

Abstract
The NMDA ionotropic glutamate receptor is ubiquitous in mammalian central neurons. Because partial agonists bind to the same site as glutamate but induce less channel activation, these compounds provide an opportunity to probe the mechanism of activation of NMDA-type glutamate receptors. Molecular dynamics simulations and site-directed mutagenesis demonstrate that the partial agonist homoquinolinate interacts differently with binding pocket residues than glutamate. Homoquinolinate and glutamate induce distinct changes in the binding pocket, and the binding pocket exhibits significantly more motion with homoquinolinate bound than with glutamate. Patch-clamp recording demonstrates that single-channel activity induced by glutamate or by homoquinolinate has identical single-channel current amplitude and mean open-channel duration but that homoquinolinate slows activation of channel opening relative to glutamate. We hypothesize that agonist-induced conformational changes in the binding pocket control the efficacy of a subunit-specific activation step that precedes the concerted global change in the receptor-channel complex associated with ion channel opening.
Keywords: ion channel, glutamate, NMDA, molecular dynamics, structure, gating
Introduction
The NMDA subtype of ionotropic glutamate receptors comprises both NR1 and NR2 receptor subunits and plays a major role in both physiological and pathophysiological processes in the brain (for review, see Dingledine et al., 1999). The co-agonists glycine and glutamate are both required for channel activation, and the binding sites for glycine and glutamate are contained within the NR1 and NR2 subunits, respectively. The crystallization of the agonist-binding S1S2 domain for both the AMPA receptor subunit GluR2 (Armstrong et al., 1998; Armstrong and Gouaux, 2000; Jin et al., 2002) and, more recently, the NMDA receptor subunit NR1 (Furukawa and Gouaux, 2003) has provided direct insights into the molecular basis for agonist binding (for review, see Gouaux, 2004; Mayer and Armstrong, 2004). Crystallization of the agonist binding domain for GluR2 with a series of agonists demonstrated that the two lobes of the agonist binding domain display a greater degree of domain closure when the binding site is occupied by glutamate than they do when a partial agonist occupies this site (Jin and Gouaux, 2003; Jin et al., 2003). Previous work by Jin et al. (2003) relating structure to agonist efficacy suggests that the degree of domain closure correlates with functional activity. This study also provided additional evidence for the hypothesis that individual AMPA receptor subunits make incremental contributions to the gating of the ion permeation pathway (Rosenmund et al., 1998; Jin et al., 2003). However, NMDA receptors differ significantly from AMPA receptors in their functional properties and perhaps even in fundamental aspects of activation and gating (for review, see Erreger et al., 2004). Previous results with NR1/NR2B NMDA receptors suggest that there are subunit-specific pregating steps that can be modulated specifically by partial agonists acting at the glutamate site on NR2B or the glycine site on NR1 (Banke et al., 2005).
In this study, we evaluate the mechanism by which the conformationally restricted partial agonist homoquinolinate activates NR1/NR2A receptors. We use molecular dynamics (MD) to predict how homoquinolinate interacts with the binding pocket in NR2A and test these predictions using site-directed mutagenesis. Additionally, we use MD to predict how the position and mobility of residues of the binding pocket are influenced by homoquinolinate compared with glutamate. We use patch-clamp recording to determine in detail the effects of the partial agonist homoquinolinate on channel activity. We propose a model of channel activation and gating based on independent subunit pregating steps, followed by concerted pore dilation. Our analyses suggest that there are multiple activating conformational changes preceding channel gating and that at least one of these pregating steps is faster for channels activated by glutamate than by the partial agonist homoquinolinate because of differences in how these agonists interact with the binding pocket.
Materials and Methods
Electrophysiological recording from human embryonic kidney 293 cells and Xenopus oocytes. Human embryonic kidney 293 (HEK293) cells were maintained and transiently transfected by the calcium phosphate method with cDNA encoding NR1-1a (GenBank accession numbers U11418 and U08261; pCIneo vector; hereafter NR1), NR2A (D13211; pCIneo), and green fluorescent protein at a ratio of 1:2:1 (0.2 μg/ml NR1) for 4-12 h, as described previously (Zheng et al., 1998). Currents from outside-out patches were digitally recorded with pClamp8 software using an Axopatch 200B amplifier (Molecular Devices, Union City, CA). Single-channel records were filtered at 5 kHz using an eight-pole Bessel filter (-3 dB; Frequency Devices, Haverhill, MA) and digitized at 40 kHz. Macroscopic currents were filtered at 2-5 kHz and digitized at 20 kHz. The extracellular solution consisted of (in mm) 150 NaCl, 10 HEPES, 0.5 CaCl2, 3 KCl, and 0.01 EDTA supplemented with 50 μm glycine and 1 mm glutamate, unless noted otherwise (pH 7.3, 23°C). For most experiments, the agonist-containing extracellular solution was made from ultra-pure salts with an Mg2+ concentration of <0.2 μm measured using inductively coupled plasma-mass spectrometry by the Laboratory for Environmental Analysis at the University of Georgia (Athens, GA). single-channel data recorded with normal salts was included in the final analysis because the measured Mg2+ concentration was <1 μm, and the mean channel open time was not significantly different in paired comparisons of normal and ultrapure salts in the same patches expressing NR1/NR2A (p > 0.05; n = 6). The internal solution consisted of the following (in mm): 110 Cs gluconate, 30 CsCl2, 5 HEPES, 4 NaCl, 0.5 CaCl2, 2 MgCl2, 5 BAPTA, 2 Na-ATP, and 0.3 Na-GTP, pH adjusted to 7.35 with CsOH. Rapid solution exchange was achieved with a two-barrel theta glass pipette controlled by a piezoelectric translator (Burleigh Instruments, Fishers, NY); junction currents were used to estimate speed of solution exchange after recordings and typically had 10-90% rise times of 0.3-1.0 ms (7- to 21-fold faster than NMDA receptor rise times).
Oocytes from Xenopus laevis were isolated, injected with NMDA receptor cRNA synthesized in vitro, and recorded under voltage clamp as described previously (Traynelis et al., 1998). Briefly, oocytes were injected with 5-10 ng of cRNA at a 1:2 ratio of NR1:NR2A. Glutamate- or homoquinolinate-evoked membrane currents were recorded at -40 mV in the presence of 50 μm glycine 24-72 h after injection. The recording solution contained (in mm) 90 NaCl, 3 KCl, 10 HEPES, 0.01 EDTA, and 0.5 BaCl2 (23°C); the pH was adjusted with NaOH, and recording electrodes were filled with 0.3 m KCl.
