

Abstract
Neuronal cell death occurs during many neurodegenerative disorders and stroke. The aberrant, excessive activity of matrix metalloproteinases (MMPs), especially MMP-9, contributes directly to neuron apoptosis and brain damage (Rosenberg et al., 1996; Asahi et al., 2001; Gu et al., 2002; Horstmann et al., 2003). We determined that MMP-9 degrades the extracellular matrix protein laminin and that this degradation induces neuronal apoptosis in a transient focal cerebral ischemia model in mice. We also determined that the highly specific thiirane gelatinase inhibitor SB-3CT blocks MMP-9 activity, including MMP-9-mediated laminin cleavage, thus rescuing neurons from apoptosis. We conclude that MMP-9 is a highly promising drug target and that SB-3CT derivatives have significant therapeutic potential in stroke patients.
Keywords: matrix metalloproteinases, thiirane inhibitor, laminin, proteolysis, neurons, apoptosis
Introduction
Matrix metalloproteinases (MMPs) constitute a family of extracellular soluble or membrane-bound proteases that collectively can degrade or proteolytically modify essentially all of the extracellular matrix (ECM) main components, including collagens, laminin, and proteoglycans. A role of MMPs, MMP-9 in particular, has been suggested in the pathogenesis of neurologic disorders, including stroke (Rosenberg et al., 1996; Yong et al., 2001). MMP-9 is significantly elevated in humans after stroke (Castellanos et al., 2003; Horstmann et al., 2003), and MMP-2 levels have been reported to be acutely increased in the brains of baboons after stroke (Heo et al., 1999). Our group demonstrated recently that a novel extracellular proteolytic cascade, in which S-nitrosylation (transfer of nitric oxide to a critical cysteine thiol group) and subsequent oxidation activates MMP-9 during cerebral ischemia, contributes to cortical neuronal apoptosis (Gu et al., 2002). Mice deficient in MMP-9 manifest a smaller cerebral infarct size; in addition, treatment with broad-spectrum MMP inhibitors or antibodies also reduces infarct size and prevents blood-brain barrier breakdown (Romanic et al., 1998; Asahi et al., 2001). Unfortunately, these broad-spectrum MMP inhibitors have significant systemic negative side effects.
Stroke ranks as the third leading cause of death in the United States. The only approved medical treatment for stroke is the administration of intravenous recombinant tissue plasminogen activator (tPA) with in 3 h of stroke onset to restore cerebral blood flow (CBF) (National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995). The efficacy of tPA, however, is limited by its side effects, which include neurotoxicity and thrombolysis-associated hemorrhagic transformation (Tsirka et al., 1995; Wang et al., 1998; Castellanos et al., 2003). A recent report indicates that tPA also upregulates MMP-9 in the brain and that the subsequent matrix degradation contributes to brain damage (Wang et al., 2003).
In the past, efforts to ameliorate MMP-mediated brain damage with broad-spectrum MMP inhibitors (like sulfonamide or hydroxamate derivatives) have produced promising results in animal models of stroke (Asahi et al., 2000; Lapchak et al., 2000). However, the low specificity, and thus systemic toxicity, of hydroxamates and sulfonamides have precluded the use of these broad-spectrum inhibitors in clinical studies (Coussens et al., 2002; Overall and Lopez-Otin, 2002). These previous MMP inhibitors were designed to provide a bidentate (double coordinating) chelating ligand to the MMP catalytic zinc and thus are incapable of discriminating among the members of the MMP family and selectively targeting individual MMPs. Recently, we described a fundamentally different, “suicide type” thiirane MMP inhibitor, designated SB-3CT. SB-3CT is the first mechanism-based MMP inhibitor that is selective for the gelatinases MMP-2 and MMP-9 (Brown et al., 2000). SB-3CT coordinates the catalytic zinc ion, contributing to both slow binding and mechanism-based inhibition. This suicide type of inhibition is unique among MMP inhibitors developed to date (Brown et al., 2000; Kleifeld et al., 2001).
We took advantage of the specificity and selectivity of SB-3CT to determine whether it could protect against brain damage in a mouse model of stroke without causing negative systemic side effects and further examined the molecular consequences of inhibiting MMP-9 proteolysis in the brain.
