

Abstract
Normal functioning of the nervous system requires precise regulation of dendritic shape and synaptic connectivity. Here, we report a severe impairment of dendritic structures in the cerebellum and motor cortex of c-Jun N-terminal kinase 1 (JNK1)-deficient mice. Using an unbiased screen for candidate mediators, we identify the dendrite-specific high-molecular-weight microtubule-associated protein 2 (MAP2) as a JNK substrate in the brain. We subsequently show that MAP2 is phosphorylated by JNK in intact cells and that MAP2 proline-rich domain phosphorylation is decreased in JNK1-/- brain. We developed compartment-targeted JNK inhibitors to define whether a functional relationship exists between the physiologically active, cytosolic pool of JNK and dendritic architecture. Using these, we demonstrate that cytosolic, but not nuclear, JNK determines dendritic length and arbor complexity in cultured neurons. Moreover, we confirm that MAP2-dependent process elongation is enhanced after activation of JNK. Using JNK1-/- neurons, we reveal a dominant role for JNK1 over ERK in regulating dendritic arborization, whereas ERK only regulates dendrite shape under conditions in which JNK activity is low (JNK1-/- neurons). These results reveal a novel antagonism between JNK and ERK, potentially providing a mechanism for fine-tuning the dendritic arbor. Together, these data suggest that JNK phosphorylation of MAP2 plays an important role in defining dendritic architecture in the brain.
Keywords: JNK, MAP2, dendrite, neuron, morphology, phosphorylation
Introduction
Fine-tuning of dendritic arbors ensures both the proper connectivity of neural circuitry and the intrinsic electrical properties of neurons (Barrett and Crill, 1974; Mainen and Sejnowski, 1996; Libersat and Duch, 2004). Although structural remodeling of dendritic shape or postsynaptic plasticity underlies the physiological process of learning and memory (Lamprecht and LeDoux, 2004), abnormal dendritic development is a consistent hallmark of mental retardation syndromes (Fiala et al., 2002; Zoghbi, 2003). Despite the importance of dendritic structure to neuronal physiology, the mechanisms regulating dendrite shape formation and maintenance in the brain remain essentially unknown (Miller and Kaplan, 2003).
Microtubules are the main structural determinants of dendritic shafts (Peters et al., 1991; Matus, 1994), and microtubule integrity is maintained in neurons by microtubule-stabilizing proteins. Among these, the most abundant in the mature brain is the high-molecular-weight microtubule-associated protein 2 (MAP2) (Sanchez et al., 2000). Binding of MAP2 to protofilaments suppresses microtubule catastrophe activity (for review, see Desai and Mitchison, 1997) and promotes dendritic elongation (Harada et al., 2002). Phosphorylation of MAP2 is an integral requirement for binding to microtubules in intact cells, and a number of MAP2 kinases have been reported (Brugg and Matus, 1991; Quinlan and Halpain, 1996; Sanchez et al., 1996, 2000). However, the effect of MAP2 kinases on dendritic growth is not fully understood.
c-Jun N-terminal kinases (JNKs) contribute to stress-induced neuronal cell death (for review, see Bozyczko-Coyne et al., 2002). However, additional roles for JNK in regulating physiological responses in the nervous system have been described previously (Xu et al., 1997; Byrd et al., 2001; Xia and Karin, 2004). Of the three JNK genes expressed in the brain, neural JNK1 displays elevated constitutive activity that is not stress related (Coffey et al., 2000, 2002; Kuan et al., 2003). This activity maintains microtubule homeostasis and axonal integrity in the adult brain (Chang et al., 2003). JNK was first identified as a MAP2 kinase in vitro (Kyriakis and Avruch, 1990), yet it is only recently that MAP2 was considered as an in vivo JNK target (Chang et al., 2003). Although MAP2 is a dendrite-specific microtubule-stabilizing protein, the effect of JNK on dendritic architecture has not been explored systematically.
Here, we report that cytoplasmic JNK1 activity increases during neuronal differentiation and that compartment-specific inhibition of this pool reveals a causal role for JNK in regulating dendritic shape. A dominant role for JNK over extracellular signal-regulated kinase (ERK) in shaping dendritic structure is supported from data comparing wild-type and JNK1-/- neurons, which display an increased dendritic arbor number and decreased arbor length. We show that JNK phosphorylates MAP2 in intact cells and that C-terminal phosphorylation of MAP2 is reduced in brains from JNK1-/- mice. Moreover, brains from JNK1-deficient mice show severe abnormalities in dendritic architecture; cerebella display increased dendrite complexity, and Golgi-Cox staining revealed a 60% reduction in dendrite length in layers III, IV, and V of the motor cortex. Together these data strongly suggest that JNK phosphorylation of MAP2 plays an integral role in regulating dendritic structure in the brain.
Materials and Methods
Antibodies and reagents. Mouse anti-MAP2 (AP20; specific for high-molecular-weight MAP2) and mouse anti-β-tubulin (KMX-1) were obtained from Leinco Technologies (St. Louis, MO). Mouse anti-JNK1 (G151-333) was obtained from PharMingen (San Diego, CA), and mouse anti-striatin was obtained from Transduction Laboratories (Lexington, KY). Rabbit anti-P-JNK, mouse anti-P-ERK, and mouse anti-ERK1/2 were obtained from Cell Signaling Technology (Beverly, MA), and anti-phosphorylated threonine flanked by proline (phospho-TP) was a gift from M. Melnick (Cell Signaling Technology). Mouse anti-actin was a gift from B. Jockusch (Technical University of Braunschweig, Braunschweig, Germany). Polyclonal anti-stress-activated protein kinase (SAPK) and anti-dephospho-MAP2 (972) were gifts from J. Kyriakis (Massachusetts General Hospital, Boston, MA) and J. Avila (Universidad Autónoma de Madrid, Madrid, Spain). Purified bovine high-molecular weight (HMW)-MAP2 was obtained from Cytoskeleton (Denver, CO).
Plasmids. HMW-MAP2 was obtained by PCR from rat brain cDNA using primers recognizing rat MAP2b and was inserted into the NotI site of a modified pEGFP-C1 (Clontech, Mountain View, CA) and into the NotI site of pEBG (University of Connecticut Health Center, Farmington, CT), after subcloning in pGEMTeasy (Promega, Madison, WI). pEGFP-JIP-JBD, pEGFP-NES-JBD, pEGFP-NLS-JBD, and pEGFP-NES-cJun(1-146) were constructed by PCR-based methods from pcDNA3-mJIP1a. pcDNA3-dnJNK1 was prepared from pSRα-JNK1APF by excising with HindIII/BamHI and ligating into pcDNA3 cut with HindIII/BamHI. pEBG-JNK, pDsRed-δMEKK1, and pcDNA3-GAL4-Jun(5-105) constructs were described previously (Coffey et al., 2002; Hongisto et al., 2003). pEBG-ERK1, pEBG-p38, and pGL3-G5E4Δ38 were gifts from B. Mayer (University of Connecticut) and P. Shaw (Nottingham University, Nottingham, UK). SEK1KD, a kinase dead mutant of SEK1 (MKK4), was a gift from J. Kyriakis. pRL-CMV was obtained from Promega.
Cell culture. Cerebellar granule neurons were prepared from postnatal day 7 (P7) Sprague Dawley rats as described previously (Coffey et al., 2000). Cells were cultured in minimal essential medium (MEM) supplemented with 10% (v/v) fetal calf serum (Invitrogen, San Diego, CA), 33 mm glucose, 2 mm glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin, and 20 mm supplementary KCl (final, 25.4 mm KCl). Cells were plated at 250,000/cm2 onto culture surfaces coated with poly-l-lysine (50 μg/ml): 24-well plates (Cellstar, Greiner, Germany) for GAL4-reporter assay and immunoblotting and 10.5 × 10.5 mm coverslips for immunofluorescent staining. Culture medium was replaced after 24 h with the inclusion of 10 μm cytosine arabinofuranoside (Sigma, St. Louis, MO) to reduce non-neuronal proliferation. After this time, fresh culture medium was not readded to the cells, to avoid serum glutamate-associated toxicity. Cortical neuron cultures were prepared from P0 rats as described previously (Hetman et al., 1999) and maintained in Eagle's basal medium (Worthington, Freehold, NJ) supplemented with 10% bovine calf serum (HyClone, Logan, UT), 2 mm glutamine, 35 mm glucose, 15 mm KCl, 50 U/ml penicillin, and 50 μm streptomycin. Cytosine arabinofuranoside (2.5 μm) was added 2 d after plating to inhibit proliferation of dividing cells. At 24 h after plating, cortical neurons were transfected using Lipofectamine 2000 according to the manufacturer's instructions. COS-7 cells were cultured in MEM supplemented with 10% (v/v) fetal calf serum, 2 mm glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. Neuro-2A cells were cultured in MEM supplemented with 10% fetal calf serum, nonessential amino acids (Sigma), 2 mm glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. All cells were cultured in a humidified 5% CO2 atmosphere at 37°C.
Transfections and morphological analysis of cells. For morphological analysis, cerebellar granule neurons were plated on 10.5 × 10.5 mm coverslips. Cells were transiently transfected at 4 d after plating with 1.4 μg of pEGFP-MAP2 together with 0.6 μg of pcDNA3, empty vector, pcDNA3-dnJNK1, pcDNA3-JIP-JBD, or compartment-targeted nuclear export sequence (NES)-JNK-binding domain (JBD) and nuclear localization sequence (NLS)-JBD, as described previously (Coffey et al., 2000). For COS-7 and Neuro-2A cell transfections, 75% of DNA was pEGFP-MAP2 or pEGFP-C1, and 5% was pDsRed-δMEKK1 as indicated. Empty vector pCMV was used to normalize DNA levels between samples. Transfections were performed using Lipofectamine (COS-7 cells) or Polyfect (Neuro-2A) according to the manufacturer's instructions (Invitrogen). Cells were fixed 48 h after transfection, and green fluorescent protein (GFP) fluorescence was analyzed using a Leica (Nussloch, Germany) DMRE microscope equipped with a Hamamatsu (Hamamatsu City, Japan) Orca CCD camera. Neuronal cell dendritic length and branch length was measured from size-calibrated images using MetaMorph software version 6.1 (Universal Imaging Corporation, West Chester, PA). The number of GFP-MAP2-expressing neurites that originated at the cell soma and were equal or greater in length to 1 nuclear diameter (main dendritic processes) were counted manually from digitized images. Processes that separated from the main dendrite, distal to the cell soma, were counted as branches. For analysis of COS-7 and Neuro-2A cell extensions, similar criteria were used, processes that were ≥1 nuclear diameter in length were counted. Cells were stained with Hoechst-33342 for measurements of nuclear diameter and viability. Dead cells with pyknotic nuclei were not analyzed.
