

Abstract
PolyADP-ribosylation is a transient posttranslational modification of proteins, mainly catalyzed by poly(ADP-ribose)polymerase-1 (PARP-1). This highly conserved nuclear protein is activated rapidly in response to DNA nick formation and promotes a fast DNA repair. Here, we examine a possible association between polyADP-ribosylation and the activity of neurotrophins and neuroprotective peptides taking part in life-or-death decisions in mammalian neurons. The presented results indicate an alternative mode of PARP-1 activation in the absence of DNA damage by neurotrophin-induced signaling mechanisms. PARP-1 was activated in rat cerebral cortical neurons briefly exposed to NGF-related nerve growth factors and to the neuroprotective peptides NAP (the peptide NAPVSIPQ, derived from the activity-dependent neuroprotective protein ADNP) and ADNF-9 (the peptide SALLRSIPA, derived from the activity-dependent neurotrophic factor ADNF) In addition, polyADP-ribosylation was involved in the neurotrophic activity of NGF-induced and NAP-induced neurite outgrowth in differentiating pheochromocytoma 12 cells as well as in the neuroprotective activity of NAP in neurons treated with the Alzheimer's disease neurotoxin β-amyloid. A fast loosening of the highly condensed chromatin structure by polyADP-ribosylation of histone H1, which renders DNA accessible to transcription and repair, may underlie the role of polyADP-ribosylation in neurotrophic activity.
Keywords: PARP-1, polyADP-ribosylation, Ca2+ signaling, nerve growth factors, neuroprotective peptides, NAP, ADNF-9
Introduction
The development, survival, and function of the mammalian nervous system are widely influenced by the presence of neurotrophins (Chao, 1992; Thoenen, 1995; Bonhoeffer, 1996; Nakajima and Kohasaka, 1998; Riccio et al., 1999; Finkbeiner, 2000; Schinder and Poo, 2000; Huang and Reichardt, 2001; Segal, 2003). Nerve growth factor (NGF)-related neurotrophins activate two different receptor classes: the tropomyosin-related kinase (Trk) family of receptor tyrosine kinases and the p75 receptor, a member of the tumor necrosis factor receptor super-family (Kaplan and Miller, 2000; Patapoutian and Reichardt, 2001). Stimulation of Trk receptors by NGF-related neurotrophins promotes survival, differentiation, and synaptogenesis in the nervous system (Majdan, et al., 1997; Kaplan and Miller, 2000; Huang and Reichardt, 2003), whereas neurotrophin binding to p75 receptors mediates mainly apoptotic responses (Majdan et al., 1997; Kaplan and Miller, 2000; Barker, 2004). Binding of neurotrophins to Trk receptors (Schlessinger, 2000; Patapoutian and Reichardt, 2001; Huang and Reichardt, 2003) couples their autophosphorylation to a variety of signaling pathways (Pawson and Nash, 2000; Patapoutian and Reichardt, 2001; Huang and Reichardt, 2003). Known members of the NGF family of neurotrophins, NGF, brain-derived neurotrophic factor (BDNF), and neurotrophin-3 (NT-3), activate TrkA, TrkB, and mainly TrkC receptor tyrosine kinases, respectively (Barbacid, 1995; Kaplan and Miller, 2000; Patapoutian and Reichardt, 2001).
Alongside with neurotrophins, two recently discovered glial-derived peptides, ADNF-9 (the peptide SALLRSIPA, derived from the activity-dependent neurotrophic factor ADNF) and NAP (the peptide NAPVSIPQ, derived from the activity-dependent neuroprotective protein ADNP), express very potent neuroprotective activities (Brenneman et al., 1998; Gozes, 2001, Gozes et al., 2004; Hashimoto et al., 2004). These two structurally related peptides were derived by peptide activity scanning from two glial proteins, ADNF (Gozes and Brenneman, 1996; Brenneman et al., 1998) and activity-dependent neuroprotective protein (Bassan et al., 1999). Whereas NGF, BDNF, and NT-3 act via phosphorylation of Trk receptor tyrosine kinases, NAP and ADNF-9 may not need surface receptors and rather act like pore-forming peptides (Brenneman et al., 2004; Chiba et al., 2004; Divinski et al., 2004).
