

Abstract
Models of Parkinson's disease (PD) based on selective neuronal death have been used to study pathogenic mechanisms underlying nigral cell death and in some instances to develop symptomatic therapies. For validation of putative neuroprotectants, a model is desirable in which the events leading to neurodegeneration replicate those occurring in the disease. We developed a human in vitro model of PD based on the assumption that dysregulated cytoplasmic dopamine levels trigger cell loss in this disorder. Differentiated human mesencephalic neuron-derived cells were exposed to methamphetamine (METH) to promote cytoplasmic dopamine accumulation. In the presence of elevated iron concentrations, as observed in PD, increased cytosolic dopamine led to oxidative stress, c-Jun N-terminal kinase (JNK) pathway activation, neurite degeneration, and eventually apoptosis. We examined the role of the mixed-lineage kinases (MLKs) in this complex degenerative cascade by using the potent inhibitor 3,9-bis[(ethylthio)methyl]-K-252a (CEP1347). Inhibition of MLKs not only prevented FeCl2+/METH-induced JNK activation and apoptosis but also early events such as neurite degeneration and oxidative stress. This broad neuroprotective action of CEP1347 was associated with increased expression of an oxidative stress-response modulator, activating transcription factor 4. As a functional consequence, transcription of the cystine/glutamate and glycine transporters, cellular cystine uptake and intracellular levels of the redox buffer glutathione were augmented. In conclusion, this new human model of parkinsonian neurodegeneration has the potential to yield new insights into neurorestorative therapeutics and suggests that enhancement of cytoprotective mechanisms, in addition to blockade of apoptosis, may be essential for disease modulation.
Keywords: in vitro, reactive oxygen species, SAPK, neurodegeneration, apoptosis, MLK
Introduction
Nigral dopaminergic neurons are under continuous oxidative stress because of the unstable nature of their neurotransmitter dopamine (Graham, 1978; Maker et al., 1981), the adverse effects of which are further enhanced by high concentrations of iron in the substantia nigra (SN) (for review, see Zecca et al., 2004). Experimental studies using methamphetamine (METH) suggest that abnormal accumulation of cytoplasmic dopamine could play an important role in disease pathogenesis by inducing oxidative stress (for review, see Lotharius and Brundin, 2002). Furthermore, nigrostriatal degeneration in mice treated with METH or the parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), whose active metabolite can also induce dopamine-dependent oxidative stress (Lotharius and O'Malley, 2000), has been associated with an increase in the phosphorylation of members of the c-jun N-terminal kinase (JNK) pathway, including c-Jun, JNK, and its upstream-activating kinase, mitogen-activated protein kinase kinase 4 (MKK4) (Saporito et al., 2000; Jayanthi et al., 2002; Willesen et al., 2002). In agreement with an active involvement of the JNK pathway in cell death, MPTP-induced neurodegeneration and motor deficits were significantly reduced in JNK2/JNK3 knock-out mice (Hunot et al., 2004), in mice overexpressing the inhibitory JNK-interacting protein 1 (Xia et al., 2001), and in mice given the JNK inhibitor SP600125 (anthra[1,9-cd]pyrazol-6(2H)-one) (W. Wang et al., 2004). Similarly, reduced c-Jun expression leads to increased resistance to METH-induced apoptosis (Deng et al., 2002).
Inhibition of the mixed-lineage kinases (MLKs), which activate JNK indirectly via MKK4 and MKK7, can protect neurons from various insults. The potent MLK inhibitor 3,9-bis[(ethylthio)methyl]-K-252a (CEP1347) reduces JNK activation and, consequently, neuronal death induced by trophic factor withdrawal (Maroney et al., 1999; Harris et al., 2002), β-amyloid (Bozyczko-Coyne et al., 2001; Troy et al., 2001), axotomy (Glicksman et al., 1998), and MPTP exposure (Saporito et al., 1999). This is accomplished without inhibiting JNK directly, allowing for physiological activation of JNK by other upstream kinases (Maroney et al., 1998, 1999, 2001; Bozyczko-Coyne et al., 2001; Troy et al., 2001; Harris et al., 2002). In addition to its antiapoptotic properties, CEP1347 exerts potent neurotrophic effects in injured neurons, such as induction of neurite outgrowth, maintenance of neurotransmitter synthesis, and preservation of cellular metabolism (for review, see L. H. Wang et al., 2004).
To predict whether CEP1347 could be beneficial for treating neurodegenerative disorders such as Parkinson's disease (PD), a broad range of experimental systems modeling disease pathogenesis is desirable. Thus, we developed a new human cellular model of neurodegeneration based on the premise that cytosolic dopamine contributes to a loss of nigral neurons in PD. We exposed cultured human mesencephalic-derived cells (Lotharius et al., 2002) to METH and iron (FeCl2) to trigger endogenous dopamine-dependent oxidative stress and cell death. We further investigated whether CEP1347 could block neurotoxicity as well as early detrimental events involved in the collapse of neuronal function. Our data suggest that CEP1347 is not only antiapoptotic but also preserves neurite integrity by increasing cellular resistance to oxidative stress.
Materials and Methods
Chemicals
Methamphetamine was obtained from Lipomed (Arleshein, Switzerland). Acridine orange (catalog #A3568) was from Molecular Probes (Eugene, OR); Mn(III)tetrakis(4-benzoic acid)porphyrin chloride (MnTBAP) was from OxisResearch (Portland, OR); 2-(4-morpholinyl)-8-phenyl-4 H-1-benzopyran-4-one (LY294002) was purchased from Calbiochem/EMD Biosciences (San Diego, CA); CEP1347 was provided by Cephalon (West Chester, PA). All other chemicals, including FeCl2 (catalog #F2130), N-acetyl-l-cysteine (NAC) (catalog #A9165), superoxide dismutase (SOD) (from bovine erythrocytes; catalog #S5395), deferoxamine mesylate (DM) (Desferal; catalog #D9533), catalase (from human erythrocytes; catalog #C3556), and α-methyl-l-tyrosine (α-MPT) (catalog #M8131) were obtained from Sigma (Brøndby, Denmark) unless otherwise specified. FeCl2 was freshly prepared in double-distilled water for every experiment and used immediately.
Cell cultures
Lund human mesencephalic (LUHMES) cells are a subclone of the tetracycline-controlled, v-myc-overexpressing human mesencephalic-derived cell line MESC2.10, characterized at and originating from Lund University (Lund, Sweden) (Lotharius et al., 2002). LUHMES cells can be differentiated into morphologically and biochemically mature dopamine-like neurons after the addition of tetracycline, glial cell line-derived neurotrophic factor (GDNF), and db-cAMP and exhibit the same dopaminergic and neuronal characteristics as MESC2.10 cells [e.g., intense β-III-tubulin immunoreactivity, extensive neuritic processes, time-dependent induction of tyrosine hydroxylase (TH), and extracellular dopamine release], as measured by cyclic voltametry (data not shown). The culturing and handling procedure of LUHMES cells was as described previously (Lotharius et al., 2002), with the exception that all plasticware was precoated overnight with 50 μg/ml poly-l-ornithine (Sigma) at room temperature (RT) followed by a 3 h incubation with 1 μg/ml fibronectin (Sigma) at 37°C to promote cell attachment. Plates/dishes were washed three times with water and completely air dried before plating. All experiments were conducted after 4 d of differentiation in Ham's F-12 high-glucose medium (Irvine Scientific, Santa Ana, CA) containing N2 supplement (Invitrogen, Glostrup, Denmark), 2 ng/ml human recombinant GDNF (R & D Systems, Wiesbaden-Nordenstadt, Germany), 1 mm dibutyryl cAMP (Sigma), and 1 μg/ml tetracycline (Sigma) after 4 d of differentiation. All experiments were vehicle controlled.
Primary ventral mesencephalic cultures
Primary rat ventral mesencephalic (VM) cultures were prepared as described previously (Boll et al., 2004). Briefly, VM tissue was dissected from 14-d-old rat embryos and kept in ice-cold HBSS. The tissue was trypsinized in a solution containing 0.1% (w/v) trypsin and 0.05% (w/v) DNase dissolved in HBSS. After 20 min of incubation at 37°C, the tissue was rinsed four times in a 0.05% DNase-HBSS solution and mechanically dissociated. The cells were resuspended in DMEM containing 1 mm sodium pyruvate, 15 mm HEPES, 1% penicillin/streptomycin, and 10% (v/v) heat-inactivated fetal bovine serum and seeded on poly-l-lysine (50 mg/L)-coated 48-well plates at a density of 200,000 cells/well. Medium was replaced with Neurobasal medium supplemented with B27, 0.5 mm l-glutamine, and 1% penicillin/streptomycin after 16 h, and one-half of the medium was replaced every other day with fresh Neurobasal medium. Cultures were pretreated with 100-1000 nm CEP1347 and then exposed to 250 μm METH for 3 d (dose responses and time courses were performed previously to establish toxicity of METH). Cultures were fixed and processed for TH immunocytochemistry. Experiments were conducted after 6 d in vitro.
Cell viability assays
MTT reduction. Cells were plated in 48-well plates (Nunc, Roskilde, Denmark) at a density of 45,000 cells/well and treated with Fe2+/METH in the presence or absence of various compounds for 72 h. To determine cell viability, the medium was removed, and cells were incubated with 5 mg/ml 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2 H-tetrazolium bromide (MTT) in fresh differentiation medium for 2 h at 37°C. At this point, the medium was removed, and cells were lysed in 95% isopropanol/5% formic acid under vigorous shaking for 15 min at RT. An aliquot was transferred to a 96-well plate, and the optical density was measured using a Multiscan Ascent spectrophotometer equipped with Ascent software version 2.4.2 (570 nm/690 nm filter pair; Titertek, Huntsville, AL). Data are expressed as mean ± SEM of quadruplicate determinations and are shown as a percentage of control cultures.
Lactate dehydrogenase release. The activity of lactate dehydrogenase (LDH) released into the medium was measured using the Cytotoxicity Detection kit (LDH) from Roche Diagnostics (Mannheim, Germany) according to the instructions of the manufacturer. Briefly, the entire medium from each well was transferred to an Eppendorf (Hamburg, Germany) tube and centrifuged for 2 min at 15,000 × g to remove cellular debris. A 50 μl aliquot of the supernatant was transferred to a 96-well plate, and 50 μl of freshly prepared reaction mixture was added. The plate was incubated for 25 min at RT while shaking, and the optical density was measured using the Multiscan Ascent spectrophotometer (492 nm/690 nm filter pair). Total LDH content was determined by lysing the cells in 0.3% Triton X-100 and measuring LDH in the lysate. Data are expressed as mean ± SEM of quadruplicate determinations and shown as a percentage of control cultures.
Calcein-AM staining. Membrane integrity was measured at the single-cell level using the LIVE/DEAD viability/cytotoxicity kit from Molecular Probes. Briefly, cells were incubated with 1 μm calcein AM for 20 min at 37°C and visualized by fluorescence microscopy. The number of calcein AM-positive cells was counted using the Target Activation BioApplication in a Cellomics (Old Amersham, UK) ArrayScan high-content screening (HCS) automated microscope. Data are expressed as mean ± SEM of quadruplicate determinations and shown as a percentage of control cultures.
Apoptosis. Assessment of nuclear condensation/fragmentation and plasma membrane integrity was conducted using the DNA-intercalating dye Hoechst 33342 and the membrane-impermeable marker SYTOX green (both from Molecular Probes). After drug exposure, LUHMES cells were incubated with 1 μg/ml Hoechst 33342 and 1 μm SYTOX green for 15 min at 37°C. Fluorescence was then visualized with a Zeiss (Oberkochen, Germany) Axiovert S100 TV microscope using excitation/emission filter pairs 365/420 nm (Hoechst) and 450/565 nm (SYTOX green). Images were taken using MetaMorph software version 5.0 (Universal Imaging, Downingtown, PA).
TH cell counting. The number of surviving dopaminergic neurons in ventral mesencephalic cultures was assessed by immunostaining cultures for TH using a 1:1000 dilution of monoclonal anti-TH antibody (catalog #MAB318; Chemicon, Temecula, CA), the ABComplex/HRP detection system (Dako, High Wycombe, UK), and DAB as a chromogen. The number of TH-positive neurons per well was counted by a blinded examiner using a cast grid system. Data are expressed as mean ± SEM of quadruplicate wells and shown as a percentage of control cultures.