Immunostaining. Immunocytochemical staining was performed as follows. Coverslips with neurons at 6 d in vitro (DIV) were fixed with 4% paraformaldehyde for 20 min at 37°C, followed by permeabilization in PBS/Triton X-100 (1%) for 3 min. After washing with PBS, cells were blocked with 10% serum, 0.2% Tween 20, and PBS for 1 h at room temperature. Incubation with primary antibodies was overnight at 4°C using 1:100 anti-SAPK, 2 μg/ml α-MAP2, or 1:2000 anti-β-galactosidase (5 Prime → 3 Prime, Boulder, CO). Immunoreactivity was detected using 1:800 anti-rabbit biotin (Sigma), followed by 1:2000 Streptavidin Alexa-488 (Molecular Probes, Eugene, OR) for SAPK and β-galactosidase, or using 1:400 anti-mouse Alexa-568 (Molecular Probes) for MAP2. Before mounting, nuclei were stained with 2 μg/ml Hoechst-33342. Slides were scanned under a 63× objective with 488 nm argon and 543 nm HeNe lasers using a Zeiss (Oberkochen, Germany) LSM 510 confocal microscope. Mouse brain (4 months) was fixed for 48 h in 4% paraformaldehyde, impregnated in 30% sucrose, and frozen in isopentane. Cryostat sections (30 μm) were blocked with Vectastain blocking solution (Vector Laboratories, Burlingame, CA). Sections were incubated with 2 μg/ml anti-MAP2 overnight, followed by anti-mouse Alexa-488 (1:1000), and examined using the argon laser of a Zeiss 510 confocal microscope.
Golgi staining and morphological analysis of tissues. Modified Golgi-Cox impregnation of 4-month-old mouse brain was performed using the rapid Golgi staining method (FD NeuroTechnologies, Ellicott City, MD). Brains were fixed in solutions A and B for 3 weeks and transferred to solution C for 2 d at 4°C according to the manufacturer's instructions. Sections (120 μm) were cut with a cryostat and stained with silver nitrate solution (solutions D and E) before dehydration and mounting on slides with Permount. Slices were examined under a 4× objective using an Olympus (Melville, NY) BX60 microscope, and digitized images were acquired using a U-CMAD-2 CCD camera. Motor cortex thickness was measured as the distance between the inner boundary of the corpus callosum directly above the hippocampal CA1 region and the outer cortical surface. For measurement of dendrite length, 15 cells were chosen randomly and analyzed in each of layers III, IV, and V of the motor cortex in four corresponding sets (360 cells in total were measured). MetaMorph 6.1 was used for morphometric measurements (dendrite length and regional thickness).
Reporter assays. For reporter assays, cerebellar granule neurons on 12-well plates were transfected at 6 DIV with 0.5 μg of pGL3-G5E4Δ38, a firefly luciferase reporter plasmid driven by five GAL4 elements in tandem, 0.5 μg of pcDNA3-GAL4-Jun(5-105), 0.5 μg of pRL-CMV sea pansy luciferase as an internal standard against which signals were normalized, and 0.5 μg of pEGFP-C1 as a marker of transfection efficiency, as described previously (Coffey et al., 2002). In addition, cells were transfected with 2 μg of pEGFP-JIP-JBD, pEGFP-NES-JBD, or pEGFP-NLS-JBD as indicated. Twenty hours after transfection, cells were switched to low-KCl (5 mm) medium (trophic withdrawal) for 4 h and lysed in 70 μl of passive lysis buffer (Promega). Firefly (reporter) and Renilla (internal standard) luciferase activities were assayed with the dual luciferase assay kit (Promega) according to the manufacturer's instructions.
Tissue extract preparation. The forebrain from adult mice or cerebellum from P7 wild-type and JNK1-/- mice was rapidly extracted after decapitation and snap-frozen in liquid N2. Frozen tissues were homogenized using an Ultra Turrax homogenizer in ice-cold lysis buffer [20 mm HEPES, pH 7.4, 2 mm EGTA, 50 mm β-glycerophosphate, 1 mm dithiothreitol (DTT), 1 mm Na3VO4, 1% Triton X-100, 10% glycerol, 1 mm benzamidine, 50 mm NaF, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 μg/ml aprotinin, and 100 μg/ml PMSF]. Lysates were normalized for protein using the Bradford method, and a 0.25 volume of 4× concentrated Laemmli sample buffer was added.
Immunoblot analysis and quantification. Cells were stimulated as indicated, washed in PBS, and lysed with Laemmli sample buffer. Samples were resolved on 5% (MAP2) or 10% SDS-PAGE and transferred by semi-dry transfer to nitrocellulose. Blots were developed using the enhanced chemiluminescence detection method. Films were preflashed, and nonsaturated exposures were digitized by flatbed scanning and quantified by densitometry.
Immune-complex kinase assays. Cytosolic and nuclear fractions were prepared from 3.5 cm dishes of cerebellar granule neurons at 1, 3, or 6 DIV, as described previously (Coffey et al., 2000). Fractions were incubated with 0.5 μl of anti-JNK1 for 2 h, followed by 1 h with 10 μl of 50% protein G-Sepharose. Immobilized kinase complexes were washed three times with lysis buffer, three times with LiCl buffer (500 mm LiCl, 100 mm Tris, pH 7.6, 0.1% Triton X-100, and 1 mm DTT), and three times with kinase buffer [20 mm 4-morpholinepropanesulfonic acid, pH 7.2, 2 mm EGTA, 10 mm MgCl2, 1 mm DTT, and 0.1% (v/v) Triton X-100]. Kinase assays were performed in kinase buffer supplemented with 50 μm ATP, 5 μCi of [γ-32P]ATP, and 4 μg of GST-c-Jun(5-89)/sample for 30 min at 30°C. Reactions were stopped by the addition of Laemmli sample buffer. Samples were resolved by SDS-PAGE gels and analyzed by phosphorimaging.
Kinetic analysis. For examination of kinase specificity toward MAP2, active recombinant pEBG-JNK, pEBG-p38, and pEBG-ERK1 were prepared as described previously (Hongisto et al., 2003). Kinase assays were performed exactly as described above, except that 0.05-1.0 μm bovine HMW-MAP2 (Cytoskeleton) was substituted for GST-c-Jun. Incubations were performed for 30 min at 30°C. Samples were resolved on 5% SDS-PAGE, and 32P incorporation was measured by phosphorimaging. Michaelis-Menten constant (Km) values were calculated from Lineweaver Burk plots of calibrated data.
Separation of phosphorylated protein by two-dimensional electrophoresis. Mouse brain extract was homogenized in kinase buffer and phosphorylated using active recombinant JNK1 as described previously (Hongisto et al., 2003). Protein extract was loaded onto a dry polyacrylamide gel strip with an immobilized pH gradient of 4-7, according to the manufacturer's instructions (Amersham Biosciences, Uppsala, Sweden). Proteins were separated in the first dimension by isoelectric focusing overnight at 3500 V, followed by two-dimensional separation on 12% SDS-PAGE. Two-dimensional electrophoresis gels were silver stained according to O'Connell and Stults (1997) and analyzed by autoradiography.
Protein identification by mass spectrometry. Reduction, alkylation, and in-gel digestion of the silver-stained proteins were performed as described previously (Shevchenko et al., 1996) using sequence grade-modified porcine trypsin (Promega). Digested protein was desalted on a C-18 nano-precolumn (0.3 × 5 mm; LC Packings, Amsterdam, The Netherlands) and separated on an analytical (150 mm × 75 μm inner diameter) nano-LC, C18 column. Bound peptides were eluted with 5-60% acetonitrile in 0.1% formic acid (200 nl/min) into the mass spectrometer (Q-StarPulsar; Applied Biosystems, Foster City, CA) for peptide mass determination and sequencing in positive ion mode. The Time-of-Flight survey scan (1 s) selected for doubly and triply charged peptides. The two most-intense peaks were selected and fragmented by collision-induced dissociation, and the product ion spectra were collected. Data were processed by Analyst QS software (Applied Biosystems) and matched to the SwissProt protein database using the MASCOT algorithm with fixed modification, carbamidomethyl (C) and variable modification, and oxidation. Peptide and mass spectrometry/mass spectrometry (MS/MS) tolerance were ±0.2 Da. Peptide charge was 2+ and 3+, using monoisotopic masses allowing for up to one missed cleaved site.
Statistical analysis. Statistical ANOVA was done using SPSS for Windows version 11.0 (SPSS, Chicago, IL). One-way ANOVA followed by Fisher's least significant difference post hoc test was used for analysis of significance in samples with more than two variable groups.
Results
JNK activity increases sharply during differentiation of cultured neurons (Coffey et al., 2000) and embryonic midbrain cells (Park et al., 2004). This elevated activity predominates in the cytoplasm where the molecular actions of JNK are not clearly defined (Coffey et al., 2002). The aim of this study was to investigate the mechanism of JNK regulation of neuronal architecture.
JNK1 activity is developmentally upregulated during neuronal differentiation and contributes to physiological JNK activity in the cerebellum
Identifying the JNK isoform that is upregulated during neuronal differentiation has been hindered by the lack of isoform-specific JNK antibodies. However, an antibody specific for the JNK1 isoform does exist and has been characterized previously (Coffey et al., 2002). Using this antibody, we isolated JNK1 from neurons at 1, 3, and 6 d after plating and measured kinase activity in vitro toward GST-c-Jun(5-89) by immune-complex kinase assay (Fig. 1A). JNK1 activity increased sharply in the cytoplasmic compartment of cerebellar granule neurons differentiating in culture. To determine the contribution of JNK1 to total JNK activity in the cerebellum at a corresponding stage of development, cerebellar cortices from wild-type and JNK1-deficient mice were normalized for protein and kinase activity measured by immunoblotting with an antibody detecting the active form of JNK (Fig. 1B). JNK activity was reduced by 70% in the JNK1-/- cerebellum.