The modification of nuclear proteins by polyADP-ribosylation has been associated with life-or-death decisions as well (Ha and Snyder, 1999; Pieper et al., 2000; Chiarugi, 2002). PolyADP-ribosylation is catalyzed mainly by poly(ADP-ribose)polymerase-1 (PARP-1), an abundant highly conserved nuclear protein (Lautier et al., 1993; Amé et al., 2004). PolyADP-ribosylation is initiated by transferring nicotinamide-adenindinucleotide, di(tri-ethyl-ammonium) (NAD)-derived ADP-riboses to gluthamic and aspartic residues and proceeds by a fast and transient polymerization of ADP-riboses into large polymers (Lautier et al., 1993; Lindahl et al., 1995; Amé et al., 2000). These polymers interfere with the protein binding to DNA (Satoh et al., 1994; D'Amours et al., 1999; Amé et al., 2000), thereby modifying the chromatin structure and affecting DNA transcription and repair (Li Oei et al., 1998; D'Amours et al., 1999).
Recent data imply a possible role for PARP-1 and polyADP-ribosylation in the nervous system. PARP-1 activation was involved in gene expression underlying differentiation of neural stem cells (Ju et al., 2004). PARP-1 was activated by electrical stimulation of mammalian cortical neurons (Homburg et al., 2000) and by stimulations inducing long-term facilitation in sensory-to-motor neuron synapses of the marine snail Aplysia (Cohen-Armon et al., 2004). Here, we examine a possible involvement of polyADP-ribosylation in neurotrophic activity.
Materials and Methods
Materials. [Adenylate-32P]NAD (1000 Ci/mmol) and DNase-I (RNase free) were purchased from Amersham Biosciences (Piscataway, NJ). Ethane-N, N,N′,N′-tetraacetic acid tetrakis (acetoxymethyl) ester (BAPTA/AM) and rhod-2/AM were from Molecular Probes (Eugene, OR). Anti-human PARP-1 monoclonal antibody (MCA1522), anti-human histone H1 monoclonal antibody (catalog #05-457), and secondary antibodies were from Serotec (Oxford, UK). Polyclonal antibody directed against ADP-ribose polymers (anti-PAR) was from Alexis Biochemicals (San Diego, CA). The neurotrophins NGF, BDNF, and NT-3 were from Alomone Labs (Jerusalem, Israel). Tyrphostin AG879 [α-cyano-(3,5-di-t-butyl-4-hydroxy)thiocinnamide] was synthesized by Dr. A. Gazit in collaboration with Prof. A. Levitzky in The Hebrew University of Jerusalem (Jerusalem, Israel). The peptides NAP and ADNF-9 were obtained as described previously (Brenneman et al., 1998; Bassan et al., 1999; Leker et al., 2002). Phorbol 12-myristate, 13-acetate (PMA) was from Biomol (Plymouth Meeting, PA). General inhibitors of Ca2+-calmodulin-dependent kinases (CAMKs) and protein kinase C (PKC), respectively, 2-[N-(2-hydroxyethyl)-N-(4methoxybenzenesul-fonyl)]amino-N-(-4-chloro-cinnamyl)-N-methylbenzylamine (KN-93) and 3-[1(3-dimethyl-aminopropyl)-indo-3-yl]-3-(indol-3-yl)-maleimide, HCl (Bim-I), were from Calbiochem (Darmstadt, Germany). Dibutyryl cAMP (dbc-AMP) and the PARP-1 inhibitor 6(5H)-phenanthridinone (Phen.) were from Alexis Biochemicals. Antibodies directed against phosphorylated phospholipase C γ (PLCγ) were from Cell Signaling Technology (Beverly, MA). Other materials were from Sigma (St. Louis, MO).
