

Abstract
The whole-cell patch-clamp technique was used to record current responses to nucleotides and nucleosides in human embryonic kidney HEK293 cells transfected with the human purinergic P2X3 receptor. When guanosine 5′-O-(3-thiodiphosphate) was included into the pipette solution, UTP at concentrations that did not alter the holding current facilitated the α,β-methylene ATP (α,β-meATP)-induced current. ATP and GTP, but not UDP or uridine, had an effect similar to that of UTP. Compounds known to activate protein kinase C (PKC) acted like the nucleoside triphosphates investigated, whereas various PKC inhibitors invariably reduced the effects of both PKC activators and UTP. The substitution by Ala of Ser/Thr residues situated within PKC consensus sites of the P2X3 receptor ectodomain either abolished (PKC2 and PKC3; T134A, S178A) or did not alter (PKC4 and PKC6; T196A, S269A) the UTP-induced potentiation of the α,β-meATP current. Both the blockade of ecto-protein kinase C activity and the substitution of Thr-134 or Ser-178 by Ala depressed the maximum of the concentration-response curve for α,β-meATP without altering the EC50 values. Molecular simulation of the P2X3 receptor structure indicated no overlap between assumed nucleotide binding domains and the relevant phosphorylation sites PKC2 and PKC3. α,β-meATP-induced currents through native homomeric P2X3 receptors of rat dorsal root ganglia were also facilitated by UTP. In conclusion, it is suggested that low concentrations of endogenous nucleotides in the extracellular space may prime the sensitivity of P2X3 receptors toward the effect of subsequently applied (released) higher agonistic concentrations. The priming effect of nucleotides might be attributable to a phosphorylation of PKC sites at the ectodomain of P2X3 receptors.
Keywords: nucleotides, ecto-protein kinase C, P2X3 receptor phosphorylation, site-directed mutagenesis, mutant, analgesia
Introduction
It has been known for a number of years that the conductance of ionotropic excitatory and inhibitory amino acid receptors is modulated by a range of protein kinases (Moss and Smart, 1996; Swope et al., 1999; Köles et al., 2001). Although these effects are mostly attributable to phosphorylation of intracellular receptor sites, the additional involvement of ecto-protein kinases by phosphorylating extracellular receptor sites or accessory membrane proteins has also been discussed repeatedly (Wieraszko and Ehrlich, 1994; Ehrlich and Kornecki, 1999). Hence, external and especially internal ATP may fulfill its classic function as a phosphate donor. However, extracellular ATP is also an agonist at purinergic P2 receptors of the P2X (ligand-gated cationic channel) and P2Y (G-protein-coupled receptor) types (Abbracchio and Burnstock, 1994). P2X receptors form a family of seven subunits (P2X1-P2X7) that have two transmembrane domains, a large extracellular loop containing the ATP binding site, as well as intracellular N- and C-terminal tails (Illes and Ribeiro, 2004). In consequence, a dual regulation of P2X receptors by ATP acting both as an agonist and a phosphate donor has to be considered as a likely possibility.
Primary sensory neurons are endowed with homomeric P2X3 and heteromeric P2X2/3 receptors, of which only the P2X3 receptor channels respond to ATP or its structure analog α,β-methylene ATP (α,β-meATP) with rapidly desensitizing membrane currents (Chen et al., 1995; Chizh and Illes, 2001). Hence, ATP released by noxious stimuli into the extracellular space activates P2X3 and P2X2/3 receptors situated at the peripheral terminals of dorsal root ganglion (DRG) neurons and thereby initiates pain sensation (Chizh and Illes, 2001). It has been demonstrated that P2X3 receptors may be positively modulated by the inflammatory mediators bradykinin and substance P (Paukert et al., 2001). The supposed mechanism of action is the phosphorylation of an N-terminal protein kinase C (PKC) site or alternatively of an associated protein.
The N-terminal phosphorylation of P2X receptors may, on the one hand, enhance the rate of desensitization and thereby also modulate the peak current amplitude [P2X2 (Boue-Grabot et al., 2000)], and, on the other hand, it may decrease the membrane expression of this receptor by interfering with its internalization [P2X5 (Jensik and Thomas, 2002)]. It has been hypothesized that the completely conserved PKC phosphorylation site of the P2X3 signaling peptide within the subsequence 1-44 is essential to pass the endoplasmatic reticulum or the plasma membrane (Mager et al., 2004).
The aim of the present study was to find out whether ecto-PKCs are able to modulate human (h) recombinant P2X3 receptor channels by phosphorylating consensus PKC sites at the extracellular loop of this receptor (Mager et al., 2004). hP2X3 receptors transfected into human embryonic kidney HEK293 cells were superfused with relatively low concentrations of UTP, which fail to activate the receptor but supply terminal phosphate residues for phosphorylation reactions. Under conditions when the crosstalk between the transfected P2X and the endogenous P2Y receptors was inhibited, UTP potentiated the α,β-meATP current via involvement of an ecto-PKC.
Materials and Methods
Site-directed mutagenesis and transfection procedures. Methods of maintenance of HEK293 cells and stable transfection of them with hP2X3 receptor cDNA have been described previously (Fischer et al., 2003). The cDNA of the wild-type (WT) P2X3 receptor (Dr. J. N. Wood, University College London, London, UK) was cloned into the bicistronic vector pIRES2/EGFP (Clontech, Heidelberg, Germany), containing the cDNA of enhanced green fluorescence protein to enable the identification of efficiently transfected cells under a fluorescence microscope. This P2X3 receptor construct was used as a template for production of plasmids containing the described amino acid residue mutations. Transient transfection of HEK293 cells with the prepared plasmids was performed with Polyfect transfection reagent (Qiagen, Hilden, Germany) according to the guidelines of the manufacturer. For this purpose, native HEK293 cells were cultured without the supply of geneticin.
Preparation of DRG neuronal cultures. One-day-old Wistar rats (own breed; WIST/Lei) were killed under CO2 and decapitated to obtain cell cultures of DRG neurons for electrophysiology. The isolation and culturing procedures of thoracic and lumbar DRG cells have been described in detail previously (Himmel et al., 2002). These cells were plated at a density of 2 × 104 cells/ml onto poly-l-lysine-coated (25 μg/ml) glass coverslips.
Whole-cell patch-clamp recordings. Whole-cell patch-clamp recordings were performed 2-6 d after the splitting of stable transfected HEK293 cells and 2-3 d after their transient transfection, at room temperature (20-22°C), using an Axopatch 200B patch-clamp amplifier (Molecular Devices, Union City, CA). Primary cultures of rat DRG neurons were maintained for 2-4 d in a humidified atmosphere with 5% CO2 before experimentation. The pipette solution contained the following for both HEK293 cells and DRG neurons (in mm): 140 CsCl, 1 MgCl2, 2 CaCl2,10 HEPES, and 11 EGTA, pH was adjusted to 7.4 using CsOH. As stated below, Li-GTP (300 μm), guanosine 5′-O-(3-thiotriphosphate) (GTP-γ-S) (300 μm), or guanosine 5′-O-(3-thiodiphosphate) (GDP-β-S) (300 μm), respectively, were added to the pipette solution. The pipette resistances were 3-6 MΩ. After the whole-cell configuration was established, an equilibrium period of 10 min was allowed to elapse for establishing adequate solution exchange between the patch pipette and the cell. All recordings were made at a holding potential of -60 mV. Data were filtered at 2 kHz with the inbuilt filter of the Axopatch 200B, digitized at 5 kHz, and stored on a laboratory computer using a Digidata 1200 interface and pClamp 8.0 software (Molecular Devices).