Western blotting
Cells were lysed in 20 mm HEPES, 100 mm NaCl, 100 mm NaF, 1 mm Na3VO4, 5 mm EDTA, 1% Triton X-100, and 1× complete protease inhibitors (Roche Diagnostics) for 15 min on ice. Protein determinations were conducted using a BCA Protein Assay kit from Pierce (Rockford, IL). Thirty micrograms of protein, heated for 10 min at 70°C, were electrophoresed on 4-12% bis-Tris NuPage gels (Invitrogen) using 3-(N-morpholino)propanesulfonic acid SDS running buffer for ∼1.5 h at 200 V. Proteins were transferred onto an activated polyvinylidene difluoride membrane (Immobilon-P; Millipore, Glostrup, Denmark) using the XCell II blot module from Invitrogen. Membranes were blocked in 5% milk in TBS/T (2.42 g/L Tris base, 8 g/L NaCl, and 0.1% Tween 20, pH 7.6), washed with TBS/T, and incubated overnight at 4°C with primary antibody dissolved in 5% milk in TBS/T. The following primary antibodies and their respective dilutions were used [all from Cell Signaling Technology (Beverly, MA) unless otherwise specified]: 1:1000 polyclonal anti-phosphorylated AKT (Thr308), 1:2000 monoclonal anti-β-III-tubulin antibody (clone SDL.3D10; Sigma), 1:1000 polyclonal anti-phosphorylated c-Jun (Ser63), 1:100 polyclonal anti-cAMP response element-binding protein 2 [CREB2; activating transcription factor 4 (ATF4); Santa Cruz Biotechnology, Santa Cruz, CA], 1:500 monoclonal anti-phosphorylated JNK (Thr183/Thr185; clone G-7; Santa Cruz Biotechnology), 1:1000 polyclonal anti-phosphorylated MKK4 (Thr261), and 1:2000 monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (clone 6C5; Novus Biologicals, Littleton, CO). Blots were washed and incubated with appropriate horseradish peroxidase-conjugated secondary antibodies [1:2000 rabbit anti-mouse and 1:1000 goat anti-rabbit from DakoCytomation (Glostrup, Denmark)] for 1 h at RT and developed using ECL or ECL Plus detection reagents (Amersham Biosciences, Arlington Heights, IL).
pH measurements
Cells were plated at a density of 45,000 cells/well in 48-well plates and differentiated for 4 d. On day 4, cells were loaded with a 25 nm concentration of the pH-sensitive dye acridine orange (catalog #A3568; Molecular Probes) and 0.2 μg/ml Hoechst for 40 min, followed by a 40 min incubation with 1 mm METH. The ring-spot fluorescence intensity in the cytoplasm was measured using a Cellomics ArrayScan HCS automated microscope (excitation/emission filter pair, 475/515 nm) using the Compartmental Analysis BioApplication. All experiments were performed in quadruplicate.
Oxidative stress
Superoxide production. Cells were plated at a density of 200,000 cells/well in 12-well plates and differentiated for 4 d. On day 4, cells were exposed for 2-60 h to FeCl2 and METH in the presence or absence of CEP1347, loaded for 30 min at 37°C with 3 μm dihydroethidium (which is oxidized to ethidium by superoxide anions), and lysed in 150 μl DMSO by freezing/thawing. The extract was transferred to a black 96-well plate, and fluorescence was measured with the Multiscan Ascent FL Fluoroscan (excitation/emission filter pair, 485/612 nm). Relative fluorescence units per sample were normalized to micrograms of protein. Protein determinations were conducted with a 5 μl aliquot from each sample using the BCA Protein Assay kit from Pierce.
Hydrogen peroxide formation. Cells were plated at a density of 400,000 cells per 5 cm dish, allowed to proliferate for 48 h, and differentiated for 4 d. Cells were exposed to METH in the presence or absence of CEP1347, rinsed with ice-cold PBS, and scraped off in 150 μl of PBS with a rubber policeman. FeCl2 was omitted from this experiment because of interference of Fe2+ with the assay reaction. Cells were homogenized by passing the resuspended cells 15 times through a 27 ga needle. The lysate was centrifuged at 1500 × g for 10 min, and a 50 μl aliquot of the supernatant was used to measure hydrogen peroxide (H2O2) using a commercial, colorimetric assay based on the oxidation of Fe2+ to Fe3+ by H2O2 under acidic conditions (Bioxytech MDA-586; OxisResearch). An H2O2 standard curve was used to determine the absolute concentrations of H2O2 in the samples. Values were standardized to micrograms of protein as determined by a BCA Protein Assay kit from Pierce.
Lipid peroxidation. Cells were plated at a density of 400,000 cells per 5 cm dish, allowed to proliferate for 48 h, and differentiated for 4 d. After drug exposure, cells were rinsed with ice-cold PBS and scraped off in 250 μl of PBS containing 5 mm butylated hydroxytoluene using a rubber policeman. Cells were homogenized by passing the lysate 15 times through a 27 ga needle and once through a 30 ga needle. The lysate was centrifuged at 1000 × g for 10 min, and a 200 μl aliquot of the supernatant was used to measure malondialdehyde (MDA) levels using a kit from OxisResearch (Bioxytech MDA-586) according to the instructions of the manufacturer. This assay is based on the reaction of a chromogenic reagent, N-methyl-2-phenylindole, with MDA at 45°C, and measures specifically MDA and not other lipid peroxidation products. An MDA standard curve was used to determine the absolute concentration of MDA in the samples using the stable MDA precursor tetramethoxypropane. Values were standardized to micrograms of protein, as determined by a BCA Protein Assay kit from Pierce.
Glutathione measurements
For glutathione (GSH) measurements, LUHMES cells were plated at a density of 400,000 cells in 5 cm dishes and differentiated for 4 d. After drug exposure, LUHMES cells were rinsed twice with ice-cold PBS and lysed in 200 μl of 1% sulfosalicylic acid (w/v) on ice. The lysates were scraped off of the dish and centrifuged for 1 min at 15,000 × g. Total glutathione content [reduced glutathione (GSH) plus oxidized glutathione (GSSG)] was determined in the supernatants using a microtiter plate assay as described previously (Dringen and Hamprecht, 1996), with a slight modification of the photometric method originally described by Tietze (1969). The pellet obtained from the centrifugation of samples was dissolved in 0.5 m NaOH, and the protein content was determined by the method of Lowry. The amount of GSH per sample was standardized to the protein content of each respective precipitate. All samples contained <1% GSSG, so that total GSH concentrations were almost identical to that of GSH in the cells.
Cystine uptake
A total of 80,000 cells/well were plated in 24-well plates and differentiated for 6 d. On day 6, cells were treated for 16 h with Fe2+/METH, 250 nm CEP1347, or both. Cells were incubated for 20 min in 0.2 μCi/ml [14C]cystine (9.25 GBq/mmol; PerkinElmer, Wellesley, MA) dissolved in prewarmed PBSG (10 mm PBS with 137 mm NaCl and 3 mm KCl, pH 7.4, containing 0.01% CaCl2, 0.01% MgCl2·6 H2O, and 0.1% glucose). Uptake was terminated by rapidly rinsing the cells three times in ice-cold PBS. Cells were then lysed in 0.5N NaOH by vigorous shaking. The lysate was transferred to a scintillation tube, and radioactivity was counted with a Packard Tri-Carb 2100TR counter (Global Medical Instrumentation, Ramsey, MN). MTT reduction assays were performed in parallel wells to ensure that Fe2+/METH or CEP1347 did not enhance cellular metabolism after the 16 h incubation period, which could possibly affect cystine uptake. Assays were run in six replicates.
Immunocytochemistry
Cells treated with 1 mm METH or 75 μm FeCl2 and 1 mm METH in the presence or absence of 250 nm CEP1347 were fixed with 4% fresh paraformaldehyde for 20 min at 37°C and rinsed three times in PBS. Cells were blocked for 1 h with 5% goat serum in 0.3% Triton X-100/PBS and incubated overnight at 4°C with a 1:1000 dilution of a rabbit anti-mitogen-activated protein 2 (MAP2) antibody (catalog #AB5622; Chemicon) or a 1:2000 dilution of a monoclonal anti-β-III-tubulin antibody (catalog #T0198; Sigma) dissolved in 0.3% Triton X-100/PBS. Cells were rinsed three times in PBS and incubated for 1 h with 1:500 Alexa Fluor 488-coupled goat anti-mouse IgG (Molecular Probes) dissolved in 0.3% Triton X-100/PBS. Cells were rinsed three times in PBS and visualized with a Zeiss Axiovert S100 TV microscope. In some cases, cells were incubated with 1 μg/ml Hoechst 33342 for 20 min in the second-to-last wash. Images were taken using MetaMorph software version 5.0.
Quantification of neurite degeneration
To quantify neurite degeneration resulting from Fe2+/METH exposure, a Cellomics ArrayScan HCS System was used. Cells were plated in 48-well plates at a density of 55,000 cells/well, differentiated for 4 d, and exposed to Fe2+/METH for 48 h. Cells were fixed with 4% fresh paraformaldehyde and processed for β-III-tubulin immunoreactivity using an Alexa Fluor 488-coupled secondary antibody and Hoechst 33342 (see above, Immunocytochemistry). Fluorescence was quantified using the GPCR (G-protein-coupled receptor) BioApplication from Cellomics, which can automatically subtract the fluorescence intensity of the cell body from the total fluorescence signal of the field being imaged to yield the neuritic intensity only. The size of the cell body was estimated using a modified mask (nuclear diameter plus 2 μm) of the nucleus of the neuron, which was defined by Hoechst 33342-positive pixels. Fifty fields/well were captured using a 10× lens. The “neurite mass” of these 50 fields was then averaged and normalized to that of control cultures. Each condition was run in quadruplicate.
Caspase-3 activation
Immunocytochemistry for activated caspase-3 was performed using an antibody specific for the cleaved protein [rabbit anti-human caspase-3 (Asp175) antibody; catalog #9661; Cell Signaling Technology]. Cells were incubated overnight with anti-activated caspase-3 antibody (1:500) at 4°C in a humid chamber. After washing, cells were incubated with Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody (1:500; catalog #A11008; Molecular Probes) and 1 μg/ml Hoechst 33342 for 1 h at RT. Cells were visualized with an Axiovert S100 TV Zeiss microscope using excitation/emission filter pairs 365/458 nm (Hoechst) and 494/517 nm (Alexa Fluor 488).
Enzymatic activity was measured as described previously (Hentze et al., 2001). Briefly, cells were plated in 24-well plates at a density of 65,000 cells/well, differentiated for 4 d, and treated with CEP1347 for 6-72 h. Fe2+/METH was added at staggered time points, and cells were lysed at the same time with 80 μl of lysis buffer containing 20 mm HEPES, 5 mm MgCl2, 1 mm EGTA, 0.4% Triton X-100, 4 mm DTT, and 1 mm 4-(2-aminoethyl)benzenesulfonylfluoride, pH 7.4 (all from Sigma). Cells were frozen at -80°C and then thawed on ice to ensure lysis. The supernatant was removed from the wells and centrifuged at 15,000 × g for 2 min. Subsequently, 50 μl of lysate was transferred to a black 96-well plate (Nunc), and the reaction was initiated by adding 50 μl of assay buffer containing 50 mm HEPES, 10 mm DTT, 1% sucrose, 0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, and 100 μm Ac-Asp-Glu-Val-Asp-7-Amino-4-trifluoromethylcoumarin (Ac-DEVD-AFC) substrate (Biomol, Plymouth Meeting, PA) to each well. The plate was shaken vigorously for 15 s, and fluorescence was measured with a Multiscan Ascent FL Fluoroscan using Ascent software version 2.4.2 [excitation/emission filter pair, 390/510 nm; kinetic interval, 20 ms; 90 scans (30 min)]. For analysis, the fluorescence increase between 10 and 20 min was measured and expressed as fluorescence units per minute. All values were normalized to those of control cells.