Primary cultures of rat brain cortical neurons were prepared from rat brain cortex of 19- to 20-d-old embryos, as described previously (Homburg et al., 2000). Glial cells proliferation was blocked 2 d after plating. Experiments were performed a week after plating. Cells were stimulated for at least 5 min with nerve growth factors. For serum deprivation, the neurons were washed and incubated in serum-free Eagle's essential medium (MEM) for 24 h before testing the effect of nerve growth factors on PARP-1 activation.
Brain cortical slices. NGF-related nerve growth factors were examined in cortical slices as well. The effect of the neuroprotective peptides NAP and ADNF on PARP-1 activity was examined in cortical slices (Nakajima et al., 1998; Gozes et al., 2003). Brain cortical slices (200-300 μm) were washed thoroughly and incubated in Krebs'-Henseleit-buffered solution containing the following (mm): 118 NaCl, 4.7 KCl, 1.18 MgCl2, 24.9 NaHCO3, 10 glucose, 1.18 KH2PO4, pH 7.4, at 25°C, 95% O2/5% CO2, as described in detail previously (Cohen-Armon et al., 1996).
Neuronal survival assays with β-amyloid and NAP. Mixed neuronal and glial cultured cells derived from newborn rat cerebral cortex were used for the neuronal survival assay. The preparation of this cell culture was described previously (Bassan et al., 1999). Nine days after plating, β-amyloid (amino acids 25-35; 25 μm) (Gozes et al., 1996) was added to each plate together with increasing concentrations of NAP (Bassan et al., 1999). Neuronal survival was assayed after a 5 d incubation period. Cells were counted after treatment with glutaraldehyde (3% in cacodylate buffer, pH 7.2 for 2 h) (Brenneman et al., 1987; Zemlyak et al., 2000). Nerve cells were identified by staining with antibodies directed against neuron-specific enolase (Brenneman and Gozes, 1996).
Crude nuclei. Nuclei with perinuclear membranes were isolated from brain cortical slices or from cultured cortical cells, as described in detail previously (Homburg et al., 2000).
Activation of PARP-1 assayed by measuring its auto-polyADP-ribosylation and polyADP-ribosylation of nuclear proteins. Activated PARP-1 is automodified by polyADP-ribosylation (Lautier et al., 1993; D'Amours et al., 1999; Amé et al., 2000). PARP-1, like other DNA binding proteins, is a basic protein (Gorg, 1999). Negatively charged ADP-ribosyl moieties added to PARP-1 by polyADP-ribosylation shift its basic isoelectric point (pI) toward more acidic pH values (Lautier et al., 1993; Amé et al., 2000). The shift in the pI of PARP-1 was related to polyADP-ribosylation only when it was prevented by applying PARP inhibitors, 3-aminobenzamide (3-AB; 0.5-1 mm) (Rankin et al., 1989) and Phen. (25 μm) (Richardson et al., 1999). Shifts in pI were measured by two-dimensional gel electrophoresis. For the first dimension, we used Immobiline DryStrip kit (Amersham Biosciences, Uppsala, Sweden) (Gorg, 1999) and polyacrylamide slab gels in the second dimension. PolyADP-ribosylated proteins were detected by immunolabeling with antibody directed against ADP-ribose polymers (Shah et al., 1995). The relative amount of polyADP-ribosylated PARP-1 in each sample was estimated by densitometry (Cohen-Armon et al., 2004).
Simultaneous recording of rhod-2 fluorescence. This technique enabled tracing stimulation-induced Ca2+ mobilization in living cells. Cultured cortical neurons on polylysine-coated coverslips were loaded with the fluorescent Ca2+ indicator/chelator rhod-2/AM (10 μm, 30 min, 25°C, in the dark) and washed. Neurons were treated under a confocal microscope with agents inducing intracellular Ca2+ mobilization. The fluorescent signal of Ca2+-bound rhod-2 (excitation, 540 nm; emission, >570 nm) was collected through appropriate filters above 520 nm and monitored.