Figure 1.
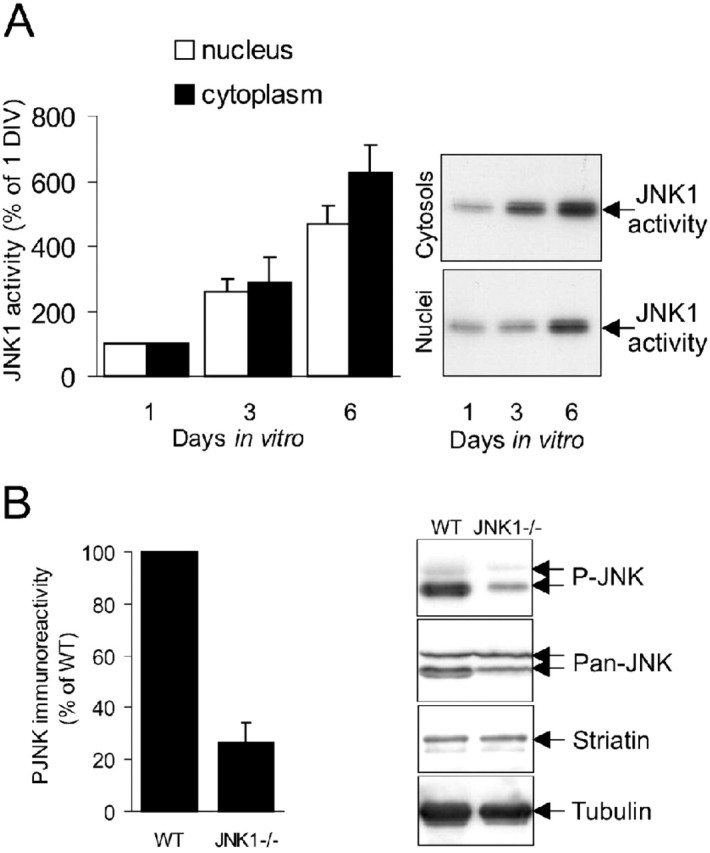
JNK1 activity is upregulated during differentiation of neurons in culture. A, Cytoplasmic and nuclear fractions from cerebellar granule neurons at 1, 3, and 6 DIV were analyzed for JNK1 activity using an isoform-specific JNK1 antibody. Immune-complex kinase activity was quantified by phosphorimaging and expressed as arbitrary units. Representative autoradiographs and mean ± SEM from four sets are shown. B, JNK activity from the P7 wild-type (WT) and JNK1-/- cerebellum was measured by immunoblotting with an antibody recognizing the active form of JNK (P-JNK). JNK activity was expressed as a percentage of wild type. The mean ± SEM are shown. Representative immunoblots from three sets depicting immunoreactivity of P-JNK, pan-JNK, striatin, and tubulin (loading controls) are shown.
JNK1 phosphorylates brain-derived MAP2
To identify potential targets for elevated JNK1 activity in the brain, we phosphorylated P7 mouse brain extract with recombinant active JNK1. Proteins were separated by two-dimensional SDS-PAGE, and phosphorylated proteins were visualized by autoradiography. JNK1 induced strong phosphorylation of a protein migrating at 250 kDa with an isoelectric point of 4.8 (Fig. 2A; the inset shows a silver-stained gel and corresponding autoradiograph; for a magnified view, see supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The prominently phosphorylated spot was excised from the gel and digested with trypsin. MS/MS sequencing revealed nine complete peptide sequences that corresponded to mouse HMW-MAP2b (GenBank accession number P20357). The total ion chromatogram of selected peptides and MS/MS spectra of one of the identified peptides is shown (Fig. 2A). These data unambiguously identified the JNK phosphorylated protein as MAP2. Lower-molecular-weight proteins, also phosphorylated by JNK1, were identified by MS as degradation products of MAP2 (Fig. 2A, arrows). If MAP2 is a bona fide target for JNK, both proteins are expected to reside in the same subcellular compartment. To test this, we immunostained cerebellar granule neurons at 6 d after plating with antibodies specific for high-molecular-weight MAP2 and JNK (Fig. 2B). Confocal sections revealed colocalization of MAP2 and JNK immunoreactivity in the somatodendritic compartments of differentiating neurons.
Figure 2.
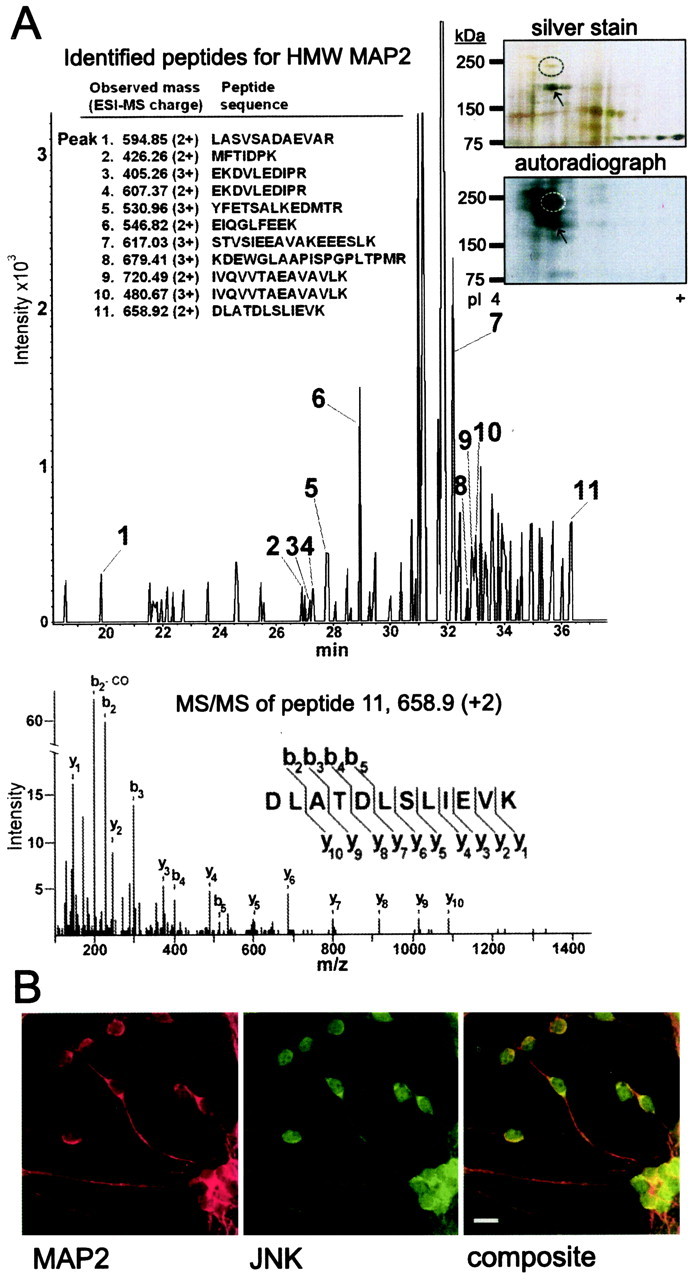
JNK1 phosphorylates and colocalizes with HMW-MAP2 in the somatodendritic compartment. A, Brain-derived HMW-MAP2 was identified as a JNK1 substrate using MS. P7 mouse brain homogenate phosphorylated by JNK1 was separated by two-dimensional gel electrophoresis. Corresponding sections of the silver-stained gel and autoradiograph are shown. JNK1 phosphorylated protein (circled) was analyzed using electrospray analysis. Nine complete peptide sequences matching mouse HMW-MAP2 were obtained. B, To determine whether JNK colocalized with MAP2 in neurons, cerebellar granule neurons at 6 DIV were immunostained for MAP2 (red) and pan-JNK (green). Representative confocal micrographs are shown. The composite image shows strong overlap (yellow) in JNK and MAP2 localization in the cell soma and dendrites. Scale bar, 10 μm.
JNK phosphorylates MAP2 somewhat more efficiently than ERK in vitro
Our screen identified MAP2 as a JNK substrate using subphysiological concentrations of MAP2 from brain extract, and furthermore, MAP2 was the most highly phosphorylated protein when JNK1 was used as kinase (Fig. 2A, two-dimensional gel). ERK and JNK mitogen-activated protein kinases (MAPKs) were originally identified as kinases that phosphorylated purified MAP2 in vitro on threonine residues (Kyriakis and Avruch, 1990; Boulton et al., 1991). To determine which of the MAPKs, JNK, ERK or p38, showed preferential phosphorylation of MAP2, we analyzed MAP2 phosphorylation using recombinant active kinases that had been first normalized for activity as described previously (Hongisto et al., 2003). JNK preferentially phosphorylated MAP2 compared with ERK with a Michaelis-Menten constant (Km) of 0.32 and 0.57 μm, respectively (Fig. 3A). p38 phosphorylation of MAP2 was very weak compared with ERK and JNK.
Figure 3.