Single-channel and macroscopic analysis. Single-channel analysis was performed using QUB software (www.qub.buffalo.edu) to idealize records and fit kinetic models to idealized single-channel data. For histogram fitting, the data were divided into uniform segments (0.5 s), and any segment with simultaneous multiple channel openings was discarded. Records were idealized with the segmentation k-means algorithm (Qin, 2004). Dwell-time distributions were fit to multiple exponential kinetic components using ChanneLab (http://www.pharm.emory.edu/straynelis/Downloads). Each individual patch contained, on average, 5796 events for glutamate and 2673 events for homoquinolinate. For maximum interval likelihood (MIL) (Qin et al., 1996), fitting data were idealized and segmented by QUB software using a critical gap time (Tcrit) that minimized the total number of misclassified events (Jackson et al., 1983) (EKDIST software provided by D. Colquhoun, University College London, London, UK). The Tcrit calculated from patches that contained a single active NR1/NR2A channel was 30 ms (Erreger et al., 2005). Idealized activations were then fit by hidden Markov models using the MIL method. Macroscopic response waveforms obtained in outside-out patches were averaged among patches. The average macroscopic glutamate response waveform was then normalized to the measured peak open probability determined from brief application of maximally effective concentration of agonist in patches that contain one active channel (Erreger et al., 2005). The homoquinolinate waveform was scaled relative to glutamate using the relative peak currents observed for glutamate and homoquinolinate in the same patches. Hidden Markov models were simultaneously fitted (ChanneLab; least squares criterion) to multiple waveforms obtained for both short and long pulses of agonist at both high and low concentrations. This involved determination of the sum of squares difference between simulation and data for all fitted waveforms at each iteration within the simplex algorithm. In other experiments, the mean macroscopic time course was fitted by the sum of two exponential functions:
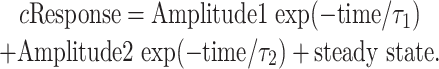 |
The ratio F test was used to determine whether the mean macroscopic relaxation (200 data points selected at a fixed interval) was fitted significantly better by one or by the sum of two exponential functions.
Homology modeling and MD. A homology model of glutamate docked into the S1-S2 region of NR2A (GenBank accession number D13211) has been described previously (Chen et al., 2005). Glutamate was removed from the binding site, and homoquinolinate was built in its place, with the nitrogen of the aromatic ring left unprotonated. The pKa for this ionizable group was calculated to be 5.82 using Solaris software (version 4.67; Advanced Chemistry Development, Toronto, Ontario, Canada). All three structures (glutamate docked in S1-S2, homoquinolinate docked in S1-S2, and the nonliganded/apo S1-S2) were simulated using the Gromos96 force field and solvated with the simple point charge water model. A hydrated cube (7 nm on each side; volume, ∼345 nm3) containing ∼11,000 water molecules was built around each structure. Each hydrated complex was energy minimized to remove any poor contacts, subjected to 20 ps of position-restrained MD at 300 K to allow water to soak into the structure. To resolve any high-energy contacts, the complexes were subjected to low-temperature unrestrained MD over very small time steps using the GROMACS package (Berendsen et al., 1995; Lindahl et al., 2001). Each system was run at 20 K for 3 ps, using a time step of 1 fs. A frame was then selected as the starting point for a subsequent 20 K simulation. Repeated iterations allowed the structures to find relaxed states without propagating large forces (i.e., from steric clashes) through the entire complex. Further MD was then performed on each of the three structures at 200 K using a time step of 2 fs. These simulations were extended to 500 ps of unrestrained MD. Structures for equilibrated complexes were calculated by averaging the final 150 ps of the simulation.
Statistical analysis. Student's t test (two tailed) was performed in Excel software for statistical comparisons (p < 0.05 was considered significant). Data are expressed as mean ± SEM.
Results
Homoquinolinate is a partial agonist for NR1/NR2A
We used two-electrode voltage-clamp recording of steady-state current responses from recombinant NR1/NR2A receptors expressed in Xenopus oocytes to quantify the potency and efficacy of homoquinolinate relative to the maximal current evoked by a saturating concentration of glutamate. Our results indicate that homoquinolinate is a candidate partial agonist when studied in oocytes, able to evoke ∼70% of the maximal response produced by glutamate (n = 19) (Fig. 1A, Table 1). This submaximal response by 1 mm homoquinolinate could be converted to a maximal response by coapplication of homoquinolinate with increasing concentrations of glutamate (n = 5) (Fig. 1A), confirming that homoquinolinate is a partial agonist. We subsequently evaluated the efficacy of rapidly applied homoquinolinate relative to glutamate for activation of NR1/NR2A receptors in outside-out patches isolated from HEK293 cells (Fig. 1B). Comparison of the peak responses for a maximally effective concentration of homoquinolinate to the average of control and recovery responses to a maximally effective concentration of glutamate showed that homoquinolinate evoked 63 ± 4% (n = 4) of the glutamate-evoked NR1/NR2A current response in mammalian cells. The relative efficacy of homoquinolinate for peak currents in outside-out patches from HEK293 cells was similar to that for steady-state currents in oocytes. We have shown previously that NR1/NR2A receptors in outside-out patches have a peak open probability of 0.50 when activated by a maximally effective concentration of glutamate (Erreger et al., 2005). From this measurement and the relative efficacy of homoquinolinate to glutamate, we estimate that the maximum open probability for homoquinolinate activation of NR1/NR2A receptors is 0.31. The EC50 value for glutamate (4.6 μm) was lower than for homoquinolinate (24.4 μm) (Table 1).
Figure 1.

Homoquinolinate is a partial agonist for NR1/NR2A NMDA receptors. A, Left, Steady-state, dose-response relationship for NR1/NR2A currents in Xenopus oocytes normalized to 100 μm glutamate (50 μm glycine; -40 mV). For glutamate, the EC50 value is 4.6 μm, and the Hill slope is 1.4 (n = 11). For homoquinolinate, the EC50 value is 24.4 μm, and the Hill slope is 1.2 (n = 19). Right, The decreased current induced by homoquinolinate can be restored to full activity by coapplication of increasing concentrations (in millimolar) of glutamate, indicating that the reduced efficacy of homoquinolinate is attributable to an effect at the glutamate binding site (n = 5). The inset shows a raw current trace from one representative oocyte. B, Example of NR1/NR2A currents recorded under voltage clamp evoked in response to rapid application of 1 mm glutamate or 1 mm homoquinolinate to outside-out patches isolated from HEK293 cells (VHOLD, -60 mV; 1 s application duration). The relative amplitude of peak currents evoked in the same patch for homoquinolinate relative to glutamate is 62.8 ± 4.3% (n = 4). C, The rise times are significantly slower for homoquinolinate-activated currents compared with those activated by glutamate. The inset shows the mean normalized rise waveforms averaged among all patches. The number in parentheses indicates the number of cells. *p < 0.001, unpaired t test. GLU, Glutamate; HQ and Homoquin, homoquinolinate.