Single-strand DNA breaks examined by alkaline gel electrophoresis. This method provides sensitive, rapid, and direct quantitation of nicks in DNA single strands (Sutherland et al., 1999). DNA was isolated from the nuclei of cortical neurons or cortical slices using the Hirt procedure (Hirt, 1967). The migration rate of equivalent amounts of single-stranded DNA was measured by electrophoresis on 1% alkaline agarose gel (Sutherland et al., 1999). The DNA single strands, stained with ethidium bromide (1 μg/ml), are visualized under UV illumination.
Neurite outgrowth in rat pheochromocytoma 12 cells. Cells were grown in DMEM supplemented with 8% fetal calf serum, 8% horse donor serum, 2 mm glutamine, and 1% penicillin streptomycin solution for 48 h (Steingart et al., 2000; Das et al., 2004). When neurotrophic activity was tested, NGF (50 ng/ml) or NAP (10-12 m) was added after seeding. A possible involvement of polyADP-ribosylation in neurite outgrowth was tested after adding PARP-1 inhibitors, 3-AB (0.5-1 mm) or Phen. (20-25 μm). PARP-1 inhibitors were added 30 min before adding NGF or NAP.
Results
Changes in the activity of PARP-1 were measured in neurons treated by nerve growth factors. Rat cortical neurons and cortical slices were exposed to NGF-related nerve growth factors, NGF, BDNF, and NT-3. A marked increase in polyADP-ribosylated proteins was detected by confocal microscopy in cortical neurons in culture by immunolabeling with antibody directed against ADP-ribose polymers (see Materials and Methods) (Fig. 1a). Activation of PARP-1 in neurons treated by nerve growth factors was also assessed by its auto-polyADP-ribosylation (Lautier et al., 1993; Lindahl et al., 1995). We assayed the activity of PARP-1 by the shift in its pI, caused by the addition of negatively charged ADP-ribose moieties (see Materials and Methods) (Cohen-Armon et al., 2004). The shift in pI was prevented when the activity of PARP-1 was specifically suppressed (Fig. 1b,c). It was therefore attributed to auto-polyADP-ribosylation rather than to other possible modifications of the protein (see Materials and Methods) (Cohen-Armon et al., 2004). Results indicated activation of PARP-1 in cerebral cortical neurons and in cortical slices briefly exposed to NGF, BDNF, or NT-3 (Fig. 1b,c) and in cortical slices incubated with the neuroprotective peptides ADNF-9 and NAP (see Materials and Methods) (Fig. 1c). The percentage of polyADP-ribosylated PARP-1 in the samples was estimated by using densitometric scanning (Cohen-Armon et al., 2004). In NGF-treated neurons, the pI of PARP-1 was shifted in 37.7 ± 2.5% of the total amount of PARP-1 in the loaded samples. Treatment with BDNF, NT-3, NAP, and ADNF-9 also induced a shift in the pI of PARP-1; the pI of 35.8 ± 3.7, 32.4 ± 4.2, 33 ± 4.2, and 26 ± 2.1% of the total amount of PARP-1 in the loaded samples was shifted, respectively. In untreated neurons, the pI of only 7.± 0.5% of the total amount of PARP-1 in the loaded samples was shifted (Fig. 1b,c).
Figure 1.