Quantitative PCR
Cells stimulated in 10 cm dishes were washed once with PBS, and total RNA was extracted using TRIzol reagent (Invitrogen) according to the protocol of the manufacturer. A DNase-free, DNase-1 kit was purchased through Ambion (Huntingdon, UK), and the purified RNA was treated according to the instructions of the manufacturer. Total RNA (1 μg) was used for additional analysis, as described previously (Falsig et al., 2004). RNA was reverse transcribed with TaqMan reverse transcription Reagent (Applied Biosystems, Naerum, Denmark), using random hexamers in a 100 μl reaction mixture with a PTC-200 DNA Engine Thermal Cycler (VWR International, Albertslund, Denmark). The program consisted of 10 min of annealing at 25°C, 30 min of reverse transcription at 48°C, and 5 min of inactivation at 95°C. The cDNA was quantified using the SYBR Green PCR Master Mix kit (PE Applied Biosystems, Foster City, CA). Each quantitative PCR (qPCR) contained 2.5 μl of cDNA of the 100 μl reverse transcription product, 300 nm forward and reverse primers, 12.5 μl of master mix, and 7 μl of water in a total volume of 25 μl. PCR amplification was run in a 96-well experimental plate format on an iCycler Thermal Cycler equipped with the iCycler Optical System (Bio-Rad, Hercules, CA). The program setup was 10 min at 95°C, 40 cycles of 15 s at 95°C, and 1 min at 60°C. A melting curve was obtained to verify the measured signal, and the product was run on a 4% agarose gel to verify the presence of the expected band. Quantification was performed as follows: using the iCycler data analysis software (Bio-Rad), the threshold cycle (Tc) was determined for each sample. Tc was defined as the cycle at which the level of fluorescence increased significantly above the background levels of fluorescence. The concentration of cDNA was calculated by comparing the Tc of samples to Tcs of a standard curve. The standard curve was obtained by a serial dilution of cDNA. Each sample was run in two reactions, one with the primer set of interest and one with a GAPDH primer set, and all data are displayed as the ratio between the calculated starting concentration of the cDNA of interest and GAPDH. All primers, except for the housekeeping gene GAPDH, were intron spanning to distinguish cDNA from genomic DNA. For GAPDH, the order of magnitude between the samples and the samples without reverse transcriptase was >106. Primers used were as follows: GAPDH sense (GenBank accession number NM_008084), 5′-TCGACAGTCAGCCGCATCTTCTT-3′; antisense, 5′-GCGCCCAATACGACCAAATCC-3′; ATF4 sense (accession number NM_009716), 5′-GTC TGC CCG TCC CAA ACC TTA C-3′; antisense, 5′-TCC TGC TCC GCC CTC TTC TTC-3′. Cystine-glutamate (Cys-Glu) transporter sense (accession number NM_014331), 5′-CCC TGG TCC GCC CTC TTC TTC-3′; antisense, 5′-TCG CAA GTT CAG GGA TTT CAC ATT-3′; glycine transporter (GlyT1) sense (accession number NM_006934), 5′-AGG CCC TCA CAC TAC TTC CCA TCT C-3′; antisense, 5′-ACT CAT TCC CCA CCT CAT CCA CA-3′. Primers were designed using the DNAstar software package (DNASTAR, Madison, WI), and all primers were blasted using BLAST (basic local alignment search tool; www.ncbi.nlm.nih.gov/BLAST/).
Statistics
Descriptive statistics (mean ± SEM) of all quantitative assays performed were calculated with statistical software [GraphPad (San Diego, CA) Prism Software]. The mean of three to four individual experiments conducted in quadruplicate determinations is shown. The significance of effects between control cultures and Fe2+/METH or CEP1347 treatments was determined by one-way ANOVA and post hoc Student's t tests (GraphPad Prism Software) (*p < 0.05; **p < 0.01; ***p < 0.001).
Results
Progressive, dopamine-dependent cell death in differentiated LUHMES cultures by combined exposure to iron and methamphetamine (Fe2+/METH)
Amphetamines, including METH, lead to an accumulation of cytoplasmic dopamine (Sulzer et al., 1995; Mosharov et al., 2003), which can, in turn, promote oxidative stress (Cubells et al., 1994; Lotharius and O'Malley, 2001) and neurite degeneration in rodent neurons (Larsen et al., 2002). In the present study, we tested whether METH could be used as a tool compound to trigger dopamine-dependent oxidative stress in a human cell culture system. Differentiated LUHMES cells were exposed to 1 mm METH for 1-5 d, and their dendritic morphology was assessed by MAP2 and β-III-tubulin immunocytochemistry. After a 5 d incubation period, cells exhibited a dramatic loss of neurites, as shown by both β-III-tubulin and MAP2 immunocytochemistry (Fig. 1A). LUHMES cultures treated with up to 1 mm METH did not display a loss of cell viability, as assessed by a battery of cell toxicity/survival assays, including quantification of MTT reduction capacity (an assay for mitochondrial dehydrogenase activity), LDH release, ethidium homodimer uptake, and calcein AM retention, the latter three of which measure a loss in plasma membrane integrity (Fig. 1B). These data are in agreement with previous reports showing that a 7 d exposure to 100 μm METH selectively destroys neurites but spares dopaminergic cell bodies in primary mesencephalic cultures (Cubells et al., 1994; Larsen et al., 2002). The inability of METH to induce dopaminergic cell loss in vitro could indicate an inherent capacity of dopaminergic neurons to combat dopamine-dependent oxidative stress, i.e., the threshold necessary to trigger apoptosis is not reached. Alternatively, because the majority of intracellular dopamine is stored in synaptic vesicles (Sulzer et al., 1996), oxidative stress in this model could be restricted to the neurites, sparing the neuronal somata.
Figure 1.
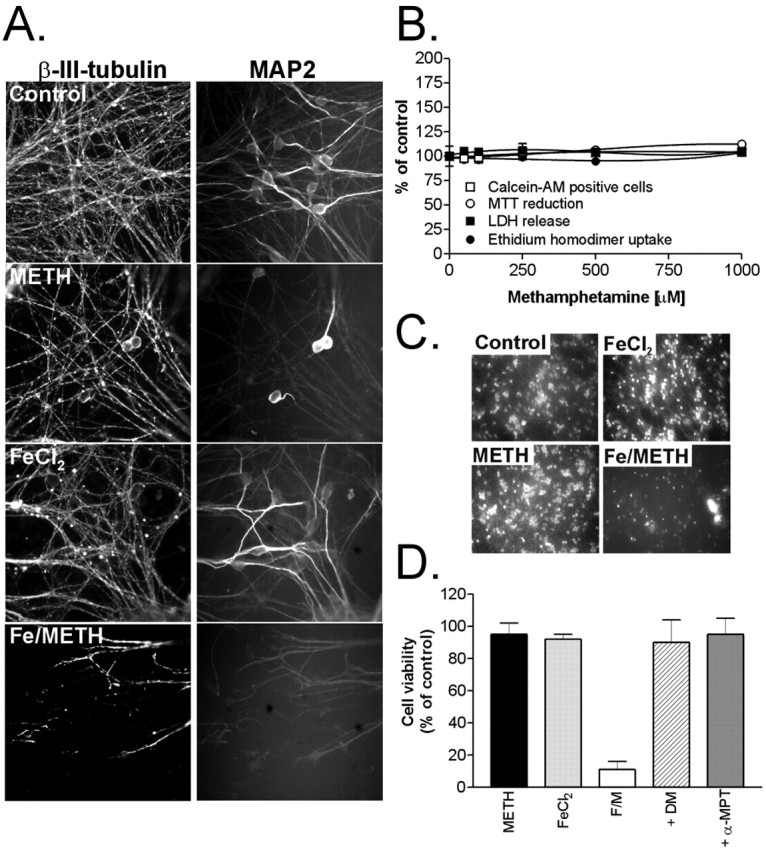
Iron (FeCl2) significantly potentiates METH toxicity in differentiated LUHMES cells. A, Differentiated LUHMES cells were treated with 1 mm METH or 75 μm FeCl2 alone or FeCl2 plus METH (Fe/METH) for 5 d, and neurite integrity was qualitatively assessed by both β-III-tubulin and MAP2 immunocytochemistry. B, Cell viability was measured in cultures exposed to 50-1000 μm METH for 5 d by a battery of cell death/viability assays. C, Potentiation of METH toxicity was examined in cultures treated with 75 μm FeCl2, 1 mm METH, or both (Fe/METH). Cell survival was assessed after 72 h by visual inspection of calcein AM-positive cells. D, A set of Fe2+/METH-treated cells (F/M) was cotreated for 72 h with 250 μm DM or 100 μm α-MPT, and cell viability was quantified by counting the number of calcein AM-positive cells. All data are expressed as the mean ± SEM of quadruplicate determinations and are shown as a percentage of control cultures. Error bars represent SEM.
To tip the balance toward neuronal death, LUHMES cells were cotreated with METH (increasing cytosolic dopamine) and nontoxic concentrations of iron to catalyze the formation of cytotoxic hydroxyl radicals from hydrogen peroxide via the Fenton reaction. In the SN of PD patients, redox-active iron levels are increased by 30-35% compared with age-matched controls (for review, see Zecca et al., 2004; Götz et al., 2004). Neuromelanin, a dark pigment produced in catecholaminergic neurons of the human SN and locus ceruleus, acts as a storage system for iron in dopaminergic neurons (Zecca et al., 2002) and has been found to associate with large amounts of iron in diseased nigral tissue (Jellinger et al., 1992, 1993). Neuromelanin could potentially serve a neuroprotective function by sequestering free intracellular iron. However, if the iron-buffering capacity of neuromelanin is exhausted, as could be the case in PD, increased leakage of iron into the cytoplasm could act synergistically with elevated cytoplasmic dopamine to promote oxidative stress. To study the potential toxic interaction between increased cytosolic dopamine levels and redox-active iron, we cotreated LUHMES cells with METH and FeCl2 and determined their combinatorial effect on neuronal toxicity. Exposure to 75 μm FeCl2 was nontoxic on its own and had minimal effects on neurite integrity (Fig. 1A,C,D); however, cotreatment with 1 mm METH led to complete neurite disintegration (Fig. 1A) and a dramatic loss in cell viability seen after 3 d of drug exposure (Fig. 1C,D).
To determine whether Fe2+/METH-induced cell death is dependent on cytosolic dopamine and iron-mediated oxidative stress, LUHMES cells were treated with either the iron chelator DM (250 μm) or the tyrosine hydroxylase inhibitor α-MPT (100 μm) in the presence of Fe2+/METH. After 3 d, cell viability was assessed by measuring both MTT reduction capacity and by counting the number of calcein AM-positive neurons. Both Desferal and α-MPT completely protected LUHMES cells from Fe2+/METH-induced cell death (Fig. 1D), supporting the notion that the toxicity conferred by this insult is dependent on both iron and dopamine.
The mode of cell death triggered by Fe2+/METH
LUHMES cells exposed to Fe2+/METH exhibited widespread nuclear fragmentation, which first became evident after 12 h of drug exposure (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). At 12-36 h, cells with fragmented nuclei still retained their plasma membrane integrity, as shown by the absence of labeling with SYTOX green, a cell-impermeable nucleic acid stain. After 48 h, cells with fragmented nuclei began to stain positively for SYTOX green, and, after 60 and 72 h, all apoptotic cells displayed broken plasma membranes. In agreement with this pattern of nuclear fragmentation, LUHMES cells treated with Fe2+/METH were strongly immunoreactive for processed caspase-3. Activated caspase-3 was readily detectable after 24 h of Fe2+/METH exposure in neurons exhibiting a significant degree of nuclear fragmentation but still had an intact plasma membrane (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Activated caspase-3 immunoreactivity could be seen as late as 60 and 72 h after Fe2+/METH exposure, when cell bodies had become shrunken and neuritic processes had disintegrated. Caspase-3-like activity was measured enzymatically using the fluorogenic caspase-3, caspase-6, caspase-7, caspase-8, and caspase-10 substrate Ac-DEVD-AFC. After 6 h, no change in Ac-DEVD-AFC cleavage was observed; however, between 12 and 60 h after Fe2+/METH treatment, cells displayed a twofold to threefold increase in caspase-3-like activity. The increase in activated caspase-3 temporally correlated with the progression of nuclear fragmentation. Notably, the level of caspase-3-like activity induced by Fe2+/METH in this culture system was relatively moderate compared with staurosporine- and colchicine-treated cells, which displayed a 16- and 24-fold increase in caspase-3-like activity, respectively, after 24 h of treatment (data not shown). To examine the causal contribution of caspases to Fe2+/METH-induced cell death, the pan-caspase inhibitor N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (zVAD-fmk) was used. Although zVAD-fmk effectively blocked nuclear fragmentation assessed 48 h after Fe2+/METH treatment, it did not protect cells from Fe2+/METH-induced cell death when used at 10-100 μm (data not shown). Although Fe2+/METH induced classical apoptotic changes, the slow nature of the insult, the mild activation of caspase-3, and the inability of caspase inhibitors to inhibit neuronal death suggest that this is not a classical apoptosis model.