Table 1.
Potency for glutamate and homoquinolinate at wild-type and mutant receptors
|
Receptor |
Glu EC50 (μm) |
Glu Hill slope |
n |
HQ EC50 (μm) |
HQ Hill slope |
n |
IMAX HQ/Glu |
Glu EC50 mut/wt |
HQ EC50 mut/wt |
|---|---|---|---|---|---|---|---|---|---|
| NR1/NRZA | 4.6 | 1.4 | 11 | 24.4 | 1.2 | 19 | 0.72 | ||
| NR1/NRZA-S670G | 421 | 1.4 | 12 | 151 | 1.1 | 9 | 0.51 | 92 | 6 |
| NR1/NRZA-T671A | 2967 | 1.3 | 5 | 384 | 0.7 | 9 | 0.10 | 645 | 16 |
| NR1/NRZA-S492A |
146 |
1.2 |
6 |
202 |
1.2 |
6 |
0.67 |
32 |
8 |
Parameters measured from steady-state recording of indicated subunits expressed in Xenopus oocytes are shown. n is the number of cells. IMAXHQ/Glu is the ratio of maximal current induced by homoquinolinate relative to glutamate at a saturating agonist concentration in the same cell. Glu, Glutamate; HQ, homoquinolinate; wt, wild type; mut, mutant.
Activation rate of NR1/NR2A by homoquinolinate is slower than for glutamate
To better understand the mechanism by which NR2A-containing receptors become activated by agonist, we performed recordings under voltage clamp of macroscopic NR1/NR2A current responses in excised outside-out patches. Current responses to prolonged application of both glutamate and homoquinolinate decayed with a mean time course that could be described by one or two exponential components, depending on the patch. However, it should be noted that NMDA channels in excised patches exhibit substantially greater desensitization (independent of glycine, calcium, or zinc) than NMDA channels recorded in whole-cell or perforated-patch recording mode (Zheng et al., 2001; Erreger et al., 2005). When a single exponential time constant is fitted to the relaxation for all patches, the mean time constant for glutamate (187 ± 64 ms; n = 14 patches) was not significantly different than for homoquinolinate (193 ± 20 ms; n = 14). The time course was significantly better fitted in the majority of patches (seven of nine patches tested) by the sum of two exponential components (p < 0.001). The averaged composite time course decayed with time constants (and relative amplitudes) of 60 ms (38%) and 257 ms (62%) for glutamate, which were similar to the time constants of 82 ms (28%) and 229 ms (72%) describing homoquinolinate-induced desensitization. In addition, the steady state-to-peak current ratio was not significantly different between glutamate (0.15 ± 0.03; n = 14) and homoquinolinate (0.13 ± 0.02; n = 12). These data suggest that the rate and degree of desensitization induced by the two agonists is similar. We also measured the time course for deactivation after a brief (5-15 ms) pulse of maximal concentration of agonist. We found that the time constant describing deactivation after brief glutamate application (36.1 ± 2.6 ms; n = 8) was significantly slower than that describing deactivation from homoquinolinate (21.7 ± 4.0 ms; n = 4; p = 0.011; unpaired t test). In addition, the 10-90% rise time of macroscopic currents (Fig. 1C) in response to rapid application of a saturating (1 mm, >40-fold EC50 value) concentration of homoquinolinate (12.2 ± 0.8 ms; n = 14) was significantly slower than for saturating glutamate (6.8 ± 0.8 ms; n = 16; p = 0.0003; unpaired t test). Identical results were obtained in a subset of patches in which rise times were measured for both glutamate- and homoquinolinate-evoked currents in the same patch (homoquinolinate, 12.3 ± 1.4 ms; glutamate, 8.0 ± 0.7 ms; p = 0.016; paired t test; n = 8). These data are consistent with the hypothesis that the main determinant of partial agonism is not a difference in desensitization but rather a specific effect on conformational changes that precede or control channel opening.
Modeling of glutamate and homoquinolinate interaction with the agonist binding pocket
To better understand the nature of the reduced potency and reduced efficacy of homoquinolinate, we used MD applied to a homology model of the S1S2 domain of NR2A docked to either glutamate or homoquinolinate (see Materials and Methods). Figure 2A illustrates the alignment used to generate a homology model of the glutamate binding domain of NR2A (Chen et al., 2005) based on a crystal structure of the NR1 glycine binding domain (Furukawa and Gouaux, 2003). The α-helices and β-sheets were labeled according to the NR1 structure (Furukawa and Gouaux, 2003). Figure 2B-D superimposes the model of the NR2A S1S2 binding domain with either glutamate or homoquinolinate docked using MD. Figure 3A shows the glutamate α-carboxyl anchored by Arg499 plus a backbone nitrogen from Ser670 (Chen et al., 2005). Arg499 also makes an important hydrogen bond with the backbone carbonyl of Thr494 (data not shown). The α-amino group of glutamate is hydrogen bonded by the hydroxyl of Tyr742 (Fig. 3B) and has the potential for favorable interactions with the hydroxyls of Ser492 and Thr494 and the carboxylate of Asp712 (data not shown). The γ-carboxyl interacts with the hydroxyl groups from Ser670 and Thr671, with additional support from the Thr671 backbone nitrogen (Fig. 3C). Homoquinolinate is a conformationally restricted glutamate analog that contains two carboxyl groups and a secondary amine embedded in an aromatic ring with side-chain bond angles of 120° compared with 109° for glutamate. Figure 2, B and C, illustrates the superposition of the docked agonists, which show similar positioning for the three functional groups. However, several differences between simulated homoquinolinate and glutamate binding are apparent. For example, the aromatic ring of homoquinolinate takes up more space than glutamate, with resulting displacement of residues in the pocket. This can be visualized by the apparent displacement of Tyr742 by homoquinolinate (Fig. 3B). In addition, Ser670 changes its primary hydrogen-bonding partner from the γ-carboxyl of glutamate to the α-carboxyl in homoquinolinate (Fig. 3C). This change in hydrogen bonding of the γ-carboxyl identified by MD analysis, if correct, would be predicted to alter homoquinolinate sensitivity to mutation of S2 contact residues by comparison with glutamate. We directly tested this idea by generating concentration-response curves for two mutant NR2A receptors, NR2A(S670G) and NR2A(T671A). Consistent with predictions from MD modeling, the NR2A mutation S670G in the S2 domain shifted the EC50 value for homoquinolinate by 15-fold, much less than the 92-fold potency shift for glutamate (Table 1). Similarly, the S2 NR2A mutation T671A only shifted homoquinolinate EC50 by 6-fold compared with 645-fold for glutamate (Table 1) (Chen et al., 2005). These data support the idea that the contact residues for the carboxylates of homoquinolinate differ from those for glutamate, lessening the contribution of Ser670 and Thr671 to the stabilization of homoquinolinate in the binding pocket. MD modeling also predicts that Ser492 makes intermittent hydrogen bonds with the protonated amine of glutamate. However, no such hydrogen bonds are observed between Ser492 and the unprotonated nitrogen of homoquinolinate, suggesting that mutation of Ser492 should have differential effects on glutamate and homoquinolinate EC50. As predicted by MD analysis, the receptors comprising NR2A(S492A) showed a 32-fold increase in glutamate EC50 but only an 8-fold increase in homoquinolinate EC50. These functional data are thus consistent with the results of MD modeling.
