) were generated by anatomically standardizing the T1-SPGR MRI of each subject to the ICBM stereotactic atlas coordinates, with subsequent application of this transformation to the μ-opioid receptor binding maps (Meyer et al., 1997). The accuracy of coregistration and nonlinear warping algorithms was confirmed for each subject individually by comparing the transformed MRI and PET images to each other and the ICBM atlas template.
Differences between conditions and groups were then mapped into stereotactic space using z-maps of statistical significance with statistical parametric mapping (SPM) (SPM 99; Wellcome Department of Cognitive Neurology, London, UK) and Matlab (WaveMetrics, Lake Oswego, OR) software, with a general linear model and correction for multiple comparisons (Friston et al., 1995). No global normalization was applied to the data, and therefore the calculations presented are based on absolute Bmax/Kd estimates. Only regions with specific μ-opioid receptor binding were included in the analyses (voxels with DVR values > 1.2 times the mean global image value for μ-opioid receptor images as calculated with SMP 99). To compensate for small residual anatomic variations across subjects and to improve signal-to-noise ratios, a three-dimensional Gaussian filter (FWHM, 6 mm) was applied to each scan.
Comparisons between conditions, within subjects were then performed using paired, two-tailed t tests, on a voxel-by-voxel basis. Areas of significant differences were detected using a statistical threshold that controls a type-I error rate at p = 0.05 for multiple comparisons. These statistical thresholds were estimated using the Euler characteristic (Worsley, 1994) based on the number of voxels in the gray matter, image smoothness, and the extent of local changes (correction for cluster volume) (Friston et al., 1991). The only region for which a lower statistical threshold was allowed (p < 0.0001 uncorrected) was the periaqueductal gray, because of its small size, making the finding of significant effects difficult, and because of its central involvement in endogenous opioid modulation of pain. Numerical values for the graphs presented in Figures 2 and 3 and for the correlations described in the text were extracted from the image data by averaging the values of voxels contained in an area where significant differences were obtained in the voxel-by-voxel analysis, down to a threshold of p = 0.01. Correlations between activation of neurotransmission and the changes in psychophysical measures were calculated with one-tailed Pearson correlations at p < 0.05, given the known directionality of these relationships (Zubieta et al., 2001).
Figure 2.

Individual data points for the magnitude of regional μ-opioid system activation in response to the placebo intervention. Individual data points for the change in the binding potential measure (BP; μ-opioid receptor availability in vivo; Bmax/Kd) from the pain condition to the pain plus placebo condition are shown. A threshold of 10% increase in the activation of this neurotransmitter system, evidenced as a reduction in the BP measure during the placebo condition, was used to identify individuals that responded with a robust placebo effect on the activation of this system in each of the regions (dashed line). ACing_Rostral, Rostral (pregenual) region of the anterior cingulate; DLPFC_Left, left dorsolateral prefrontal cortex; Insula_Right, right insular cortex; N.Acc._Left, left nucleus accumbens.
Figure 3.

Relationship between placebo-induced regional μ-opioid system activation and expectation of analgesia. Negative correlation between the expected analgesic effects as rated by the volunteers before the study and the placebo-induced activation of dorsolateral prefrontal cortex (DLPFC) μ-opioid neurotransmission in placebo responders (black circles) and nonresponders (gray circles) are shown. BP, Binding potential.
The within-subject, interexperimental variability in the binding measures was calculated previously from a separate sample of five healthy individuals, 20-30 years of age, studied twice without interventions. For the regions in which placebo effects were detected in this study (anterior cingulate, prefrontal cortex, insular cortex, nucleus accumbens), inter-experimental differences ranged from 7.7 to -8.4% (mean, -3.2 ±6.4%). A conservative cutoff of 10% was then used to assess the formation of a substantial neurochemical placebo effect for the analyses of its psychophysical correlates.
Results
Effects of sustained pain on μ-opioid receptor-mediated neurotransmission
Consistent with previous work (Zubieta et al., 2001), the sustained pain stimulus, which was always applied on the left side, was associated with a significant activation of endogenous opioid transmission and μ-opioid receptors (n = 14). The areas involved included the dorsal anterior cingulate, medial prefrontal cortex, right insular cortex, ventral basal ganglia, bilaterally (nucleus accumbens extending to the ventral pallidum), medial thalamus, right amygdala, left subamygdalar temporal cortex, and periaqueductal gray (Fig. 1).
Figure 1.
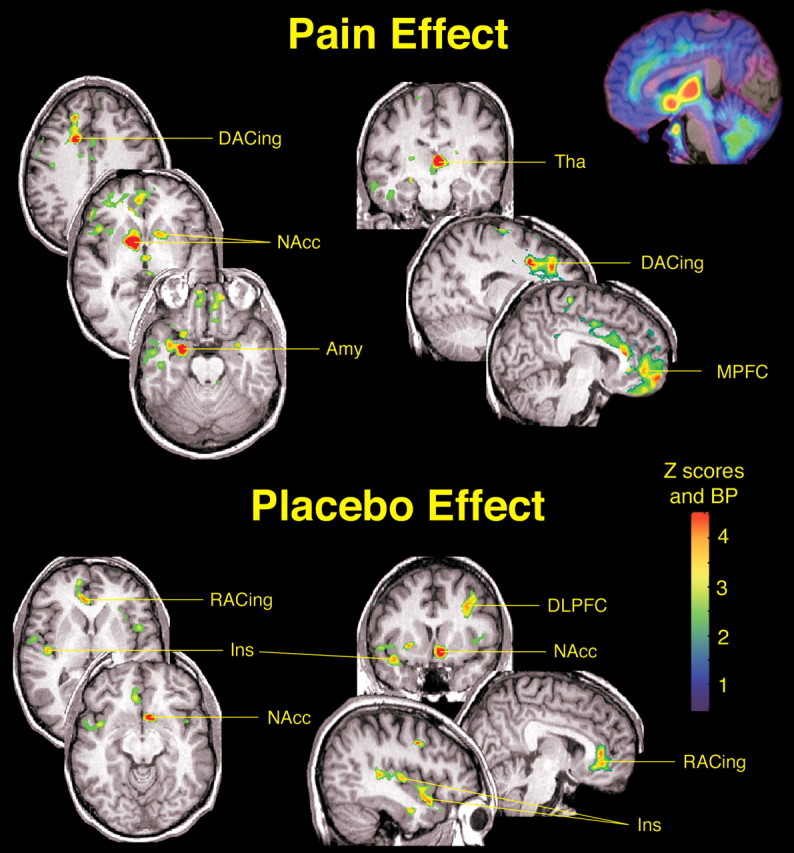