Materials and Methods
In vivo transient focal cerebral ischemia. Following a protocol approved by The Burnham Institute Animal Usage Committee, C57BL/6J mice weighing 25-30 g were housed in a 12 h light/dark cycle and permitted food and water intake ad libitum. These mice were subjected to transient focal cerebral ischemia under isoflurane anesthesia using a 6.0 monofilament suture to intraluminally occlude the right middle cerebral artery (MCA) for 2 h. The filament was then removed for reperfusion (R) for 24 h (Wang et al., 1998; Gu et al., 2002). A laser Doppler flowmeter (Perimed, North Royalton, OH) with the probe fixed on the skull surface (3 mm lateral to midline and 2 mm posterior to the bregma), located at the distal arterial supply of the middle cerebral artery, measured regional CBF (rCBF), as described previously (Wang et al., 1998). The initial reading of rCBF was assigned a value of 100%, and subsequent readings were expressed relative to this value. SB-3CT was designed as a highly selective, mechanism-based inhibitor to MMP-2 and MMP-9. The Ki values of MMP-2 and MMP-9 are in the nanomolar range, which are similar to the Ki values of endogenous TIMPs (tissue inhibitors of metalloproteinases). In contrast, the Ki values of SB-3CT against other MMPs (MMP-1, MMP-3, and MMP-7) are in the micromolar range (Brown et al., 2000). SB-3CT (25 mg/kg body weight) was injected intraperitoneally as a suspension in a vehicle solution (10% DMSO in normal saline). Mice were divided into four groups for administration of SB-3CT. One group was initially treated 30 min before ischemia, followed by a second injection immediately before reperfusion. The other three groups were treated at different time points after ischemia and received the first injection 2, 6, or 10 h after occlusion, followed by a second injection 3 h later. The control groups received only vehicle in each case. Mice were killed 24 h after reperfusion, and brains were dissected to prepare unfixed tissue OCT blocks for an in situ MMP gelatinolytic activity assay or storage at -80°C for later analysis. Infarct volumes were quantified with NIH Image software (version 1.62) on 1.0-mm-thick coronal sections stained with 2,3,5-triphenyltetrazolium chloride (TTC) (Wang et al., 1998). To minimize the effect of brain edema, the infarct volume was determined by subtracting the volume of the contralateral noninfarcted hemisphere (left) from the ipsilateral hemisphere (right). The right femoral artery was cannulated to monitor blood pressure and sample arterial blood gases and glucose. Arterial blood pressure was continually recorded before ischemia, during ischemia, and at reperfusion with a blood-pressure transducer, a bridge amplifier, and a computerized data acquisition system (MacLabs 8s; ADInstruments, Castle Hill, New South Wales, Australia). Arterial blood gases and glucose were measured before ischemia and 15 min after reperfusion with a blood gas and glucose analyzer (Stat Profile Ultra C; Nova Biomedical, Waltham, MA).
Assessment of neurological deficits. After the MCA is occluded, an animal usually exhibits marked thorax twisting when suspended by the tail. Contralateral forepaw flexure is also usually observed. We assessed the severity of this neurological deficit (referred to as postural asymmetry) using a five-point scale (Wang et al., 1998; Asahi et al., 2001): 0, no postural asymmetry (animal reaches down with both forepaws); 1, failure to extend the contralateral forepaw; 2, twisting the entire body toward the contralateral side; 3, falling to the contralateral side; 4, inability to walk spontaneously.
Gelatin zymography and in situ zymography. The concentration and activation of MMP-9 or MMP-2 in brain homogenates were determined by gelatin zymography (Zhang and Gottschall, 1997; Gu et al., 2002). Brain tissues were homogenized in Tris-buffered saline (TBS), pH 7.6, containing 5 mm CaCl2, 150 mm NaCl, 0.05% Brij 35, 0.02% NaN3, 1% Triton X-100, 100 μm PMSF, and a protein inhibitor cocktail (PIC; Roche Diagnostics, Mannheim, Germany) and centrifuged at 10,000 × g for 30 min. Aliquots of the supernatant, containing 1.5 mg of protein, were subjected to affinity precipitation with gelatin-conjugated Sepharose beads (Gelatin Sepharose 4B; Amersham Biosciences, Piscataway, NJ). The bound material was released from the beads in 1% SDS, and the samples were analyzed by electrophoresis in a 10% gelatin zymogram gel (Invitrogen, Carlsbad, CA). For in situ zymography, 8-μm-thick sections were cut from OCT-embedded fresh mouse brain and incubated overnight at 37°C with 10 μg/ml DQ-gelatin-FITC (DQ-gel; Molecular Probes, Eugene, OR) in TBS (Gu et al., 2002).
Ex vivo laminin degradation and Western blots. Brain lysates (50 mg of total protein in TBS with 0.05% Brij 35) were incubated overnight at 37°C with activated MMP-9, latent proform of MMP-9 (proMMP-9), or the catalytic domain of membrane type 1 (MT1)-MMP. The digested samples were subjected to electrophoresis on a bis-Tris 4-12% acrylamide gradient gel (Invitrogen). Laminin and MMP-9 were analyzed by immunoblotting using rabbit pan-laminin (pan-Ln) and MMP-9 antibodies [from Sigma (St. Louis, MO) and Calbiochem (La Jolla, CA), respectively], followed by peroxidase-conjugated anti-rabbit IgG antibody. Peroxidase was visualized by enhanced chemiluminescence (Amersham Biosciences).
Immunohistochemistry. Brain sections were immunostained with antibodies to NeuN (a well known neuronal marker; Chemicon, Temecula, CA), pan-Ln, laminin α-2 (generated by Dr. Eva Engvall, The Burnham Institute, La Jolla, CA), and α-5 (from Dr. Jeffrey Minor, Washington University, St. Louis, MO) and visualized with fluorescent chromatin-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) (Indyk et al., 2003).
Apoptosis detection. After intracardiac perfusion with 4% paraformaldehyde, brains were dissected, 16 μm coronal sections were cut, and apoptotic-like cell nuclei were detected by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL; Roche Diagnostics) and Hoechst dye 33342 (Sigma) to identify characteristic condensed apoptotic bodies (Gu et al., 2002).