Drugs were dissolved in an external solution containing the following (in mm): 135 NaCl, 4.5 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, and 10 glucose, pH was adjusted to 7.4 using NaOH and applied by pressure, locally to single cells, using a DAD12 superfusion system (Adams and List, Westbury, NY). To block the firing of DRG neurons during agonist application, tetrodotoxin (0.5 μm) was added to the extracellular solution. Between drug application, cells were continuously superfused with the standard external solution by one pressure-independent valve of this system. In most experiments, α,β-meATP was applied six times for 1 s each with an interval of 5 min between two applications, with the exception of the second and third application, which were spaced apart by 10 min. Unless otherwise stated, UTP [UDP, uridine, GTP, phorbol 12-myristate 13-acetate (PMA), and diacylglycerol-lactone (DAG-lactone)] superfusion started 5 min after the second α,β-meATP application and lasted for a total of 10 min with subsequent washout. Superfusion with PKC inhibitors [12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo(2,3-a)pyrrolo(3,4-c)-carbazole (Gö 6976), staurosporine, K252b, and PKC inhibitor peptide] and procedures known to block the PKC-mediated phosphorylation reaction (Mg2+-free external medium) started 10 min before the first application of α,β-meATP and continued for the whole duration of the experiment. Because the amplitudes of the α,β-meATP-induced currents showed great variability, the results were expressed as a percentage of the second control current (the last current before UTP application).
Concentration-response curves for α,β-meATP were fitted using the following logistic function (SigmaPlot; SPSS, Erkrath, Germany): I = Imax - Imin)[1 + (IC50 + agonist)n], where I is the steady-state inhibition produced by the agonist, Imax and Imin are the maximal and minimal inhibition, respectively, n is the Hill coefficient, and IC50 is the concentration of agonist producing 50% of Imax.
Structural bioinformatics. The amino acid sequences of the species-dependent P2X receptor subunits have been taken from the Swiss Protein Sequence Database (Swiss-Prot; Swiss Institute of Bioinformatics and European Bioinformatics Institute, EMBL; primary accession number of rat P2X3, P49654; primary accession number of human P2X3, P56373). The input coordinates of the spatial structure of rat P2X3 were obtained by profile-based neural network prediction. The resulting structure was geometry optimized using the quantum chemistry RHF/3-21G minimal basic set (Gaussian94; Gaussian, Pittsburgh, PA) and the all-atom molecular mechanics AMBER force field. All hydrogen bonds have been included into the calculation. It was assumed that P2X3 has five disulfide bonds (C117-C165, C126-C149, C132-C159, C217-C227, and C261-C270). A dielectric constant of ϵ = 3.5 was used to simulate an apolar environment, the behavior of the membrane-embedded P2X3 subunit, and the interior of the protein. The template rat P2X3 protein was converted to human P2X3 by the replacement of 25 residues. To avoid interactions between the nonbonded atoms by a sharp cutoff that can cause discontinuities in the potential surface, a smoothing applied from the inner radius (10 Å) to the outer radius (14 Å) was used. The structure was refined using a conjugate gradient minimizer (Fletcher-Reeves modification of the Polak-Ribiere method). Convergence was obtained when the gradient root mean square (RMS) was RMS ≤ 0.05 kcal/Å × mol (HyperChem; Hypercube, Waterloo, Ontario, Canada). The results of the geometry optimization were checked by the Ramanchandran plot, rotamer analysis, all-atom contact dots, and the C(β) deviation. UTP was docked into a putative nucleotide binding domain (NBD). Naturally occurring attachment sites were predicted by the ScanPROSITE regular expression search via the interface of PROSITE. Additional details of the molecular modeling techniques have been described previously (Mager et al., 2004).
Materials and drugs. The following pharmacological agents were used: ATP, α,β-meATP, UTP, UDP, uridine, GTP, GDP-β-S, GTP-γ-S, PMA (Sigma, Deisenhofen, Germany), Gö 6976, K252b, protein kinase C inhibitor peptide 19-36, staurosporine, and DAG-lactone (Calbiochem, Schwalbach, Germany).
All drugs were prepared as a concentrated stock solution in distilled water or DMSO (DAG-lactone, Gö 6976, K252b, PMA, and staurosporine) and were diluted to final concentration in external medium. The final concentration of DMSO did not exceed 0.1%.
Data analysis. Data were analyzed off-line using pClamp 8.0 software (Molecular Devices). Figures show mean ± SEM values of n experiments. Kruskal-Wallis ANOVA on ranks followed by either the Student's t test or the Bonferroni's t test were used for statistical analysis. p values of <0.05 were considered to reflect a significant difference.
Results
Potentiation by UTP of the α,β-meATP current amplitude in HEK293-hP2X3 cells and interaction with protein kinase C inhibitors
Although in HEK293-hP2X3 cells, ATP, α,β-meATP, and UTP caused, at a holding potential of -60 mV, inward currents, the threshold concentrations of ATP and α,β-meATP were >0.3 μm, whereas the threshold concentration of UTP was considerably higher (>10 μm) (Fischer et al., 2003). In accordance with these results, UTP (3 μm) did not evoke a current response by itself and also failed to alter the effect of α,β-meATP (3 μm; 1 s superfusion time) (Fig. 1Aa,Ab). However, when the usual GTP (300 μm) in the pipette solution was replaced by its enzymatically stable analog GDP-β-S (300 μm), UTP (3 μm) still continued of being ineffective on the holding current but markedly potentiated the response to α,β-meATP (Fig. 1Ba,Bb). Furthermore, α,β-meATP on its second application after a 10 min superfusion with UTP caused considerably less inward current than on its first application after a 5 min superfusion with UTP. Although we did not investigate the reasons for this phenomenon, it may be attributable to an accelerated desensitization rate of the α,β-meATP effect after a longer-lasting contact with UTP. The effect of UTP was in the range of 0.3-3000 nm, clearly concentration dependent with a threshold at 300 nm and a maximum at 3 μm (Fig. 1Ca). Based on these results, we have chosen for all additional experiments 3 μm UTP.
Figure 1.

Effects of extracellular UTP on α,β-meATP-induced currents in HEK293 cells permanently transfected with the human P2X3 receptor (HEK293-hP2X3 cells). The whole-cell variant of the patch-clamp method was used to record membrane currents. Cells were held at -60 mV. Both α,β-meATP and UTP were locally applied by means of a rapid superfusion system. α,β-meATP was applied six times for 1 s each with an interval of 5 min between two applications, with the exception of the second and third application, which were spaced apart by 10 min. Superfusion with UTP started 5 min (A, B, Ca) or variable intervals before the third application of α,β-meATP (1-5 min; Cb) and was stopped immediately after the fourth application of α,β-meATP. Aa, Original tracing recorded with a patch pipette filled with GTP (300 μm)-containing solution. UTP (3 μm) did not alter the α,β-meATP (3 μm) current. Ab, Mean ± SEM of eight experiments similar to that shown in Aa. Ba, Original tracing recorded with a patch pipette filled with solution containing GDP-β-S (3 μm) instead of GTP. UTP (3 μm) markedly facilitated the α,β-meATP (3 μm)-induced current. Bb, Mean ± SEM of seven experiments similar to that shown in Ba. *p < 0.05, statistically significant difference from the preceding column. Ca, Concentration-response relationship for the potentiating effect of UTP on the α,β-meATP (3 μm)-induced current. Mean ± SEM of four to eight experiments. Cb, Dependence of the potentiating effect of UTP (3 μm) on its superfusion time before the application of α,β-meATP. Mean ± SEM of four to seven experiments. *p < 0.05, statistically significant difference from 100%.