The kinetics of neurite degeneration versus cell death
As mentioned above, treatment with METH alone induced neurite degeneration but not cell death. In LUHMES cells treated with FeCl2 in combination with METH, neurite beading and disintegration were clearly evident before any changes in MTT reduction or LDH release could be seen, i.e., after 24-48 h (Fig. 2A). FeCl2 had only a minimal effect on dendritic morphology, as determined by MAP2 staining, but induced a loss in overall neuritic density, as shown by β-III-tubulin immunocytochemistry (Fig. 1A). After 24 h of Fe2+/METH exposure, LUHMES cells began to exhibit significant beading of neuronal processes, which ultimately culminated in extensive neurite disintegration, detected by an overall decrease in β-III-tubulin and MAP2-labeled structures (Figs. 1A, 2A). Because in our cultures, β-III-tubulin appeared to label axons and also dendrites in ∼50% of neurons, as opposed to MAP2, which labeled primarily dendritic structures and neuronal perikaya, we opted to use β-III-tubulin as a general neuritic marker for quantification of degeneration of neuronal processes. LUHMES cultures treated with Fe2+/METH were immunoprocessed for β-III-tubulin, and an imaging algorithm was applied to selectively capture neuritic β-III-tubulin immunofluorescence (cell body staining was automatically excluded), referred to as the neurite mass of the cultures (see Materials and Methods).
Figure 2.
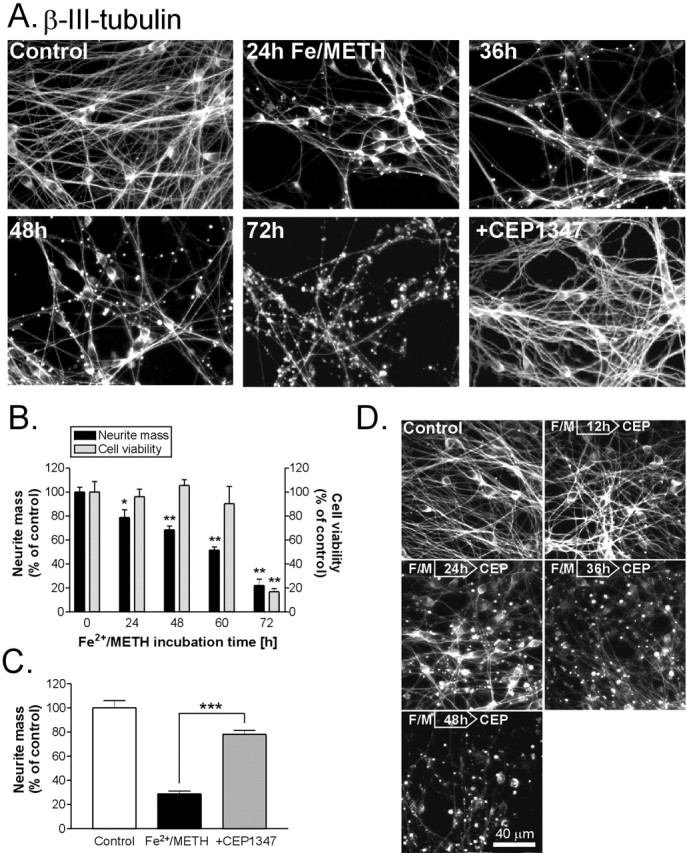
Fe2+/METH leads to progressive neurite degeneration, which is significantly attenuated by CEP1347. A, LUHMES cells were treated with Fe2+/METH for 24-72 h, fixed, and immunoprocessed for β-III-tubulin. Cells were also cotreated with Fe2+/METH in the presence of 250 nm CEP1347 for 72 h. B, Neurite mass and cell viability were quantified in cultures immunostained for β-III-tubulin 0-72 h after exposure to Fe2+/METH, using a Cellomics ArrayScan instrument. Neuritic β-III-tubulin immunofluorescence was used as a measure of neurite mass. Number of cells, as assessed by neurons with normal nuclear morphology, was used as an indicator of cell survival. Data are expressed as mean ± SEM of quadruplicate determinations and are shown as a percentage of control. C, LUHMES cells were exposed to Fe2+/METH for 72 h in the presence or absence of 250 nm CEP1347, and neurite mass was quantified with the Cellomics ArrayScan. Values are expressed as mean ± SEM of quadruplicate determinations and are shown as a percentage of control. D, LUHMES cultures were treated with 250 nm CEP1347 (CEP) 6-48 h after Fe2+/METH (F/M) addition. All cultures were fixed and immunostained for β-III-tubulin 72 h after the initial insult. Error bars represent SEM.
In agreement with visual assessments, cells exposed to Fe2+/METH for 24-72 h exhibited a gradual, significant decrease in neuritic β-III-tubulin fluorescence. After 24 h of exposure, cells already demonstrated a 21% decrease in neurite mass (Fig. 2B) 2 d before changes in MTT reduction, LDH release, or calcein AM-positive cells could be detected (data not shown). Neurite degeneration was progressive, with LUHMES cells showing a gradual 45-65% reduction in their neurite mass between 48-60 h of Fe2+/METH exposure, still with no evident decrease in the number of surviving cells (Fig. 2B). This assay therefore proved to be a more sensitive indicator of neuronal damage than traditional viability assays such as MTT reduction, calcein AM staining, or LDH release and showed that Fe2+/METH targets neurites days before it can damage neuronal somata.
JNK pathway activation and its blockade by the MLK inhibitor CEP1347
In addition to its well established role in the execution of apoptosis (Lin and Dibling, 2002), recent evidence suggests that the stress-induced JNK pathway could be involved in the neurodegenerative cascade leading to cell loss in PD (for review, see Saporito et al., 2002; Peng and Andersen, 2003). LUHMES cells exposed to Fe2+/METH exhibited an increase in the phosphorylated forms of various kinases belonging to the JNK signaling pathway. Increased levels of the phosphorylated JNK-activating kinase MKK4 were already seen 6 h after Fe2+/METH exposure and peaked after 36 h of treatment (Fig. 3A); changes in MKK7 phosphorylation were not observed (data not shown). An increase in Fe2+/METH-induced JNK phosphorylation was detected 24 h after Fe2+/METH exposure, ∼6 h after the onset of MKK4 activation, reaching peak levels after 36 h. Basal levels of phosphorylated c-Jun in LUHMES cells were high, but an additional induction in phosphorylation was seen after 36 h of Fe2+/METH exposure. We examined the possible role of MLKs as upstream activators of Fe2+/METH-induced toxicity by using the potent MLK inhibitor CEP1347. Fe2+/METH-induced MKK4, JNK, and c-Jun phosphorylation was significantly reduced after 36 h of exposure by cotreatment with 250 nm CEP1347 (Fig. 3B), suggesting a role for MLK in the activation of the JNK signaling pathway in this model.
Figure 3.
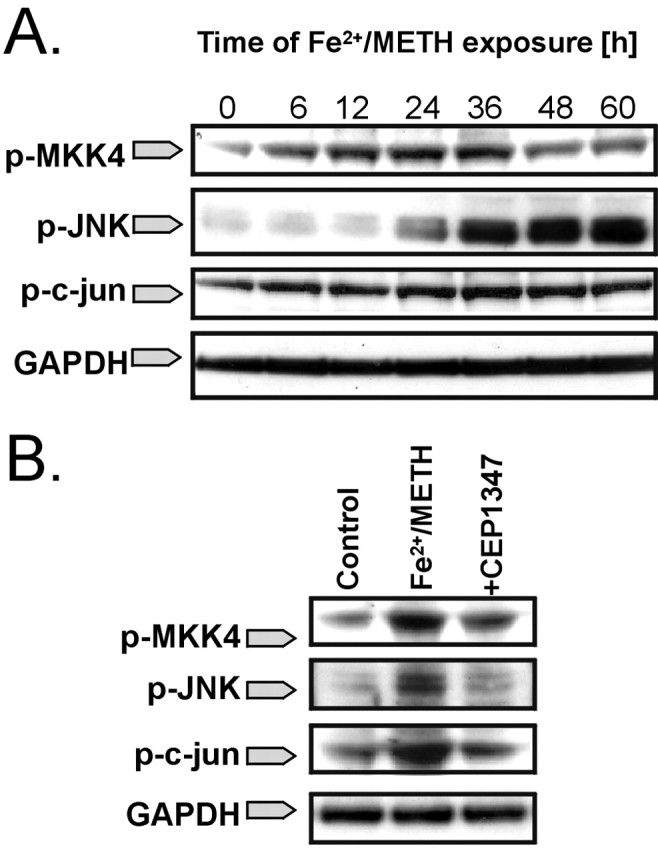
Fe2+/METH induces activation of the JNK signaling pathway, which is blocked by CEP1347. A, LUHMES cells were exposed to Fe2+/METH for 6-60 h, and the induction of JNK pathway intermediates was assessed by Western blotting using antibodies recognizing the phosphorylated forms of MKK4 (p-MKK4), JNK (p-JNK), and c-Jun (p-c-Jun). GAPDH was used as a loading control. B, LUHMES cells were cotreated with Fe2+/METH and 250 nm CEP1347 for 36 h, and the levels of p-MKK4, p-JNK, and p-c-Jun were measured by Western blotting. Blots shown represent three independent experiments.
Attenuation of Fe2+/METH-induced neurite degeneration, caspase activation, and cell death by the MLK inhibitor CEP1347
Because CEP1347 blocked JNK pathway activation in cells treated with Fe2+/METH, we assessed the ability of the MLK inhibitor to block cell death and/or attenuate earlier pathogenic events preceding the loss of cell viability. LUHMES cells were preincubated with CEP1347 for 1 h before the addition of Fe2+/METH. Viability was assessed by measuring LDH release into the media and MTT reduction capacity and by counting the number of calcein AM-positive cells after 72 h of Fe2+/METH exposure. CEP1347 completely prevented Fe2+/METH-induced cell death with an EC50 of ∼100 nm according to all measurement parameters examined (only MTT reduction and calcein AM-positive cell counts are shown) (Fig. 4A). The neuroprotective effect of CEP1347 did not appear to be transient, because robust neuroprotection was still seen 5 d after the initial insult. To confirm that the neuroprotective properties of CEP1347 were not restricted to this cell culture system, we examined the ability of the MLK inhibitor to block METH-induced toxicity in primary rat ventral mesencephalic cultures, which contain ∼5% dopaminergic neurons. These neurons, which were identified by TH immunocytochemistry, were, to our surprise, vulnerable to METH such that cotreatment with FeCl2 was not necessary. A 3 d exposure to 250 μm METH led to a 50% decrease in TH-positive neurons, which could be completely rescued by pretreatment with CEP1347, with an EC50 <100 nm (Fig. 4C).
Figure 4.