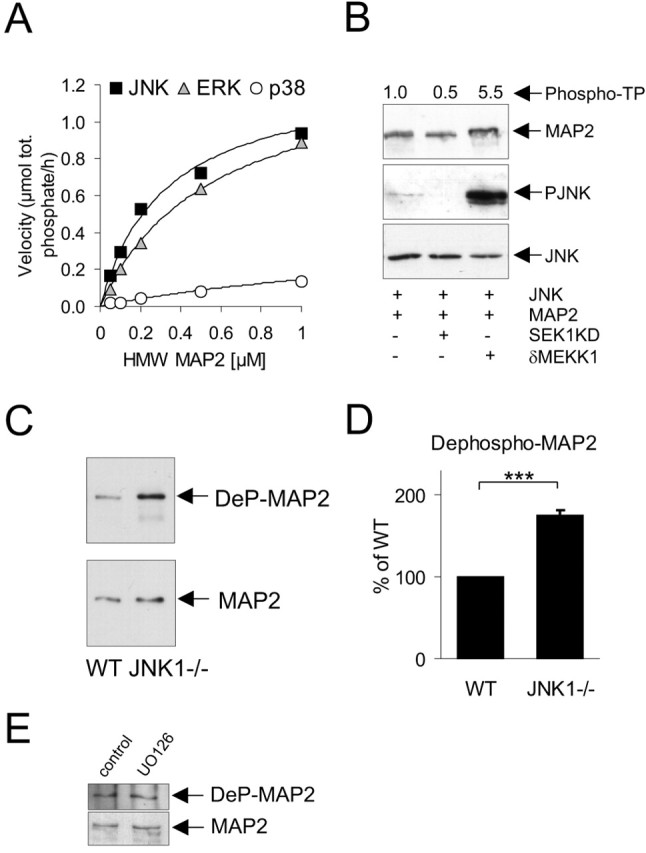
JNK1 phosphorylates the proline-rich C-terminal domain of MAP2 in the brain. A, Kinetic analysis of JNK, ERK, and p38 phosphorylation of MAP2 in vitro. Active recombinant kinases were used to phosphorylate increasing concentrations of MAP2. B, To evaluate the ability of JNK to phosphorylate MAP2 in intact cells, COS-7 cells were transfected with GFP-MAP2 and δMEKK1 to activate coexpressed JNK or with SEK1KD to prevent JNK activation. JNK activation was visualized by immunoblotting for active JNK (PJNK). MAP2 displayed retarded mobility on SDS-PAGE after activation of JNK. Representative blots from five repeats are shown. Phosphate incorporation to Thr-Pro motifs on MAP2 was evaluated by repeating the transfections substituting GST-MAP2. GST-MAP2 sequestered on glutathione-Sepharose was immunoblotted with antibodies recognizing phosphorylated Thr-Pro motifs (Phospho-TP) and quantified by densitometry. Fold increase in MAP2 phospho-TP immunoreactivity above control is depicted above the gel panels. C, Phosphorylation of MAP2 was examined in the wild-type and JNK1-/--deficient cortex from P7 mice using an antibody recognizing the dephosphorylated form of the C-terminal proline-rich domain (RTPGTPGTPSY) of MAP2 (Ab972). Tissue lysates were normalized for MAP2 expression (bottom) and probed with Ab972 (DeP-MAP2). D, Mean data from three sets of animals ± SEM is shown. E, Cerebellar granule neurons at 6 DIV were treated with U0126 (10 μm) for 24 h and lysates blotted for dephospho-MAP2 (DeP-MAP2) or MAP2 as indicated. Inhibition of ERK did not alter MAP2 phosphorylation in this domain. ***p < 0.001 (ANOVA). WT, Wild type.
JNK1 phosphorylates MAP2 in intact cells and in the brain
We subsequently examined whether JNK phosphorylated MAP2 in intact cells. COS-7 cells were transfected with GFP-MAP2 and dsRed-δMEKK1, to activate coexpressed glutathione S-transferase (GST)-JNK, or with SEK1KD, to prevent JNK activation (Fig. 3B). Activation of JNK led to a retarded migration of GFP-MAP2 on SDS-PAGE, consistent with increased MAP2 phosphorylation (Fig. 3B, lane 3). This was blocked by coexpression of a dominant-negative inhibitor of JNK signaling, SEK1KD. JNK phosphorylates Thr residues on MAP2 in vitro (Kyriakis and Avruch, 1990). To directly measure MAP2 phosphorylation in intact cells, we used antibodies that recognize phospho-TP. Cells were transfected as above, this time substituting GST-MAP2 for GFP-MAP2. GST-MAP2 was isolated using glutathione-Sepharose and phospho-TP immunoreactivity detected by immunoblotting. Active JNK induced a 5.5-fold increase in specific phosphorylation on Thr-Pro motifs of MAP2 in intact cells (Fig. 3B). To examine JNK phosphorylation of MAP2 in vivo, we used an antibody that recognizes the proline-rich C-terminal domain of MAP2 in its dephosphorylated state (Arg1616-Thr-Pro-Gly-Thr-Pro-Gly-Thr-Pro-Ser-Tyr1626). This domain contains three consensus sites for JNK phosphorylation and is highly phosphorylated in the adult brain (Sanchez et al., 1996). Immunoblotting of the cortex from JNK1-/- mice revealed a significant increase in dephospho-MAP2 immunoreactivity compared with wild-type (Fig. 3C,D). This suggests that JNK1 phosphorylates the proline-rich C-terminal domain of MAP2 in the brain. Because ERK phosphorylated MAP2 to a similar extent as JNK in vitro, we examined the effect of ERK inhibition on MAP2 proline-rich domain phosphorylation in neurons. Cerebellar granule neurons were treated with U0126, a pharmacological inhibitor of MAPK/ERK kinase 1/2 (MEK1/2), which we demonstrated effectively inhibited ERK activation (see Fig. 7D). However, ERK inhibition did not affect MAP2 phosphorylation in the proline-rich domain (Fig. 3E). This is not surprising because ERK is reported to phosphorylate MAP2 predominantly in the N-terminal projection domain (Silliman and Sturgill, 1989; Berling et al., 1994).
Figure 7.
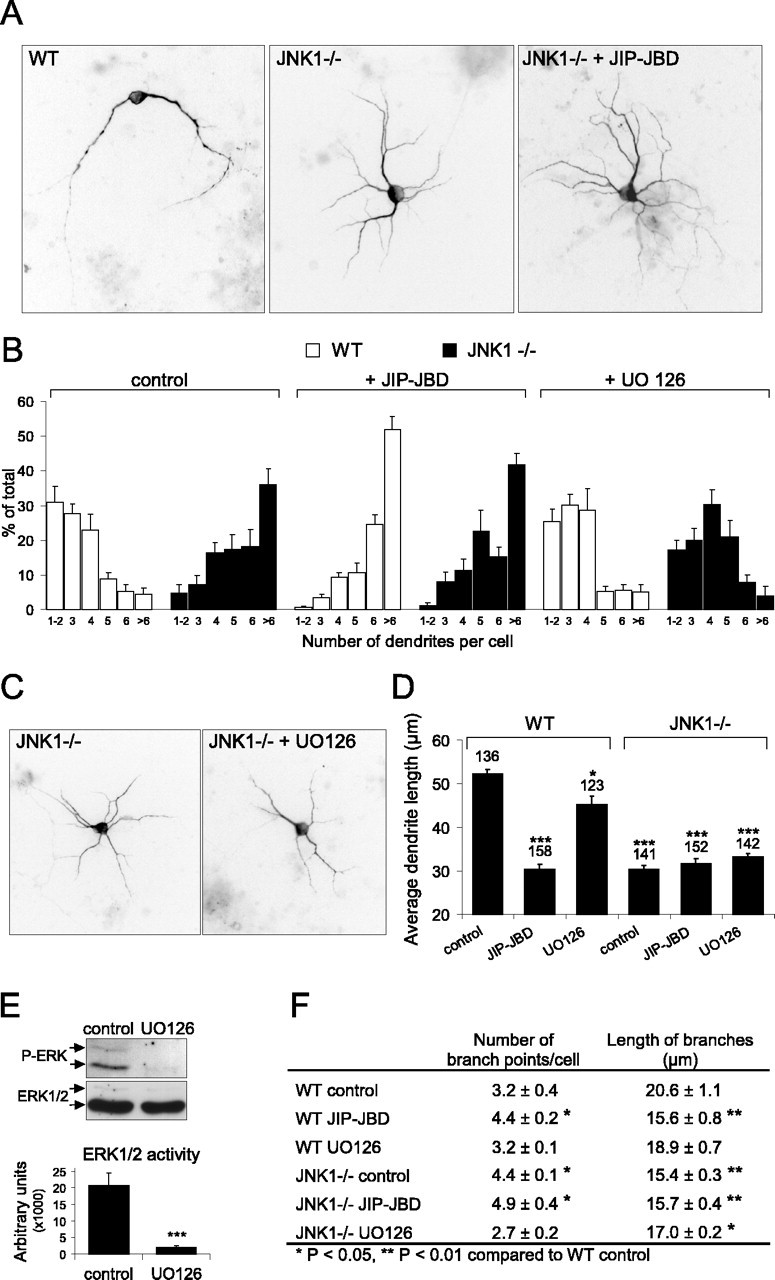
Dendritic architecture is dramatically altered in JNK1-/- neurons. A, Cerebellar granule neurons isolated from wild-type and JNK1-/- cerebella were transfected 5 d after plating with GFP-MAP2 together with pcDNA3 empty vector (control) or pcDNA3-JIP-JBD as indicated. Cells were fixed after 7 d in culture, and inverted images of representative fluorescence micrographs of GFP-MAP2 are shown. B, The dendritic architecture of neurons cultured as described in A were analyzed. The number of main dendrites extending from the cell body was counted and presented as distribution plots. Wild-type and JNK1-/- cells (left), wild-type and JNK1-/- cells with coexpressed JIP-JBD (middle), and wild-type and JNK1-/- cells treated for 48 h with U0126 (10 μm)(right) were compared. C, Representative images of GFP-MAP2 fluorescence in transfected JNK1-/- neurons with and without U0126 (10 μm). D, Average dendrite length was measured from the same population of neurons as described in B. Dendrites from JNK1-/- neurons were significantly shorter in length than wild-type neurons. Inhibition of ERK with U0126 had only a minor influence on dendrite length. Neurons lacking JNK1 display dendritic architecture changes that are indistinguishable from JBD-treated neurons. Coexpression of JBD in JNK1-/- neurons exerts no additional morphology changes. The number of cells counted for each condition is depicted above the histogram bar. The averaged data ± SEM are shown for five to six data sets. E, To determine whether U0126 effectively inhibited ERK activity under these conditions, cerebellar neurons treated with and without U0126 (10 μm) were immunoblotted with antibodies detecting active ERK1/2 (P-ERK) or ERK1/2 protein. The mean data from three sets are shown. F, The number of branch points and dendritic branch length were measured from wild-type and JNK1-/- cells expressing GFP-MAP2 together with JIP-JBD or U0126 as indicated. The mean ± SEM from five to six separate sets are shown. *p < 0.05, **p < 0.01, ***p < 0.001 (ANOVA). WT, Wild type.