Finally, we asked ourselves about the length of the time period required for the action of UTP (3 μm) to develop. For this reason, we gradually prolonged the superfusion time of UTP before the first application of α,β-meATP (3 μm) and found that the UTP effect increased from 3 to 5 min (Fig. 1Cb). Hence, the time course of the UTP effect appeared to exclude the classic P2X and P2Y receptor-mediated reactions, which usually evolve within the range of milliseconds and seconds rather than minutes.
HEK293 cells have been shown to possess a range of endogenous P2Y receptors that preferentially react either to ADP (P2Y1, P2Y13) or UTP (P2Y4) (Fischer et al., 2003). Because GDP-β-S is known to inhibit G-protein-mediated second-messenger mechanisms (Sternweis and Pang, 1990) of, for example, P2Y receptors (Ralevic and Burnstock, 1998), it has been suggested that endogenous P2Y receptors of HEK293 cells may negatively interact with the exogenous hP2X3 receptor (Fischer et al., 2003). In the present study, UTP may exert both a P2Y4 receptor-mediated depression and a hitherto unknown facilitation of P2X3 receptors. Hence, in all subsequent experiments, GDP-β-S was routinely included into the pipette solution to exclude a possible P2Y/P2X receptor interaction.
Then, we attempted to roughly clarify the structural requirements for the observed potentiation of the α,β-meATP-induced current by testing identical concentrations (3 μm) of selected nucleotides and nucleosides. Apparently, only UTP and GTP were equally facilitatory, whereas UDP and uridine were without effect (Fig. 2Aa-Ac,B). Thus, the complete nucleoside triphosphate structure of UTP and GTP appears to be required for potentiating the α,β-meATP current in HEK293-hP2X3 cells. Eventually, ATP in a low concentration (10 nm), much below that used to activate hP2X3 receptors (>300 nm) (Fischer et al., 2003), also potentiated the α,β-meATP (3 μm) current (Fig. 2Ad,B). These findings show that possibly all nucleotides investigated may prime P2X3 receptors toward agonist effects.
Figure 2.
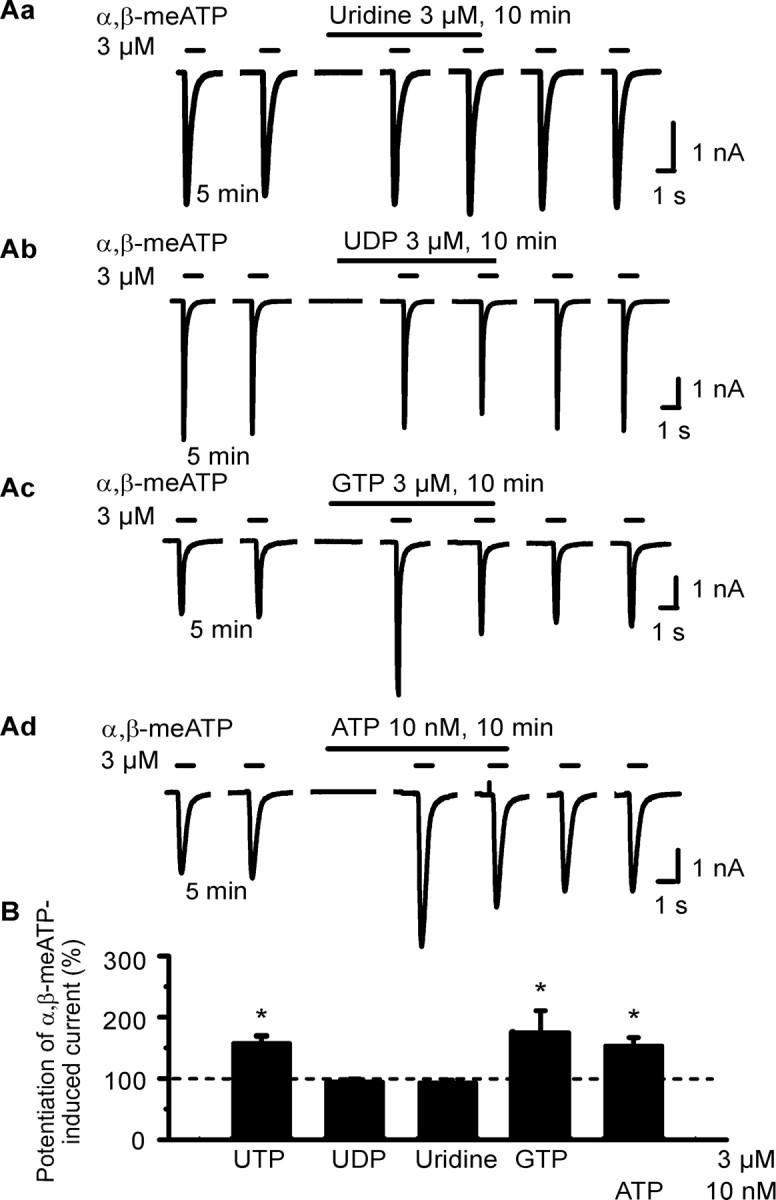
Effects of extracellular nucleotides and nucleosides on α,β-meATP-induced currents in HEK293-hP2X3 cells. The experimental procedures were similar to those described for Figure 1 B. Patch pipettes filled with GDP-β-S (300 μm)-containing solution were used. Superfusion with agonists started 5 min before the third application of α,β-meATP. A, Original tracings show that the α,β-meATP currents were potentiated by GTP (3 μm; Ac) and ATP (10 nm; Ad) but not uridine (Aa) or UDP (3 μm each; Ab). B, Mean ± SEM of 4-11 experiments similar to those shown in A.*p < 0.05, statistically significant difference from 100%.
In view of the presence of consensus phosphorylation sites in the extracellular loop of the P2X3 receptor (Mager et al., 2004), we hypothesized about the functional significance of these sites as potential targets of ecto-PKC-induced phosphorylation. Initially, two selective PKC activators were superfused with a protocol also used for the application of nucleotides. Both DAG-lactone (1 μm) and PMA (0.1 μm) facilitated the current response to α,β-meATP (Fig. 3Aa,B). In contrast to the UTP- and GTP-induced potentiation, this effect was not reversible on washout for 10 min, in good agreement with the pronounced lipophilic character of these compounds. Furthermore, superfusion with the selective PKC inhibitor Gö 6976 (0.3 μm), 10 min before the first application of α,β-meATP and throughout the whole experiment, abolished the effect of DAG-lactone (Fig. 3Ab,B). The bisindolylmaleimide Gö 6976 has been described previously to exhibit exceptionally high potency in blocking the PKCβ and PKCγ isoforms, which belong to the conventional and novel groups of PKCs, respectively (Way et al., 2000). These results strongly suggest that a PKC isoform is involved in the potentiation of P2X3 receptor-mediated currents, but, considering the capability of PMA, DAG-lactone, and Gö 6976 to pass the cell membrane, they give no information about the extracellular or intracellular localization of the phosphorylation site.