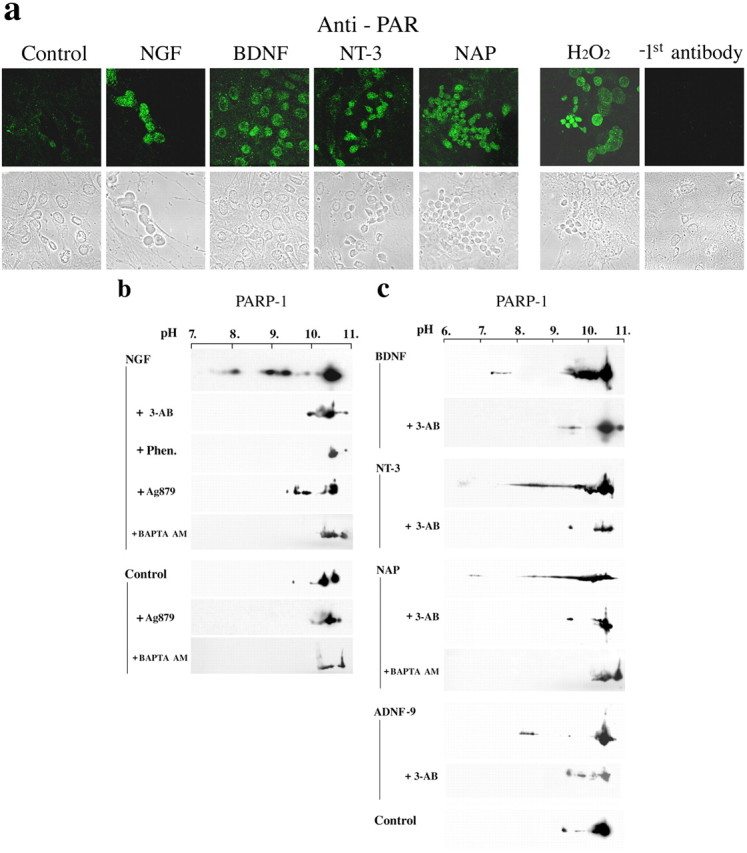
Activated PARP-1 and polyADP-ribosylated nuclear proteins in rat brain cortical neurons treated with nerve growth factors and neuroprotective peptides. a, PolyADP-ribosylated proteins in the nuclei of prefixed cultured embryonic rat cortical neurons immunolabeled in situ with anti-PAR. Before fixation, cells were exposed for 5 min to 100 ng/ml of the indicated nerve growth factors or to the neuropeptide NAP (10-12 m). PolyADP-ribosylated proteins in cells treated with H2O2 (1 mm, 5 min, 25°C), which causes nicks in DNA, were used as a positive control. Confocal images in randomly chosen fields show neuronal nuclei labeled with fluorescein-conjugated secondary antibody in the treated neurons (top). The cells were also visualized by phase contrast in transmitted light (bottom). Various cell types were immunolabeled by the different treatments (n = 3). b, PolyADP-ribosylation of active PARP-1 in nuclei isolated from brain cortical neurons treated as indicated. PolyADP-ribosylation of PARP-1 is shown by the shift in its pI toward lower pH values using two-dimensional gel electrophoresis. Brain cortical neurons were incubated for 5 min with 100 ng/ml NGF in a serum-depleted MEM (see Materials and Methods) (n = 5). Some of the samples were pretreated with tyrphostin Ag879 (100 μm, 10 min, 25°C; n = 3) or with the permeable Ca2+ chelator BAPTA/AM (10 μm, 30 min, in the dark; n = 3). The shift in pI was prevented by two inhibitors of PARP-1, 3-AB (0.5 mm; added 15 min before adding NGF) or Phen. (25 μm; added 30 min before adding NGF). Nuclear proteins were extracted by high salt (0.5 m NaCl, 30 min, 4°C), separated by two-dimensional gel electrophoresis, electrotransfered (Western blot), and immunolabeled for PARP-1 by anti-human PARP-1 antibody (MCA1522). c, Cortical neurons in culture were incubated for 5 min with 100 ng/ml BDNF or 100 ng/ml NT-3 in serum-depleted MEM (see Materials and Methods) (n = 3 for each sample). Brain cortical slices were incubated (5 min, 25°C, Krebs'-Henseleit 95% O2/5% CO2; n = 3) with NAP (10-15 m) or ANDF-9 (1 nm). The shift in pI toward acidic pH was prevented by inhibiting the activity of PARP-1 with 3-AB (0.5 mm) or after application of the Ca2+ chelator BAPTA/AM (10 μm, 30 min 25°C, in the dark). Nuclear proteins were extracted from the treated cortical neurons or slices as described in b.