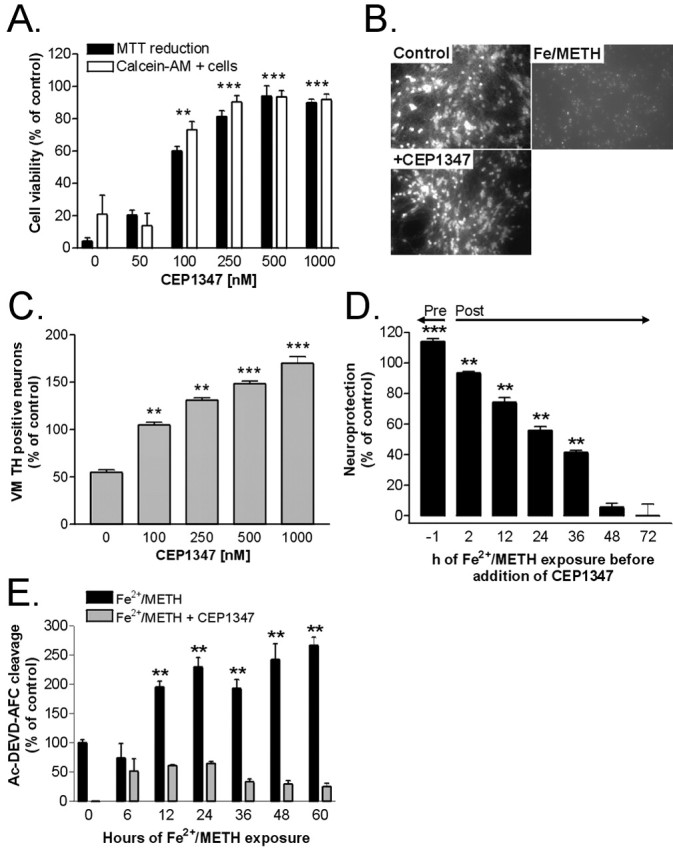
The MLK inhibitor CEP1347 rescues LUHMES cells and primary rat ventral mesencephalic dopaminergic neurons from Fe2+/METH-induced toxicity. A, LUHMES cells were treated with Fe2+/METH for 72 h in the presence of 50-1000 μm CEP1347, and cell viability was assessed by counting the number of calcein AM-positive cells and by measuring MTT reduction capacity. B, Micrographs showing cultures treated with Fe2+/METH in the presence or absence of 250 nm CEP1347 stained with calcein AM. C, Primary rat ventral mesencephalic cultures were treated with METH alone or in the presence of 100-1000 μm CEP1347, and dopaminergic cell survival was assessed by counting the number of TH-positive neurons. Values are expressed as mean ± SEM of quadruplicate determinations and are shown as a percentage of control cultures. C, CEP1347 was added to LUHMES cells either 1 h before or 2-72 h after Fe2+/METH addition. The toxicity of Fe2+/METH was measured after 72 h by quantification of LDH release and expressed as a percentage of neuroprotection compared with control cultures. All data are expressed as the mean ± SEM of quadruplicate determinations. D, Caspase-3-like enzyme activity in LUHMES cells treated with Fe2+/METH in the presence or absence of 250 nm CEP1347 was quantified for the ability to cleave a fluorogenic tetrapeptide, Ac-DEVD-AFC, which is a substrate for caspase-3, caspase-6, caspase-7, caspase-8, and caspase-10, after 6-60 h of drug treatment. The fluorescence emitted by cleavage of the substrate was quantified during a 20 min incubation period by kinetic fluorimetric measurements. Values were standardized to micrograms of protein, expressed as mean ± SEM of quadruplicate determinations, and shown as a percentage of control cultures. Error bars represent SEM.
Because a primary aim of antiparkinsonian medications would be to halt disease progression after the onset of neurodegeneration, we examined whether CEP1347 could rescue LUHMES cells if given hours to days after the initiation of the cell death cascade. LUHMES cells were exposed to Fe2+/METH 2-48 h before the addition of CEP1347, and viability was assayed 72 h after the addition of the death-inducing stimulus by measuring LDH release. A minimum effective concentration of CEP1347 (250 nm) rescued the majority of LUHMES cells from Fe2+/METH-induced cell death, even when given as late as 24 h after initial exposure to the insult (Fig. 4D). This suggests that MLK inhibitors can halt the progression of an ongoing pathogenic process. Notably, coincubation of Fe2+/METH-treated cells with 250 nm CEP1347 completely blocked nuclear fragmentation (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), assessed after 72 h of drug exposure, as well as caspase-3-like activation (Fig. 4E), measured after 6-60 h.
As mentioned above, LUHMES cells exposed to Fe2+/METH exhibited significant neurite beading and disintegration. This early event was completely blocked by 250 nm CEP1347, as assessed by visual inspection of the cultures 72 h after cotreatment with the MLK inhibitor (Fig. 2 A). In agreement with qualitative assessments of CEP1347-mediated neurite rescue, CEP1347 significantly attenuated Fe2+/METH-induced neuritic dystrophy in a novel quantitative neurite degeneration assay. The total neurite mass in cells cotreated with CEP1347 and Fe2+/METH was 270% higher than in cells treated with Fe2+/METH alone (Fig. 2C). CEP1347 retained its ability to prevent neurite degeneration even when added 12-24 h after Fe2+/METH exposure (Fig. 2 D). Although extensive beading could be seen in some cells given CEP1347 24 h after Fe2+/METH, the majority of neurites remained intact. Nevertheless, these results suggest that the commitment for neurite loss lies somewhere between 12-24 h and clearly demonstrate that CEP1347 is not only neuroprotective but also prevents the occurrence of earlier dystrophic changes such as neurite degeneration in human mesencephalic-derived cells.
No evidence for involvement of AKT activation in the neuroprotective effects of CEP1347
A recent study by Roux et al. (2002) showed that CEP1347 could promote the activation of AKT, a prosurvival kinase, in primary cortical neurons, an effect most likely independent of its inhibitory effect on the MLKs. To determine whether CEP1347 blocked Fe2+/METH-induced neurite degeneration and cell death by activating AKT, protein levels of Thr308 phosphorylated AKT (p-AKT) were measured in LUHMES cells treated with Fe2+/METH in the presence or absence of CEP1347. Although Fe2+/METH treatment alone did not change protein levels of p-AKT measured after 6, 24, and 36 h (a transient 50% decrease was observed only at the 12 h time point), cotreatment with CEP1347 led to a persistent upregulation of the activated form of the kinase first detected after 6 h of drug addition (Fig. 5A). This increase in p-AKT expression induced by Fe2+/METH and CEP1347 was blocked by treatment with 10 μm LY294002 (Fig. 5B), suggesting that upregulation of p-AKT by CEP1347 involves the phosphoinositide 3-kinase (PI3K) pathway.
Figure 5.

Activation of AKT and its role in the neuroprotective effects of CEP1347. A, Protein levels of phosphorylated AKT (Thr308) were measured by Western blotting in whole-cell lysates from LUHMES cultures treated with Fe2+/METH for 6-36 h in the presence or absence of 250 nm CEP1347. B, Levels of p-AKT were measured by Western blotting after 24 h of treatment with CEP1347, Fe2+/METH with CEP1347 (F/M + CEP), or Fe2+/METH with CEP1347 in the presence or absence of a 10 μm concentration of the PI3 kinase inhibitor LY294002. For A and B,β-actin was used as a loading control. Blots shown are representative of three independent experiments, and numbers under the blots correspond to densitometric quantification of band densities. C, LUHMES cells were treated with Fe2+/METH in the presence or absence of 250 nm CEP1347 and 10 μm LY294002. MTT reduction was used to assess cell survival after 72 h. Values are expressed as mean ± SEM of quadruplicate determinations and are shown as a percentage of control. D, To exclude the possibility that LY294002 may be inefficacious in blocking PI3K at later time points during >6 h of CEP1347 exposure, the inhibitor was added 6-60 h after the addition of Fe2+/METH and CEP1347. Viability was assayed after 72 h by measuring the release of LDH into the medium. Values are expressed as mean ± SEM of quadruplicate determinations and are shown as a percentage of control. Error bars represent SEM.
To determine whether activation of the PI3K-AKT pathway was involved in the neuroprotective effects of CEP1347, cells were cotreated for 72 h with Fe2+/METH and CEP1347 in the presence or absence of the PI3 kinase inhibitor LY294002 (10 μm), and cell survival was assayed by the MTT reduction assay. Fe2+/METH treatment led to a 60% decrease in cell viability, which was completely blocked by CEP1347 (Fig. 5C). The PI3K inhibitor alone decreased cell viability by ∼25% and significantly potentiated the degree of cell death induced by Fe2+/METH. However, CEP1347 retained its ability to reduce cell death in the presence of LY294002, suggesting that the PI3K pathway is not necessary for the ability of CEP1347 to rescue cells from Fe2+/METH. One flaw of the experiment is the relatively low stability of LY294002 (t1/2 < 12 h). Given the late onset of AKT activation (>6 h), LY294022 may not have adequately blocked PI3K at later time points. Therefore, LY294002 was added in a parallel set of experiments several hours after combined Fe2+/METH and CEP1347 addition to ensure that PI3K was adequately inhibited. Even when added 6-60 h after Fe2+/METH and CEP1347 addition, LY294002 did not decrease the ability of CEP1347 to rescue cells from Fe2+/METH-induced toxicity as measured by LDH release into the media (Fig. 5D). These results suggest that, although AKT is activated in response to CEP1347, it is not involved in the rescue of Fe2+/METH-induced toxicity.
Blockade of Fe2+/METH-induced oxidative stress by CEP1347
METH is known to induce dopamine-dependent oxidative stress in mesencephalic neurons (Cubells et al., 1994; Larsen et al., 2002) by collapsing intracellular pH gradients and redistributing vesicular dopamine to the cytoplasm (Sulzer and Rayport, 1990; Mosharov et al., 2003). Indeed, in our model, METH led to a dose-dependent collapse in pH as indicated by a decrease in the intensity of fluorescent puncta in the cytoplasm after loading with the fluorescent acidotropic dye acridine orange. This decrease in pH occurred within minutes of psychostimulant exposure and reached 51.4 ± 3.9% of control levels in cells treated with 1 mm METH (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). After a collapse in pH gradients, an increase in cytoplasmic dopamine and subsequent elevation in oxidative stress would be expected. Although we currently have no available methods for measuring cytoplasmic dopamine in neurons, dopamine-dependent oxidative stress can be measured indirectly. In the cytosol, dopamine can auto-oxidize into dopamine-quinone species, superoxide radicals, and hydrogen peroxide (Graham, 1978) or can be enzymatically deaminated by monoamine oxidase into the nontoxic metabolite 3,4-dihydroxyphenylacetic acid and hydrogen peroxide (Maker et al., 1981). Superoxide and hydrogen peroxide can be measured by various methods. The involvement of oxidative stress in a given injury model can also be assessed indirectly by examining whether antioxidants block toxicity.
To test whether Fe2+/METH-induced cell death is oxidative stress dependent, treated cells were exposed to various antioxidants including NAC (0.05-1 mm), which acts as a source of cysteine and stimulates the production of the endogenous cellular antioxidant GSH, SOD (200 U/ml), an antioxidant enzyme that catalyzes the dismutation of superoxide radicals into hydrogen peroxide and molecular oxygen, MnTBAP (1-25 μm), a cell-permeable SOD mimetic, catalase (200-400 U/ml), and Desferal (100-500 μm). All antioxidants tested prevented or significantly reduced Fe2+/METH-induced cell death after 3 d of exposure, as assessed by counting the number of calcein AM-positive cells (Fig. 6A). To obtain indirect evidence for dopamine-dependent oxidative reactions, several markers of oxidative stress were measured, including levels of the lipid peroxidation product MDA, H2O2, and superoxide. Exposure of LUHMES cells to Fe2+/METH led to a progressive increase in MDA, already evident after 48 h, which culminated in 4.5-fold higher levels, compared with control, after 72 h of exposure (Fig. 6B). Levels of hydrogen peroxide, a major by-product of dopamine oxidation and a primary source of cytotoxic hydroxyl radicals, were increased by 230% in the presence of METH after 6 h (control levels were ≈1 μm) and reached maximal levels after 12 h (260% of control) (Fig. 6C). FeCl2 had to be omitted from these experiments because of interference with the assay reaction. However, treatment of cells with METH alone was an important condition to examine given that METH alone leads to extensive neurite degeneration, presumably by increasing oxidative stress. The TH inhibitor α-MPT reduced METH-induced hydrogen peroxide formation to control levels (270.1 ± 10.5 and 123.6 ± 11.8), suggesting that oxidative stress in our model is indeed dopamine dependent. Furthermore, LUHMES cells treated with FeCl2 and METH exhibited a 1.7-fold increase in superoxide levels (Fig. 6D) and a 35% decrease in intracellular levels of the major cellular antioxidant GSH (Fig. 7A). These biochemical data, together with the capacity of NAC, catalase, MnTBAP, and SOD to confer complete protection against Fe2+/METH-induced toxicity, support the role of reactive oxygen species (ROS) as primary mediators of damage in our system.
Figure 6.