Figure 2.

MD simulation of glutamate or homoquinolinate docking to a homology model of the NR2A S1S2 agonist binding pocket. A, Alignment used for construction of the homology model for NR2A. B, Superposition of MD simulation of glutamate (orange) or homoquinolinate (gray) docked to S1S2. ATD, N-terminal domain; M1-M4, membrane domains 1-4. C, Superposition of glutamate and homoquinolinate shown without amino acid residue side chains emphasizes superposition of functional groups. D, An expanded view shows differential displacement of helix F when glutamate versus homoquinolinate is docked in the binding pocket. Letters F-I indicate helices. PDB files of the coordinates for both models can be found at http://www.pharm.emory.edu/straynelis/StructuralModels/.
Figure 3.

Comparison of glutamate and homoquinolinate binding. A, Glutamate and homoquinolinate docking are shown with α-carboxyl and γ-carboxyl hydrogen bond contacts. B, Displacement of Tyr742 illustrates the steric clash produced by the aromatic ring of homoquinolinate when docked into the glutamate binding pocket. C, Ser670 is reoriented to interact with the α-carboxyl of homoquinolinate compared with the hydrogen bond it forms with the γ-carboxyl of glutamate.
The bulkier size of homoquinolinate and its displacement of residues such as Tyr742 suggest that homoquinolinate may not be held as tightly in the pocket as glutamate. If this is the case, we predict that the EC50 value determined from functional data and the molecular motion of side chains comprising the pocket will both increase. To evaluate this hypothesis, we analyzed the root mean square (rms) deviations of the atomic positions of 22 residues comprising the binding pocket over the final 150 pS of MD. With the exception of Tyr742, which fluctuates in and out of a hydrogen bond with the amino group of glutamate, these residues showed considerably more motion around docked homoquinolinate (mean ratio rmsHQ/rmsGLU for each residue, 1.33) than around both glutamate-bound and apo structures during MD runs (Fig. 4) (apo data not shown). The greatest contribution to pocket motion came from the S2 residues 669-674 (rmsHQ/rmsGLU, 1.73) that secure the carboxylates of homoquinolinate and more distant residues that make up the top of helix F. This is the same region that shows wide variation in hydrogen binding between glutamate and homoquinolinate γ-carboxyl, which act as a bridge across the binding pocket (Figs. 3C, 5). Unlike glutamate, homoquinolinate binding in this region involves a water bridge to Asp712 that will increase intrapocket motion (Fig. 5C). Additional determinants of the increased rms values may also reflect increased side-chain motion that results from ongoing steric contacts between homoquinolinate and residues within the binding pocket. This increased motion suggests a decreased binding stability for homoquinolinate compared with glutamate. Increased motion for homoquinolinate also suggests an increased KD value, consistent with the experimentally determined increased EC50 value for homoquinolinate compared with glutamate (Table 1).
Figure 4.

Increased atomic motion within the binding pocket for homoquinolinate. The SD (i.e., rms) of atomic position is shown in pseudocolor for residues that comprise the agonist binding pocket, with cold colors (blue) representing low rms and hot colors (red) representing high rms. Results are shown after MD runs for glutamate and homoquinolinate. The range of rms values for residues comprising the binding pocket were 0.0257-0.1045 nm for glutamate and 0.0316-0.0823 nm for homoquinolinate.
Figure 5.

γ-Carboxyl bridges the agonist binding pocket. A shows the MD run of the apo protein without agonist. Note the hydrogen bonds between Ser670 and Thr671 with Asp712 and a backbone nitrogen. B shows how the γ-carboxyl inserts itself into this region, interrupting these hydrogen bonds to form a hydrogen-bonded bridge between Ser670/Thr671 and Asp712. C shows how the orientation of homoquinolinate requires waters to form this same bridge, which results in increased mobility in this region of the pocket (Fig. 4). Only the bridging water molecules are shown, although all models were fully hydrated during MD. Letters F and G indicate helices.
Translation of agonist binding to channel opening
A number of interesting features of the S2 backbone helix F are apparent in our simulations. First, the displacement of helix F from helix G does not change considerably between the apo state and the glutamate-bound state (Fig. 5). In addition, homoquinolinate appears not only to displace helix F (Fig. 2 D), compared with glutamate, but to open up more space within the binding pocket, suggesting that homoquinolinate can propagate longer range perturbations through the domain structure. Furthermore, contact between helix F and other elements of the secondary structure is lost with homoquinolinate in the pocket (Fig. 5 B, C). These contacts are formed through three hydrogen bonds in the apo-NR2A that connect helix F to β-sheet 12 and a loop between β-sheets 5 and 6. The hydrogen bond between Ser670 and Asp712 is lost after glutamate or homoquinolinate binding. Homoquinolinate binding further disrupts the hydrogen bonds between Thr671 and the backbone oxygen of Ile710 as well as between the backbones of Asn668 and Gly467 (Fig. 5C). In the NR2A-glutamate complex, these contacts are essentially retained by preservation of the backbone hydrogen bond between Asn668 and Gly467 and a frequent hydrogen bond between the γ-carboxyl of glutamate and the backbone hydrogen of Asp712. The homoquinolinate system substitutes the latter interaction with a trio of water molecules (Fig. 5C). The direct contacts between helix F and the neighboring secondary structure are not found in the averaged structure with homoquinolinate. One intriguing, yet speculative, hypothesis is that this uncoupling of helix F to the rest of the protein may play a role in the reduced translation of binding energy to displacement of gating elements by homoquinolinate, which may in part contribute to its reduced ability to open the channel (see below).