PARP-1 activation in cortical neurons treated with NGF was suppressed by the tyrphostin AG879 (see Materials and Methods) (Fig. 1b). Tyrphostins, a family of tyrosine kinase blockers, selectively and competitively inhibit the autophosphorylation of receptor tyrosine kinases (Lyall et al., 1989; Ohmichi et al., 1992). Tyrphostin AG879 specifically inhibits NGF-induced autophosphorylation of TrkA receptors (Ohmichi et al., 1993). We examined signal transduction cascades that are triggered by Trk phosphorylation for their effect on PARP-1 activation.
Nerve growth factors inducing phosphorylation of receptor tyrosine kinases also induce phosphorylation of PLCγ-1 (Ohmichi et al., 1992, 1993). A possible pattern of PARP-1 activation by neurotrophins via activation of PLC and phosphatidylinositol 4,5-bisphosphate (PIP2) hydrolysis, forming diacylglycerol (DAG) and inositol 1,4,5 trisphosphate (IP3) (Huang and Reichardt, 2003), was therefore tested. Our previous results indicated a dose-dependent activation of PARP-1 by IP3 and Ca2+ applied to isolated nuclei (Homburg et al., 2000), suggesting the involvement of intracellular IP3-induced Ca2+ mobilization in the activation of PARP-1. Here, polyADP-ribosylation in neurons treated by nerve growth factors and by the neuroprotective peptides NAP and ADNF-9 was suppressed completely by the permeable Ca2+ chelator BAPTA/AM (Fig. 1b,c). Blocking Ca2+ channels and NMDA-dependent Ca2+ influx (Homburg et al., 2000) did not prevent the activation of PARP-1 in the treated neurons (data not shown). Intracellular Ca2+ mobilization in the treated cortical neurons was further assessed by confocal microscopy (Fig. 2). In these experiments, neurons were exposed to neurotrophic agents, and Ca2+ mobilization was traced by the fluorescent emission of Ca2+ bound to the fluorescent permeable Ca2+ chelator rhod-2/AM (see Materials and Methods). Results indicated Ca2+ release into the cytoplasm and nucleoplasm in the treated neurons (Fig. 2).
Figure 2.

Intracellular Ca2+ mobilization in neurons treated by nerve growth factors and NAP. Ca2+ mobilization into the cytoplasm and nucleoplasm of treated cortical neurons monitored by the fluorescence of Ca2+-bound rhod-2/AM (top). Cortical neurons were incubated (5 min, 25°C, in serum-depleted MEM) with NGF (100 ng/ml), NAP (1012 m), NT-3 (100 ng/ml), and BDNF (100 ng/ml) under a confocal microscope. Bottom, The same neurons visualized by phase contrast in transmitted light (n = 3).
Because PARP-1 was activated by extranuclear IP3 (Homburg et al., 2000), PLCγ-1 was phosphorylated in the treated neurons (Fig. 3a), and intracellular Ca2+ mobilization occurred (Fig. 2), we examined a possible involvement of Ca2+- and DAG-dependent activation of PKC (Poole et al., 2004) in PARP-1 activation by neurotrophins. Results show that PKC inhibitors suppressed PARP-1 activation in cortical slices treated by NGF-related growth factors (Fig. 3b). Bim-I, a general inhibitor of PKC (Toullec et al., 1991), and Gő6976 [12-(2-cyanoethyl)- 6,7,12,13-tetrahydro-13-methyl-S-oxo-5H-indolo(2,3a)pyrrolo (3,4-c)-carbazole], which mainly suppresses the activity of Ca2+-dependent PKC isozymes (Martiny-Baron et al., 1993), also suppressed PARP-1 activation in neurons treated by NGF-related nerve growth factors and NAP (Fig. 3b). In support, PARP-1 was activated when PKC activity was enhanced by a short treatment with phorbol esther (PMA) (Gerwien et al., 1998; Schlessinger, 2000) (Fig. 3b).