Figure 3.

Effects of PKC activators on α,β-meATP-induced currents in HEK293-hP2X3 cells; interaction of PKC inhibitors with PKC activators and UTP. The experimental procedures were similar to those described for Figure 1 B. Patch pipettes filled with GDP-β-S (300 μm) containing solution were used. Superfusion with PKC activators (PMA and DAG-lactone) started 5 min before the third application of α,β-meATP. Superfusion with PKC inhibitors (Gö 6976, staurosporine, K252b, and PKC inhibitor peptide) and procedures known to block the PKC-mediated phosphorylation reaction (Mg2+-free external medium) started 10 min before the first application of α,β-meATP and continued for the duration of the whole experiment. A, Original tracings show that the α,β-meATP (3 μm) currents were potentiated by DAG-lactone (1 μm;Aa) in the absence of Gö 6976 (0.3 μm) but not in its presence (Ab). B, Potentiating effect of PMA (0.1 μm) and DAG-lactone alone, but not DAG-lactone in combination with Gö 6976, on the α,β-meATP current. Mean ± SEM of 7-12 experiments similar to those shown in A. C, Blockade of the potentiating effect of UTP (3 μm) on the α,β-meATP (3 μm) current by staurosporine (0.1 μm), K252b (0.2 μm), PKC inhibitor peptide (10 μm), Gö 6976 (0.3 μm), and a nominally Mg2+-free external medium. Mean ± SEM of 6-10 experiments similar to those shown in A. *p < 0.05, statistically significant difference from 100% both in B and C.
In the following experiments, we investigated the interaction of UTP with a number of compounds known to nonselectively or selectively inhibit the activity of PKC. Because polar substances such as UTP are unable to pass the cell membrane by simple diffusion and inward nucleotide transporters were not described to operate in HEK293 cells, it is highly probable that UTP acts at an extracellular site. Regardless of this fact, staurosporine (0.1 μm), a wide-range inhibitor of various protein kinases, and a nominally Mg2+-free medium, which is known to interfere with protein kinase-induced phosphorylation reactions (Christie et al., 1992), abolished the UTP effect (Fig. 3C). The selective, membrane-permeable, PKC inhibitor Gö 6976, the nonselective ecto-PKC-inhibitor K252b (0.2 μm) (Kase et al., 1987), and the selective, membrane-impermeable PKC inhibitor peptide (10 μm) all depressed the UTP-induced potentiation of the response to α,β-meATP (Fig. 3C). In conclusion, UTP appears to act via an ecto-PKC-mediated transfer of its terminal phosphate group onto extracellular phosphorylation sites of the P2X3 receptor.
Effects of mutations of Ser and Thr residues of the hP2X3 receptor on the UTP-induced potentiation of the current response to α,β-meATP
In contrast to the previous experiments, HEK293 cells were not permanently but transiently transfected with the hP2X3 receptor or its mutants. To identify the functionally relevant PKC phosphorylation site(s) at the hP2X3 receptor, Ser and Thr residues situated within five of the seven consensus sites (see Fig. 7A) were consecutively mutated to the neutral amino acid Ala. An intracellular PKC site highly conserved in all P2X subunits is located N terminally (PKC1, 12-14), whereas additional sites are extracellularly localized (PKC2, 134-136; PKC3, 178-180; PKC4, 196-198; and PKC6, 269-271) (Mager et al., 2004). Furthermore, two Ser residues (S110, S267) that are present in the human but not in the rat P2X3 receptor were also mutated to Ala.
Figure 7.
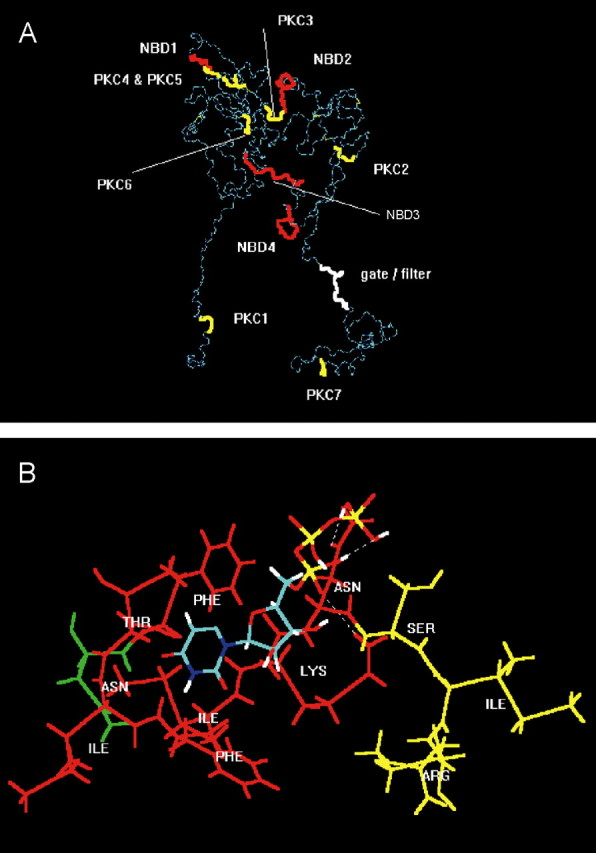
Molecular simulation of nucleotide binding sites and consensus PKC phosphorylation sites at the P2X3 receptor. A, For visual clarity, the backbone structure (in dark and light blue) of the geometry optimized, monomeric, charged, membrane-embedded, unoccupied human P2X3 glycoprotein receptor is shown. The structure is viewed parallel to the plane of a membrane. A five-disulfide core (thin yellow lines) has been assumed. The extracellular domain forms a twisted loop that is solvent dependent. The intracellularly occurring N- and C-terminal PKC phosphorylation sites (thick yellow) are denoted by PKC1 (amino acids in positions 12-14) and PKC7 (365-367); the extracellular PKC phosphorylation sites are denoted by PKC2-PKC6 (134-136, 178-180, 196-198, 202-204, and 269-271). The extracellularly occurring potential NBDs (red) are denoted by NBD1-NBD4 (62-66, 171-177, 279-286, and 295-302). The potential filter (white) is probably formed by the residues in positions 333-341. The fully functional receptor is a 3n homomeric (n = 1, 2,...) ion channel. B, Model of a putative interaction of UTP (colors: green, carbon; red, oxygen; white, hydrogen; blue, nitrogen; yellow, phosphorus) with the second putative nucleotide binding domain NBD2 of human P2X3 (Phe-Thr-Ile-Phe-Lys-Asn, 171-177; red), which is in steric neighborhood of the PKC phosphorylation site PKC3 (amino acids Ser-Ile-Arg in positions 178-180; yellow). The first residue (Asn, green) of the N-glycosylation attachment site (Asn-Phe-Thr-Ile, 170-173) is also shown. The uridine ring of UTP is bound into a lipophilic pocket (Phe171, Ile175); the oxygen atoms of the phosphate groups form hydrogen bonds with the hydrogen donating groups of the residues of Lys-176 and Asn-177.