Intracortical infusions. Mice were anesthetized as described above and placed in a stereotaxic apparatus. An Alzet micro-osmotic pump (Durect, Cupertino, CA), containing 100 μl of 1% BSA in PBS, normal rabbit preimmune serum, or affinity-purified rabbit pan-Ln antibodies (0.25 mg/ml in 1% BSA/PBS; Sigma) was then placed subcutaneously on the backs of the animals (Chen and Strickland, 1997). A brain infusion cannula connected to the pump was positioned at the following coordinates: bregma, 1.0 mm; mediolateral, 1.5 mm; and dorsoventral, 1.6 mm. The infusion rate was 0.5 μl/h, and the pump was allowed to infuse the designated solution for 2 d before MCA occlusion (MCAO). The mice were killed 1 d after MCA occlusion/reperfusion; the brains were processed for histochemical staining with cresyl violet and acid fuchsin and assessed for neuronal survival.
Results
SB-3CT protects against brain damage and ameliorates neurological deficits after transient focal cerebral ischemia
As a model of focal ischemia in C57BL/6 mice, we occluded the right middle cerebral artery for 2 h, followed by 24 h of reperfusion (MCAO/R) (Wang et al., 1998). To ensure successful placement of the intraluminal suture for occlusion and subsequent reperfusion, we monitored the rCBF in the area of the right middle cerebral artery in all animals. All mice subjected to MCAO/R met the criteria that the rCBF was reduced to <25% of the baseline during ischemia and recovered to >50% of the baseline during reperfusion (Fig. 1A). We measured infarct volume by staining brain sections with TTC. Animals treated with vehicle alone (10% DSMO in normal saline) developed extensive brain damage in the ischemic cortex, the striatum, and the hippocampus (Fig. 1B). The mean volume of the infarction was 55% of the total hemisphere after accounting for brain edema (Wang et al., 1998). Cresyl violet staining and Fluoro-Jade B staining, which are specific for degenerating cells, confirmed neuronal cell death in the ischemic region (data not shown). Both preischemic and 2 h postischemic injections of SB-3CT reduced infarct volume to ∼30% of control. Administration of this thiirane MMP inhibitor 6 h after ischemia was still potently protective histologically, with an infarct volume of <40% compared with 55% in vehicle-treated animals, thereby providing significant protection against brain ischemia (Fig. 1C). We also evaluated the severity of the neurobehavioral deficits of these mice 24 h after reperfusion, quantifying our findings using a five-point scale (see Materials and Methods) (Asahi et al., 2001). Mice treated with SB-3CT either before ischemia or 2 h after ischemia manifested significant improvement in behavioral scores compared with vehicle-treated controls (Fig. 1D). Treatment with SB-3CT did not affect physiological parameters including arterial blood gases and glucose compared with vehicle-treated controls (Table 1).
Figure 1.

Thiirane inhibitor SB-3CT protects against brain damage and ameliorates neurological outcome after transient focal cerebral ischemia in mice. A, Laser Doppler flowmetry of rCBF. rCBF was measured over the ischemic core of the right MCA region, and the preischemic rCBF was assigned a value of 100% at baseline. For each mouse, rCBF decreased to < 25% of the baseline value during a 2 h period of ischemia and recovered to >50% of baseline during reperfusion. There were no significant differences in rCBF between vehicle-treated controls (solid squares; n = 11) and SB-3CT-treated animals (solid triangles represent mean ± SEM; n = 13). B, Representative TTC staining of stroke in mouse-brain sections after SB-3CT treatment versus vehicle-treated control (Vehicle). SB-3CT (25 mg/kg body weight per treatment) was administrated intraperitoneally as a suspension in a vehicle solution (10% DMSO in saline). SB-3CT was administered in four groups plus parallel vehicle-treated control groups: a preischemic group treated 0.5 h before insult (-0.5 h) and groups treated 2, 6, or 10 h after ischemia (labeled +2, +6, and +10 h; see Materials and Methods). Coronal sections, 1 mm in thickness, were prepared and stained with TTC. C, Quantification of infarct volume by TTC staining. Infarct volumes were determined 24 h after reperfusion. SB-3CT decreased infarct volume compared with vehicle-treated controls (Ctrl). Data represent mean ± SEM. The numbers of animals in each group are as follows: Ctrl, n = 19; SB-3CT, n = 23. *p < 0.001 by ANOVA. D, Neurological behavioral score (see Materials and Methods) 24 h after MCA occlusion/reperfusion. Treatment with SB-3CT (n = 31) significantly improved neurological function compared with vehicle-treated controls (n = 22); *p < 0.02 by ANOVA. Error bars represent SEM.
Table 1.
Physiological variables
|
|
Before MCAO |
During MCAO |
After MCAO |
||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
Vehicle |
SB-3CT |
Vehicle |
SB-3CT |
Vehicle |
SB-3CT |
|||
| MABP (mmHg) | 76.17 ± 4.65 | 78.68 ± 3.17 | 76.22 ± 4.90 | 79.49 ± 3.48 | 76.91 ± 4.43 | 76.12 ± 2.89 | |||
| pH | 7.274 ± 0.020 | 7.313 ± 0.060 | 7.177 ± 0.078 | 7.106 ± 0.025 | 7.139 ± 0.078 | 7.170 ± 0.027 | |||
| PCO2 (mmHg) | 37.27 ± 1.28 | 40.70 ± 7.828 | 50.50 ± 11.33 | 44.95 ± 14.55 | 51.93 ± 10.58 | 43.90 ± 5.28 | |||
| PO2 (mmHg) | 252.8 ± 6.7 | 230.8 ± 31.7 | 259.2 ± 21.9 | 214.5 ± 10.3 | 242.5 ± 6.0 | 245.4 ± 24.0 | |||
| Na+ (mmol/L) | 154.9 ± 1.1 | 155.7 ± 1.8 | 158.4 ± 2.4 | 157.20 ± 0.5 | 158.7 ± 2.6 | 157.7 ± 2.0 | |||
| Ca2+ (mmol/L) | 1.113 ± 0.003 | 1.150 ± 0.021 | 1.133 ± 0.014 | 1.117 ± 0.081 | 1.157 ± 0.023 | 1.100 ± 0.074 | |||
| Glucose (mg/dl) |
147.7 ± 7.5 |
160.7 ± 13.2 |
148.0 ± 11.5 |
162.70 ± 13.3 |
147.7 ± 11.7 |
154.3 ± 12.6 |
|||
Note that we observed no significant changes in the physiological values of blood gases and electrolytes in animals with or without MCAO. Blood was drawn 5 min before ischemia, 15 min after ischemia, and 15 min after reperfusion. Data are mean ± SEM (n = 5). MABP Mean arterial blood pressure.