Figure 3.
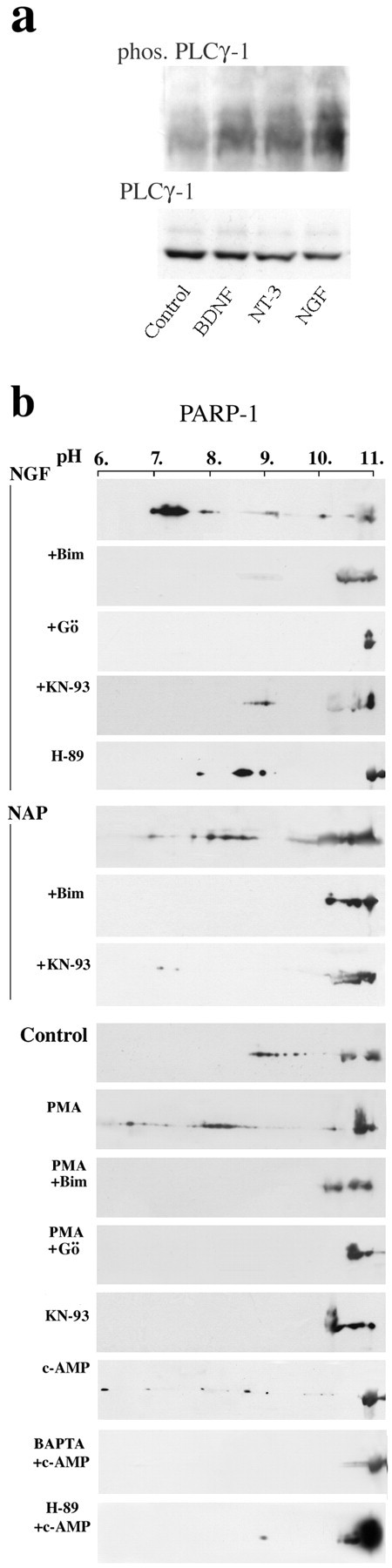
Mediators affecting neurotrophin induced PARP-1 activation. a, Phosphorylation of PLCγ-1 in rat brain cortical neurons in response to stimulation by NGF-related nerve growth factors. Immunolabeling of PLCγ-1 and phosphorylated PLCγ-1 (phos. PLCγ-1; on Tyr783; see Materials and Methods) are presented. b, Changes in polyADP-ribosylation of PARP-1 assayed by the shift in its isoelectric point. PARP-1 was activated in brain cortical slices after a 5 min incubation (at room temperature, 5% O2, 95% CO2) with NGF (100 ng/ml) or NAP (10-15 m), by cAMP (150 μm dbc-AMP), and by PKC activation after a 30 min incubation of cortical slices with PMA (200 nm). NGF-induced PARP-1 activation was suppressed by blockers of PKC [1 μm Bim-I (Bim) or 0.5 μm Gő6976 (Gö)] and by a blocker of CAMK (10 μm KN-93) added 40 min before stimulation with NGF or NAP. cAMP-induced PARP-1 activation was suppressed by the PKA inhibitor H-89 (20 μm; applied for 40 min before stimulation). Nuclei were isolated from cortical slices treated by the indicated compounds. Nuclear proteins were extracted and analyzed by two-dimensional gel electrophoresis. Immunolabeled PARP-1 (MCA1522) in the nuclear protein extracts is displayed for each treatment (n = 3 for each treatment).
PARP-1 activation in NGF-treated cortical slices was not prevented by N-[2-(Bromocinnamylamino)ethyl]-5-isoquinoline-sulfonamide (H-89), which acts as a PKA inhibitor (Kavalali et al., 1997; Wong et al., 2004) (Fig. 3b). However, PARP-1 was activated by the permeable cAMP dbc-AMP (Fig. 3b), and this effect was suppressed by H-89 (Fig. 3b). cAMP promotes Ca2+ release from internal stores including IP3-gated Ca2+ stores as a result of PKA activation (Cameron et al., 1995; Haug et al., 1999; DeSouza et al., 2002). Because PARP-1 was activated by extranuclear IP3 and Ca2+ (Homburg et al., 2000), cAMP-induced PARP-1 activation could be attributed to PKA-induced Ca2+ leakage from IP3-gated Ca2+ stores. However, other mechanisms may operate as well.