Effects of pain and placebo on the activation of μ-opioid receptor-mediated neurotransmission. After correction for multiple comparisons, significant μ-opioid system activations during the sustained pain challenge (n = 14) were obtained in the dorsal anterior cingulate [DACing; x,y,z coordinates (in millimeters), 17, 10, 40; cluster size, 715 mm3; z score = 5.54; p < 0.005 after correction for multiple comparisons], medial prefrontal cortex (MPFC; x,y,z, 8, 45, -5; cluster size, 1064 mm3; z score = 3.90;p < 0.001), right (contralateral to pain) insular cortex (Ins; x,y,z, 49, 16, 6; cluster size, 257 mm3; zscore = 4.32; p < 0.005), ventral basal ganglia, bilaterally [nucleus accumbens (NAcc) extending to the ventral pallidum; right, x,y,z, 12, 4, -2, cluster size, 1700 mm3, z score = 9.26, p < 0.0001; left, x,y,z, -19, 9, 4, cluster size, 2177 mm3, z score = 4.32, p < 0.005], medial thalamus (Tha; x,y,z, -3, -15, 7; cluster size, 2283 mm3; z score = 6.49; p < 0.0001), right amygdala (Amy; x,y,z, 25, -2, -21; cluster size, 464 mm3; z score = 6.34; p < 0.0001), left subamygdalar temporal cortex (x,y,z, -28, 10, -38; cluster size, 560 mm3; zscore = 5.36; p < 0.005), and periaqueductal gray (x,y,z, -5, -28, -3; cluster size, 122 mm3; zscore = 3.56; p < 0.05). Significant effects of placebo on the activation of the μ-opioid system (n = 14) were detected in the left dorsolateral prefrontal cortex (DLPFC; x,y,z peak coordinates, -36, 13, 39; cluster size, 1403 mm3; z score = 4.27; p < 0.0001), rostral anterior cingulate (RACing; x,y,z, 14, 49, 13; cluster size, 3193 mm3; z score = 4.18; p < 0.0001), left NAcc (x,y,z, -7, 11, -11; cluster size, 1332 mm3; z score = 4.83; p < 0.0001), and right anterior insula (Ins; x,y,z, 41, 10, -17; cluster size, 844 mm3; z score = 4.15; p < 0.05). The posterior right insula achieved subthreshold levels of significance (x,y,z, 44, -15, 4; cluster size, 732 mm3; z score = 3.81; p < 0.0001 uncorrected for multiple comparisons). z scores of statistical significance are represented by the pseudocolor scale on the right side of the image and are superimposed over an anatomically standardized MRI image in coronal views. The left side of the axial and coronal images corresponds to the right side of the body (contralateral to pain; radiological convention). A map of μ-opioid receptor distribution is shown in the top right corner of the figure in a sagittal view, with binding potential (BP) values (receptor availability in vivo; Bmax/Kd) depicted by the same pseudocolor scale.
Psychophysical responses to placebo administration
Expectation of analgesia before the introduction of the placebo was rated at 50 ± 18% VAS units (range, 10-75%), with end-points denoted as “no effect at all” and “complete pain relief,” respectively. The analgesic effectiveness of the placebo was subjectively rated at 54 ± 24% VAS units (range, 10-90%) after completion of the study. This information confirmed that there was expectation of analgesia induced by our clinical trial type of instructions.
Introduction of the placebo (1 ml of isotonic saline, i.v.; every 4 min) was associated with an increase in the average rate of algesic stimulus required to maintain pain, an objective assessment of the activation of antinociceptive mechanisms (Table 1, top). Pain intensity, as rated every 15 s by the volunteers, was maintained for the duration of the study, as expected given the adaptive nature of the algesic infusion system used. However, a small mean reduction of 4 points in the 0-100 scale used to rate momentary pain was noted after the introduction of the placebo, which reached statistical significance (Table 1, top). This was attributed to five subjects reaching the maximum allowable infusion rate to maintain pain (250 μl/min) when the placebo was administered, an unexpected occurrence. This maximum, safety-bound rate had been originally established to avoid tissue swelling and damage. However, because the subjective ratings of the pain experience were related to the volume of algesic substance introduced, the ceiling effect reached in some subjects was not felt to compromise these assessments. Just the opposite, reductions in the pain signal would tend to bias the experiments toward finding lower placebo effects as well.
Table 1.
Psychophysiological responses during pain and pain plus placebo conditions, all subjects
|
|
Pain |
Pain + Plbo |
t |
p |
|---|---|---|---|---|
| Measure | ||||
| Mean q 15 s momentary VAS intensity | 28.3 ± 13.1 | 24.0 ± 13.5 | 2.01 | 0.03 |
| Volume 5% intramuscular saline (μl) | 2796 ± 956 | 3025 ± 100 | 1.10 | 0.14 |
| VAS overall intensity | 37.5 ± 19.9 | 27.4 ± 15.7 | 3.40 | 0.002 |
| VAS overall unpleasantness | 40.4 ± 25.5 | 32.9 ± 21.6 | 2.05 | 0.03 |
| MPQ sensory subscale | 14.3 ± 4.5 | 13.7 ± 5.4 | 0.44 | 0.33 |
| MPQ affective subscale | 0.9 ± 1.0 | 0.4 ± 0.6 | 2.83 | 0.007 |
| MPQ total | 19.4 ± 5.5 | 18.5 ± 7.4 | 0.40 | 0.35 |
| PANAS negative affect | 2.1 ± 2.1 | 2.6 ± 3.5 | 0.97 | 0.44 |
| PANAS positive affect | 13.4 ± 7.4 | 14.1 ± 6.1 | 0.87 | 0.20 |
| POMS-TMD score | 9.7 ± 7.9 | 10.7 ± 9.5 | 0.99 | 0.17 |
| Rating to stimulus (milliliters of algesic substance required) ratio | ||||
| Mean q 15 s momentary VAS intensity | 14.7 ± 16.1 | 10.8 ± 1.0 | 1.40 | 0.09 |
| VAS overall intensity | 19.5 ± 21.5 | 12.3 ± 11.4 | 1.98 | 0.03 |
| VAS overall unpleasantness | 18.5 ± 18.6 | 13.2 ± 10.6 | 1.76 | 0.05 |
| MPQ sensory subscale | 6.0 ± 3.3 | 5.5 ± 4.4 | 0.43 | 0.38 |
| MPQ affective subscale | 0.5 ± 0.6 | 0.2 ± 0.4 | 2.32 | 0.02 |
| MPQ total | 8.4 ± 5.1 | 7.6 ± 6.6 | 0.41 | 0.34 |
| PANAS negative affect | 0.8 ± 1.0 | 1.5 ± 2.9 | 0.97 | 0.17 |
| PANAS positive affect | 5.8 ± 4.6 | 5.7 ± 4.4 | 0.15 | 0.44 |
| POMS-TMD score |
4.7 ± 4.6 |
5.3 ± 6.2 |
0.36 |
0.37 |
Mean ± 1 SD of psychophysical measures of pain during the sustained pain challenge in the absence (Pain) and presence (Pain + Plbo) of placebo in 14 males 20-30 years of age. Mean q 15 s momentary VAS intensity refers to the average ratings of momentary pain acquired every 15 s for the duration of the pain challenge (20 min). Volume 5% intramuscular saline (in microliters) is the total volume of algesic substance introduced by the adaptive pain control system to maintain pain. The remainder of the scales (VAS, 0-100 of overall pain intensity and unpleasantness; MPQ; PANAS; and POMS-TMD) were obtained immediately after completion of the pain challenge. Data are also expressed as the relationship between the subject's ratings and the stimulus needed to maintain pain during the experimental period [rating to stimulus (in milliliters) ratio]. These ratios are used as the subjective assessment of placebo effects. Paired, one-tailed t test, p < 0.05.