SB-3CT inhibits MMP-9 activity after transient focal cerebral ischemia
To determine whether administration of SB-3CT inhibited total gelatinolytic MMP activity after ischemia, we used in situ zymography together with Hoechst dye counterstaining in the mouse stroke model. We compared gelatinolytic MMP activity in the ischemic cortex of samples treated with the broad-spectrum MMP inhibitors 1,10-phenanthroline or GM6001 (Ilomastat; Chemicon) versus control. In the ischemic cortex, the broad-spectrum MMP inhibitors abrogated gelatinolytic MMP activity. In contrast, a non-MMP PIC had no effect on gelatinolytic activity (Fig. 2A). Deconvolution microscopy revealed that SB-3CT also significantly reduced the gelatinolytic activity in the ischemic region compared with vehicle-treated controls (Fig. 2B). Consistent with these findings, gelatin zymography and Western blot assays demonstrated that the levels of active MMP-9 significantly decreased in ischemic brains after SB-3CT treatment (Fig. 2C-D). As noted previously, proMMP-9 levels increased after stroke (Rosenberg et al., 1996; Romanic et al., 1998; Asahi et al., 2000; Gu et al., 2002). Intriguingly, the level of proMMP-9 also decreased after SB-3CT treatment. The reason is readily explained by a positive feedback mechanism that links MMP-9 activity to the efficacy of MMP-9 gene transcription, with low levels of active MMP-9 resulting in less transcription of proMMP-9 (Opdenakker et al., 2001). Under our conditions, we did not see changes in MMP-2 levels after stroke in these rodents (Gu et al., 2002).
Figure 2.
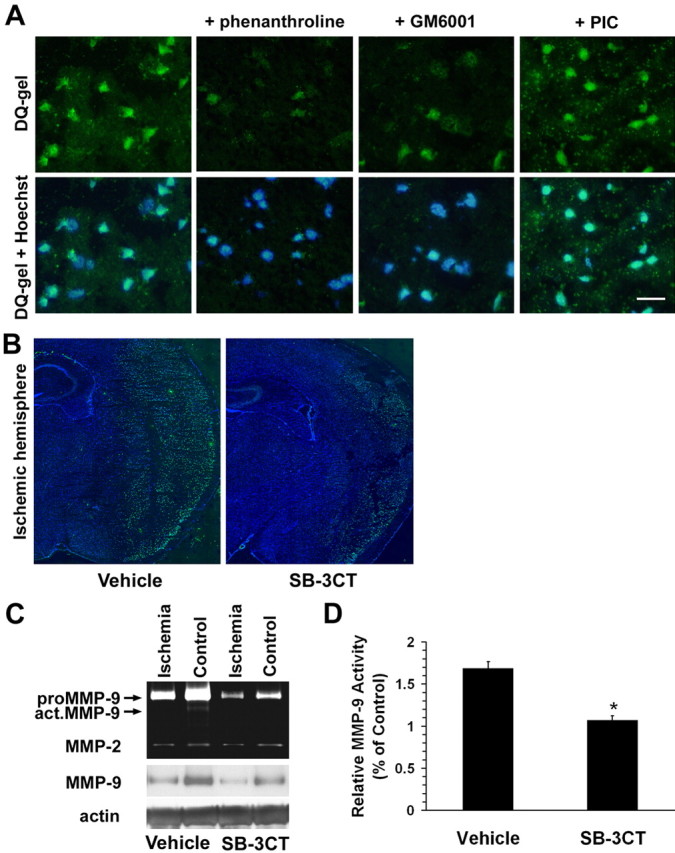
Thiirane inhibitor SB-3CT inhibits MMP-9 activity and consequent increased expression of MMP-9 in the ischemic mouse brain after transient middle cerebral artery occlusion. A, In situ zymography with the MMP fluorogenic substrate DQ-gel (green in top panels) merged with nuclear DNA staining by Hoechst dye (blue plus green in bottom panels). The broad-spectrum MMP inhibitors 1,10-phenanthroline and GM6001, but not a non-MMP PIC, abrogated MMP gelatinolytic activity in the ischemic cortex after MCA occlusion/reperfusion. Scale bar, 25 μm. B, SB-3CT significantly reduced MMP gelatinolytic activity in the ischemic region compared with the vehicle-treated control, as demonstrated by deconvolution microscopy. C, Gelatin zymography and Western blotting reveal upregulation of proMMP-9 (92 kDa) and activation of MMP-9 (act.MMP-9) in the ischemic brain compared with the contralateral hemisphere. In contrast, MMP-2 was not affected. SB-3CT attenuated the increase in proMMP-9 and act.MMP-9. Actin was used as a loading control. D, Quantification of relative MMP-9 activity by densitometry of gelatin zymography. Vehicle, n = 8; SB-3CT, n = 6; *p < 0.0001. Error bars represent SEM.