JNK regulates dendritic architecture in cerebellar granule neurons
MAP2 plays a critical role in regulating dendritic elongation (Harada et al., 2002). Similarly, elevated basal JNK activity regulates neuronal cell shape (Coffey et al., 2000). However, this previous study did not distinguish between dendritic and axonal structures. To determine whether the effects of JNK on cytoarchitecture occurred in dendritic structures, cerebellar granule neurons at 5 DIV were transfected with GFP-MAP2 together with the JBD of JIP1 (JIP-JBD) (Fig. 4), a fragment of JIP1 that inhibits JNK substrate phosphorylation (Dickens et al., 1997; Coffey et al., 2000). A dominant-negative JNK inhibitory protein (dnJNK1) was used as an additional inhibitor of JNK substrate phosphorylation (Kallunki et al., 1994). Control cells expressing GFP-MAP2 alone projected an average of two or three dendrites from the cell soma, whereas there was a dramatic and significant increase in dendrite number from cells in which JNK substrate phosphorylation was inhibited (Fig. 4A,B). The average length of dendrites was also significantly reduced after expression of JNK inhibitory proteins (Fig. 4C). It is notable that the changes measured in cytoarchitecture were in the dendritic compartment, because GFP-MAP2 was excluded from the axonal compartment, whereas the soluble β-galactosidase was present in both axons and dendrites (Fig. 4D).
Figure 4.

Inhibition of JNK leads to shorter and more numerous dendritic processes. A, To determine the effect of JNK inhibition on dendritic length, cerebellar granule neurons at 5 DIV were transfected with GFP-MAP2 together with the JNK inhibitory protein JIP-JBD or dnJNK1. Scale bar, 10 μm. B, After a 48 h expression, the number of dendrites, extending from the cell soma, that were >1 nuclear diameter in length were counted. The percentage of cells with a given number of dendrites was plotted from six to nine data sets. C, Dendrite length was measured from the same cells. The number of cells counted for each condition is shown above the corresponding histogram bars. D, Cerebellar granule neuron transfected at 5 DIV with GFP-MAP2 (green) and β-galactosidase (β-gal; red). β-Galactosidase is expressed in both axonal and dendritic compartments, whereas GFP-MAP2 localization is restricted to the dendrites. The mean ± SEM for six data sets is shown. **p < 0.01, ***p < 0.001 (ANOVA).
MAP2-dependent process elongation is regulated by JNK
Having demonstrated that JNK regulated dendrite length and number in GFP-MAP2-expressing cerebellar granule neurons (Fig. 4), we wanted to establish whether MAP2 phosphorylation by JNK played a causal role in this event. To avoid possible interference from the neuron-specific JNK targets tau, neurofilament, and doublecortin (O'Ferrall et al., 2000; Gdalyahu et al., 2004; Yoshida et al., 2004), we chose to use a non-neuronal cell model in which production of cell protrusions is entirely dependent on exogenously expressed GFP-MAP2. It is well established that expression of MAP2 induces “neurite-like” extensions in cells that otherwise do not develop processes (Berling et al., 1994; Boucher et al., 1999; Sanchez et al., 2000). We observed that expression of GFP-MAP2 in nonprocess-bearing cells induced projections from the cell soma that were >1 nuclear diameter in length (Fig. 5A). Resting JNK activity in COS-7 cells is low, therefore the JNK activator δMEKK1 was coexpressed to activate endogenous JNK. Expression of δMEKK1 led to an increase in GFP-MAP2-dependent process length, whereas expression of δMEKK1 alone had no effect (Fig. 5A,B). Given that MAP2 is believed to play a role in microtubule stability, these data suggested that JNK phosphorylation of MAP2 is involved in MAP2-dependent stabilization of microtubules and process outgrowth and that this event is independent of other known JNK targets.
Figure 5.

JNK increases MAP2-dependent process length. A, To examine the influence of JNK on MAP2-dependent process growth, COS-7 cells were transfected with GFP or GFP-MAP2. GFP-expressing cells did not extend processes, whereas GFP-MAP2-expressing cells generated processes that were >1 nuclear diameter in length. Nuclear diameter was assessed by Hoechst-33342 staining. B, The effect of JNK on MAP2-dependent process growth was measured by coexpressing the JNK activator δMEKK1. Process length was measured and defined as short (≥1 nuclear diameter), medium (2-3 nuclear diameters), or long (>3 nuclear diameters). Nuclear diameters were measured from Hoechst-33342-stained nuclei. The mean ± SEM from three separate sets is shown. C, A similar analysis was performed in Neuro-2A cells. Expression of the JNK activator δMEKK1 greatly enhanced GFP-MAP2-dependent process elongation, whereas expression of δMEKK1 alone did not induce elongation. Representative images of GFP-MAP2-expressing Neuro-2A cells are shown. D, Process length was measured as described in B. The mean ± SEM from three sets are shown. *p < 0.05, ***p < 0.001 (ANOVA).
We then tested the ability of JNK to regulate MAP2-dependent process outgrowth in the Neuro-2A neuroblastoma model. These cells produce large amounts of microtubules and are classically used for studies of tubulin-based mechanisms (Stamer et al., 2002). Expression of GFP-MAP2 alone in Neuro-2A induced only short protrusions from the cell body, whereas expression of the JNK activator δMEKK1 led to a dramatic increase in GFP-MAP2-generated process length (Fig. 5C,D). Together, these data indicate that JNK phosphorylation of MAP2 is genuinely involved in facilitating MAP2-dependent extension of projections.
Characterization of compartment-specific JNK inhibitors
JNK activity predominates in the cytoplasm in developing neurons, although residual nuclear JNK activity is also detectable (Fig. 1A) (Coffey et al., 2000). Therefore, we could not exclude the possibility that the regulation of dendritic architecture observed (Fig. 4) resulted from JNK regulation of nuclear targets. To determine whether cytoplasmic or nuclear JNK was responsible for maintaining cell shape homeostasis, we prepared compartment-specific inhibitors of JNK. An NES from MEK1 or three NLSs from SV40 large T antigen were fused in tandem upstream of the JNK inhibitor protein JIP-JBD. As expected, these targeted proteins localized to cytoplasmic and nuclear compartments, respectively, when expressed in COS-7 cells (Fig. 6A) and in cerebellar granule neurons (Fig. 6B). The functional specificity of NES-JBD and NLS-JBD toward JNK activity in the nuclear compartment was tested using a gene reporter assay. NLS-JBD, but not NES-JBD, prevented JNK-dependent induction of GAL4-Jun-driven firefly luciferase activity, induced by withdrawing trophic support from cerebellar granule neurons (Fig. 6C). This demonstrated that NES-JBD was not functional in the nuclear compartment. To verify that NES-JBD inhibited cytoplasmic JNK action, we tested the ability of NES-JBD to prevent phosphorylation of a cytoplasmic JNK reporter; NES-cJun(1-146). Because expression of NES-cJun(1-146) in cerebellar granule neurons was not detectable by immunoblotting, we used cortical neurons where a higher transfection efficiency was obtained. Phosphorylation of exogenously expressed NES-cJun(1-146) was entirely blocked by coexpression of NES-JBD (Fig. 6D).
Figure 6.

Cytoplasmic JNK regulates dendritic architecture in cerebellar neurons. A, To selectively target cytoplasmic and nuclear JNK activity, compartment-specific JNK inhibitors (NES-JBD and NLS-JBD) were prepared. GFP-tagged (green) NES-JBD localized to the cytoplasm and NLS-JBD localized to the nucleus when expressed in COS-7 cells (top). Nuclei were stained with Hoechst-33342 (blue), and composite images are shown (bottom). B, GFP-NES-JBD localized to the cytoplasm and GFP-NLS-JBD localized to the nucleus in cerebellar granule neurons. C, The ability of NES-JBD and NLS-JBD to inhibit nuclear JNK activity was tested by reporter assay. Cerebellar granule neurons were transfected at 6 DIV with a GAL4-driven luciferase reporter and GAL4-Jun. Trophic withdrawal (low-KCl medium) induced JNK-dependent GAL-4-Jun activity. This was inhibited by coexpression of NLS-JBD but not of NES-JBD. Firefly luciferase activity was normalized to Renilla luciferase internal standard. Reporter activity is expressed as fold response from controls without trophic withdrawal. The mean ± SEM from three sets are shown. D, The ability of NES-JBD to block cytoplasmic JNK activity was tested by measuring the phosphorylation of cotransfected NES-c-Jun(1-146). Because of detection problems from transfected cerebellar granule neurons, these transfections were performed in cortical neurons. Coexpression of NES-JBD efficiently blocked phosphorylation of NES-Jun(1-146). E, Cerebellar granule neurons were transfected with NES-JBD or NLS-JBD together with GFP-MAP2 as shown. The number of dendrites extending from the cell soma was counted as before (Fig. 4). NES-JBD induced a dramatic increase in dendrite number, whereas NLS-JBD had no effect. The mean ± SEM from four to seven sets is shown. F, Dendrite length was measured from the same cells. NES-JBD significantly reduced dendrite length. The mean ± SEM from four sets is shown. G, Total dendritic network length was measured from cells expressing GFP-MAP2 together with JIP-JBD, NES-JBD, or NLS-JBD as shown. Inhibition of cytoplasmic JNK selectively increased the dendritic network length. The mean ± SEM from four to six sets is shown. *p < 0.05, **p < 0.01, ***p < 0.001 (ANOVA).
Cytoplasmic JNK regulates dendritic complexity and length in neurons
To examine whether cytoplasmic or nuclear JNK was responsible for the regulation of dendritic architecture, cerebellar neurons were transfected with GFP-MAP2 in the presence or absence of NES-JBD or NLS-JBD. Inhibition of cytoplasmic JNK activity with NES-JBD evoked a dramatic increase in dendrite number (Fig. 6E) and concomitant decrease in dendrite length (Fig. 6F). Interestingly, inhibition of nuclear JNK with NLS-JBD had no effect on dendrite number or length. Because the overall extent of dendrites has an impact on firing pattern (Mainen and Sejnowski, 1996), we analyzed the influence of JNK activity on dendritic structure as a whole by measuring total dendrite length (Fig. 6G). The total dendritic network length was defined as the combined length of main dendrites and branches for a given cell. Inhibition of cytoplasmic JNK induced an increase in the total length of the dendritic network (Fig. 6G). This is not surprising given the dramatic increase in dendrite number observed after JNK inhibition (Fig. 6E). These data provide the first evidence that cytoplasmic JNK activity regulates dendritic architecture in cultured neurons. Moreover, it implies that the JNK effectors regulating dendritic architecture reside in the cytoplasm.