The additional psychophysical variables used to assess the subjective experience of sustained pain with and without placebo administration are shown in Table 1 (bottom). With the exception of the momentary VAS ratings of pain intensity, which was acquired every 15 s for the duration of the studies, other rating scales (overall VAS ratings of pain intensity and unpleasantness, MPQ, PANAS, POMS-TMD) were administered immediately after completion of each of the challenges. For the entire sample, significant effects of placebo on the pain experience in terms of stimulus magnitude to stimulus response were observed for the ratings of pain intensity and unpleasantness, as well as for the affective information content of the pain, as measured with the MPQ (Table 1).
We then contrasted the response properties of high and low placebo responders using a 20% difference in the response-to-stimulus ratios (average of every 15 s VAS intensity ratings/volume infused) as the cutoff to classify placebo responders. In this manner, nine subjects were classified as high placebo responders, whereas five were classified as low placebo responders. High placebo responders demonstrated significant reductions in rating to stimulus ratios for VAS ratings of overall pain intensity (t = 2.12) and pain unpleasantness (t = 2.21), MPQ sensory subscale scores (t = 3.13), MPQ affective subscale scores (t = 2.66), and MPQ total scores (t = 3.16) and presented lower PANAS negative affect (t = 2.02) and higher PANAS positive affect scores during placebo administration (t = 2.25) (df = 12; unpaired, one-tailed t tests; p = 0.05). Trend effects were observed for POMS-TMD scores, which were also lower in the placebo responders, albeit below statistically significant thresholds (t = 1.62; p = 0.06).
Effect of placebo on μ-opioid receptor-mediated neurotransmission
The activation of the endogenous opioid system and μ-opioid receptors was then compared between sustained pain and sustained pain plus placebo conditions for all subjects (n = 14). Significantly higher levels of activation were obtained for the condition in which placebo was administered. After correction for multiple comparisons, statistically significant effects of placebo on μ-opioid system activation were obtained in the left (ipsilateral to pain) dorsolateral prefrontal cortex (Brodmann areas 8 and 9), pregenual rostral right (contralateral) anterior cingulate (Brodmann areas 24 and 25), right (contralateral) anterior insular cortex, and left (ipsilateral) nucleus accumbens (Fig. 1). A second area within the contralateral insular cortex, in its posterior region, also showed changes in neurotransmission; however, it did no longer reach statistical significance after correction for multiple comparisons (Fig. 1). Data points for significant regions are presented in Figure 2, confirming robust effects of the placebo on this neurotransmitter system. However, examination of the individual datasets showed that the degree of placebo-induced μ-opioid system activation was not of the same magnitude across all the brain sites in which these effects were detected. Overall, these data pointed to the fact that an unselected sample of young healthy volunteers, when subjected to clinical trials instructions, showed robust placebo-induced activations of μ-opioid receptor-mediated neurotransmission that was relatively regionally specific for individual subjects.
Psychophysical correlates of placebo-induced activation of μ-opioid neurotransmission
In view of the differences in individual placebo-induced neurotransmitter activation patterns, regional psychophysical correlates were examined using a threshold of 10% change in the receptor binding measure to capture data points for which the formation of a substantial regional placebo effect could be ascertained (Fig. 2). This threshold clearly exceeded the typical variability of receptor binding measures obtained in separate experiments (see Materials and Methods). Correlations were conducted between these regional changes in receptor availability, the algesic infusion volume changes after placebo, and the placebo-induced changes in subjective reports of pain (overall VAS intensity and unpleasantness scores, MPQ sensory and affective subscales, PANAS negative and positive affect scales, and POMS-TMD scores). No correction for multiple comparisons was applied to these correlations and should be considered exploratory in nature.
In the pregenual anterior cingulate, placebo-induced μ-opioid system activation above those levels (n = 7) was correlated with the changes in stimulus-to-response ratios for overall ratings of VAS pain intensity (r = -0.87; p < 0.01) and pain unpleasantness (r = -0.74; p = -0.05), MPQ sensory subscale (r = -0.84; p < 0.01), and total MPQ scores (r = -0.89; p < 0.01). Placebo-induced activation of endogenous opioid neurotransmission in this region was also highly and positively correlated with the increases in the volume of algesic substance required to maintain pain during the placebo condition (r = 0.96; p < 0.0001).
In the right anterior insular cortex (n = 9), significant correlations were obtained with the changes in VAS ratings of pain intensity (r = -0.58; p < 0.05), MPQ sensory (r = -0.60; p < 0.05), and total MPQ scores (r = -0.58; p < 0.05). At the level of same direction, with the change in VAS pain intensity ratings (r = -0.80; p < 0.01), MPQ affective subscale (r = -0.81; p < 0.01), and reductions in the POMS-TMD scores (r = -0.71; p < 0.05). No significant correlations were obtained between placebo-induced changes in these psychophysical measures and left dorsolateral prefrontal cortex (n = 10) μ-opioid neurotransmission. However, and uniquely for this region, μ-opioid system activation was negatively correlated with the expected analgesic effect as rated by the subjects before placebo administration, whether only placebo responders or all study participants were included in the analysis (r = -0.55 and -0.65, respectively; p < 0.05) (Fig. 3).