Homoquinolinate alters NR1/NR2A single-channel properties
To examine the mechanism of activation by glutamate and homoquinolinate, single-channel recordings were made from outside-out patches. Channel activity was evoked by steady-state application of a maximally effective concentration of either glutamate (1 mm) or homoquinolinate (1 mm) (Fig. 6). The single-channel chord conductance and the mean channel open duration were the same for both agonists. These two measures suggest that the properties of the open-channel conformation are not directly dependent on the identity of the agonist. The primary difference among the functional properties of the single-channel activations caused by the two agonists was within the distribution of the shut times, which was described by at least four exponential components. There were no significant differences in the relative area of any of the four shut-time components between glutamate and homoquinolinate (p > 0.05; paired t test; n = 7). The shortest shut-time component, which we estimate to be <0.1 ms, was too near the limit of our recording resolution to be measured precisely in some patches. The next fastest time constant (shut time τ2) (Fig. 6, gray arrow) was not significantly different between agonists. Shut time τ3 (Fig. 6, black arrow) was significantly longer for homoquinolinate-induced activity than for glutamate-induced activity. A similar shift in the analogous shut-time component has been described previously with partial agonists for the glutamate site for NR1/NR2B and was hypothesized to reflect a slowing of a single conformational change within the NR2 subunit (Banke and Traynelis, 2003). Changes in the fourth shut-time component (τ4) could not be interpreted, because this component likely contained time periods between closure of one channel and opening of another channel, given that the total number of channels contained within the patches we recorded from was unknown and was unlikely to be equal for all patches. The relative area of this fourth component of the shut-time distribution is substantial (glutamate, 10 ± 3%; homoquinolinate, 16 ± 3%; n = 7). Nonetheless, we suggest that this kinetic component reflects in part the time course of a desensitization-related process and not the fundamental mechanism of channel opening. We consider the fitting of shut-time distributions as a first-pass model-independent analysis of agonist-specific features of channel gating. We subsequently fitted the sequence of single-channel events with an explicit kinetic model to attempt to draw mechanistic conclusions from the data.
Figure 6.

Rate of activation of by homoquinolinate is slower than for glutamate. A, Left, Single-channel current activity elicited in an outside-out patch in response to steady-state application of 1 mm glutamate. The recording was made in the presence of 50 μm glycine and 10 μm EDTA at a holding potential of -80 mV, and downward deflections reflect channel opening. Right, The distribution of shut times from the same patch as the sample trace is shown and can be fit by four exponential components. B, The corresponding data are plotted for 1 mm homoquinolinate in the same patch. The single-channel chord conductance was identical for both agonists (glutamate, 62.6 ± 1.7 pS; homoquinolinate, 60.9 ± 2.3 pS; n = 7), as was the mean channel open duration (glutamate, 3.72 ± 0.39 ms; homoquinolinate, 3.62 ± 0.31 ms; n = 7). The time constant for the second shut-time component (τ2; gray arrow) was not significantly different between agonists (glutamate, 0.489 ± 0.025 ms; homoquinolinate, 0.455 ± 0.074 ms; n = 7). Shut time τ3 (black arrow) was significantly longer for homoquinolinate-induced activity than for glutamate-induced activity (glutamate, 4.69 ± 0.43 ms; homoquinolinate, 6.17 ± 0.50 ms; p = 0.0060; paired t test; n = 7). SQRT, Square root.
Homoquinolinate slows a specific pregating step
As indicated above, the only consistent feature of single-channel open and closed time histograms that changed when channel activity was evoked by homoquinolinate or glutamate was the value of the third exponential component in the shut-time distribution. Such a change could reflect any number of features of the NMDA receptor activation process. To circumvent the indirect nature of inferences made from histogram fitting, we used maximum likelihood fitting of explicit models that encapsulated our working hypothesis of NMDA receptor activation to single-channel records (Banke and Traynelis, 2003; Erreger et al., 2005). A number of previous studies have convincingly argued that at least two pregating kinetically distinct conformation changes are required before NMDA receptors can open (Gibb and Colquhoun, 1991; Wyllie et al., 1998; Banke and Traynelis, 2003; Popescu and Auerbach, 2003; Popescu et al., 2004; Erreger et al., 2005). We therefore used hidden Markov modeling of our single-channel data to explore whether differences exist between glutamate-evoked or homoquinolinate-evoked channel transition rates for the conformational changes that precede NMDA channel opening. Data were analyzed by subdividing records on the basis of a critical time (30 ms) that was calculated to allow identification of individual activations (Erreger et al., 2005). That is, the critical closed times were used to separate open and closed times within an activation from closed times between two different activations (see Materials and Methods). The idealized sequence of channel events was then fitted with a model postulating two pregating conformational changes (a fast step for NR1, a slow step for NR2), followed by rapid pore dilation. The hypothesized assignment of individual subunits to the kinetic steps is based on previous work demonstrating the pharmacological sensitivities of these steps to agonists acting at the glycine or glutamate site in NR1/NR2B receptors, as well as the sensitivity of these steps to the identity of the NR2 subunit (Banke and Traynelis, 2003; Erreger et al., 2005). Two directly interconverting open states were included to better describe the distribution of open-channel dwell times (Popescu et al., 2004).
We directly fit this explicit kinetic model to the specific sequence of channel openings and closings. Scheme 1 (Fig. 7) postulates that the NR1 and NR2 subunits activate independently and that both must be active before the channel pore opens. The rates “f” (fast) and “s” (slow) that are shown are for the hypothesized transition between an agonist-bound “inactive” state and an agonist-bound “active” state for the NR1 (f) and NR2 (s) subunits (Banke and Traynelis, 2003; Erreger et al., 2005). Because the recordings were performed in the continuous presence of a saturating concentration of both glutamate and glycine, no explicit binding steps are included and full occupancy of ligand binding sites is assumed. Desensitized states are also omitted because data were segmented with a critical shut time to remove desensitized shut durations. We fitted scheme 1 to the sequence of channel openings in each patch for sequential application of both glutamate and homoquinolinate and found that our working model fit data from each patch well, with limited variability between rate constants among patches. The average log likelihood (a measure of the quality of the fit of the model to the data) value was identical when the model was fitted to either single-channel activations evoked by glutamate or homoquinolinate (log likelihood per event: glutamate, 4.97 ± 0.03; homoquinolinate, 4.94 ± 0.03). Table 2 compares the mean (±SEM) rate constants for activations by glutamate or homoquinolinate fitted to scheme 1. The fitted rate constants show that only the “slow” (hypothesized NR2-dependent) activation rate (Banke and Traynelis, 2003; Erreger et al., 2005) changes significantly for scheme 1, consistent with the hypothesis that the identity of the agonist bound to the NR2A subunit controls the rate of a slower conformational change required for gating.