Increased MMP gelatinolytic activity is spatially associated with neuronal laminin after transient focal cerebral ischemia
We showed that activated MMP-9 directly induces neuronal apoptosis both in vitro and in vivo after focal cerebral ischemia/reperfusion (Gu et al., 2002). An additional recent report states that MMP-9 contributes to delayed neuronal cell death in the hippocampus after transient global ischemia (Lee et al., 2004). Although several reports (Asahi et al., 2001; Castellanos et al., 2003; Chen et al., 2003; Hamann, 2003; Horstmann et al., 2003) suggest that basement membrane proteins are involved in an MMP-9 proteolytic pathway, it is unclear how MMP substrates contribute to neuronal cell death. To identify potential targets of MMP-9 proteolysis in ischemic cortex, we examined several basement membrane proteins, including laminin, by immunohistochemistry. Pan-Ln antibody staining, followed by deconvolution microscopy, detected high immunoreactivity in proximity to NeuN-positive cells (Fig. 3, left panels), and microvascular structures. Excess laminin (purified from the mouse Engelbreth-Holm-Swarm sarcoma) completely blocked anti-Ln immunostaining in these brain sections (data not shown), indicating the high specificity of the pan-Ln immunoreactivity. We confirmed this neuronal laminin immunoreactivity by immunostaining with a polyclonal antibody against the neuron-associated α-5 laminin subunit (Fig. 3, top left, inset). Ln immunoreactivity was also detected in regions adjacent to laminin α-2 subunit-positive microvascular structures (data not shown). According to these immunostaining data and in situ zymography, gelatinolytic activity was primarily colocalized with Ln-positive neurons in the ischemic cortex, within 2 h of reperfusion (Fig. 3, right panels). These data suggest colocalization of MMP-9 activity with neuronal laminin in the early stages of brain damage after transient ischemia.
Figure 3.

Increased MMP gelatinolytic activity is spatially associated with neuronal laminin in the ischemic cortex of mouse brains after transient middle cerebral artery occlusion. Left panels, Double-immunofluorescent staining revealed two types of morphology, representing Ln (red) on elongated microvascular structures and on the neuronal surface (neurons labeled with the neuron-specific marker anti-NeuN; green). Top left, Inset, Immunolabeling of the α-5 subunit of laminin-10 (Lnα5), which is specific to neurons (Indyk et al., 2003). Right panels, Increased MMP gelatinolytic activity (DQ-gel; green) colocalized with laminin detected by immunostaining with a pan-Ln antibody (red) in the ischemic cortex 2 h after reperfusion. Bottom panels, Merged images counterstained with Hoechst dye to visualize nuclei (blue). Scale bar, 25 μm.
Exogenous MMP-9 degrades laminin in mouse brain
To corroborate these observations, we tested whether purified activated MMP-9 can cleave laminin. For this purpose, we coincubated activated MMP-9 with brain lysates and followed laminin cleavage by analyzing digested samples with SDS-PAGE and Western blotting. We found that MMP-9, in a dose-dependent manner, cleaved laminin subunits (Fig. 4A, top bands) to generate a 51 kDa fragment. As controls, latent proMMP-9 or catalytically active MT1-MMP did not degrade laminin (Fig. 4A,B). A broad-spectrum MMP inhibitor, GM6001, unlike a cocktail of non-MMP protease inhibitors, inhibited MMP-9 proteolysis of neuronal laminin (Fig. 4B). Together, the data indicate that MMP-9 can cleave laminin on the neuronal surface.
Figure 4.

Exogenous MMP-9 degrades lamininin the extracellular matrix protein of mouse brain. A, Western blot with a pan-Ln polyclonal antibody reveals degradation of laminin (especially the 360 and 170 kDa subunits) to a 51 kDa fragment (frag.) in brain lysates treated with activated MMP-9 but not with latent proMMP-9 or catalytic MT1-MMP (50 μg of total protein per lane). Purified mouse Engelbreth-Holm-Swarm laminin (ms EHS Ln) served as a molecular marker for laminin immunoblotting. The membrane was reblotted with anti-actin antibody to ensure equal protein loading in each lane. B, Ex vivo degradation of neuronal laminin by exogenous MMP-9 in mouse-brain sections. Double immunolabeling of laminin by pan-Ln polyclonal antibody (Ln; red) and neurons with NeuN antibody (green) reveals that activated MMP-9 degraded neuronal laminin. A broad-spectrum MMP inhibitor, GM6001, significantly reduced laminin degradation, whereas a non-MMP PIC did not. Latent proMMP-9 or catalytic MT1-MMP could not degrade neuronal laminin. Merged images were counterstained with Hoechst dye to visualize nuclei (blue). Scale bar, 25 μm.