Fe2+/METH triggers oxidative stress, which is blocked by CEP1347. A, LUHMES cells were exposed to Fe2+/METH (F/M) and varying concentrations of antioxidants for 72 h. Results for treatment with 250 nm CEP1347 (as a positive control), 200 U/ml SOD, 1 μm MnTBAP, 250 μm DM, 200 U/ml catalase, and 100 μm NAC are shown. Cell survival was assessed by counting calcein AM-positive cells. Data are expressed as mean ± SEM of quadruplicate determinations and are shown as a percentage of control. B, LUHMES cells were treated with Fe2+/METH with or without 250 nm CEP1347, and the levels of MDA, a lipid peroxidation product, were measured using a Bioxytech MDA-596 kit from OxisResearch. C, The effect of 1 mm METH on H2O2 production was examined over 24 h using the Bioxytech H2O2-560 kit from OxisResearch (see Materials and Methods). FeCl2 was not added in the cultures for H2O2 measurements because of interference of Fe2+ with the assay reaction. D, Levels of H2O2 and superoxide (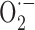 ), the latter of which was assayed fluorimetrically by measuring the oxidation of dihydroethidium to fluorescent ethidium, were measured after 12 h (H2O2) or 36 h (
), the latter of which was assayed fluorimetrically by measuring the oxidation of dihydroethidium to fluorescent ethidium, were measured after 12 h (H2O2) or 36 h (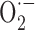 ) of Fe2+/METH treatment in the presence or absence of 250 nm CEP1347. For B and C, values were standardized to protein concentration and expressed as mean ± SEM of quadruplicate determinations. Data are shown as a percentage of control. Error bars represent SEM.
) of Fe2+/METH treatment in the presence or absence of 250 nm CEP1347. For B and C, values were standardized to protein concentration and expressed as mean ± SEM of quadruplicate determinations. Data are shown as a percentage of control. Error bars represent SEM.
Figure 7.
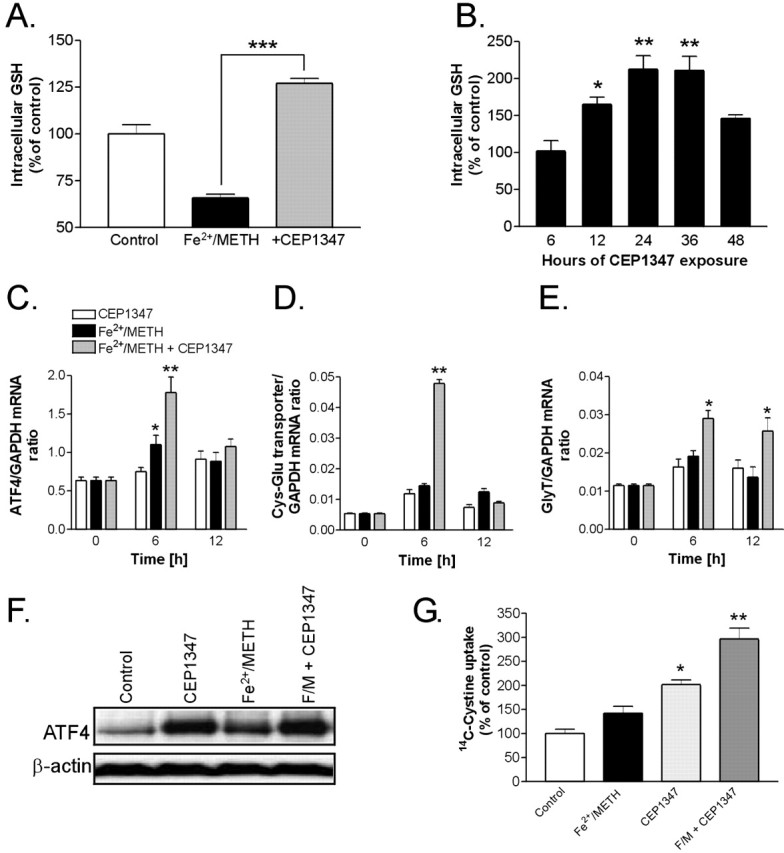
CEP1347 increases cellular antioxidant capacity. A, LUHMES cells were treated with Fe2+/METH in the presence or absence of 250 nm CEP1347 for 24 h, and total intracellular glutathione levels were measured by a photometric assay. Values were standardized to protein concentration, normalized to control cultures, and expressed as mean ± SEM of quadruplicate determinations. B, LUHMES cells were treated with 250 nm CEP1347 for 6-48 h, and GSH levels were measured. C, Using qPCR, mRNA levels of the transcription factor ATF4 were assessed after 6 or 12 h of Fe2+/METH exposure in the presence or absence of 250 nm CEP1347. Cells exposed to 250 nm CEP1347 were also tested. Values were standardized to GAPDH mRNA and are expressed as a ratio of the gene of interest to GAPDH mRNA ± SEM of quadruplicate determinations. D, mRNA levels of the Cys-Glu transporter were also measured by qPCR after 6 or 12 h of drug exposure. E, mRNA levels of the Gly transporter were assessed as in D. F, Protein levels of ATF4 were examined by Western blotting 12 h after exposure to 250 nm CEP1347, Fe2+/METH, Fe2+/METH in the presence of CEP1347, or Fe2+/METH in the presence of CEP1347 and a 10 μm concentration of the PI3 kinase inhibitor LY294002. Blots shown are a representation of three independent experiments. G, [14C] cystine uptake was measured in LUHMES cells treated for 16 h with Fe2+/METH in the presence or absence of 250 nm CEP1347. Values were standardized to protein concentration, normalized to control, and expressed as mean ± SEM of quadruplicate determinations. Error bars represent SEM.
To determine whether CEP1347 could decrease oxidative stress in LUHMES cells exposed to Fe2+/METH, MDA, hydrogen peroxide, and superoxide levels were measured in the absence or presence of 250 nm CEP1347. As shown in Figure 6B, CEP1347 blocked Fe2+/METH-induced MDA production at all time points tested (48-72 h). Although it had no effect on METH-induced H2O2 formation, it abrogated Fe2+/METH-induced superoxide production measured at 12 h (Fig. 6D). These results suggest that, although CEP1347 is unable to block METH-induced H2O2 production, it can effectively attenuate other aspects of oxidative stress such as free radical production and lipid peroxidation.
Enhancement of intracellular GSH levels by CEP1347 via an ATF4-dependent mechanism
Published reports, as well as data shown in this study (Fig. 6), show that METH promotes oxidative stress, a process that can be significantly enhanced by coadministration of iron. GSH is the most abundant soluble antioxidant in the brain and is synthesized by both neurons and glia as the major pathway for counteracting oxidative stress. Previous studies have shown that increased GSH levels in ventral mesencephalic neurons induced by bFGF (basic fibroblast growth factor) exposure or treatment with cell-permeable GSH analogs protects cells from 6-OHDA, MPP+, and NO (nitric oxide)-induced toxicity. Furthermore, it was shown that depleting cells of GSH can be neurotoxic (for review, see Mytilineou et al., 2002). We examined whether GSH depletion was an early event in the Fe2+/METH-triggered neurodegenerative cascade and whether GSH levels could be affected by CEP1347. Consistent with enhanced levels of oxidative stress and increased antioxidant use, exposure of LUHMES cells to Fe2+/METH already led to a 30% decrease in cellular levels of GSH after 24 h (Fig. 7A). This is in agreement with in vivo studies, in which METH has been shown to deplete striatal GSH shortly after drug administration (Moszczynska et al., 1998; Acikgoz et al., 2001). We tested whether the broad neuroprotective effect of CEP1347 on early damage events was based on its ability to increase the antioxidant capacity of the cells. Consistent with its ability to attenuate markers of oxidative stress, CEP1347 completely blocked the Fe2+/METH-induced depletion of intracellular GSH and even enhanced the level of the endogenous antioxidant above that of control cultures. Notably, the net increase in GSH was controlled for both the number of cells and the protein content. This observation suggested that CEP1347, when given alone, could promote GSH synthesis. Indeed, 250 nm CEP1347 alone led to a twofold increase in GSH after 24-36 h of treatment, returning again to baseline after 48 h (Fig. 7B). CEP1347 treatment alone also led to a similar increase in GSH content in other cell systems, including primary murine astrocyte cultures, which exhibited a 56.3 ± 8.6% increase in GSH content after 24 h of CEP1347 exposure (data not shown).
A recent study by Harding et al. (2003) showed that the transcription factor ATF4 (CREB2) is responsible for the clearance of peroxides after endoplasmic reticulum stress, apparently by controlling the expression of both the cystine/glutamate and glycine 1 transporters, which are responsible for the cellular uptake of the three key GSH substrates: glutamate, cysteine, and glycine. We previously identified ATF4 as being upregulated in cultures of primary murine astrocytes treated with CEP1347 (Falsig et al., 2004). Therefore, we determined whether CEP1347 was capable of upregulating the transcription of ATF4 as well as the cystine/glutamate and glycine 1 transporter mRNAs. LUHMES cells were treated with CEP1347, Fe2+/METH, or Fe2+/METH plus CEP1347 for 6 or 12 h. The mRNA expression of ATF4 and the amino acid transporters was assayed by qPCR. The mRNA levels of ATF4 were increased 1.5-fold by Fe2+/METH and 2.8-fold by Fe2+/METH given in combination with 250 nm CEP1347 (Fig. 7C). An increase in ATF4 mRNA was also observed in a microarray chip experiment (data not shown). At the protein level, treatment with Fe2+/METH, CEP1347, or a combination of both also resulted in a substantial upregulation of ATF4 (Fig. 7F). Similarly to ATF4, cystine/glutamate transporter mRNA was elevated 10-fold by Fe2+/METH given together with CEP1347 (Fig. 7D). No effect of Fe2+/METH alone was seen. Both ATF4 and the cystine/glutamate transporter were upregulated at 6 h, but the increase in mRNA expression was lost again after 12 h of drug exposure. In contrast, the threefold upregulation of GlyT1 mRNA by combined Fe2+/METH and CEP1347 exposure persisted after 12 h of drug treatment (Fig. 7E). Again, no effect of Fe2+/METH alone could be detected. These results suggest that, under conditions of oxidative stress induced by Fe2+/METH, CEP1347 promotes the transcription of ATF4 and both amino acid transporters. The transcriptional regulation of ATF4 and the cystine/glutamate transporter is fast but transient. Nonetheless, the increased GSH levels persisted for days.
To determine whether increased mRNA expression of the cystine/glutamate transporter resulted in a functional effect, cellular uptake of [14C]cystine was measured after a 16 h exposure to Fe2+/METH, CEP1347, or a combination of both. Although Fe2+/METH only slightly increased [14C]cystine uptake into cells, CEP1347 alone increased [14C]cystine uptake twofold, and CEP1347 in combination with Fe2+/METH elevated cystine uptake to 300% of control levels (Fig. 7G). Thus, increased transcription of the cystine/glutamate transporter mRNA after combined Fe2+/METH and CEP1347 exposure appears to be responsible for enhanced transport activity as measured by increased [14C]cystine uptake at 16 h. These data suggest that CEP1347 promotes ATF4 transcription and translation (within 6 h), leading to a functionally relevant increase in the expression of the cystine/glutamate transporter that, in turn, can be correlated to elevated cystine uptake (evident at 16 h) and increased GSH synthesis (peak levels at 24 h). This complex sequence of events results in an enhanced cellular antioxidant capacity that could underlie the capacity of CEP1347 to block the detrimental effects of increased oxidative stress, such as lipid peroxidation, as well as subsequent downstream events.
Discussion
METH promotes the accumulation of cytosolic dopamine (Sulzer et al., 1995; Lotharius and O'Malley, 2001; Mosharov et al., 2003), by both blocking vesicular dopamine uptake and redistributing vesicular dopamine to the cytoplasm (Sulzer and Rayport, 1990; Brown et al., 2002). This results in increased metabolism of cytoplasmic dopamine associated with an increase in oxidative stress (for review, see Cadet and Brannock, 1998), neurite degeneration (Cubells et al., 1994; Larsen et al., 2002), and, in some cases, neurodegeneration (Ricaurte et al., 1982, 1983; Seiden, 1985; Sonsalla et al., 1996). In agreement with these studies, we showed that exposure of differentiated LUHMES cells to METH led to a collapse in intracellular pH gradients in vesicular structures (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), an increase in H2O2 and superoxide anions, and significant neurite degeneration. However, METH alone did not lead to a loss in LUHMES cell viability. This is in agreement with METH toxicity studies performed on postnatal rat primary mesencephalic cultures (Cubells et al., 1994; Larsen et al., 2002) but differs from studies showing that METH may directly induce cell death in some cell lines (e.g., CATH.a or CSM14.1 cells) at higher concentrations and, in our hands, in embryonic rat primary mesencephalic cultures (Cadet et al., 1997; Choi et al., 2002) (Fig. 4C). The different degrees of toxicity observed in these in vitro models could be linked to differences in cellular dopamine concentrations or factors enhancing dopamine toxicity in the respective experimental systems.