None of the mutations abolished the membrane expression of the hP2X3 receptor in HEK293 cells or its sensitivity to extracellular nucleotides, because the application of α,β-meATP (3 μm) caused reproducible current amplitudes in all cases (for the second current response in the usual series, see Fig. 4B). T13A, S267A, and S269A increased the α,β-meATP-induced current amplitudes, whereas T134A and N177V decreased them when compared with the wild-type receptor (Fig. 4B). However, these changes should not be over-interpreted in view of the variable expression of receptor protein after transient transfection procedures. It is noteworthy that the substitution of Thr at position 12 with Ala was previously reported to result in no measurable current, by probably interfering with the P2X3 channel expression in the cell membrane (Paukert et al., 2001). Therefore, we mutated Thr only at position 13 to Ala; this procedure did not alter the current amplitude.
Figure 4.

Mutation of Ser and Thr residues in consensus PKC phosphorylation sites of hP2X3 receptors alters the sensitivity of these receptors toward α,β-meATP in HEK293-P2X3 cells. A transient transfection protocol was used. The experimental procedures were similar to those described for Figure 1 B. Patch pipettes filled with GDP-β-S (300 μm)-containing solution were used. Superfusion with UTP (300 μm) started 5 min before the third application of α,β-meATP. A, Original tracings show that the α,β-meATP-induced currents were potentiated by UTP (3 μm) in the wild-type hP2X3 receptor (Aa) but not in its mutant T134A (Ab). B, Amplitudes of α,β-meATP (3 μm) currents recorded from HEK293 cells transfected with the wild-type hP2X3 receptor or its mutants. Mean ± SEM of 7-14 experiments. C, Potentiation of α,β-meATP (3 μm) current amplitudes by UTP (3 μm) evoked at wild-type hP2X3 receptors but not at some of their mutants. Mean ± SEM of seven to eight experiments similar to those shown in Aa and Ab. Substitution of Ser and Thr residues localized within the extracellular consensus PKC sites near transmembrane domain 2 with Ala (T196A, S269A) failed to interfere with the facilitatory effect of UTP. In contrast, identical mutations in the Ser and Thr residues localized within the two extracellular consensus PKC sites near transmembrane domain 1 (T134A, S178A) abolished the action of UTP. *p < 0.05, statistically significant difference from the wild-type receptor (B) or from 100% (C). The comparison of A and B indicates that all mutants react to agonist application with stable current responses, and there is no correlation between the magnitude of these currents and the ability of UTP to facilitate them.
When α,β-meATP (3 μm) was superfused six times for 1 s each and according to the usual protocol onto HEK293 cells transiently transfected with the hP2X3 receptor (Fig. 4Aa), UTP (3 μm) tended to cause less potentiation of current amplitudes after an application time of 5 min (43.6 ± 21.1%; n = 7) (Fig. 4B) than in HEK293 cells permanently transfected with the same receptor (66.4 ± 17.1%; n = 7) (Figs. 1Bb, 5A,B). UTP continued to facilitate the α,β-meATP current at the mutants S110A and S267A as well as at the mutants T196A and S269A (Fig. 4C). Hence, mutations in the Ser residues present in the human but not in the rat P2X3 receptor (S110A, S267A) or in the Thr and Ser residues localized within the extracellular consensus PKC sites PKC4 and PKC6 (T196A, S269A) failed to interfere with the facilitatory effect of UTP. In contrast, mutations in the Thr and Ser residues localized within the two extracellular consensus PKC sites PKC2 and PKC3 (T134A, S178A) abolished the action of UTP. Both the double substitution of Thr-134 and Ser-178 with Ala (T134A+S178A) as well as the substitution of Asn-177 besides the critical Ser-178 with Val (N177V) resulted in UTP-resistant mutants (Fig. 4C).
Figure 5.

Depression of α,β-meATP effects at wild-type hP2X3 receptor after ecto-PKC inhibition or mutation of consensus ecto-PKC phosphorylation sites. Although lower concentrations (0.03-0.3 μm) of α,β-meATP were superfused every 2 min, the higher concentrations (3-300 μm) were superfused every 5-7 min for 1 s each and onto the same HEK293 cell permanently or transiently transfected with hP2X3 receptors or their mutants T134A, S178A, and T134A+S178A. Mean ± SEM of five to seven experiments. A, Concentration-response curve for α,β-meATP in the absence and presence of K252b (2 μm). HEK293 cells permanently transfected with the wild-type hP2X3 receptor. Superfusion with K252b started 10 min before the first application of α,β-meATP and continued for the duration of the whole experiment. •, Control (Emax, 7127 ± 516 pA; EC50, 1.6 ± 0.6 μm; Hill coefficient, 1.5). ▪, K252b (0.2 μm) (Emax, 5411 ± 395 pA; EC50, 2.6 ± 0.8 μm; Hill coefficient, 1.2). *p < 0.05, statistically significant difference from the Emax value of the control curve. B, Concentration-response curve for α,β-meATP. HEK293 cells were transiently transfected with the hP2X3 receptor or its mutants T134A, S178A, and T134A+S178A. •, Wild-type (Emax, 8682 ± 858 pA; EC50, 7.3 ± 2.7 μm; Hill coefficient, 1.3). ▴, T134A (Emax, 6600 ± 1607 pA; EC50, 3.3 ± 1.4 μm; Hill coefficient, 0.7). ▾, T134A+S178A (Emax, 3889 ± 538 pA; EC50, 4.9 ± 3.0 μm; Hill coefficient, 1.3). ▪, S178A (Emax, 2842 ± 217 pA; EC50, 4.0 ± 1.3 μm; Hill coefficient, 1.1). *p < 0.05, statistically significant difference from the Emax value of the wild-type curve (comparison of all 4 groups). +p < 0.05, statistically significant difference from the Emax value of the wild-type curve (comparison of two groups). The EC50 values of the individual concentration-response curves in A and B did not differ from each other in a statistically significant manner.
Interestingly, the intracellular N-terminal mutant T13A did not react to UTP either. This is an unexpected finding, because UTP is expected to phosphorylate Thr residues of the extracellular loop only (Fig. 4C). In this case, either the missing basal phosphorylation of this site or alterations in receptor structure and a consequently decreased affinity/accessibility for UTP may be possible reasons.
Depression of the maximum current response to α,β-meATP by protein kinase C inhibitors or by mutation of Ser and/or Thr residues of the hP2X3 receptor
Previous experiments documented that the amplitude of current responses to α,β-meATP at 3 μm measured in the WT P2X3 receptor and its five Ala mutants exhibited a considerable variability (Fig. 4B). The important observation was, however, that all mutants reacted to agonist application with stable current responses, and there was no correlation between the magnitude of these currents and the ability of UTP to facilitate them. Now we constructed complete concentration-response curves for α,β-meATP (0.03-300 μm) by using HEK293 cells permanently transfected with the WT P2X3 receptor, in both the absence and presence of the ecto-PKC inhibitor K252b (0.2 μm) (Fig. 5A). K252b depressed the maximum current response to α,β-meATP but left its EC50 value unaffected, suggesting that a basal ecto-phosphorylation of P2X3 receptors is necessary for full agonistic activity, despite similar agonist potency.
Then, concentration-response curves for α,β-meATP were constructed in HEK293 cells transiently transfected with the WT P2X3 receptor or its mutants T134A, S178A, and T134A+S178A (Fig. 5B). Previous experiments identified these Thr and Ser residues as phosphorylation sites for ecto-protein kinase C. In fact, α,β-meATP caused lower maximum current amplitudes accompanied by similar EC50 values at the two single mutants or the double mutant than at WT P2X3 receptors. Hence, both the blockade of an ecto-PKC and mutation of the relevant phosphorylation sites of P2X3 receptors equally decreased the maximum effect of α,β-meATP but did not alter its agonist potency. Thus, it is most likely that a missing basal phosphorylation of the receptor protein leads to a restricted agonist responsibility.