MMP-9 activation is essential for degradation of laminin after transient focal cerebral ischemia
Previously, we reported (Gu et al., 2002) that S-nitrosylation of MMP-9 leads to its activation and that increased MMP-9 activity in the ischemic cortex does not occur in neuronal NOS (nNOS) knock-out (KO) mice or in wild-type mice treated with the specific nNOS inhibitor 3-bromo-7-nitroindazole. Here, we demonstrated blockage of laminin degradation in the ischemic cortex during stroke in nNOS KO mice and in wild-type mice treated with the specific nNOS inhibitor (Fig. 5). These new results provide evidence for a link between MMP-9 activation by S-nitrosylation and laminin cleavage.
Figure 5.

NO-activated MMP-9 leads to laminin degradation in the ischemic cortex after MCA occlusion/reperfusion. A, Laminin immunoreactivity (red) and Hoechst DNA stain (blue). Deconvolution microscopy revealed that laminin immunoreactivity was significantly reduced in the ischemic cortex of wild-type mice (top right) compared with the contralateral nonischemic control hemisphere (top left). Laminin degradation in the ischemic cortex was attenuated after MCA occlusion/reperfusion in either wild-type mice treated with the specific nNOS inhibitor 3-bromo-7-nitroindazole (3br7NI; bottom left) or in nNOS KO mice (bottom right). Scale bar, 25 μm. B, Quantification of Ln-positive cells in cortex was determined 24 h after reperfusion. Data represent mean ± SEM on 600-1000 cells counted from each brain section (n = 5 in each group; *p < 0.001 compared with control group and #p < 0.001 compared with ischemic group by ANOVA). Error bars represent SEM.
Increased MMP gelatinolytic activity induces laminin degradation before apoptotic cell death in the ischemic cortex
We examined the time course of laminin degradation and apoptotic cell death in the ischemic cortex. Increased gelatinolytic activity was associated with apoptotic cells 24 h after reperfusion (Fig. 6A-C). We also found decreased laminin immunoreactivity with moderate cell death 3 h after reperfusion and increasing apoptotic cell death 24 h later (Fig. 6D-I). These results indicate that laminin degradation in the ischemic cortex occurs before cell death and after MMP activation.
Figure 6.

Time course of laminin degradation and apoptotic cell death in the ischemic cortex after transient MCAO/R in mice. A-C, In situ zymography reveals that increased MMP gelatinolytic activity (A; green) is associated with apoptotic cell death detected by TUNEL (B; red). Merged images were counterstained with Hoechst dye to visualize nuclei (C; blue). D-F, After 2 h focal cerebral ischemia plus 3 h reperfusion (E) or 24 h reperfusion (F), animals were killed, and the brains were processed for immunohistochemistry. Coronal brain sections were stained for laminin immunoreactivity (red) and nuclear DNA staining with Hoechst dye (blue). D, Immunostaining with anti-pan-Ln antibody (red; indicated by arrowheads) was decreased in the ischemic cortex as early as 3 h after reperfusion compared with the control contralateral cortex. The remaining laminin was mostly associated with elongated microvascular structures rather than neurons. Modest neuronal cell death occurred at 3 h but more massive death at 24 h (arrows), as evidenced by condensed nuclei. G, Quantification of Ln-positive and apoptotic cells in the cortex 24 h after reperfusion. Data represent mean ± SEM on 600-1000 cells counted from each brain section; n = 5 in each group; *p < 0.001 by ANOVA compared with Ln-positive cells in the control contralateral hemisphere (Ctrl; filled bar), and #p < 0.001 compared with apoptotic cells in the control contralateral hemisphere (Ctrl; open bar). H, I, Coronal brain sections were stained for laminin immunoreactivity (green) and TUNEL (red) to demonstrate the reduction in laminin and increase in apoptosis in the ischemic cortex (I) compared with the contralateral control cortex (H). Brain sections were counterstained with Hoechst dye to show nuclei (blue). Together, the data in this figure suggest that MMP-induced laminin degradation occurs before neuronal apoptotic-like cell death. Scale bar, 25 μm. Error bars represent SEM.
SB-3CT attenuates laminin degradation after transient focal cerebral ischemia
We next examined the effects of the specific MMP-9/MMP-2 inhibitor SB-3CT on MMP-mediated proteolysis of laminin in vivo in the mouse stroke model. Western blots of brain extracts demonstrated partial degradation of laminin to a 51 kDa fragment in the ischemic hemisphere after transient MCAO (Fig. 7). Degradation of laminin and formation of the 51 kDa laminin fragment were reduced in the ischemic hemisphere of mice injected with SB-3CT, indicating that specific gelatinase inhibition significantly protected laminin from proteolysis.
Figure 7.

SB-3CT attenuates laminin degradation in the ischemic hemisphere after MCAO/R. Western blot demonstrates laminin proteolysis (especially of the 360 and 170 kDa subunits) to a 51 kDa fragment in the ischemic brain (arrowhead at bottom of gel), whereas treatment with SB-3CT decreased laminin degradation after transient MCAO/R. The ∼60 kDa fragment may represent an additional proteolytic derivative of the γ subunit (bottom molecular band) lacking NH2-terminal residues, as reported previously (Giannelli et al., 1997). The membrane was reprobed with anti-actin antibody to ensure equal loading.