Neurons from JNK1-/- mice display increased dendritic arbor number and decreased arbor length
JNK1 activity contributes to 70% of the constitutive activity existing in the developing cerebellum (Fig. 1B) and adult cerebral cortex (Kuan et al., 2003). If JNK1 is primarily responsible for regulating dendritic architecture, neurons from JNK1 knock-out mice should show disrupted dendritic shape similar to that obtained after inhibition of JNK action with JIP-JBD or dnJNK1 (Figs. 4, 6). To closely examine dendritic structure, neurons from the JNK1-/- cerebellum were transfected with GFP-MAP2, allowing single-cell morphology analysis to be performed against a dense network of fasiculated neurites from the nontransfected cell population. Cerebellar granule neurons from JNK1-/- mice exhibited a striking increase in the number of main dendrites extending from the cell body (Fig. 7A,B). The majority of JNK1-/- cells extended more than six dendritic processes, whereas most of the wild-type cells extended only two to three dendrites. Similarly, the dendrite length in JNK1-/- cells was significantly reduced and indistinguishable from that of cells in which JNK activity was inhibited with JIP-JBD (Fig. 7D). Moreover, expression of JIP-JBD in JNK1-/- neurons did not further alter dendrite number or length, indicating that JNK1 plays a nonredundant role in regulating dendritic architecture.
JNK1 dominates over ERK in regulating dendritic shape in cerebellar granule neurons
The closely related protein kinase ERK also phosphorylates MAP2 in vitro (Fig. 3), albeit at distinct sites from the JNK phosphorylated proline-rich domain. We therefore examined whether ERK regulated dendritic architecture. Treatment of neurons with U0126 (10 μm) elicited long-term inhibition of ERK activity (Fig. 7E); however, this did not induce any significant change in dendritic process number in wild-type cerebellar granule neurons. Interestingly, blocking ERK activity in JNK1-/- neurons resulted in decreased process number (Fig. 7B,C, compare JNK1-/- cells with and without U0126). ERK activity was unaltered in JNK1-/- neurons, indicating that JNK was not acting via ERK (data not shown). These data suggest that although ERK has the capacity to increase dendritic complexity, this does not occur in differentiating cerebellar granule neurons in which JNK1 is active.
The function of dendrites is to transmit electrical signals from incoming synaptic contacts. Increased branching and increased dendritic length attenuate electrical spread and thereby affect signal integration and the firing properties of neurons (Mainen and Sejnowski, 1996; Shepherd, 1999; Hausser et al., 2000). To examine how loss of JNK1 expression affected dendritic network complexity, we measured dendritic branch number and length in wild-type and JNK1-/- neurons (Fig. 7F). There was a significant increase in branch points and a simultaneous decrease in branch length in cells expressing JIP-JBD and in cells from JNK1-/- mice. This suggests that JNK negatively regulates dendritic complexity in cerebellar granule neurons. Conversely, inhibition of ERK in a JNK1-negative background caused a significant decrease in branch number and increase in branch length. These data suggest that ERK can positively regulate dendritic branch complexity if steps are taken to lower JNK1 activity.
Dendrite arborization is deregulated in the cerebellum and motor cortex of the JNK1-/- brain
Having demonstrated that MAP2 phosphorylation was decreased in the JNK1-/- brain (Fig. 3C,D), we subsequently determined whether dendritic structure was altered. Dendrite arborization was examined in the wild-type and knock-out cerebellum stained with antibodies specific for MAP2 (Fig. 8A). The wild-type cerebellum displayed a uniform dendritic architecture, most clearly visualized in the Purkinje cells of the molecular layer. In contrast, the JNK1-/- cerebellum exhibited a notable increase in dendritic complexity in the molecular layer, dendrites appearing more twisted and branched than in the wild-type cerebellum. This striking disturbance of Purkinje cell dendritic architecture was observed consistently in the JNK1-/- but not in the wild-type cerebellum. MAP2 expression levels were not altered in the adult JNK1-/- brain (Fig. 8B). We then inspected dendritic structure in the cortex using the classical Golgi-Cox impregnation method (Fig. 8C). Sagittal sections through the medial cortex revealed a remarkable decrease in dendritic arbor length in the motor cortex of JNK1-/- mice compared with wild type. Digital images of corresponding sections from the JNK1-/- and wild-type motor cortex were collected. From these, dendrite lengths were measured from neurons in layers III, IV, and V, where Golgi-Cox staining is most clearly visible. The average dendrite length in the motor cortex of knock-out mice decreased by 60-70% compared with wild type (Fig. 8F). The majority of these dendrites, even in the deeper layers, were <150 μm long (Fig. 8E). Such overt changes in dendritic morphology were not apparent in the surrounding neocortex. Another conspicuous feature of the JNK1-deficient motor cortex was the decrease in thickness (Fig. 8C,D). Although the motor cortex was dramatically reduced in thickness by >50%, there was only a minor reduction in the size of the prefrontal cortex (Fig. 8D), indicating that there is a relatively selective regional deterioration in the absence of JNK1. An additional phenomenon was consistently observed in the cortex of JNK1-/- mice. There was a dramatic increase in thick Golgi-impregnated structures in JNK1-/- brains compared with wild type (Fig. 8C, white arrow). This feature has previously been attributed to Golgi-Cox staining of blood vessel epithelial cells (Kolb et al., 1999). This is unlikely to be an artifact because brains from wild-type and knock-out mice were extracted and fixed under identical conditions, and this phenomenon was observed only in knock-out mice. This was widespread throughout the cortex and cerebellum of knock-out mice, suggesting a difference in vascularization of JNK1-/- brains.
Figure 8.

Dendritic architecture is substantially altered in the cerebellum and motor cortex of JNK1-/- mice. A, To examine dendritic architecture in the cerebellum, tissues were cut laterally at the midline, and comparable sagittal sections (30 μm) were immunostained for MAP2. Purkinje cell dendritic shape was dramatically altered in the molecular layer (ML) of JNK1-/- mice. Scale bar, 100 μm. The images shown are representative of four sets of animals. PL, Purkinje cell layer; GL, granule layer; WM, white matter. B, Brain lysates from wild-type and JNK1-/- mice were normalized for protein and immunoblotted with antibodies detecting JNK, MAP2, tubulin, and actin as indicated. Expression of MAP2 was not notably altered in the JNK1-/- cortex. C, Golgi-Cox staining of sagittal sections (120 μm) through the medial motor cortex revealed a dramatic reduction in dendrite length and cortex thickness in JNK1-/- mice. Regions CA1 and CA2 of the hippocampus are labeled. CC, Corpus callosum. Scale bar, 500 μm. D, Regional thickness of the motor cortex and prefrontal cortex was measured. Motor cortex thickness was distinctly reduced in the JNK1-/- brain. E, Dendritic length distribution in layer III, IV, and V of the motor cortex. Fifteen cells per layer were measured from each of four sets of wild-type and knock-out tissues. F, Data from E are presented as average dendrite length in layers III, IV, and V of the medial motor cortex. The mean ± SEM are shown for four sets of animals. *p < 0.05, **p < 0.01, **p < 0.001 (ANOVA). WT, Wild type.
Discussion
Maintenance of dendrite homeostasis is important for normal neuronal physiology, and dysregulation of dendritic structure is a feature of schizophrenia and mental retardation disorders such as autism, Rett syndrome, and Down's syndrome, in which reduction in length and branching of dendritic arbors is observed (Rosoklija et al., 2000; Broadbelt et al., 2002; Fiala et al., 2002; Zoghbi, 2003; Mukaetova-Ladinska et al., 2004). Understanding the mechanisms of dendrite formation and homeostasis may provide important clues to the etiology of such diseases. We and others have shown previously that JNK regulates neuronal cell shape (Coffey et al., 2000; Waetzig and Herdegen, 2003). In this study, we tested the importance of physiologically active JNK for dendritic architecture in cultured neurons and in the brain. We studied the dendrites of cerebellar granule neurons in vitro, because the maturation of these cells in culture closely mimics that in the brain (Burgoyne and Cambray-Deakin, 1988) and granule cell JNK activity is known to increase during differentiation in culture in parallel to JNK upregulation in the developing cerebellum (Coffey et al., 2000). Analysis of dendrite structure in brains lacking the physiologically active form of JNK (JNK1) provided an in vivo model in which to assess the significance of JNK activity for dendrite structure.
A conspicuous feature of elevated JNK activity in neurons is its almost exclusive localization to the cytoplasmic compartment (Coffey et al. 2000), contrasting with the expectation of activity-dependent translocation to the nucleus. Consistent with this, a growing list of cytoplasmic targets have emerged for JNK, suggesting that this pool of activity may be of physiological importance (McDonald et al., 2000; Huang et al., 2003; Inomata et al., 2003; Gdalyahu et al., 2004). Nonetheless, a minor pool of nuclear JNK activity also exists in differentiating neurons, and the most comprehensively studied JNK effectors are transcriptional regulators that are localized to the nucleus (Coffey et al., 2000; Hazzalin and Mahadevan, 2002; Shaulian and Karin, 2002). A major concern during this study was to determine whether nuclear or cytosolic JNK signaling was responsible for refining neuronal shape. Here, we describe for the first time compartment-targeted inhibition of JNK. Our data establish that cytoplasmic and not nuclear JNK is the dominant regulator of dendritic form in neurons. MAP2 would appear to be the most likely effector because it is the only candidate JNK target known to regulate dendritic structure, and more importantly, we demonstrate that JNK phosphorylates MAP2 in intact cells and regulates its ability to induce process outgrowth.