Overwhelming evidence suggests that iron could be a key vulnerability factor in nigral cells succumbing to degeneration in PD by enhancing oxidative stress resulting from intracellular dopamine metabolism (for review, see Sulzer and Zecca, 2000; Zecca et al., 2004; Götz et al., 2004). We administered a subtoxic concentration of FeCl2, along with METH, tipping the balance toward neuronal death. Combined addition of METH and FeCl2 led to neurite disintegration and eventually resulted in cell death (Figs. 1, 2). The temporal sequence of events characterizing the resultant progressive human model of parkinsonian-like neurodegeneration is compiled for an overview in supplemental Figure 4 (available at www.jneurosci.org as supplemental material). FeCl2/METH-induced toxicity was blocked by iron chelation, dopamine depletion, and antioxidant treatment (Fig. 6), suggesting that cell death in our model is not only dependent on enhanced cytosolic dopamine metabolism but on iron-mediated oxidative stress. These results are in agreement with previous studies showing that METH toxicity in mice can be blocked by the tyrosine hydroxylase inhibitor α-methyl-p-tyrosine (Wagner et al., 1983; Schmidt et al., 1985) by overexpression of superoxide dismutase (Cadet et al., 1994) or by pretreatment with antioxidants (Wagner et al., 1985; De Vito and Wagner, 1989). In this respect, our in vitro model bears great resemblance to in vivo models of METH toxicity.
Previous studies have shown that METH leads to an induction in MKK4/MKK7, JNK1/JNK3, and c-Jun at the mRNA and protein level and increases AP-1 DNA-binding activity (Sheng et al., 1996; Jayanthi et al., 2002; Deng et al., 2002). These biochemical findings, together with transgenic animal studies showing that heterozygous c-Jun knock-out mice are more resistant to METH-induced toxicity (Deng et al., 2002), suggest that activation of the JNK pathway is important for the toxic effects of METH. The JNK signaling pathway was also activated in LUHMES cells treated with FeCl2/METH, allowing us to investigate which kinases were involved in the death-signaling cascade. In our system, the MLK inhibitor CEP1347 completely blocked phosphorylation of all members of the JNK signaling pathway, including MKK4, JNK, and c-Jun and at similar concentrations (IC50 ≈ 100 nm) completely abrogated FeCl2/METH-induced cell death (Figs. 3, 4). A nearly identical neuroprotective effect of the drug was seen in primary cultures of ventral mesencephalic dopamine neurons (Fig. 4). Moreover, CEP1347 retained most of its neuroprotective capacity even when added as late as 24 h after the insult (Fig. 4). Neuroprotective concentrations are well within the range of specific MLK1-MLK3 enzyme inhibition (Maroney et al., 2001). Our results are consistent with many other studies on neuroprotection by CEP1347 (for review, see L. H. Wang et al., 2004) implying that similar damaging cascades could play a causal role both in this and other models of neurodegeneration.
In addition to its well known antiapoptotic properties, neurotrophic-like effects of CEP1347 have been described previously in several systems (Borasio et al., 1990; Glicksman et al., 1993, 1998; Maroney et al., 1995; Saporito et al., 1998, 1999; Harris et al., 2002; Boll et al., 2004). In our LUHMES model, CEP1347 also preserved neurite integrity, superoxide formation, lipid peroxidation, and depletion of GSH in response to FeCl2/METH (Figs. 2, 6, 7). This novel observation suggested that CEP1347 had additional neurorestorative properties not linked previously to its known effects on the JNK cascade. Data showing that CEP1347 not only completely prevented FeCl2/METH-induced depletion of reduced GSH but also increased the levels of the antioxidant above those of control cells led us to test whether CEP1347 alone could enhance GSH synthesis. In vivo, GSH is normally synthesized by astrocytes and released into the extracellular space, where it is cleaved into the dipeptide cysteinylglycine and glutamate, which are then are taken up by neurons via amino acid transporters, allowing them to subsequently resynthesize GSH. Although astrocytes would be expected to supply neurons with GSH precursors in complex culture systems, neurons have the machinery to synthesize the GSH if such precursors are present in the media, as they were in our experimental setup (Dringen and Hirrlinger, 2003). Both in pure LUHMES and primary murine astrocyte cultures, CEP1347 significantly elevated intracellular GSH for up to 36 h after a single exposure (Fig. 7E), suggesting that this property is not restricted to neurons. CEP1347 promoted GSH synthesis in stressed cells by enhancing the expression of amino acid transporters and, subsequently, cystine uptake (Fig. 7). This effect was mediated by ATF4 (CREB2), a transcription factor shown recently to mediate the clearance of peroxides after endoplasmic reticulum stress by controlling the expression of both the cystine/glutamate and glycine 1 transporters (Harding et al., 2003). Whether this new action triggered by CEP1347 is linked to MLK inhibition is not clarified at the present stage of research, but it could explain why its protective action lies far upstream in the cell death cascade (supplemental Fig. 5, available at www.jneurosci.org as supplemental material).
In our model, METH and FeCl2/METH triggered a significant increase in both H2O2 and superoxide radicals, respectively, as early as 6 h after drug exposure, which preceded JNK activation by several hours (Figs. 2, 6). Thus JNK pathway activation could be directly mediated by oxidative stress. Although an increase in caspase-3-like activity was observed in our model, and the morphological features of cell death resembled caspase-dependent apoptosis (Fig. 4) (supplemental Figs. 1, 2, available at www.jneurosci.org as supplemental material), caspase-3-like processing was quite modest compared with that induced by staurosporine or colchicine (data not shown), and the onset of cell death was late compared with other apoptotic insults. This might be attributable to the ability of ROS to inhibit caspase-3 activity (Ueda et al., 1998), which could lead to decreased caspase activity resulting in either significantly delayed apoptosis or a change in the mode of cell death (Hirsch et al., 1997). In diseases with a strong oxidative stress component, the full execution of an apoptotic program may be hampered by the oxidative status of the cells, as could be the case in PD, in which the involvement of apoptosis in the cell death process is still debated (for review, see Blum et al., 2001).
In vivo administration of METH leads to a robust upregulation of Src both at the mRNA and protein level, which precedes the induction of MKK7 and JNK by several hours (Jayanthi et al., 2002). Src, in turn, has been shown to activate PI3K (Pleiman et al., 1994). Thus, it appears that oxidative stress triggers a transcription-dependent mechanism aimed at enhancing cell survival, which is mediated by Src and potentially by AKT. In agreement with this notion, LUHMES cells treated with FeCl2/METH exhibited a significant increase in AKT activation, an event that has been suggested as the primary trophic action of CEP1347 (Roux et al., 2002). However, blockade of this pathway with PI3 kinase inhibitors did not abrogate the ability of CEP1347 to protect cells from FeCl2/METH-induced toxicity (Fig. 5). It should be noted that LY294002 has been reported to also inhibit kinases other than PI3K. However, the neuroprotective effect of CEP1347 correlates much better with MLK inhibition than with PI3K activation.
In conclusion, we presented a new in vitro model of dopamine-dependent oxidative stress in human mesencephalic-derived cells, in which the succession of events that leads to cell death closely resembles some of the pathogenic features present in PD. The psychostimulant METH was used as a tool for elevating cytoplasmic dopamine levels, which may be increased in PD nigral neurons as a functional consequence of either environmental or genetic factors. Cytosolic dopamine-dependent oxidative stress was enhanced by the addition of iron, likely to be a major player in nigral degeneration in PD. This insult resulted in activation of the JNK cell death pathway, progressive neurite degeneration, and, eventually, apoptosis (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). The MLK inhibitor CEP1347 exerted not only neuroprotective but also neurorestorative properties linked to its ability to enhance cellular protective mechanisms including cellular antioxidant capacity (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Thus, this new model has provided us with a unique tool for testing the efficacy and mechanism of action of new PD therapies.
Footnotes
We thank Trine Marie Nielsen and Andreas Rassov for excellent technical assistance, Tobias Minich for the GSH measurements, and Cephalon for CEP1347.
Correspondence should be addressed to Dr. Julie Lotharius, H. Lundbeck A/S, Ottiliavej 9, 2500 Valby, Denmark. E-mail: mjl@lundbeck.com.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256329-14$15.00/0
References
- Acikgoz O, Gonenc S, Gezer S, Kayatekin BM, Uysal N, Semin I, Gure A (2001) Methamphetamine causes depletion of glutathione and an increase in oxidized glutathione in the rat striatum and prefrontal cortex. Neurotox Res 3: 277-280. [DOI] [PubMed] [Google Scholar]
- Blum D, Torch S, Lambeng N, Nissou M, Benabid AL, Sadoul R, Verna JM (2001) Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson's disease. Prog Neurobiol 65: 135-172. [DOI] [PubMed] [Google Scholar]
- Boll JB, Geist MA, Kaminski Schierle GS, Petersen K, Leist M, Vaudano E (2004) Improvement of embryonic dopaminergic neurone survival in culture and after grafting into the striatum of hemiparkinsonian rats by CEP-1347. J Neurochem 88: 698-707. [DOI] [PubMed] [Google Scholar]
- Borasio GD, Horstman S, Anneser JM, Neff NT, Glicksman MA (1990) CEP-1347/KT7515, a JNK pathway inhibitor, supports the in vitro survival of chick embryonic neurons. NeuroReport 9: 1435-1439. [DOI] [PubMed] [Google Scholar]
- Bozyczko-Coyne D, O'Kane TM, Wu ZL, Dobrzanski P, Murthy S, Vaught JL, Scott RW (2001) CEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, promotes survival and blocks multiple events associated with Abeta-induced cortical neuron apoptosis. J Neurochem 77: 849-863. [DOI] [PubMed] [Google Scholar]
- Brown JM, Riddle EL, Sandoval V, Weston RK, Hanson JE, Crosby MJ, Ugarte YV, Gibb JW, Hanson GR, Fleckenstein AE (2002) A single methamphetamine administration rapidly decreases vesicular dopamine uptake. J Pharmacol Exp Ther 302: 497-501. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Ali S, Epstein C (1994) Involvement of oxygen-based radicals in methamphetamine-induced neurotoxicity: evidence from the use of CuZnSOD transgenic mice. Ann NY Acad Sci 738: 388-391. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Ordonez SV, Ordonez JV (1997) Methamphetamine induces apoptosis in immortalized neural cells: protection by the proto-oncogene, bcl-2. Synapse 25: 176-184. [DOI] [PubMed] [Google Scholar]
- Choi HJ, Yoo TM, Chung SY, Yang JS, Kim JI, Ha ES, Hwang O (2002) Methamphetamine-induced apoptosis in a CNS-derived catecholaminergic cell line. Mol Cells 13: 221-327. [PubMed] [Google Scholar]
- Cubells JF, Rayport S, Rajendran G, Sulzer D (1994) Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J Neurosci 14: 2260-2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Jayanthi S, Ladenheim B, Krasnova IN, Cadet JL (2002) Mice with partial deficiency of c-Jun show attenuation of methamphetamine-induced neuronal apoptosis. Mol Pharmacol 62: 993-1000. [DOI] [PubMed] [Google Scholar]
- De Vito MJ, Wagner GC (1989) Methamphetamine-induced neuronal damage: a possible role for free radicals. Neuropharmacology 28: 1145-1150. [DOI] [PubMed] [Google Scholar]