In conclusion, we did not investigate whether Ala substitution of Ser or Thr residues in two consensus PKC phosphorylation sites (T134, S178) of hP2X3 receptors interferes with either receptor synthesis or the insertion of the receptor into the plasma membrane. However, the standard concentration of α,β-meATP used by us (3 μm) was for both the WT receptor and all of its mutants investigated submaximal, and therefore the conclusions drawn for the UTP-induced potentiation of current amplitudes are certainly unaffected by these possible complicating factors.
Potentiation by UTP of the α,β-meATP current amplitude in rat DRG neurons and interaction with protein kinase C inhibitors
Small-diameter nociceptive neurons of the rat DRG possess either rapidly desensitizing P2X3 homomeric receptors or slowly desensitizing P2X2/3 heteromeric receptors (Chizh and Illes, 2001). For the following experiments, we have chosen cells that reacted to α,β-meATP (3 μm) with current responses that belong to the transient (Fig. 6Ba) or biphasic (Fig. 6Aa, P2X3 each), but not the sustained (P2X2/3), types (Dunn et al., 2001). UTP at concentrations of >3 μm induced rapidly desensitizing inward current (data not shown) (Robertson et al., 1996). However, UTP at a lower concentration of 30 nm invariably potentiated the effect of α,β-meATP (Fig. 6Aa,Ab) regardless of whether the current response completely or only partially desensitized during a 1 s superfusion period. In the presence of the PKC inhibitor Gö 6976 (0.3 μm), UTP failed to produce the expected potentiation (Fig. 6Ba,Bb). Hence, UTP may act by phosphorylating native P2X3 receptor subunits in a manner similar to that described previously for their recombinant counterparts. It is noteworthy that the relatively few DRG neurons assumedly endowed with P2X2/3 heteromeric receptors and not included into the above evaluation also reacted to UTP (3 μm) with a potentiation of α,β-meATP (3 μm)-induced currents (data not shown).
Figure 6.

Effects of extracellular UTP on α,β-meATP-induced currents in rat cultured DRG neurons. The whole-cell variant of the patch-clamp method was used to record membrane currents. Cells were held at -60 mV. α,β-meATP and UTP were locally applied by an experimental protocol similar to that used in Figure 1 B. The patch pipettes were filled with a GDP-β-S (300 μm)-containing solution. Aa, Facilitation by UTP (3 μm) of the α,β-meATP (3 μm)-induced current; original tracing. Ab, Mean ± SEM of seven experiments similar to that shown in Aa. Ba, Failure of UTP (3 μm) to facilitate the α,β-meATP (3 μm)-induced current in the presence of the PKC-inhibitor Gö 6976; original tracing. Superfusion with Gö 6976 started 10 min before the first application of α,β-meATP and continued for the duration of the whole experiment. Bb, Mean ± SEM of eight experiments similar to that shown in Ba. *p < 0.05, statistically significant difference from the preceding column.
Molecular simulation of the binding of UTP to a putative nucleotide binding domain of hP2X3 in the immediate neighborhood of a consensus PKC site; differential location of additional nucleotide binding domains and consensus PKC sites
The purpose of the following studies was to find out whether the consensus ecto-PKC sites (PKC2 and PKC3) that definitely appear to be involved in the potentiating effect of UTP may immediately interfere with the ligand recognition site(s) of the P2X3 receptor. Many modern tools of homology-based molecular modeling are Web server based as opposed to installable software for local use key. Unfortunately, sufficiently close targets (to confidently model the sequence of P2X receptor proteins) were not available (e.g., ProModII of the SwissModel Server, 3D-JIGSAW, PSI-BLAST, CPHmodels, SDSC, Superfamily HMM Library and Genome Assignments, 3D-PSSM, and mGenTHREADER). As expected from these “negative results,” state-of-the-art homology modeling techniques such as Modeler or the respective modules of SYBYL or MOE could also not solve the problem. Therefore, the predicted conformation of the rat P2X3 was applied as template protein and converted to human P2X3.
The resulting predicted backbone structure of the geometry optimized, monomeric, charged, membrane-embedded, unoccupied human P2X3 glycoprotein receptor is shown in Figure 7A. Putative N-glycosylation addition sites of the ectodomain are in amino acid positions 139-142, 170-173, 194-197, 290-293, and 362-365. Distance geometry analysis of the ectodomain indicates that long-ranging intramolecular distances may be more strongly changed by disulfide bonding than short-ranging distances. The ectodomain would than form a twisted loop that is significantly solvent exposed (Mager et al., 2004). The intracellularly occurring N- and C-terminal PKC phosphorylation sites are in positions 12-14 and 365-367, and the extracellular PKC phosphorylation sites are in positions 134-136, 178-180, 196-198, 202-204, and 269-271 (Fig. 7A). Putative extracellularly occurring potential NBDs are in positions 62-66, 171-177, 279-286, and 295-302 (Mager et al., 2004). The mass centers between the PKC phosphorylation sites and NBDs may vary from short-ranging (<20 Å) to long-ranging (up to 115 Å) distances. A remarkable exception is NBD2 and PKC3 (see below).
Because S178 of the extracellular PKC phosphorylation site (termed PKC3, amino acid fragment Ser-Ile-Arg in positions 178-180) are of particular interest (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), it appeared to be useful to visualize the neighboring residues (Fig. 7B). They contain the potential nucleotide binding domain NBD2 (Phe-Thr-Ile-Phe-Lys-Asn, 171-177). The NBD2 includes also the putative glycopeptide addition site NFTI (Asn-Phe-Thr-Ile, 170-173). The uridine ring of UTP would be bound into a lipophilic pocket (Phe-171, Ile-175) of the NBD2; the oxygen atoms of the phosphate groups may form hydrogen bond with the hydrogen donating groups of the residues of Asn-177 and Lys-176. It is hypothesized that, if Asn-170 is not glycosylated, barely detectable currents in response to nucleotides might be found. Furthermore, the results support the hypothesis that nucleotides are bound to NBD2 in higher concentrations, whereas in lower concentrations, they may serve in the Mg2+ nucleotide form as substrates of ecto-PKC to transfer their terminal phosphate group onto S-178 within PKC3. The second functionally relevant PKC phosphorylation site (PKC2) may be located in considerable distance from the residual nucleotide binding domains NBD1, NBD3, and NBD4 (Fig. 7A).
Discussion
The main finding of this study is the identification of a novel regulatory function of nucleotides exerted on P2X3 receptor function via the phosphorylation of PKC sites at the extracellular loop of this receptor. Ecto-protein kinases and the corelease of ATP with glutamate (Chen et al., 1996) have been suggested to be involved in events associated with hippocampal synaptic plasticity [long-term potentiation (LTP)] (Wieraszko and Ehrlich, 1994; Ehrlich and Kornecki, 1999; Fujii, 2004). Because low- (but not high-) frequency stimulation of Schaffer collateral-commissural afferents produced LTP in CA1 pyramidal neurons of the hippocampus in the presence of exogenous ATP only, it was concluded that this nucleotide has a permissive action on the generation of LTP. ATP was supposed to phosphorylate a surface protein substrate rather than activate P2 receptors; enzymatically stable structural analogs of ATP or ATP in the presence of the ecto-protein kinase inhibitor K252b failed to favor the induction of LTP (Wieraszko and Ehrlich, 1994). There is no information available about the source of ecto-protein kinase, although it may be assumed that the enzyme is synthesized intracellularly and is subsequently delivered to the extracellular space either by a transporter or through the discontinuous plasma membrane of damaged cells. Thereby, the sensitivity of, for example, endo- and ecto-protein kinases C versus various pharmacological inhibitors may be practically identical (see below).