Morphogenesis defects have been reported in brains from JNK knock-out mice. JNK1 is required for fiber tract formation, and JNK3 is required for optic fissure closure (Chang et al., 2003; Weston et al., 2003). Here, we report a striking dysregulation of dendritic structure in the motor cortex of JNK1-/- mice, where a substantial reduction in dendrite length was accompanied by localized atrophy and thinning as is reported during aging and in multiple sclerosis (Nakamura et al., 1985; Sailer et al., 2003). The degeneration of the motor cortex in JNK1-deficient mice indicates a physiological requirement for JNK in maintaining this center of movement control. There are no published studies demonstrating behavioral defects in these mice, and it is not known whether the absence of JNK1 results in diminished motor function. Moreover, whether disruption of JNK signaling underlies the pathology of diseases such as mental retardation syndromes and mood disorders that are characterized by dendritic anomalies is also unknown. Interestingly, however, a translocation of the JNK3 gene has been described in a patient suffering from severe mental retardation syndrome (Shoichet, 2004). This translocation results in expression of a truncated form of JNK3 that could conceivably have a dominant-negative influence on JNK signaling. Notably, this patient showed early progressive loss of motor coordination skills, as would be expected if JNK is critical for maintaining motor cortex integrity, as our data suggest. It is also worth noting that PAK3 (p21-activated kinase), which is mutated in several families affected with mental retardation disorders (Bienvenu et al., 2000), is a member of the Ste20-related kinase group, proposed upstream JNK regulators (Brown et al., 1996).
Metric changes in dendrite length and number leading to increased complexity were observed in cultured cerebellar granule neurons after inhibition of cytoplasmic JNK. Similarly, in the JNK1-/- cerebellum, there was a prominent increase in dendritic arbor complexity in the molecular layer (Fig. 8A), which consists primarily of Purkinje cell dendrites. Purkinje cells display a characteristically complex and planar dendritic tree that extends like a branched candelabrum toward the surface of the cerebellum. An interesting possibility is that the planarity of Purkinje dendrites is lost in the JNK1-/- brain, because the staining observed is consistent with what may be expected if this tree was three-dimensional. The extensive foliation of the cerebellum makes equivalent orientation of the tissue during sectioning difficult. However, we observed a distinct pattern of dendritic architecture in each of four sets of knock-out mice compared with wild-type mice, suggesting that the differences observed were not attributable to random sectioning through the Purkinje cell arbors and instead represent a genuine disparity in dendritic architecture in the JNK1-/- cerebellum.
External cues reported to regulate dendritic maturation include neurotrophins, semaphorins, Reelin, and electrical activity (McAllister et al., 1999; Whitford et al., 2002; Libersat and Duch, 2004; Niu et al., 2004). It is feasible that JNK indirectly modifies dendritic architecture in vivo by modulating such signals. However, we favor a model in which JNK phosphorylation of MAP2 directly regulates microtubule rigidity and, subsequently, dendritic architecture. It has been reported that JNK phosphorylation of MAPs regulates binding to and stabilization of microtubules in vitro (Chang et al., 2003). We show that JNK promotes MAP2-dependent process elongation in non-neuronal cells in which the receptors for neurotrophins, semaphorins, and Reelin are not known to be expressed. Likewise, if JNK regulated dendritic architecture only by modifying the electrical properties of neurons, we would not anticipate a regulation of MAP2 architecture in nonexcitable COS-7 cells after JNK activation. Thus, JNK induction of MAP2-generated process length appears to occur in the absence of extrinsic, neuronal factors. Importantly, JNK regulation of dendrite elongation and complexity was independent of classical JNK transcriptional regulatory events involving c-Jun or ATF2 activation (Fig. 6). JNK facilitation of MAP2-dependent process elongation was observed in COS-7 cells, neuroblastoma, and cerebellar granule neurons. This supports our proposal that MAP2 mediates JNK regulation of process length.
There is good evidence that the related MAPK ERK regulates activity-dependent dendrite formation in neuronal systems (Wu et al., 2001; Vaillant et al., 2002; Miller and Kaplan, 2003). Interestingly, we find that in cerebellar granule neurons, inhibition of ERK1/2 activity only affects dendritic shape in a JNK1-/- context. This suggests that under conditions when JNK activity is low, ERK may play an important role in promoting dendritic plasticity, being required for increased dendritic arborization, although in differentiating cerebellar granule neurons ERK does not appear to contribute to dendritic shape. These results reveal a novel down-stream antagonism between JNK and ERK, potentially providing a mechanism for fine-tuning the dendritic arbor.
MAP2 is a highly phosphorylated brain protein incorporating 46 mol phosphate/mol (Tsuyama et al., 1987). MAP2 phosphorylation increases developmentally, correlating with increased arborization (Riederer et al., 1995). Attributing JNK phosphorylation sites to MAP2 is hindered by the fact that MAP2 contains 43 Ser-Pro/Thr-Pro motifs, potential sites for JNK phosphorylation. We unambiguously identified MAP2 as a JNK substrate by MS. Furthermore, we demonstrated that the C-terminal proline-rich domain of MAP2 is phosphorylated by JNK in the brain. This domain of MAP2 is phosphorylated in cultured neurons during dendrite formation (Sanchez et al., 2000). However, the state of phosphorylation of MAP2 on this site in the brain throughout development is not known. We observed a clear deficit in dendritic morphology in 4-month-old JNK1-/- mice, indicating that the JNK requirement must have occurred earlier. Indeed, we first identified reduced phosphorylation of the proline-rich domain of MAP2 in the P7 cortex from JNK1-/- mice. This is the first report identifying a JNK substrate motif on MAP2. It is plausible that JNK1 phosphorylates additional sites among the 40 remaining candidate sites on MAP2. A systematic analysis of these sites is a necessary prerequisite to identification of the functionally important site. Other cytoskeletal regulatory proteins have been reported as JNK targets (e.g., tau, neurofilament, and doublecortin) (O'Ferrall et al., 2000; Gdalyahu et al., 2004; Yoshida et al., 2004). In addition, using our methodology, we have identified an additional candidate JNK target that is a neuron-specific protein. For this reason, the morphology analysis in COS-7 cells (Fig. 5) is critical because it establishes that MAP2-dependent process growth is regulated by JNK in the absence of known neuron-specific JNK targets. The alternative targets are classically known for their function in axons and growth cones and therefore are unlikely to influence dendritic changes, making MAP2 the most likely effector of dendritic alterations by JNK in vivo.
In conclusion, we have demonstrated that JNK plays a causal role in regulating MAP2-dependent process length, dendrite number, and elongation in cultured cells and in the brain. Together, these data suggest that JNK phosphorylation of MAP2 is of genuine importance for normal dendrite homeostasis.
Footnotes
This work was supported by Finnish Academy Grants 47536, 49949, 206497 (E.T.C.), and 203520 (M.J.C.) and by Åbo Akademi University, the National Graduate School in Informational and Structural Biology, the Turku Graduate School of Biomedical Sciences, the Finnish Graduate School for Neurosciences, and the Svenska Kulturfonden. We are grateful to the Cell Imaging Core and Proteomics Unit at the Turku Centre for Biotechnology for the use of equipment.
Correspondence should be addressed to Dr. Eleanor T. Coffey, Turku Centre for Biotechnology, Åbo Akademi and Turku University, BioCity, Tykistokatu 6, FIN-20521 Turku, Finland. E-mail: ecoffey@btk.fi.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256350-12$15.00/0
References
- Barrett JN, Crill WE (1974) Influence of dendritic location and membrane properties on the effectiveness of synapses on cat motoneurones. J Physiol (Lond) 239: 325-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berling B, Wille H, Roll B, Mandelkow EM, Garner C, Mandelkow E (1994) Phosphorylation of microtubule-associated proteins MAP2a,b and MAP2c at Ser136 by proline-directed kinases in vivo and in vitro. Eur J Cell Biol 64: 120-130. [PubMed] [Google Scholar]
- Bienvenu T, des Portes V, McDonell N, Carrie A, Zemni R, Couvert P, Ropers HH, Moraine C, van Bokhoven H, Fryns JP, Allen K, Walsh CA, Boue J, Kahn A, Chelly J, Beldjord C (2000) Missense mutation in PAK3, R67C, causes X-linked nonspecific mental retardation. Am J Med Genet 93: 294-298. [DOI] [PubMed] [Google Scholar]
- Boucher M, Belanger D, Beaulieu C, Leclerc N (1999) Tau-mediated process outgrowth is differentially altered by the expression of MAP2b and MAP2c in Sf9 cells. Cell Motil Cytoskeleton 42: 257-273. [DOI] [PubMed] [Google Scholar]
- Boulton TG, Gregory JS, Cobb MH (1991) Purification and properties of extracellular signal-regulated kinase 1, an insulin-stimulated microtubule-associated protein 2 kinase. Biochemistry 30: 278-286. [DOI] [PubMed] [Google Scholar]
- Bozyczko-Coyne D, Saporito MS, Hudkins RL (2002) Targeting the JNK pathway for therapeutic benefit in CNS disease. Curr Drug Targets CNS Neurol Disord 1: 31-49. [DOI] [PubMed] [Google Scholar]
- Broadbelt K, Byne W, Jones LB (2002) Evidence for a decrease in basilar dendrites of pyramidal cells in schizophrenic medial prefrontal cortex. Schizophr Res 58: 75-81. [DOI] [PubMed] [Google Scholar]
- Brown JL, Stowers L, Baer M, Trejo J, Coughlin S, Chant J (1996) Human Ste20 homologue hPAK1 links GTPases to the JNK MAP kinase pathway. Curr Biol 6: 598-605. [DOI] [PubMed] [Google Scholar]
- Brugg B, Matus A (1991) Phosphorylation determines the binding of microtubule-associated protein 2 (MAP2) to microtubules in living cells. J Cell Biol 114: 735-743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne RD, Cambray-Deakin MA (1988) The cellular neurobiology of neuronal development: the cerebellar granule cell. Brain Res 472: 77-101. [DOI] [PubMed] [Google Scholar]