Discussion
This report provides the first direct evidence that the administration of a placebo with implied analgesic properties regionally activates a pain and stress inhibitory neurotransmitter system, the endogenous opioid system, through direct effects on the μ-opioid receptors. Furthermore, that this activation is associated with quantifiable reductions in the physical and emotional attributes of a sustained pain challenge. We also observe that the respective neurotransmitter activity took place directly in associative, higher-order brain regions, namely the pregenual and subgenual area of the rostral anterior cingulate, the dorsolateral prefrontal and insular cortex, and in the nucleus accumbens, correlating with various aspects of the experience of pain. In the case of the rostral anterior cingulate, activation of this neurotransmitter system was highly associated with the algesic infusion requirements to maintain pain for the duration of the study, an objective measure of the activation of antinociceptive mechanisms. Dorsolateral prefrontal cortex endogenous opioid activity, on the other hand, was associated with the magnitude of analgesia expected by the volunteers before placebo administration.
For the regions in which placebo administration increased the endogenous opioid neurotransmission, with exception of the nucleus accumbens, their localization primarily coincided with that observed by Wager et al. (2004) as reductions in pain-induced metabolic demands as measured by blood oxygenation level-dependent fMRI (BOLD-fMRI) during placebo administration (i.e., prefrontal cortex, pregenual anterior cingulate, and insular cortex). Basal ganglia signals are often difficult to obtain with BOLD-fMRI techniques, which may explain the difference with respect to this region between their data and ours. The regions implicated in the placebo effect are, as would be expected, part of those where prominent endogenous opioid neurotransmission and μ-opioid receptor populations are present in rodent and human brains (e.g., cingulate, prefrontal, temporal and insular cortices, thalamus, basal ganglia, amygdala, hypothalamus, brainstem) (Gross-Isseroff et al., 1990; Gabilondo et al., 1995; Mansour et al., 1995). These are the areas in which increases in regional blood flow are registered after the exogenous administration of μ-opioid receptor agonists (Firestone et al., 1996; Adler et al., 1997; Schlaepfer et al., 1998; Casey et al., 2000; Wagner et al., 2001); some of them (i.e., rostral anterior cingulate) had also been noted to be more prominently activated in high placebo responders after μ-opioid agonist administration (Petrovic et al., 2002).
Our work takes the investigation of placebo effects directly into the realm of human brain neurotransmission, addressing the function of a single neurotransmitter system. In this regard, μ-opioid receptor-mediated neurotransmission is one of the principal systems involved in the modulation of pain (Matthes et al., 1996), stress responses (Akil et al., 1984), and stress-induced analgesia in animal models (Mogil et al., 1994; Rubinstein et al., 1996) as well as in humans (Zubieta et al., 2001, 2002, 2003a). The μ-opioid system has also been implicated in the regulation of behavioral responses to novel environments and stimulus-reward associations in studies using animal models devoid of these receptors (Filliol et al., 2000; Moles et al., 2004), as well as in the regulation of affective responses in humans (Zubieta et al., 2003b). As such, this neurotransmitter system appears to support the neurobiologies of pain, stress, and their modulation by cognitive-emotional influences.
Prominent effects of the placebo intervention on μ-opioid receptor-mediated neurotransmission were observed in Brodmann areas 24 and 25 of the pregenual and subgenual anterior cingulate, part of the so-called affective subdivision of the anterior cingulate (Bush et al., 2000). Notably, the metabolic activation of the anterior cingulate by pain (Talbot et al., 1991; Davis et al., 1995; Vogt et al., 1996; Aziz et al., 1997; Casey, 1999; Gelnar et al., 1999; Willoch et al., 2000) and by more specific aspects of the pain experience, such as the pain intensity (Coghill et al., 1999; Hofbauer et al., 2001), its affective qualities (Rainville et al., 1997; Tolle et al., 1999), pain anticipation (Hsieh et al., 1999; Ploghaus et al., 1999), the illusion of pain (Craig et al., 1996), or even empathy to pain experienced by others (Singer et al., 2004) have typically involved more dorsal subregions of the anterior cingulate. Conversely, the metabolic activity of the pregenual and subgenual anterior cingulate has been traditionally implicated in responses to stressful challenges of emotional significance (Phan et al., 2002) and more recently, in the placebo effect (Petrovic et al., 2002; Wager et al., 2004). Of interest, reductions in its baseline metabolism and volume have been associated with familial and treatment-refractory major depression (Drevets et al., 1997; Mayberg et al., 1997), an illness thought to be mediated by alterations in homeostatic responses to salient or stressful environmental events. This illness otherwise presents with high responses to placebo administration in controlled trials (Schatzberg and Kraemer, 2000).
In the present report, placebo-induced activation of μ-opioid neurotransmission in the pregenual and subgenual anterior cingulate was not only significantly correlated with the suppression of volunteer's reports of perceived pain intensity and sensory pain qualities, but also with the increases in algesic infusion requirements for maintaining pain intensity during placebo administration. The connectivity of this region (Brodmann areas 24 and 25) with brain nuclei involved in pain, saliency-reward, affect, and autonomic and neuroendocrine regulatory (Carmichael and Price, 1995; Haber et al., 1995; An et al., 1998; Ongur et al., 1998), together with recent data on the effects of placebo on the metabolic activity (Petrovic et al., 2002; Wager et al., 2004), makes it a particularly likely site for the mediation of various placebo effects.
Placebo-induced activation of μ-opioid neurotransmission was also observed in the insular cortex, an area that becomes metabolically activated in response to painful stimuli in imaging studies in humans (Casey, 1999; Davis, 2000; Peyron et al., 2000). This region is thought to have a more general involvement in the representation and modulation of interoceptive, internal bodily responses to both physical and emotional stimuli (Casey, 1999; Cameron, 2001; Craig, 2002; Phan et al., 2002). Its posterior portions aid in the processing of somatosensory information, including muscular pain, such as is the case of the present study (Craig et al., 2000; Kupers et al., 2004). The anterior insula, however, appears implicated in the more complex, subjective processing of the affective qualities of various physical and emotional stimuli (Craig, 2003; Singer et al., 2004). We also observed placebo-induced μ-opioid system activation in both posterior and anterior areas of the right insular cortex (contralateral to pain in the present study); however, only the anterior region reached statistical thresholds of significance, correlating with pain intensity and sensory qualities of the pain.