Disruption of laminin-cell surface interactions with anti-laminin antibody increases neuronal death in the ischemic mouse brain treated with SB-3CT
To determine causality between laminin degradation and neuronal cell death in vivo, we infused a pan-laminin antibody into the brain. If laminin degradation contributes to neuronal cell death, this procedure should increase neuronal vulnerability during MCAO despite treatment with SB-3CT. First, we performed brain infusions with laminin antibody or control IgG into normal animals. We found no significant cellular damage, except for minor changes at the site of infusion (data not shown). However, if we infused anti-laminin antibody into the brains of ischemic animals treated with SB-3CT, we found significant damage and neuronal cell death in the ischemic regions, including the cortex, hippocampus, and striatum. This finding suggested that the anti-laminin antibody had abrogated the neuroprotective effect of SB-3CT; as a control, normal serum did not alter the protective effect of SB-3CT (Fig. 8).
Figure 8.

Disruption of laminin-cell surface interactions increases sensitivity to ischemic death. Mouse brains were infused with normal rabbit serum (IgG) or with a neutralizing antibody to pan-laminin (anti-Ln) in 1% BSA/PBS for 2 d before MCAO/R plus SB-3CT treatment or vehicle only. Brain sections were stained with cresyl violet and acid fuchsin. The dashed red line encircles the area of cell death. Pan-laminin antibody increased cell death in the MCAO/R mouse model despite SB-3CT treatment. This finding is consistent with the notion that the action of anti-laminin antibody is downstream to MMP-9 activation. Scale bar, 1 mm.
Discussion
MMPs have been implicated in the pathogenesis of brain injury after ischemia and a number of neurodegenerative disorders (Rosenberg et al., 1996; Yong et al., 2001). After various insults, MMPs, especially MMP-9 and MMP-2, are upregulated and lead to neuronal cell death and/or hemorrhagic consequences because of neurovascular matrix degradation (Heo et al., 1999; Asahi et al., 2001; Gu et al., 2002; Horstmann et al., 2003). In the mouse brain, MMP-9 appears to play a dominant role, because MMP-9 knock-out mice are relatively protected from ischemic and traumatic damage (Asahi et al., 2001). Broad-spectrum pharmacological inhibitors of MMPs significantly reduce brain damage after insults in animal models (Romanic et al., 1998; Asahi et al., 2000). However, previous human clinical trials with MMP inhibitors, representing hydroxamate derivatives, failed because of side effects attributed, at least in part, to their lack of specificity (Coussens et al., 2002; Overall and Lopez-Otin, 2002). Our results demonstrate that a new chemical class of MMP inhibitors, represented by thiirane derivative SB-3CT, potently decreases brain damage and can extend the window of therapeutic intervention to 6 h after insult in animal models of cerebral ischemia/reperfusion. This class of drugs represents a more specific form of MMP antagonist, targeting only MMP-9 and MMP-2, and appears to be well tolerated, at least in our animal models.
Additionally, our results show that the proteolysis of laminin by MMP-9 follows cerebral ischemia and contributes to neuronal apoptosis. These results build on our previous work that identified the molecular mechanism whereby nitric oxide (NO) activates MMP-9 in the brain (Gu et al., 2002). MMP-9 may arise from different cell types, including neutrophils and macrophages, which are known to migrate into the brain after damage to the blood-brain barrier because of ischemia/reperfusion injury (Yong et al., 2001). Although Lo and colleagues (Asahi et al., 2001) could not unambiguously demonstrate degradation of laminin in the ischemic brain, they could not rule out this possibility. Our results support a model in which production of proMMP-9, and subsequent activation by NO (Gu et al., 2002) or tPA (Zhao et al., 2004), leads to laminin cleavage and resulting neuronal cell death. In fact, the effect of MMP-9 is functionally similar to that of tPA, which is also known to contribute to laminin degradation (Chen and Strickland, 1997; Wang et al., 1998; Indyk et al., 2003). This point underscores the fact that tPA, which is frequently used as a clot buster and hence as a therapy for stroke, can also contribute to neuronal cell death (Wang et al., 1998). Nonetheless, specific inhibition of MMP-9 should be able to prevent the dire consequences of excessive activation of this gelatinase MMP either by NO or tPA.
Previously, it had been demonstrated that ECM proteins such as laminin are important for cell survival and prevention of apoptosis, representing a form of cell death known as anoikis, in which cells detach from their matrix (Frisch and Francis, 1994). If cells are prohibited from interacting with the ECM, their viability is thus impaired. The laminin antibody that we used disrupts cell-laminin interactions and can therefore contribute to neuronal cell death (Chen and Strickland, 1997). These data suggest that laminin serves as a cell-survival factor in this system. We used the anti-laminin neutralizing antibody in this case to show that the effect of laminin disruption was downstream to the action of the SB-3CT compound, because treatment with SB-3CT was unable to rescue neurons from damage initiated by the anti-laminin antibody.
In summary, we show that SB-3CT, a mechanism-based and selective gelatinase inhibitor, provides significant protection against brain damage in an experimental model of focal cerebral ischemia, consistent with a role for MMP-9 in laminin degradation and neuronal cell death in stroke. We conclude that targeting MMP-9 in stroke patients is a highly promising therapeutic approach that deserves additional exploration. The use of novel and selective MMP inhibitors such as SB-3CT or its derivatives, which exhibit selective inhibition of MMP-9/MMP-2 gelatinases, should minimize the undesired side effects caused by broad-spectrum MMP inhibitors in previous clinical trials. Our study is the first to demonstrate a significant pharmacological benefit of a new type of synthetic MMP inhibitor in an animal model of stroke. These results hold promise for the successful application of SB-3CT derivatives to stroke patients.