- Byrd DT, Kawasaki M, Walcoff M, Hisamoto N, Matsumoto K, Jin Y (2001) UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans Neuron 32: 787-800. [DOI] [PubMed] [Google Scholar]
- Chang L, Jones Y, Ellisman MH, Goldstein LS, Karin M (2003) JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev Cell 4: 521-533. [DOI] [PubMed] [Google Scholar]
- Coffey ET, Hongisto V, Dickens M, Davis RJ, Courtney MJ (2000) Dual roles for c-Jun N-terminal kinase in developmental and stress responses in cerebellar granule neurons. J Neurosci 20: 7602-7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey ET, Smiciene G, Hongisto V, Cao J, Brecht S, Herdegen T, Courtney MJ (2002) c-Jun N-terminal protein kinase (JNK) 2/3 is specifically activated by stress, mediating c-Jun activation, in the presence of constitutive JNK1 activity in cerebellar neurons. J Neurosci 22: 4335-4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai A, Mitchison TJ (1997) Microtubule polymerization dynamics. Annu Rev Cell Dev Biol 13: 83-117. [DOI] [PubMed] [Google Scholar]
- Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ (1997) A cytoplasmic inhibitor of the JNK signal transduction pathway. Science 277: 693-696. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM (2002) Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev 39: 29-54. [DOI] [PubMed] [Google Scholar]
- Gdalyahu A, Ghosh I, Levy T, Sapir T, Sapoznik S, Fishler Y, Azoulai D, Reiner O (2004) DCX, a new mediator of the JNK pathway. EMBO J 23: 823-832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A, Teng J, Takei Y, Oguchi K, Hirokawa N (2002) MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J Cell Biol 158: 541-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausser M, Spruston N, Stuart GJ (2000) Diversity and dynamics of dendritic signaling. Science 290: 739-744. [DOI] [PubMed] [Google Scholar]
- Hazzalin CA, Mahadevan LC (2002) MAPK-regulated transcription: a continuously variable gene switch? Nat Rev Mol Cell Biol 3: 30-40. [DOI] [PubMed] [Google Scholar]
- Hetman M, Kanning K, Cavanaugh JE, Xia Z (1999) Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J Biol Chem 274: 22569-22580. [DOI] [PubMed] [Google Scholar]
- Hongisto V, Smeds N, Brecht S, Herdegen T, Courtney MJ, Coffey ET (2003) Lithium blocks the c-Jun stress response and protects neurons via its action on glycogen synthase kinase 3. Mol Cell Biol 23: 6027-6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Rajfur Z, Borchers C, Schaller MD, Jacobson K (2003) JNK phosphorylates paxillin and regulates cell migration. Nature 424: 219-223. [DOI] [PubMed] [Google Scholar]
- Inomata H, Nakamura Y, Hayakawa A, Takata H, Suzuki T, Miyazawa K, Kitamura N (2003) A scaffold protein JIP-1b enhances amyloid precursor protein phosphorylation by JNK and its association with kinesin light chain 1. J Biol Chem 278: 22946-22955. [DOI] [PubMed] [Google Scholar]
- Kallunki T, Su B, Tsigelny I, Sluss HK, Derijard B, Moore G, Davis R, Karin M (1994) JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev 8: 2996-3007. [DOI] [PubMed] [Google Scholar]
- Kolb B, Pedersen B, Ballermann M, Gibb R, Whishaw IQ (1999) Embryonic and postnatal injections of bromodeoxyuridine produce age-dependent morphological and behavioral abnormalities. J Neurosci 19: 2337-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan CY, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ, Dong C, Bao J, Banasiak KJ, Haddad GG, Flavell RA, Davis RJ, Rakic P (2003) A critical role of neural-specific JNK3 for ischemic apoptosis. Proc Natl Acad Sci USA 100: 15184-15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J (1990) pp54 microtubule-associated protein 2 kinase. A novel serine/threonine protein kinase regulated by phosphorylation and stimulated by poly-l-lysine. J Biol Chem 265: 17355-17363. [PubMed] [Google Scholar]
- Lamprecht R, LeDoux J (2004) Structural plasticity and memory. Nat Rev Neurosci 5: 45-54. [DOI] [PubMed] [Google Scholar]
- Libersat F, Duch C (2004) Mechanisms of dendritic maturation. Mol Neurobiol 29: 303-320. [DOI] [PubMed] [Google Scholar]
- Mainen ZF, Sejnowski TJ (1996) Influence of dendritic structure on firing pattern in model neocortical neurons. Nature 382: 363-366. [DOI] [PubMed] [Google Scholar]
- Matus A (1994) Stiff microtubules and neuronal morphology. Trends Neurosci 17: 19-22. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC (1999) Neurotrophins and synaptic plasticity. Annu Rev Neurosci 22: 295-318. [DOI] [PubMed] [Google Scholar]
- McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ (2000) Beta-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science 290: 1574-1577. [DOI] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR (2003) Signaling mechanisms underlying dendrite formation. Curr Opin Neurobiol 13: 391-398. [DOI] [PubMed] [Google Scholar]
- Mukaetova-Ladinska EB, Arnold H, Jaros E, Perry R, Perry E (2004) Depletion of MAP2 expression and laminar cytoarchitectonic changes in dorsolateral prefrontal cortex in adult autistic individuals. Neuropathol Appl Neurobiol 30: 615-623. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Akiguchi I, Kameyama M, Mizuno N (1985) Age-related changes of pyramidal cell basal dendrites in layers III and V of human motor cortex: a quantitative Golgi study. Acta Neuropathol (Berl) 65: 281-284. [DOI] [PubMed] [Google Scholar]
- Niu S, Renfro A, Quattrocchi CC, Sheldon M, D'Arcangelo G (2004) Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron 41: 71-84. [DOI] [PubMed] [Google Scholar]
- O'Connell KL, Stults JT (1997) Identification of mouse liver proteins on two-dimensional electrophoresis gels by matrix-assisted laser desorption/ionization mass spectrometry of in situ enzymatic digests. Electrophoresis 18: 349-359. [DOI] [PubMed] [Google Scholar]
- O'Ferrall EK, Robertson J, Mushynski WE (2000) Inhibition of aberrant and constitutive phosphorylation of the high-molecular-mass neurofilament subunit by CEP-1347 (KT7515), an inhibitor of the stress-activated protein kinase signaling pathway. J Neurochem 75: 2358-2367. [DOI] [PubMed] [Google Scholar]
- Park KS, Lee RD, Kang SK, Han SY, Park KL, Yang KH, Song YS, Park HJ, Lee YM, Yun YP, Oh KW, Kim DJ, Yun YW, Hwang SJ, Lee SE, Hong JT (2004) Neuronal differentiation of embryonic midbrain cells by upregulation of peroxisome proliferator-activated receptor-gamma via the JNK-dependent pathway. Exp Cell Res 297: 424-433. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster HD (1991) The fine structure of the nervous system. Neurons and their supporting cells. New York: Oxford UP.
- Quinlan EM, Halpain S (1996) Postsynaptic mechanisms for bidirectional control of MAP2 phosphorylation by glutamate receptors. Neuron 16: 357-368. [DOI] [PubMed] [Google Scholar]
- Riederer BM, Draberova E, Viklicky V, Draber P (1995) Changes of MAP2 phosphorylation during brain development. J Histochem Cytochem 43: 1269-1284. [DOI] [PubMed] [Google Scholar]
- Rosoklija G, Toomayan G, Ellis SP, Keilp J, Mann JJ, Latov N, Hays AP, Dwork AJ (2000) Structural abnormalities of subicular dendrites in subjects with schizophrenia and mood disorders: preliminary findings. Arch Gen Psychiatry 57: 349-356. [DOI] [PubMed] [Google Scholar]
- Sailer M, Fischl B, Salat D, Tempelmann C, Schonfeld MA, Busa E, Bodammer N, Heinze HJ, Dale A (2003) Focal thinning of the cerebral cortex in multiple sclerosis. Brain 126: 1734-1744. [DOI] [PubMed] [Google Scholar]
- Sanchez C, Tompa P, Szucs K, Friedrich P, Avila J (1996) Phosphorylation and dephosphorylation in the proline-rich C-terminal domain of microtubule-associated protein 2. Eur J Biochem 241: 765-771. [DOI] [PubMed] [Google Scholar]
- Sanchez C, Diaz-Nido J, Avila J (2000) Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog Neurobiol 61: 133-168. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M (2002) AP-1 as a regulator of cell life and death. Nat Cell Biol 4: E131-E136. [DOI] [PubMed] [Google Scholar]
- Shepherd GM (1999) Electronic properties of axons and dendrites. In: Fundamental neuroscience (Zigmond MJ, Bloom FE, Landis SC, Roberts JL, Squire LR, eds), pp 107-127. Toronto: Academic.
- Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins from silver stained polyacrylamid gels. Anal Chem 68: 850-858. [DOI] [PubMed] [Google Scholar]
- Shoichet S (2004) Identification and characterisation of genes involved in cognitive function. Dissertation http://www.diss.fu-berlin.de/2004/218/indexe.html.
- Silliman CC, Sturgill TW (1989) Phosphorylation of microtubule-associated protein 2 by MAP kinase primarily involves the projection domain. Biochem Biophys Res Commun 160: 993-998. [DOI] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM (2002) Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol 156: 1051-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuyama S, Terayama Y, Matsuyama S (1987) Numerous phosphates of microtubule-associated protein 2 in living rat brain. J Biol Chem 262: 10886-10892. [PubMed] [Google Scholar]
- Vaillant AR, Zanassi P, Walsh GS, Aumont A, Alonso A, Miller FD (2002) Signaling mechanisms underlying reversible, activity-dependent dendrite formation. Neuron 34: 985-998. [DOI] [PubMed] [Google Scholar]
- Waetzig V, Herdegen T (2003) The concerted signaling of ERK1/2 and JNKs is essential for PC12 cell neuritogenesis and converges at the level of target proteins. Mol Cell Neurosci 24: 238-249. [DOI] [PubMed] [Google Scholar]
- Weston CR, Wong A, Hall JP, Goad ME, Flavell RA, Davis RJ (2003) JNK initiates a cytokine cascade that causes Pax2 expression and closure of the optic fissure. Genes Dev 17: 1271-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitford KL, Dijkhuizen P, Polleux F, Ghosh A (2002) Molecular control of cortical dendrite development. Annu Rev Neurosci 25: 127-149. [DOI] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW (2001) Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat Neurosci 4: 151-158. [DOI] [PubMed] [Google Scholar]
- Xia Y, Karin M (2004) The control of cell motility and epithelial morphogenesis by Jun kinases. Trends Cell Biol 14: 94-101. [DOI] [PubMed] [Google Scholar]
- Xu X, Raber J, Yang D, Su B, Mucke L (1997) Dynamic regulation of c-Jun N-terminal kinase activity in mouse brain by environmental stimuli. Proc Natl Acad Sci USA 94: 12655-12660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Hastie CJ, McLauchlan H, Cohen P, Goedert M (2004) Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK). J Neurochem 90: 352-358. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY (2003) Postnatal neurodevelopmental disorders: meeting at the synapse? Science 302: 826-830. [DOI] [PubMed] [Google Scholar]