In the nucleus accumbens, placebo-induced activation of μ-opioid neurotransmission was significantly correlated with the suppression of pain intensity and pain affect, as well as with the negative mood state induced by the pain challenge. This is a dopamine-rich region in which both dopamine and opioid peptides have been shown to regulate responses to painful and stressful stimuli (Gear et al., 1999; Horvitz, 2000; Zubieta et al., 2003a) but also natural rewards (Berridge and Robinson, 1998; Will et al., 2003) and drugs of abuse (Unterwald, 2001; Volkow et al., 2004), both in animals and humans. As a result, it is thought to have broader role in responding to and modulating salient stimuli with both rewarding and nonrewarding valences (Berridge and Robinson, 1998; Horvitz, 2000; Lorenz et al., 2003; Pruessner et al., 2004; Zald et al., 2004; Zink et al., 2004). Activation of dopaminergic neurotransmission in this region, and reductions in the neuronal activity of some of its efferent targets (subthalamic nucleus), have also been shown under conditions of placebo administration and expectation of improvement in Parkinson's disease (de la Fuente-Fernandez et al., 2001; Benedetti et al., 2004). Of relevance to the present results, Parkinson's disease is characterized by reductions in dopaminergic neurotransmission, which in turn has been associated with both a higher content of opioid peptides in this region (George and Kertesz, 1987; Steiner and Gerfen, 1998) and a more prominent activation of the μ-opioid system in response to pain (Zubieta et al., 2003a). These mechanisms may then underlie the particular responsiveness of this neurodegenerative illness to placebo-associated expectancies, previously observed by various authors (Shetty et al., 1999; Goetz et al., 2000).
Finally, although the activation of endogenous opioid neurotransmission in the left dorsolateral prefrontal cortex was not correlated with placebo-induced changes in the pain psychophysical measures obtained from our subjects, we did observe a negative relationship between μ-opioid system activation in this region and the magnitude of the analgesic effect expected by our subjects before the administration of the placebo. Consistent with this observation, this brain region was found bilaterally activated during placebo-induced anticipation of analgesia in an fMRI study (Wager et al., 2004). Lorenz et al. (2003) also found that the metabolic activity of this region was negatively correlated with that of other elements of the “pain matrix” (e.g., medial thalamus, midbrain, and insular cortex) during pain; it was also negatively correlated with the perceived intensity and unpleasantness of the painful stimulus. These authors then suggested a role for this region in the indirect modulation of responses to pain through other cortical (e.g., insular cortex) and subcortical (thalamus, midbrain) regions. If this were the case, a reduction in opioid inhibitory control in the dorsolateral prefrontal cortex would be associated with a permissive effect on the engagement of other pain control regions, a testable hypothesis for future studies. The hypothesized involvement of the dorsolateral prefrontal cortex in the temporal organization and adjustments in the control of behavior (Fuster, 2000; Kerns et al., 2004) may then extend into the metabolic (Wager et al., 2004) and neurotransmitter responses (as is the case in the present report) to placebo-induced expectations of analgesia. Conversely, Apkarian et al. (2004) reported progressive gray-matter atrophy in this brain region in chronic back pain patients as a function of pain duration. As more persistent forms of illness tend to present with lower placebo response rates, the contribution of variations in dorsolateral prefrontal function to the formation of a placebo effect warrants additional study.
The results presented are consistent with reports implicating the endogenous opioid system in the mediation of placebo analgesic effects, examined previously by their blockade after the systemic administration of naloxone (Gracely et al., 1983; Grevert et al., 1983; Levine and Gordon, 1984; Benedetti, 1996; Amanzio and Benedetti, 1999). Naloxone, however, is not a selective μ-opioid receptor antagonist, and it may influence other opioid receptor types. The present work, conversely, exclusively addresses the μ-opioid receptor and the psychophysical correlates of its activation, leaving open the question of whether other opioid receptor types or neurotransmitter mechanisms may also be involved in these effects. The data presented here also highlight that changes in neurochemical signaling induced by the introduction of a placebo do not represent an on-off phenomenon, but rather a graded effect that is influenced, with relative independence, by a number of brain regions with complex, “integrative-motivational” functions.
The results presented and recent data from animal models (Moles et al., 2004) are consistent with the notion that placebo-responding regions and neurochemical systems (e.g., the endogenous opioid system and μ-opioid receptors) are an intrinsic part of neuronal processes that mediate the interaction between positive environmental conditions (in the present case the suggestion of analgesia) and the corresponding physical and emotional responses of the individual. From a different perspective, disruptions in these normal regulatory processes, exemplified by the typically lower rates of placebo responding in the more persistent or severe forms of various illnesses, may then represent points of vulnerability for the expression or maintenance of various pathological states.
Footnotes
This work was supported by National Institutes of Health Grants R01 AT 001415 and R01 DA 016423 to J.-K.Z. We acknowledge the contributions of the PET Center Nuclear Medicine Technologists (Jill M. Rothley, Edward J. McKenna, Andrew R. Weeden, Paul Kison, and Shayna Huber).
Correspondence should be addressed to Dr. Jon-Kar Zubieta, Mental Health Research Institute, University of Michigan, 205 Zina Pitcher Place, Ann Arbor, MI 48109-0720. E-mail zubieta@umich.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257754-09$15.00/0
References
- Adler LJ, Gyulai FE, Diehl DJ, Mintun MA, Winter PM, Firestone LL (1997) Regional brain activity changes associated with fentanyl analgesia elucidated by positron emission tomography. Anesth Analg 84: 120-126. [DOI] [PubMed] [Google Scholar]