- Dringen R, Hamprecht B (1996) Glutathione content as an indicator for the presence of metabolic pathways of amino acids in astroglial cultures. J Neurochem 67: 1375-1382. [DOI] [PubMed] [Google Scholar]
- Dringen R, Hirrlinger J (2003) Glutathione pathways in the brain. Biol Chem 384: 505-516. [DOI] [PubMed] [Google Scholar]
- Falsig J, Porzgen P, Lotharius J, Leist M (2004) Specific modulation of astrocyte inflammation by inhibition of mixed lineage kinases with CEP-1347. J Immunol 173: 2762-2770. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619-633. [DOI] [PubMed] [Google Scholar]
- Glicksman MA, Prantner JE, Meyer SL, Forbes ME, Dasgupta M, Lewis ME, Neff N (1993) K-252a and staurosporine promote choline acetyltransferase activity in rat spinal cord cultures. J Neurochem 61: 210-221. [DOI] [PubMed] [Google Scholar]
- Glicksman MA, Chiu AY, Dionne CA, Harty M, Kaneko M, Murakata C, Oppenheim RW, Prevette D, Sengelaub DR, Vaught JL, Neff NT (1998) CEP-1347/KT7515 prevents motor neuronal programmed cell death and injury-induced dedifferentiation in vivo. J Neurobiol 35: 361-370. [PubMed] [Google Scholar]
- Götz ME, Double K, Gerlach M, Youdim MB, Riederer P (2004) The relevance of iron in the pathogenesis of Parkinson's disease. Ann NY Acad Sci 1012: 193-208. [DOI] [PubMed] [Google Scholar]
- Graham DG (1978) Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol 14: 633-643. [PubMed] [Google Scholar]
- Harris CA, Deshmukh M, Tsui-Pierchala B, Maroney AC, Johnson Jr EM (2002) Inhibition of the c-Jun N-terminal kinase signaling pathway by the mixed lineage kinase inhibitor CEP-1347 (KT7515) preserves metabolism and growth of trophic factor-deprived neurons. J Neurosci 22: 103-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze H, Schwoebel F, Lund S, Keel M, Ertel W, Wendel A, Jaattela M, Leist M (2001) In vivo and in vitro evidence for extracellular caspase activity released from apoptotic cells. Biochem Biophys Res Commun 283: 1111-1117. [DOI] [PubMed] [Google Scholar]
- Hirsch T, Marchetti P, Susin SA, Dallaporta B, Zamzami N, Marzo I, Geuskens M, Kroemer G (1997) The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene 15: 1573-1581. [DOI] [PubMed] [Google Scholar]
- Hunot S, Vila M, Teismann P, Davis RJ, Hirsch EC, Przedborski S, Rakic P, Flavell RA (2004) JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson's disease. Proc Natl Acad Sci USA 101: 665-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayanthi S, McCoy MT, Ladenheim B, Cadet JL (2002) Methamphetamine causes coordinate regulation of Src, Cas, Crk, and the Jun N-terminal kinase-Jun pathway. Mol Pharmacol 61: 1124-1131. [DOI] [PubMed] [Google Scholar]
- Jellinger K, Kienzl E, Rumpelmair G, Riederer P, Stachelberger H, Ben-Shachar D, Youdim MB (1992) Iron-melanin complex in substantia nigra of parkinsonian brains: an x-ray microanalysis. J Neurochem 59: 1168-1171. [DOI] [PubMed] [Google Scholar]
- Jellinger K, Kienzl E, Rumpelmair G, Riederer P, Stachelberger H, Ben-Shachar D, Youdim MB (1993) Neuromelanin and nigrostriatal dopamine neuron degeneration. J Neurochem 60: 1976-1977. [DOI] [PubMed] [Google Scholar]
- Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D (2002) Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci 22: 8951-8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Dibling B (2002) The true face of JNK activation in apoptosis. Aging Cell 1: 112-116. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Brundin P (2002) Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci 3: 932-942. [DOI] [PubMed] [Google Scholar]
- Lotharius J, O'Malley KL (2000) The parkinsonism-inducing drug 1-methyl-4-phenylpyridinium triggers intracellular dopamine oxidation. A novel mechanism of toxicity. J Biol Chem 275: 38581-38588. [DOI] [PubMed] [Google Scholar]
- Lotharius J, O'Malley KL (2001) Role of mitochondrial dysfunction and dopamine-dependent oxidative stress in amphetamine-induced toxicity. Ann Neurol 49: 79-89. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Barg S, Wiekop P, Lundberg C, Raymon HK, Brundin P (2002) Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J Biol Chem 277: 38884-38894. [DOI] [PubMed] [Google Scholar]
- Maker HS, Weiss C, Silides DJ, Cohen G (1981) Coupling of dopamine oxidation (monoamine oxidase activity) to glutathione oxidation via the generation of hydrogen peroxide in rat brain homogenates. J Neurochem 36: 589-593. [DOI] [PubMed] [Google Scholar]
- Maroney AC, Lipfert L, Forbes ME, Glicksman MA, Neff NT, Siman R, Dionne CA (1995) K-252a induces tyrosine phosphorylation of the focal adhesion kinase and neurite outgrowth in human neuroblastoma SHSY5Y cells. J Neurochem 64: 540-549. [DOI] [PubMed] [Google Scholar]
- Maroney AC, Glicksman MA, Basma AN, Walton KM, Knight Jr E, Murphy CA, Bartlett BA, Finn JP, Angeles T, Matsuda Y, Neff NT, Dionne CA (1998) Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J Neurosci 18: 104-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroney AC, Finn JP, Bozyczko-Coyne D, O'Kane TM, Neff NT, Tolkovsky AM, Park DS, Yan CY, Troy CM, Greene LA (1999) CEP-1347 (KT7515), an inhibitor of JNK activation, rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. J Neurochem 73: 1901-1912. [PubMed] [Google Scholar]
- Maroney AC, Finn JP, Connors TJ, Durkin JT, Angeles T, Gessner G, Xu Z, Meyer SL, Savage MJ, Greene LA, Scott RW, Vaught JL (2001) Cep-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J Biol Chem 276: 25302-25308. [DOI] [PubMed] [Google Scholar]
- Mosharov EV, Gong LW, Khanna B, Sulzer D, Lindau M (2003) Intracellular patch electrochemistry: regulation of cytosolic catecholamines in chromaffin cells. J Neurosci 23: 5835-5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moszczynska A, Turenne S, Kish SJ (1998) Rat striatal levels of the antioxidant glutathione are decreased following binge administration of methamphetamine. Neurosci Lett 255: 49-52. [DOI] [PubMed] [Google Scholar]
- Mytilineou C, Kramer BC, Yabut JA (2002) Glutathione depletion and oxidative stress. Parkinsonism Relat Disord 8: 385-387. [DOI] [PubMed] [Google Scholar]
- Peng J, Andersen JK (2003) The role of c-Jun N-terminal kinase (JNK) in Parkinson's disease. IUBMB Life 55: 267-271. [DOI] [PubMed] [Google Scholar]
- Pleiman CM, Hertz WM, Cambier JC (1994) Activation of phosphatidylinositol-3′ kinase by Src-family kinase SH3 binding to the p85 subunit. Science 263: 1609-1612. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY (1982) Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res 235: 93-103. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Seiden LS, Schuster CR (1983) Increased dopamine metabolism in the rat neostriatum after toxic doses of d-methylamphetamine. Neuropharmacology 22: 1383-1388. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Seiden LS, Schuster CR (1984) Further evidence that amphetamines produce long-lasting dopamine neurochemical deficits by destroying dopamine nerve fibers. Brain Res 303: 359-364. [DOI] [PubMed] [Google Scholar]
- Roux PP, Dorval G, Boudreau M, Angers-Loustau A, Morris SJ, Makkerh J, Barker PA (2002) K252a and CEP1347 are neuroprotective compounds that inhibit mixed-lineage kinase-3 and induce activation of Akt and ERK. J Biol Chem 277: 49473-49480. [DOI] [PubMed] [Google Scholar]
- Saporito MS, Brown ER, Carswell S, DiCamillo AM, Miller MS, Murakata C, Neff NT, Vaught JL, Haun FA (1998) Preservation of cholinergic activity and prevention of neuron death by CEP-1347/KT-7515 following excitotoxic injury of the nucleus basalis magnocellularis. Neuroscience 86: 461-472. [DOI] [PubMed] [Google Scholar]
- Saporito MS, Brown EM, Miller MS, Carswell S (1999) CEP-1347/KT-7515, an inhibitor of c-jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons In vivo. J Pharmacol Exp Ther 288: 421-427. [PubMed] [Google Scholar]
- Saporito MS, Thomas BA, Scott RW (2000) MPTP activates c-Jun NH(2)-terminal kinase (JNK) and its upstream regulatory kinase MKK4 in nigrostriatal neurons in vivo. J Neurochem 75: 1200-1208. [DOI] [PubMed] [Google Scholar]
- Saporito MS, Hudkins RL, Maroney AC (2002) Discovery of CEP-1347/KT-7515, an inhibitor of the JNK/SAPK pathway for the treatment of neurodegenerative diseases. Prog Med Chem 40: 23-62. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ, Ritter JK, Sonsalla PK, Hanson GR, Gibb JW (1985) Role of dopamine in the neurotoxic effects of methamphetamine. J Pharmacol Exp Ther 233: 539-544. [PubMed] [Google Scholar]
- Seiden LS (1985) Methamphetamine: toxicity to dopaminergic neurons. NIDA Res Monogr 62: 100-116. [PubMed] [Google Scholar]
- Sheng P, Wang XB, Ladenheim B, Epstein C, Cadet JL (1996) AP-1 DNA-binding activation by methamphetamine involves oxidative stress. Synapse 24: 213-217. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Jochnowitz ND, Zeevalk GD, Oostveen JA, Hall ED (1996) Treatment of mice with methamphetamine produces cell loss in the substantia nigra. Brain Res 738: 172-175. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Rayport S (1990) Amphetamine and other psychostimulants reduce pH gradients in midbrain dopaminergic neurons and chromaffin granules: a mechanism of action. Neuron 5: 797-808. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Zecca L (2000) Intraneuronal dopamine-quinone synthesis: a review. Neurotox Res 1: 181-195. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A (1995) Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci 15: 4102-4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, St Remy C, Rayport S (1996) Reserpine inhibits amphetamine action in ventral midbrain culture. Mol Pharmacol 49: 338-342. [PubMed] [Google Scholar]
- Tietze F (1969) Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem 27: 502-522. [DOI] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, Greene LA (2001) beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem 77: 157-164. [DOI] [PubMed] [Google Scholar]
- Ueda S, Nakamura H, Masutani H, Sasada T, Yonehara S, Takabayashi A, Yamaoka Y, Yo J (1998) Redox regulation of caspase-3(-like) protease activity: regulatory roles of thioredoxin and cytochrome c. J Immunol 161: 6689-6695. [PubMed] [Google Scholar]
- Wagner GC, Lucot JB, Schuster CR, Seiden LS (1983) Alpha-methyltyrosine attenuates and reserpine increases methamphetamine-induced neuronal changes. Brain Res 270: 285-288. [DOI] [PubMed] [Google Scholar]
- Wagner GC, Carelli RM, Jarvis MF (1985) Pretreatment with ascorbic acid attenuates the neurotoxic effects of methamphetamine in rats. Res Commun Chem Pathol Pharmacol 47: 221-228. [PubMed] [Google Scholar]
- Wang LH, Besirli CG, Johnson Jr EM (2004) Mixed-lineage kinases: a target for the prevention of neurodegeneration. Annu Rev Pharmacol Toxicol 44: 451-474. [DOI] [PubMed] [Google Scholar]
- Wang W, Shi L, Xie Y, Ma C, Li W, Su X, Huang S, Chen R, Zhu Z, Mao Z, Han Y, Li M (2004) SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson's disease. Neurosci Res 48: 195-202. [DOI] [PubMed] [Google Scholar]
- Willesen MG, Gammeltoft S, Vaudano E (2002) Activation of the c-Jun N terminal kinase pathway in an animal model of Parkinson's disease. Ann NY Acad Sci 973: 237-240. [DOI] [PubMed] [Google Scholar]
- Xia XG, Harding T, Weller M, Bieneman A, Uney JB, Schulz JB (2001) Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson's disease. Proc Natl Acad Sci USA 98: 10433-10438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca L, Tampellini D, Gatti A, Crippa R, Eisner M, Sulzer D, Ito S, Fariello R, Gallorini M (2002) The neuromelanin of human substantia nigra and its interaction with metals. J Neural Transm 109: 663-672. [DOI] [PubMed] [Google Scholar]
- Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR (2004) Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci 5: 863-873. [DOI] [PubMed] [Google Scholar]