G-protein-coupled receptors such as those for substance P, bradykinin (Paukert et al., 2001), glutamate [metabotropic glutamate receptor (Vial et al., 2004)] and ATP/UTP [P2Y2 (Molliver et al., 2002); P2Y4 (Fischer et al., 2003)] may alter P2X receptor function via intracellular protein kinases using ATP as a substrate. The endo-protein kinases may either phosphorylate an associated protein (Adinolfi et al., 2003; Vial et al., 2004) or the receptor channel itself (Paukert et al., 2001) and thereby regulate the ionic conductance. Phosphorylation of a large family of interacting proteins, which control P2X receptor turnover, may promote the trafficking of these receptors to the membrane and thereby cause sensitization under injurious conditions (Xu and Huang, 2004) or an increase of synaptic strength (Khakh et al., 2001; Bobanovic et al., 2002).
In the present experiments, UTP and GTP were used in HEK293-P2X3 cells, because these nucleotides activate hP2X3 receptors at much higher concentrations than ATP itself (Fischer et al., 2003) but may be in their Mg2+ associated forms comparable substrates of ecto-protein kinases. The facilitatory action of UTP was observed only when the negative interaction between an UTP-sensitive P2Y4 receptor and the transfected P2X3 receptor was inhibited (Fischer et al., 2003) by the intracellular application of GDP-β-S (Sternweis and Pang, 1990) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Under these conditions, low concentrations of UTP, which do not activate P2X3 receptors, increased the amplitude of the current response to α,β-meATP. The use of various compounds [staurosporine, K252b, PKC inhibitor peptide, and Gö 6976 (Kase et al., 1987; Way et al., 2000)] and manipulations [Mg2+-free external medium (Christie et al., 1992)] known to block ecto-PKC activity allowed us to conclude that either a surface protein of the cell membrane or phosphorylation sites at the extracellular loop of the P2X3 receptors are the targets of ecto-PKC. Point mutations in consensus ecto-PKC sites of the P2X3 receptor confirmed that UTP, in fact, acts as a substrate of ecto-PKC. The substitution of the critical Ser and Thr residues by Ala brought out the participation of two consensus phosphorylation sites at PKC2 and PKC3 (T134A, S178A) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
An important question relates to a possible interference with agonist recognition of the P2X3 receptor after the structural alterations made by us. Basic amino acids (Lys and Arg) contributing to ligand binding were identified in the P2X1 and P2X2 receptor, with Phe coordinating the binding of the adenine ring of ATP (Jiang et al., 2000; North, 2002; Roberts and Evans, 2004). Furthermore, mutational analysis of the significance of conserved cysteines forming disulfide bridges in the ectodomains of P2X receptors confirmed reduced agonist sensitivity in the case of Ala substitution for Cys (Clyne et al., 2002; Ennion and Evans, 2002; Nakazawa et al., 2004). Finally, molecular simulation of the P2X3 receptor structure did not indicate any overlap between nucleotide binding domains and possible ecto-PKC sites (Fig. 7). Although, the present Ala-scanning mutagenesis strategy may alter the receptor-trafficking properties of P2X3, we did not try to clarify this point, because it was irrelevant with respect to the acutely observed potentiating effect of extracellular nucleotides.
Although a phosphorylation of P2X1 receptor channels by PMA but not metabotropic glutamate receptor activation has been demonstrated by labeling with [32P]orthophosphate and subsequent immunoprecipitation (Vial et al., 2004), in the present experiments, the expression levels of the hP2X3 receptor in HEK293 cell lines were not high enough to be able to successfully measure phosphorylation of the channel (C. Vial and R. J. Evans, unpublished observation). However, our findings as a whole strongly suggest that low concentrations of UTP are able to initiate phosphorylation of PKC sites localized at the ectodomain of hP2X3 receptors.
Finally, we investigated whether native P2X3 receptors of DRG neurons known to be involved in pain transmission are also modulated by extracellular nucleotides (Robertson et al., 1996; Dunn et al., 2001). In accordance with previous findings, UTP at concentrations of >0.3 μm induced rapidly inactivating inward currents (Robertson et al., 1996). However, UTP at a lower concentration of 30 nm invariably potentiated the effect of αβ-meATP by a mechanism that depended on protein kinase C activity, because the PKC inhibitor Gö 6976 abolished the UTP-induced facilitation. Hence, both native P2X3 receptors of rat DRG neurons and recombinant human P2X3 receptors expressed in HEK293 cells were similar concerning their responses to UTP.
In conclusion, it is suggested that, under physiological conditions, low extracellular concentrations of endogenous nucleotides (Lazarowski et al., 2000) may prime the sensitivity of P2X3 receptors toward the effect of subsequently applied (released) higher agonistic concentrations. Such a priming effect may be a prerequisite for the full functionality of pain-mediating P2X3 receptors situated at both the peripheral and central processes of dorsal root ganglion neurons (Chizh and Illes, 2001). Because this priming effect may be counterbalanced by the activation of P2Y receptors depressing P2X3 receptor-mediated currents (Fischer et al., 2003), the net result of these opposing influences will define the functional outcome.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (IL 20/11-1) and the Hungarian Scientific Research Fund (OTKA 047030). The generous support to P.I. during his stay in Budapest, Hungary, by a Szent-Györgyi-Award of the Hungarian Ministry of Education, is gratefully acknowledged. We are grateful to M. Henschke and R. Wolfram for technical assistance.