- Akil H, Watson S, Young E, Lewis M, Khachaturian H, Walker J (1984) Endogenous opioids: biology and function. Annu Rev Neurosci 7: 223-255. [DOI] [PubMed] [Google Scholar]
- Amanzio M, Benedetti F (1999) Neuropharmacological dissection of placebo analgesia: expectation-activated opioid systems versus conditioning-activated specific subsystems. J Neurosci 19: 484-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An X, Bandler R, Ongur D, Price JL (1998) Prefrontal cortical projections to longitudinal columns in the midbrain periaqueductal gray in macaque monkeys. J Comp Neurol 401: 455-479. [PubMed] [Google Scholar]
- Apkarian AV, Sosa Y, Sonty S, Levy RM, Harden RN, Parrish TB, Gitelman DR (2004) Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J Neurosci 24: 10410-10415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz Q, Andersson JL, Valind S, Sundin A, Hamdy S, Jones AK, Foster ER, Langstrom B, Thompson DG (1997) Identification of human brain loci processing esophageal sensation using positron emission tomography. Gastroenterology 113: 50-59. [DOI] [PubMed] [Google Scholar]
- Bencherif B, Fuchs PN, Sheth R, Dannals RF, Campbell JN, Frost JJ (2002) Pain activation of human supraspinal opioid pathways as demonstrated by [11C]-carfentanil and positron emission tomography (PET). Pain 99: 589-598. [DOI] [PubMed] [Google Scholar]
- Benedetti F (1996) The opposite effects of the opiate antagonist naloxone and the cholecystokinin antagonist proglumide on placebo analgesia. Pain 64: 535-543. [DOI] [PubMed] [Google Scholar]
- Benedetti F, Colloca L, Torre E, Lanotte M, Melcarne A, Pesare M, Bergamasco B, Lopiano L (2004) Placebo-responsive Parkinson patients show decreased activity in single neurons of subthalamic nucleus. Nat Neurosci 7: 587-588. [DOI] [PubMed] [Google Scholar]
- Berridge KC, Robinson TE (1998) What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev 28: 309-369. [DOI] [PubMed] [Google Scholar]
- Bush G, Luu P, Posner M (2000) Cognitive and emotional influences in anterior cingulate cortex. Trends Cogn Sci 4: 215-222. [DOI] [PubMed] [Google Scholar]
- Cameron OG (2001) Interoception: the inside story-a model for psychosomatic processes. Psychosom Med 63: 697-710. [DOI] [PubMed] [Google Scholar]
- Carmichael ST, Price JL (1995) Limbic connections of the orbital and medial prefrontal cortex in macaque monkeys. J Comp Neurol 363: 615-641. [DOI] [PubMed] [Google Scholar]
- Casey K (1999) Forebrain mechanisms of nociception and pain: analysis through imaging. Proc Natl Acad Sci USA 96: 7668-7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey K, Svensson P, Morrow T, Raz J, Jone C, Minoshima S (2000) Selective opiate modulation of nociceptive processing in the human brain. J Neurophysiol 84: 525-533. [DOI] [PubMed] [Google Scholar]
- Coghill R, Sang C, Maisog J, Iadarola M (1999) Pain intensity processing within the human brain: a bilateral, distributed mechanism. J Neurophysiol 82: 1934-1943. [DOI] [PubMed] [Google Scholar]
- Craig A (2002) How do you feel? Interoception: the sense of the physiological condition of the body. Nat Rev Neurosci 3: 655-666. [DOI] [PubMed] [Google Scholar]
- Craig A, Reiman E, Evans A, Bushnell M (1996) Functional imaging of an illusion of pain. Nature 384: 258-260. [DOI] [PubMed] [Google Scholar]
- Craig AD (2003) Pain mechanisms: labeled lines versus convergence in central processing. Annu Rev Neurosci 26: 1-30. [DOI] [PubMed] [Google Scholar]
- Craig AD, Chen K, Bandy D, Reiman EM (2000) Thermosensory activation of insular cortex. Nat Neurosci 3: 184-190. [DOI] [PubMed] [Google Scholar]
- Dannals R, Ravert H, Frost J, Wilson A, Burns H, Wagner HJ (1985) Radio-synthesis of an opiate receptor binding radiotracer: [11C]carfentanil. Int J Appl Radiat Isot 36: 303-306. [DOI] [PubMed] [Google Scholar]
- Davis K, Wood M, Crawley A, Mikulis D (1995) fMRI of human somatosensory and cingulate cortex during painful electrical nerve stimulation. NeuroReport 7: 321-325. [PubMed] [Google Scholar]
- Davis KD (2000) The neural circuitry of pain as explored with functional MRI. Neurol Res 22: 313-317. [DOI] [PubMed] [Google Scholar]
- de la Fuente-Fernandez R, Ruth TJ, Sossi V, Schulzer M, Calne DB, Stoessl AJ (2001) Expectation and dopamine release: mechanism of the placebo effect in Parkinson's disease. Science 293: 1164-1166. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Price JL, Simpson Jr JR, Todd RD, Reich T, Vannier M, Raichle ME (1997) Subgenual prefrontal cortex abnormalities in mood disorders. Nature 386: 824-827. [DOI] [PubMed] [Google Scholar]
- Drolet G, Dumont EC, Gosselin I, Kinkead R, Laforest S, Trottier JF (2001) Role of endogenous opioid system in the regulation of the stress response. Prog Neuropsychopharmacol Biol Psychiatry 25: 729-741. [DOI] [PubMed] [Google Scholar]
- Filliol D, Ghozland S, Chluba J, Martin M, Matthes H, Simonin F, Befort K, Gaveriaux-Ruff C, Dierich A, Le MM, Valverde O, Maldonado R, Kieffer B (2000) Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet 25: 195-200. [DOI] [PubMed] [Google Scholar]
- Firestone L, Gyulai F, Mintun M, Adler L, Urso K, Winter P (1996) Human brain activity response to fentanyl imaged by positron emission tomography. Anesth Analg 82: 1247-1251. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Frith CD, Liddle PF, Frackowiak RSJ (1991) Comparing functional (PET) images: the assessment of significant change. J Cereb Blood Flow Metab 11: 690-699. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Holmes AP, Worsley KJ, Poline J-P, Frith CD, Frackowiak RSJ (1995) Statistical parametric maps in functional imaging: a general linear approach. Hum Brain Mapp 2: 189-210. [Google Scholar]
- Fuster JM (2000) Executive frontal functions. Exp Brain Res 133: 66-70. [DOI] [PubMed] [Google Scholar]
- Gabilondo A, Meana J, Garcia-Sevilla J (1995) Increased density of muopioid receptors in the postmortem brain of suicide victims. Brain Res 682: 245-250. [DOI] [PubMed] [Google Scholar]
- Gear R, Aley K, Levine J (1999) Pain-induced analgesia mediated by mesolimbic reward circuits. J Neurosci 19: 7175-7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelnar P, Krauss B, Sheehe P, Szeverenyi N, Apkarian A (1999) A comparative fMRI study of cortical representations for thermal painful, vibrotactile, and motor performance tasks. NeuroImage 10: 460-482. [DOI] [PubMed] [Google Scholar]
- George SR, Kertesz M (1987) Met-enkephalin concentrations in striatum respond reciprocally to alterations in dopamine neurotransmission. Peptides 8: 487-492. [DOI] [PubMed] [Google Scholar]