Figure 7.

Maximum interval likelihood fitting of steady-state activations of NR1/NR2A. Steady-state currents were idealized and fit to a kinetic model that includes two independent activation steps. A, Scheme 1 hypothesizes that these two independent pregating activation steps involve a fast NR1-dependent step f and a slow NR2-dependent step s. Recordings were performed in the presence of a saturating concentration of agonist and divided into segments to remove desensitization. Therefore, explicit agonist binding and desensitization rates (gray arrows) are not included in the model. B, Dwell-time distribution histograms are shown for open and shut times in a patch in which channel activity was evoked by 1 mm glutamate. The solid lines show probability density functions predicted by the fit of the model to the sequence of channel openings. A summary of the fitting results is given in Table 2. SQRT, Square root.
Table 2.
Hidden Markov maximum likelihood fitting of channel data and least-squares fitting of macroscopic data
|
|
Units |
Glutamate |
Homoquinolinate |
% change |
|---|---|---|---|---|
| Single channels | ||||
| kf+ | s−1 | 2559 ± 280 | 2468 ± 196 | −3.6 |
| kf− | s−1 | 1978 ± 288 | 3005 ± 696 | 51.9 |
| ks+ | s−1 | 234 ± 22 | 184 ± 12 | −21.1* |
| ks− | s−1 | 926 ± 139 | 896 ± 149 | 3.2 |
| ko+ | s−1 | 5008 ± 647 | 5822 ± 899 | 16.3 |
| ko− | s−1 | 790 ± 66 | 740 ± 54 | −6.3 |
| Macroscopic | ||||
| kd1+ | s−1 | 20 | 22 | |
| kd1− | s−1 | 0.76 | 0.71 | |
| kd2+ | s−1 | 7.5 | 68 | |
| kd2− | s−1 | 6.7 | 45 | |
| kon | μm−1 s−1 | 10.4 | 2.5 | |
| koff | s−1 | 73 | 149 | |
|
KD
|
μm
|
7.0 |
60 |
|
The sequence of openings and closing of single-channel records from individual patches were analyzed and fitted with scheme 1 (Fig. 7). f and s refer to a “fast” and “slow” pregating step, hypothesized to reflect conformational changes with NR1 and NR2 required for gating, respectively. Agonist binding and desensitization rates were not fit because these records were recorded under supramaximal agonist concentration and segmented to remove desensitized closed times. The mean ± SEM rate constant for each step is shown (n = 7); on average, 13,384 and 6410 channel dwell times per patch were fitted for glutamate and homoquinolinate, respectively. Subsequent fitting of macroscopic currents (Fig. 8) to scheme 2 with kf+, kf−, ks+, ks−, ko+, ko− all fixed to values determined above was used to determine binding and desensitization constants. KD = koff/kon.
p = 0.0085; paired t test.
Macroscopic NR1/NR2A current responses to glutamate or homoquinolinate
Partial agonists by definition produce reduced response compared with a full or prototypical agonist. If our analysis of single-channel records is correct to a first approximation, then we should be able to predict the relative degree of partial agonism from the rate constants derived from steady-state, single-channel records. To directly evaluate the ability of gating rate constants derived from fitted single-channel data to accurately describe peak open probability and the time course of macroscopic currents, we first recorded macroscopic currents in response to rapid application of glutamate or homoquinolinate to excised outside-out patches. Three sets of responses were generated for each agonist (Fig. 8B,C), including (1) saturating agonist concentration applied for 1 s, (2) saturating agonist concentration applied for 5-15 ms, and (3) subsaturating agonist concentration applied for 1 s. The response for each patch to prolonged application of the high (1 mm) agonist concentration response was normalized to the measured peak open probability of NR1/NR2A in response to glutamate in one channel patch (0.50) (Erreger et al., 2005), and responses averaged across patches for all three protocols. Homoquinolinate-evoked responses in each patch were scaled to the relative open probability (0.31) calculated from the peak open probability to glutamate (0.50) and the relative efficacy of homoquinolinate (62%) determined in outside-out patches in response to maximal concentrations of glutamate and homoquinolinate (see above) (Fig. 1B), and averaged across patches. All three curves for each agonist were then fitted simultaneously with scheme 2 (Fig. 8A) with the gating rates fixed to those determined from single-channel analysis. Scheme 2 incorporated two explicit binding steps for glutamate or homoquinolinate as well as two desensitization steps given the dual exponential time course for desensitization; binding and desensitization parameters were free to vary in these fits. Scheme 2 produced excellent fits to the macroscopic peak open probability and time course, suggesting that our single-channel measurements accurately describe the independently recorded peak macroscopic response (Fig. 8C). Table 2 summarizes the results from fitting scheme 2 to macroscopic data for both glutamate and homoquinolinate and confirms that an independent subunit gating model with two pregating steps can accurately predict the magnitude and time course of macroscopic currents over a range of agonist concentrations and application durations, as well as generate the experimentally determined peak open probability. Furthermore, simulations show that the time constant describing deactivation is virtually identical (33.3-34 ms) for 1, 5, 10, 15, and 20 ms pulses of glutamate, confirming that the application duration we chose did not confound determination of rate constants. Fitted data in Table 2 also show a reduced association rate for homoquinolinate, an increased KD, and an accelerated dissociation rate, consistent with predictions from MD simulations.
Figure 8.