Correspondence should be addressed to Dr. Peter Illes, Rudolf-Boehm-Institute of Pharmacology and Toxicology, University of Leipzig, Haertelstrasse 16-18, 04107 Leipzig, Germany. E-mail: illp@medizin.uni-leipzig.de.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257734-09$15.00/0
References
- Abbracchio MP, Burnstock G (1994) Purinoceptors: are there families of P2X and P2Y purinoceptors? Pharmacol Ther 64: 445-475. [DOI] [PubMed] [Google Scholar]
- Adinolfi E, Kim M, Young MT, Di Virgilio F, Surprenant A (2003) Tyrosine phosphorylation of HSP90 within the P2X7 receptor complex negatively regulates P2X7 receptors. J Biol Chem 278: 37344-37351. [DOI] [PubMed] [Google Scholar]
- Bobanovic LK, Royle SJ, Murrell-Lagnado RD (2002) P2X receptor trafficking in neurons is subunit specific. J Neurosci 22: 4814-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boue-Grabot E, Archambault V, Seguela P (2000) A protein kinase C site highly conserved in P2X subunits controls the desensitization kinetics of P2X2 ATP-gated channels. J Biol Chem 275: 10190-10195. [DOI] [PubMed] [Google Scholar]
- Chen CC, Akopian AN, Sivilotti L, Colquhoun D, Burnstock G, Wood JN (1995) A P2X purinoceptor expressed by a subset of sensory neurons. Nature 377: 428-431. [DOI] [PubMed] [Google Scholar]
- Chen W, Wieraszko A, Hogan MV, Yang HA, Kornecki E, Ehrlich YH (1996) Surface protein phosphorylation by ecto-protein kinase is required for the maintenance of hippocampal long-term potentiation. Proc Natl Acad Sci USA 93: 8688-8693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizh BA, Illes P (2001) P2X receptors and nociception. Pharmacol Rev 53: 553-568. [PubMed] [Google Scholar]
- Christie A, Sharma VK, Sheu S-S (1992) Mechanism of extracellular ATP-induced increase of cytosolic Ca2+ concentration in isolated rat ventricular myocytes. J Physiol (Lond) 445: 369-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyne JD, Wang L-F, Hume RI (2002) Mutational analysis of the conserved cysteines of the rat P2X2 purinoceptor. J Neurosci 22: 3873-3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn P, Zhong Y, Burnstock G (2001) P2X receptors in peripheral neurons. Prog Neurobiol 65: 107-134. [DOI] [PubMed] [Google Scholar]
- Ehrlich YH, Kornecki E (1999) Ecto-protein kinases as mediators for the action of secreted ATP in the brain. Prog Brain Res 120: 411-426. [DOI] [PubMed] [Google Scholar]
- Ennion SJ, Evans RJ (2002) Conserved cysteine residues in the extracellular loop of the human P2X1 receptor form disulfide bonds and are involved in receptor trafficking to the cell surface. Mol Pharmacol 61: 303-311. [DOI] [PubMed] [Google Scholar]
- Fischer W, Wirkner K, Weber M, Eberts C, Köles L, Reinhardt R, Franke H, Allgaier C, Gillen C, Illes P (2003) Characterization of P2X3, P2Y1 and P2Y4 receptors in cultured HEK293-hP2X3 cells and their inhibition by ethanol and trichloroethanol. J Neurochem 85: 779-790. [DOI] [PubMed] [Google Scholar]
- Fujii S (2004) ATP- and adenosine-mediated signaling in the central nervous system: the role of extracellular ATP in hippocampal long-tem potentiation. J Pharmacol Sci 94: 103-106. [DOI] [PubMed] [Google Scholar]
- Himmel HH, Kiss T, Borvendeg SJ, Gillen C, Illes P (2002) The argininerich hexapeptide R4W2 is a stereoselective antagonist at the vanilloid receptor-1: a Ca2+ imaging study in adult rat dorsal root ganglion neurons. J Pharmacol Exp Ther 301: 981-986. [DOI] [PubMed] [Google Scholar]
- Illes P, Ribeiro JA (2004) Molecular physiology of P2 receptors in the central nervous system. Eur J Pharmacol 483: 5-17. [DOI] [PubMed] [Google Scholar]
- Jensik P, Thomas C (2002) ATP-induced internalisation of amphibian epithelial P2X receptors is linked to channel opening. Pflügers Arch 444: 795-800. [DOI] [PubMed] [Google Scholar]
- Jiang L-H, Rassendren F, Surprenant A, North RA (2000) Identification of amino acid residues contributing to the ATP-binding site of a purinergic P2X receptor. J Biol Chem 44: 34190-34196. [DOI] [PubMed] [Google Scholar]
- Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M (1987) K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem Biophys Res Commun 142: 436-440. [DOI] [PubMed] [Google Scholar]
- Khakh BS, Smith WB, Chiu C-S, Ju D, Davidson N, Lester HA (2001) Activation-dependent changes in receptor distribution and dendritic morphology in hippocampal neurons expressing P2X2-green fluorescent protein receptors. Proc Natl Acad Sci USA 98: 5288-5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köles L, Wirkner K, Illes P (2001) Modulation of ionotropic glutamate receptor channels. Neurochem Res 26: 925-932. [DOI] [PubMed] [Google Scholar]
- Lazarowski ER, Boucher RC, Harden TK (2000) Constitutive release of ATP and evidence for major contribution of ecto-nucleotide pyrophosphatase and nucleoside diphosphokinase to extracellular nucleotide concentration. J Biol Chem 275: 31061-31068. [DOI] [PubMed] [Google Scholar]
- Mager PP, Weber A, Illes P (2004) Bridging the gap between structural bioinformatics and receptor research: the membrane-embedded, ligand-gated, P2X glycoprotein receptor. Curr Top Med Chem 4: 1657-1705. [DOI] [PubMed] [Google Scholar]
- Molliver DC, Cook SP, Carlsten JA, Wright DE, McCleskey EW (2002) ATP and UTP excite sensory neurons and induce CREB phosphorylation through the metabotropic receptor, P2Y2 Eur J Neurosci 16: 1850-1860. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Smart TG (1996) Modulation of amino acid-gated ion channels by protein phosphorylation. Int Rev Neurobiol 39: 1-52. [DOI] [PubMed] [Google Scholar]
- Nakazawa K, Ojima H, Ishii-Nozawa R, Takeuchi K, Ohno Y (2004) Amino acid substitutions from an indispensable disulfide bond affect P2X2 receptor activation. Eur J Pharmacol 483: 29-35. [DOI] [PubMed] [Google Scholar]
- North RA (2002) Molecular physiology of P2X receptors. Physiol Rev 82: 1013-1067. [DOI] [PubMed] [Google Scholar]
- Paukert M, Osteroth R, Geisler HS, Brändle U, Glowatzki E, Ruppersberg JP, Grunder S (2001) Inflammatory mediators potentiate ATP-gated channels through the P2X3 subunit. J Biol Chem 276: 21077-21082. [DOI] [PubMed] [Google Scholar]
- Ralevic V, Burnstock G (1998) Receptors for purines and pyrimidines. Pharmacol Rev 50: 413-492. [PubMed] [Google Scholar]
- Roberts JA, Evans RJ (2004) ATP binding at human P2X1 receptors. Contribution of aromatic and basic amino acids revealed using mutagenesis and partial agonists. J Biol Chem 279: 9043-9055. [DOI] [PubMed] [Google Scholar]
- Robertson SJ, Rae MG, Rowan EG, Kennedy C (1996) Characterization of a P2X-purinoceptor in cultured neurones of the rat dorsal root ganglia. Br J Pharmacol 118: 951-956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternweis P, Pang I-H (1990) The G protein-channel connection. Trends Neurosci 13: 122-126. [DOI] [PubMed] [Google Scholar]
- Swope SL, Moss SJ, Raymond LA, Huganir RL (1999) Regulation of ligand-gated ion channels by protein phosphorylation. Adv Second Messenger Phosphoprotein Res 33: 49-78. [DOI] [PubMed] [Google Scholar]
- Vial C, Tobin AB, Evans RJ (2004) G-protein-coupled receptor regulation of P2X1 receptors does not involve direct channel phosphorylation. Biochem J 382: 101-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way KJ, Chou E, King GL (2000) Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol Sci 21: 181-187. [DOI] [PubMed] [Google Scholar]
- Wieraszko A, Ehrlich YH (1994) On the role of extracellular ATP in the induction of long-term potentiation in the hippocampus. J Neurochem 63: 1731-1738. [DOI] [PubMed] [Google Scholar]
- Xu G-Y, Huang L-YM (2004) Ca2+/calmodulin-dependent protein kinase II potentiates ATP responses by promoting trafficking of P2X receptors. Proc Natl Acad Sci USA 101: 11868-11873. [DOI] [PMC free article] [PubMed] [Google Scholar]