- Goetz CG, Leurgans S, Raman R, Stebbins GT (2000) Objective changes in motor function during placebo treatment in PD. Neurology 54: 710-714. [DOI] [PubMed] [Google Scholar]
- Gracely RH, Dubner R, Wolskee PJ, Deeter WR (1983) Placebo and naloxone can alter post-surgical pain by separate mechanisms. Nature 306: 264-265. [DOI] [PubMed] [Google Scholar]
- Grevert P, Albert L, Goldstein A (1983) Partial antagonism of placebo analgesia by naloxone. Pain 16: 129-143. [DOI] [PubMed] [Google Scholar]
- Gross-Isseroff R, Dillon K, Israeli M, Biegon A (1990) Regionally selective increases in mu opioid receptor density in the brains of suicide victims. Brain Res 530: 312-316. [DOI] [PubMed] [Google Scholar]
- Haber SN, Kunishio K, Mizobuchi M, Lynd-Balta E (1995) The orbital and medial prefrontal circuit through the primate basal ganglia. J Neurosci 15: 4851-4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer RK, Rainville P, Duncan GH, Bushnell MC (2001) Cortical representation of the sensory dimension of pain. J Neurophysiol 86: 402-411. [DOI] [PubMed] [Google Scholar]
- Horvitz J (2000) Mesolimbic and nigrostriatal dopamine responses to salient non-rewarding stimuli. Neuroscience 96: 651-656. [DOI] [PubMed] [Google Scholar]
- Hsieh J, Stone-Elander S, Ingvar M (1999) Anticipatory coping of pain expressed in the human anterior cingulate cortex: a positron emission tomography study. Neurosci Lett 262: 61-64. [DOI] [PubMed] [Google Scholar]
- Jewett D (2001) A simple synthesis of [11C]carfentanil. Nucl Med Biol 28: 733-734. [DOI] [PubMed] [Google Scholar]
- Kalin N, Shelton S, Barksdale C (1988) Opiate modulation of separation-induced distress in non-human primates. Brain Res 440: 285-292. [DOI] [PubMed] [Google Scholar]
- Kerns JG, Cohen JD, MacDonald III AW, Cho RY, Stenger VA, Carter CS (2004) Anterior cingulate conflict monitoring and adjustments in control. Science 303: 1023-1026. [DOI] [PubMed] [Google Scholar]
- Kupers RC, Svensson P, Jensen TS (2004) Central representation of muscle pain and mechanical hyperesthesia in the orofacial region: a positron emission tomography study. Pain 108: 284-293. [DOI] [PubMed] [Google Scholar]
- Levine J, Gordon N, Jones R, Fields H (1978) The narcotic antagonist naloxone enhances clinical pain. Nature 272: 826-827. [DOI] [PubMed] [Google Scholar]
- Levine JD, Gordon NC (1984) Influence of the method of drug administration on analgesic response. Nature 312: 755-756. [DOI] [PubMed] [Google Scholar]
- Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL (1996) Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab 16: 834-840. [DOI] [PubMed] [Google Scholar]
- Lorenz J, Minoshima S, Casey KL (2003) Keeping pain out of mind: the role of the dorsolateral prefrontal cortex in pain modulation. Brain 126: 1079-1091. [DOI] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Akil H, Watson SJ (1995) Opioid-receptor mRNA expression in the rats CNS: anatomical and functional implications. Trends Neurosci 18: 22-29. [DOI] [PubMed] [Google Scholar]
- Matthes HWD, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, LeMeur M, Dolle P, Tzavara E, Hannoune J, Roques BP, Kieffer BL (1996) Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the μ-opioid-receptor gene. Nature 383: 819-823. [DOI] [PubMed] [Google Scholar]
- Mayberg H, Brannan S, Mahurin RK, Jerabek PA, Brickman J, Tekell JL, Silva JA, McGinnis S, Glass T, Martin C, Fox PT (1997) Cingulate function in depression: a potential predictor of treatment response. NeuroReport 8: 1057-1061. [DOI] [PubMed] [Google Scholar]
- McNair D, Lorr M, Droppleman L (1992) EdITS manual for the profile of mood states. San Diego: Educational and Industrial Testing Service (EdITS).
- Melzack R, Torgerson W (1971) On the language of pain. Anesthesiology 34: 50-59. [DOI] [PubMed] [Google Scholar]
- Meyer CR, Boes JL, Kim B, Bland PH, Zasadny KR, Kison PV, Koral K, Frey KA, Wahl RL (1997) Demonstration of accuracy and clinical versatility of mutual information for automatic multimodality image fusion using affine and thin-plate spline warped geometric deformations. Med Image Anal 1: 195-206. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Koeppe RA, Mintun MA, Berger KL, Taylor SF, Frey KA, Kuhl DE (1993) Automated detection of the intercommissural line for stereotactic localization of functional brain images. J Nucl Med 34: 322-329. [PubMed] [Google Scholar]
- Mogil J, Marek P, O'Toole L, Helms M, Sadowski B, Liebeskind J, Belknap J (1994) Mu-opiate receptor binding is up-regulated in mice selectively bred for high stress-induced analgesia. Brain Res 653: 16-22. [DOI] [PubMed] [Google Scholar]
- Moles A, Kieffer BL, D'Amato FR (2004) Deficit in attachment behavior in mice lacking the mu-opioid receptor gene. Science 304: 1983-1986. [DOI] [PubMed] [Google Scholar]
- Nelson EE, Panksepp J (1998) Brain substrates of infant-mother attachment: contributions of opioids, oxytocin, and norepinephrine. Neurosci Biobehav Rev 22: 437-452. [DOI] [PubMed] [Google Scholar]
- Ongur D, An X, Price JL (1998) Prefrontal cortical projections to the hypothalamus in macaque monkeys. J Comp Neurol 401: 480-505. [PubMed] [Google Scholar]
- Petrovic P, Kalso E, Petersson KM, Ingvar M (2002) Placebo and opioid analgesi-imaging a shared neuronal network. Science 295: 1737-1740. [DOI] [PubMed] [Google Scholar]
- Peyron R, Laurent B, Garcia-Larrea L (2000) Functional imaging of brain responses to pain. A review and meta-analysis. Neurophysiol Clin 30: 263-288. [DOI] [PubMed] [Google Scholar]
- Phan K, Wager T, Taylor S, Liberzon I (2002) Functional neuroanatomy of emotion: a meta-analysis of emotion activation studies in PET and fMRI. NeuroImage 16: 331-348. [DOI] [PubMed] [Google Scholar]
- Ploghaus A, Tracey I, Gati J, Clare S, Menon R, Matthews P, Rawlins J (1999) Dissociating pain from its anticipation in the human brain. Science 284: 1979-1981. [DOI] [PubMed] [Google Scholar]
- Price DD, Milling LS, Kirsch I, Duff A, Montgomery GM, Nicholls SS (1999) An analysis of factors that contribute to the magnitude of placebo analgesia in an experimental paradigm. Pain 83: 147-156. [DOI] [PubMed] [Google Scholar]