Least-squares fitting of a model of activation to macroscopic currents in response to rapid agonist application. A, Scheme 2 is an extension of scheme 1 with explicit agonist binding and desensitization rates. The rates for f+,-, s+,-, and o+,- (gray arrows) were fixed to the values determined from single-channel analysis. The rates for agonist binding (kon, koff) and desensitization (d1+,-, d2+,-) were varied by a simplex algorithm (least-squares criterion). d1+ and d2+ represent entry into desensitized states, and d1- and d2-represent recovery from desensitized states. B, Representative macroscopic currents recorded in outside-out patches excised from HEK293 cells expressing NR1/NR2A and held at a potential of -60 mV are shown for each of the three protocols: 1 mm/1 s, 1 mm/8 ms, 5 μm/1 s for glutamate (left) and 1 mm/1 s, 1 mm/8 ms, 30 μm/1 s for homoquinolinate (right). The insets show the responses evoked by brief (8 ms) 1 mm glutamate or homoquinolinate application on an expanded time scale with the junction current (top trace) used to determine the time course of solution exchange. C, The results of the simultaneous fitting of the scheme 2 shown in A to the macroscopic current data sets in B are shown as solid black lines, with the mean experimental normalized currents shown in gray (n = 10). The results of fitting scheme 2 to the macroscopic data are given in Table 2.
Discussion
There are three main findings of this study. First, homoquinolinate is a low-affinity partial agonist at NR1/NR2A receptors with predicted increased intrapocket motion relative to glutamate. Second, NR1/NR2A receptors activated by glutamate open faster and have a higher open probability than those activated by the partial agonist homoquinolinate, despite identical single-channel chord conductance and mean open time. Third, we hypothesize that helix F in the S2 domain of NR2A appears well positioned to translate the effects of agonist binding to regions of the receptor that control channel gating.
MD simulations suggest that glutamate and homoquinolinate interact differently with the agonist binding pocket of NR2A. In particular, residues interacting with the carboxylates are significantly displaced by homoquinolinate from their location when glutamate is bound. This modeling prediction was experimentally verified by the finding that the apparent affinity of homoquinolinate is less sensitive to mutations predicted to interact with the carboxylates of glutamate. The increased motion of portions of the binding pocket around homoquinolinate may explain why the experimental KD value is higher for homoquinolinate (60 μm) than for glutamate (7 μm). These KD values can be used to estimate a standard free energy change for equilibrium binding of homoquinolinate (-5.76 kcal/mol) and glutamate (-7.03 kcal/mol).
Comparison of the apo, glutamate-bound, and homoquinolinate-bound states reveals not only that helix F in the S2 domain is significantly displaced by glutamate binding but also that it becomes decoupled from local secondary structure elements after homoquinolinate binding. This decoupling does not occur with glutamate binding, suggesting that movement of helix F may exert strain on critical elements for transducing agonist binding to channel gating and that this process may be compromised by the changes in the binding pocket induced by the conformationally restrained partial agonist homoquinolinate.
Patch-clamp recording revealed that the single-channel conductance, mean channel open time, and rates governing desensitization were unaffected by homoquinolinate compared with glutamate but that channels activate more slowly in response to homoquinolinate than to glutamate. This is consistent with the hypothesis that agonist binding controls a pregating conformational change for each subunit but that stability and size of the channel pore reflects a concerted global conformational change not directly dependent on the conformation of the agonist binding domain. These results are all compatible with a physical model of activation that includes at least two obligatory conformational changes that precede gating, one of which is controlled by agonist binding to the NR2 subunit and perhaps involves the displacement of helix F in the S2 domain.
Mechanism of partial agonism at glutamate receptors
Although the concept of partial agonism was developed more than 50 years ago (Ariens, 1954; Stephenson, 1956; Del Castillo and Katz, 1957), only recently has an understanding of the structural and functional basis of partial agonism been elucidated for at least one receptor, the AMPA subtype of glutamate receptors. It has been shown for AMPA receptors that partial agonists induce only partial closure of the agonist-binding S1S2 domain purified in isolation from the receptor (Armstrong and Gouaux, 2000), suggesting that partial agonism in some ways reflects the ability of different structural agonists to induce distinct receptor domain conformations. Functionally, activation of AMPA receptors by any agonist induces multiple conductance levels, and evidence exists that the permeation properties of each conductance level may be independent of the structure of the agonist. It has been proposed that each AMPA receptor subunit can gate independently and make an incremental contribution to ion permeation (Rosenmund et al., 1998; Smith et al., 2000). This important result drove the compelling convergence of structure and function achieved by Jin et al. (2003), who demonstrated that the degree of domain closure of the S1S2 domain in GluR2 induced by partial agonists that differ by only a single atom correlates directly with the functional efficacy for coupling agonist binding to channel gating. However, there is evidence that the glycine binding domain of NR1 does not exhibit a similar correlation between domain closure and agonist efficacy (Furukawa and Gouaux 2003; Inanobe et al., 2005).
Unlike AMPA receptors, NMDA receptors must be fully liganded to open. There is evidence that multiple conformational changes are required to transduce agonist binding into channel gating (Banke and Traynelis, 2003; Popescu and Auerbach, 2003; Popescu et al., 2004). Although NMDA receptor activation is thus distinct from that of AMPA receptors (Erreger et al., 2004), there are still important precedents operating for AMPA receptors that hold implications for NMDA receptors. For example, both AMPA and NMDA receptor subunits can contribute kinetically distinct features to channel activation, suggesting that individual NMDA receptor subunits can have independent effects on gating. In addition, Armstrong and Gouaux (2000) first showed that partial agonism at AMPA receptors involves at least some degree of altered protein conformation. Whereas direct empirical structural data on agonist binding to NR2A are unavailable, our MD docking simulations intimate that homoquinolinate and glutamate induce different conformations within the binding cleft. In particular, MD analysis of agonist docking suggests that homoquinolinate, a constrained analog of glutamate, engages in a significantly different set of interactions within the binding domain and thereby stimulates a qualitatively variable response from the protein by comparison with glutamate. Thus, the different conformations induced by the glutamate- and homoquinolinate-bound agonist binding domains may control efficacy. The divergent efficacies could reflect global perturbation in the structure of the agonist binding domain when homoquinolinate is docked (Fig. 2, helix F). Alternatively, the partial agonism of homoquinolinate may imply increased intraprotein motion and decreased stability of the binding pocket (Fig. 5), which might not spend as much time in the fully closed conformation hypothesized to activate the ion channel. Although we cannot yet distinguish between these two hypotheses, the combined structural and functional approach described here helps define more clearly the molecular basis of partial agonism for NMDA receptors.