Footnotes
This work was supported in part by National Institutes of Health Grants EY05477, EY09024, NS44326, and HD29587 (S.A.L.), CA77470 (A.Y.S.), and CA100475 (R.F.). We thank Claire Gibson for technical support and Dr. Eva Engvall for helpful and valuable comments.
Correspondence should be addressed to Dr. Stuart A. Lipton, Center for Neuroscience and Aging, The Burnham Institute, La Jolla, CA 92037. E-mail: slipton@burnham.org.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256401-08$15.00/0
References
- Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH (2000) Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab 20: 1681-1689. [DOI] [PubMed] [Google Scholar]
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH (2001) Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci 21: 7724-7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S, Bernardo MM, Li ZH, Kotra LP, Tanaka Y, Fridman R, Mobashery S (2000) Potent and selective mechanism-based inhibition of gelatinases. J Am Chem Soc 122: 6799-6800. [Google Scholar]
- Castellanos M, Leira R, Serena J, Pumar JM, Lizasoain I, Castillo J, Davalos A (2003) Plasma metalloproteinase-9 concentration predicts hemorrhagic transformation in acute ischemic stroke. Stroke 34: 40-46. [PubMed] [Google Scholar]
- Chen ZL, Strickland S (1997) Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell 91: 917-925. [DOI] [PubMed] [Google Scholar]
- Chen ZL, Indyk JA, Strickland S (2003) The hippocampal laminin matrix is dynamic and critical for neuronal survival. Mol Biol Cell 14: 2665-2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Fingleton B, Matrisian LM (2002) Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295: 2387-2392. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Francis H (1994) Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol 124: 619-626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannelli G, Falk-Marzillier J, Schiraldi O, Stetler-Stevenson WG, Quaranta V (1997) Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science 277: 225-228. [DOI] [PubMed] [Google Scholar]
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA (2002) S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science 297: 1186-1190. [DOI] [PubMed] [Google Scholar]
- Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, del Zoppo GJ (1999) Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab 19: 624-633. [DOI] [PubMed] [Google Scholar]
- Horstmann S, Kalb P, Koziol J, Gardner H, Wagner S (2003) Profiles of matrix metalloproteinases, their inhibitors, and laminin in stroke patients: influence of different therapies. Stroke 34: 2165-2170. [DOI] [PubMed] [Google Scholar]
- Indyk JA, Chen ZL, Tsirka SE, Strickland S (2003) Laminin chain expression suggests that laminin-10 is a major isoform in the mouse hippocampus and is degraded by the tissue plasminogen activator/plasmin protease cascade during excitotoxic injury. Neuroscience 116: 359-371. [DOI] [PubMed] [Google Scholar]
- Kleifeld O, Kotra LP, Gervasi DC, Brown S, Bernardo MM, Fridman R, Mobashery S, Sagi I (2001) X-ray absorption studies of human matrix metalloproteinase-2 (MMP-2) bound to a highly selective mechanism-based inhibitor. Comparison with the latent and active forms of the enzyme. J Biol Chem 276: 17125-17131. [DOI] [PubMed] [Google Scholar]
- Lapchak PA, Chapman DF, Zivin JA (2000) Metalloproteinase inhibition reduces thrombolytic (tissue plasminogen activator)-induced hemorrhage after thromboembolic stroke. Stroke 31: 3034-3040. [DOI] [PubMed] [Google Scholar]
- Lee SR, Tsuji K, Lo EH (2004) Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J Neurosci 24: 671-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group (1995) Tissue plasminogen activator for acute ischemic stroke. N Engl J Med 333: 1581-1587. [DOI] [PubMed] [Google Scholar]
- Opdenakker G, Van den Steen PE, Van Damme J (2001) Gelatinase B: a tuner and amplifier of immune functions. Trends Immunol 22: 571-579. [DOI] [PubMed] [Google Scholar]
- Overall CM, Lopez-Otin C (2002) Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer 2: 657-672. [DOI] [PubMed] [Google Scholar]
- Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC (1998) Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 29: 1020-1030. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Navratil M, Barone F, Feuerstein G (1996) Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. J Cereb Blood Flow Metab 16: 360-366. [DOI] [PubMed] [Google Scholar]
- Tsirka SE, Gualandris A, Amaral DG, Strickland S (1995) Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature 377: 340-344. [DOI] [PubMed] [Google Scholar]
- Wang X, Lee SR, Arai K, Tsuji K, Rebeck GW, Lo EH (2003) Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat Med 9: 1313-1317. [DOI] [PubMed] [Google Scholar]
- Wang YF, Tsirka SE, Strickland S, Stieg PE, Soriano SG, Lipton SA (1998) Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med 4: 228-231. [DOI] [PubMed] [Google Scholar]
- Yong VW, Power C, Forsyth P, Edwards DR (2001) Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci 2: 502-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JW, Gottschall PE (1997) Zymographic measurement of gelatinase activity in brain tissue after detergent extraction and affinity-support purification. J Neurosci Methods 76: 15-20. [DOI] [PubMed] [Google Scholar]
- Zhao B-Q, Ikeda Y, Ihara H, Urano T, Fan W, Mikawa S, Suzuki Y, Kondo K, Sato K, Nagai N, Umemura K (2004) Essential role of endogenous tissue plasminogen activator through matrix metalloproteinase 9 induction and expression on heparin-produced cerebral hemorrhage after cerebral ischemia in mice. Blood 103: 2610-2616. [DOI] [PubMed] [Google Scholar]