- Pruessner JC, Champagne F, Meaney MJ, Dagher A (2004) Dopamine release in response to a psychological stress in humans and its relationship to early life maternal care: a positron emission tomography study using [11C]raclopride. J Neurosci 24: 2825-2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainville P, Duncan G, Price D, Carrier B, Bushnell M (1997) Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science 277: 968-971. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Mogil JS, Japon M, Chan EC, Allen RG, Low MJ (1996) Absence of opioid stress-induced analgesia in mice lacking μ-endorphin by site directed mutagenesis. Proc Natl Acad Sci USA 93: 3995-4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatzberg AF, Kraemer HC (2000) Use of placebo control groups in evaluating efficacy of treatment of unipolar major depression. Biol Psychiatry 47: 736-744. [DOI] [PubMed] [Google Scholar]
- Schlaepfer T, Strain E, Greenberg B, Preston K, Lancaster E, Bigelow G, Barta P, Pearlson G (1998) Site of opioid action in the human brain: mu and kappa agonists' subjective and cerebral blood flow effects. Am J Psychiatry 155: 470-473. [DOI] [PubMed] [Google Scholar]
- Shetty N, Friedman JH, Kieburtz K, Marshall FJ, Oakes D (1999) The placebo response in Parkinson's disease. Parkinson Study Group. Clin Neuropharmacol 22: 207-212. [PubMed] [Google Scholar]
- Singer T, Seymour B, O'Doherty J, Kaube H, Dolan RJ, Frith CD (2004) Empathy for pain involves the affective but not sensory components of pain. Science 303: 1157-1162. [DOI] [PubMed] [Google Scholar]
- Smith Y, Zubieta J, Del Carmen M, Dannals R, Ravert H, Zacur H, Frost J (1998) Brain mu opioid receptor measurements by positron emission tomography in normal cycling women: relationship to LH pulsatility and gonadal steroid hormones. J Clin Endocrinol Metab 83: 4498-4505. [DOI] [PubMed] [Google Scholar]
- Sora I, Takahashi N, Funada M, Ujike H, Revay RS, Donovan DM, Miner LL, Uhl GR (1997) Opiate receptor knockout mice define μ receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc Natl Acad Sci USA 94: 1544-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Gerfen CR (1998) Role of dynorphin and enkephalin in the regulation of striatal output pathways and behavior. Exp Brain Res 123: 60-76. [DOI] [PubMed] [Google Scholar]
- Stohler C, Kowalski C (1999) Spatial and temporal summation of sensory and affective dimensions of deep somatic pain. Pain 79: 165-173. [DOI] [PubMed] [Google Scholar]
- Talbot J, Marrett S, Evans A, Meyer E, Bushnell M, Duncan G (1991) Multiple representations of pain in human cerebral cortex. Science 251: 1355-1358. [DOI] [PubMed] [Google Scholar]
- Tolle T, Kaufmann T, Siessmeier T, Lautenbacher S, Berthele A, Munz F, Zieglgansberger W, Willoch F, Schwaiger M, Conrad B, Bartenstein P (1999) Region-specific encoding of sensory and affective components of pain in the human brain: a positron emission tomography correlation analysis. Ann Neurol 45: 40-47. [DOI] [PubMed] [Google Scholar]
- Unterwald EM (2001) Regulation of opioid receptors by cocaine. Ann NY Acad Sci 937: 74-92. [DOI] [PubMed] [Google Scholar]
- Vogt B, Derbyshire S, Jones A (1996) Pain processing in four regions of human cingulate cortex localized with co-registered PET and MR imaging. Eur J Neurosci 8: 1461-1473. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Swanson JM (2004) Dopamine in drug abuse and addiction: results from imaging studies and treatment implications. Mol Psychiatry 9: 557-569. [DOI] [PubMed] [Google Scholar]
- Wager TD, Rilling JK, Smith EE, Sokolik A, Casey KL, Davidson RJ, Kosslyn SM, Rose RM, Cohen JD (2004) Placebo-induced changes in FMRI in the anticipation and experience of pain. Science 303: 1162-1167. [DOI] [PubMed] [Google Scholar]
- Wagner KJ, Willoch F, Kochs EF, Siessmeier T, Tolle TR, Schwaiger M, Bartenstein P (2001) Dose-dependent regional cerebral blood flow changes during remifentanil infusion in humans: a positron emission tomography study. Anesthesiology 94: 732-739. [DOI] [PubMed] [Google Scholar]
- Watkins L, Mayer D (1982) Organization of endogenous opiate and non-opiate pain control systems. Science 216: 1185-1192. [DOI] [PubMed] [Google Scholar]
- Watson D, Clark LA, Tellegen A (1988) Development and validation of brief measures of positive and negative affect: the PANAS scales. J Pers Soc Psychol 54: 1063-1070. [DOI] [PubMed] [Google Scholar]
- Will MJ, Franzblau EB, Kelley AE (2003) Nucleus accumbens μ-opioids regulate intake of a high-fat diet via activation of a distributed brain network. J Neurosci 23: 2882-2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoch F, Rosen G, Tolle T, Oye I, Wester H, Berner N, Schwaiger M, Bartenstein P (2000) Phantom limb pain in the human brain: unraveling neural circuitries of phantom limb sensations using positron emission tomography. Ann Neurol 48: 842-849. [PubMed] [Google Scholar]
- Worsley KJ (1994) Local maxima and the expected euler characteristic of excursion sets of x2, F and t fields. Adv Appl Prob 26: 13-42. [Google Scholar]
- Zald DH, Boileau I, El-Dearedy W, Gunn R, McGlone F, Dichter GS, Dagher A (2004) Dopamine transmission in the human striatum during monetary reward tasks. J Neurosci 24: 4105-4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ashton-Miller JA, Stohler CS (1993) A closed-loop system for maintaining constant experimental muscle pain in man. IEEE Trans Biomed Eng 40: 344-352. [DOI] [PubMed] [Google Scholar]
- Zink CF, Pagnoni G, Martin-Skurski ME, Chappelow JC, Berns GS (2004) Human striatal responses to monetary reward depend on saliency. Neuron 42: 509-517. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Dannals R, Frost J (1999) Gender and age influences on human brain mu opioid receptor binding measured by PET. Am J Psychiatry 156: 842-848. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Smith Y, Bueller J, Xu Y, Kilbourn M, Meyer C, Koeppe R, Stohler C (2001) Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science 293: 311-315. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Smith Y, Bueller J, Xu Y, Woike T, Kilbourn M, Meyer C, Koeppe R, Stohler C (2002) μ-opioid receptor mediated antinociception differs in men and women. J Neurosci 22: 5100-5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubieta JK, Heitzeg MM, Smith YR, Bueller JA, Xu K, Xu Y, Koeppe RA, Stohler CS, Goldman D (2003a) COMT val158met genotype affects muopioid neurotransmitter responses to a pain stressor. Science 299: 1240-1243. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Ketter TA, Bueller JA, Xu Y, Kilbourn MR, Young EA, Koeppe RA (2003b) Regulation of human affective responses by anterior cingulate and limbic mu-opioid neurotransmission. Arch Gen Psychiatry 60: 1145-1153. [DOI] [PubMed] [Google Scholar]