Footnotes
This work was supported by The Howard Hughes Medical Institute (K.E.), National Institutes of Health-National Institute of Neurological Disorders and Stroke Grant NS36654 (S.F.T.), the National Alliance for Research on Schizophrenia and Depression (S.F.T.), and the Biotechnology and Biological Sciences Research Council (D.J.A.W.). We thank Drs. Morris Benveniste and Anders Kristensen for critical comments on this manuscript and Polina Lyuboslavsky, Phuong Le, and Antoine Almonte for excellent technical assistance. We also thank Drs. Hiro Furukawa and Eric Gouaux for sharing unpublished data. M.T.G. and J.P.S. are grateful to Prof. Dennis Liotta (Emory University, Atlanta, GA) for support and encouragement.
Correspondence should be addressed to Dr. Kevin Erreger, Department of Molecular Physiology and Biophysics, Vanderbilt University, 7124 Medical Research Building III, 465 21st Avenue South, Nashville, TN 37232. E-mail: kevin.erreger@vanderbilt.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257858-09$15.00/0
References
- Ariens EJ (1954) Affinity and intrinsic activity in the theory of competitive inhibition. I. Problems and theory. Arch Int Pharmacodyn Ther 99: 32-49. [PubMed] [Google Scholar]
- Armstrong N, Gouaux E (2000) Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron 28: 165-181. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Sun Y, Chen GQ, Gouaux E (1998) Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature 395: 913-917. [DOI] [PubMed] [Google Scholar]
- Banke TG, Traynelis SF (2003) Activation of NR1/NR2B NMDA receptors. Nat Neurosci 6: 144-152. [DOI] [PubMed] [Google Scholar]
- Banke TG, Dravid SM, Traynelis SF (2005) Protons trap NR1/NR2B NMDA receptors in a nonconducting state. J Neurosci 25: 42-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen HJC, van der Spoelvan D, Drunen R (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comp Phys Commun 91: 43-56. [Google Scholar]
- Chen PE, Geballe MT, Stansfeld PJ, Johnston AR, Yuan H, Jacob AL, Snyder JP, Traynelis SF, Wyllie DJ (2005) Structural features of the glutamate binding site in recombinant NR1/NR2A N-methyl-d-aspartate receptors determined by site-directed mutagenesis and molecular modeling. Mol Pharmacol [DOI] [PubMed]
- Del Castillo J, Katz B (1957) Interaction at end-plate receptors between different choline derivatives. Proc R Soc Lond B Biol Sci 146: 369-381. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7-61. [PubMed] [Google Scholar]
- Erreger K, Chen PE, Wyllie DJ, Traynelis SF (2004) Glutamate receptor gating. Crit Rev Neurobiol 16: 187-224. [DOI] [PubMed] [Google Scholar]
- Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF (2005) Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol (Lond) 563: 345-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa H, Gouaux E (2003) Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. EMBO J 22: 2873-2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb AJ, Colquhoun D (1991) Glutamate activation of a single NMDA receptor-channel produces a cluster of channel openings. Proc Biol Sci 243: 39-45. [DOI] [PubMed] [Google Scholar]
- Gouaux E (2004) Structure and function of AMPA receptors. J Physiol (Lond) 554: 249-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inanobe A, Furukawa H, Gouaux E (2005) Mechanism of partial agonist action at the NR1 subunit of NMDA receptors. Neuron 47: 71-84. [DOI] [PubMed] [Google Scholar]
- Jackson MB, Wong BS, Morris CE, Lecar H, Christian CN (1983) Successive openings of the same acetylcholine receptor channel are correlated in open time. Biophys J 42: 109-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R, Gouaux E (2003) Probing the function, conformational plasticity, and dimer-dimer contacts of the GluR2 ligand-binding core: studies of 5-substituted willardiines and GluR2 S1S2 in the crystal. Biochemistry 42: 5201-5213. [DOI] [PubMed] [Google Scholar]
- Jin R, Horning M, Mayer ML, Gouaux E (2002) Mechanism of activation and selectivity in a ligand-gated ion channel: structural and functional studies of GluR2 and quisqualate. Biochemistry 41: 15635-15643. [DOI] [PubMed] [Google Scholar]
- Jin R, Banke TG, Mayer ML, Traynelis SF, Gouaux E (2003) Structural basis for partial agonist action at ionotropic glutamate receptors. Nat Neurosci 6: 803-810. [DOI] [PubMed] [Google Scholar]
- Lindahl E, Hess B, van der Spoelvan D (2001) GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Mod 7: 306-317. [Google Scholar]
- Mayer ML, Armstrong N (2004) Structure and function of glutamate receptor ion channels. Annu Rev Physiol 66: 161-181. [DOI] [PubMed] [Google Scholar]
- Popescu G, Auerbach A (2003) Modal gating of NMDA receptors and the shape of their synaptic response. Nat Neurosci 6: 476-483. [DOI] [PubMed] [Google Scholar]
- Popescu G, Robert A, Howe JR, Auerbach A (2004) Reaction mechanism determines NMDA receptor response to repetitive stimulation. Nature 430: 790-793. [DOI] [PubMed] [Google Scholar]
- Qin F (2004) Restoration of single-channel currents using the segmental k-means method based on hidden Markov modeling. Biophys J 86: 1488-1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F, Auerbach A, Sachs F (1996) Estimating single-channel kinetic parameters from idealized patch-clamp data containing missed events. Biophys J 70: 264-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stern-Bach Y, Stevens CF (1998) The tetrameric structure of a glutamate receptor channel. Science 280: 1596-1599. [DOI] [PubMed] [Google Scholar]
- Smith TC, Wang LY, Howe JR (2000) Heterogeneous conductance levels of native AMPA receptors. J Neurosci 20: 2073-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson RP (1956) A modification of receptor theory. Br J Pharmacol 11: 379-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Burgess MF, Zheng F, Lyuboslavsky P, Powers JL (1998) Control of voltage-independent zinc inhibition of NMDA receptors by the NR1 subunit. J Neurosci 18: 6163-6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie DJ, Behe P, Colquhoun D (1998) Single-channel activations and concentration jumps: comparison of recombinant NR1a/NR2A and NR1a/NR2D NMDA receptors. J Physiol (Lond) 510: 1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F, Gingrich MB, Traynelis SF, Conn PJ (1998) Tyrosine kinase potentiates NMDA receptor currents by reducing tonic zinc inhibition. Nat Neurosci 1: 185-191. [DOI] [PubMed] [Google Scholar]
- Zheng F, Erreger K, Low CM, Banke T, Lee CJ, Conn PJ, Traynelis SF (2001) Allosteric interaction between the amino terminal domain and the ligand binding domain of NR2A. Nat Neurosci 4: 894-901. [DOI] [PubMed] [Google Scholar]