A possible involvement of CAMKs in the activation of PARP-1 was tested by suppressing the activity of CAMK with KN-93, a general inhibitor of CAMKs (Corcoran and Means, 2000) (Fig. 3b). NGF-induced PARP-1 activation and NAP-induced PARP-1 activation were suppressed in the presence of KN-93 (Fig. 3b). Because recent findings challenge the exclusive effect of KN-93 on CAMK activity (Smyth et al., 2002), the effect of KN-93 on neurotrophin-induced PARP-1 activation could involve other mechanisms as well. However, it should be noted that Ju et al. (2004) have recently reported evidence for PARP-1 activation by stimulation of CAMK-IIδ in differentiating neural stem cells.
None of the nerve growth factors and neuroprotective peptides produced nicks in single-stranded DNA prepared from the treated neurons (see Materials and Methods) (Fig. 4). Thus, a possible activation of PARP-1 by DNA damage (de Murcia et al., 1994; Lindahl et al., 1995) as a result of treatment with neurotrophins may be excluded.
Figure 4.

Single stranded DNA examined for nick formation in neurons treated by nerve growth factors and NAP. Alkaline gel electrophoresis of DNA was used to examine nicks in single-stranded DNA. DNA was extracted from nuclei of rat brain cortical neurons, untreated (lane 1), or incubated (5 min, 25°C) with 100 ng/ml NGF, BDNF, or NT-3 and with 10-12 m NAP, as indicated. As a positive control for nicked DNA, DNA was treated with DNase I. Isolated nuclei of cortical neurons were incubated for 5 min at 37°C with DNase I (10 U; RNase free; D7291; Sigma). Single-stranded DNA in the samples was stained with ethidium bromide (1 μg/ml) and photographed under UV illumination. MW, Molecular weight.
In view of PARP-1 activation in neurons treated by nerve growth factors and neuroprotective peptides (Figs. 1, 3), we examined the possibility that polyADP-ribosylation is involved in their neurotrophic activity. It has been reported previously that NGF-treated pheochromocytoma 12 (PC12) cells cease proliferation, extend neurites, and become electrically excitable (Tischler et al., 1983; Das et al., 2004). To examine whether polyADP-ribosylation is necessary for NGF-induced neurite outgrowth in PC12 cells (see Materials and Methods), we examined morphological changes as well as levels of microtubule-associated protein 2 (MAP2), which is expressed during differentiation (Sanchez et al., 2000). After 48 h of incubation with NGF (50 ng/ml) or NAP (10-12 m), we found neurite outgrowth and MAP2 expression in the PC12 cells (Fig. 5a). In randomly chosen fields, neurite outgrowth was observed in 55 of 103 NGF-treated cells. When the cells were treated by NAP, neurite outgrowth was observed in 57 of 134 counted cells. In untreated cells, neurite outgrowth was observed in 16 of 124 counted cells. PARP-1 was activated in the NGF-treated cells as long as they were exposed to the nerve growth factor (Fig. 5b). When the activity of PARP-1 was suppressed immediately after platting by application of 6(5H)-phenanthridinone or 3-AB, neurite outgrowth was dramatically reduced. Neurite outgrowth was observed in only four neurite-bearing cells of 118 cells treated by NGF and only two neurite-bearing cells, of 104 cells treated with NAP. These results suggest a possible role of poly-ADP-ribosylation in neurotrophin-induced differentiation of PC12 cells.
Figure 5.

PolyADP-ribosylation is involved in neurite outgrowth in rat PC12 cells. a, Neurite outgrowth in PC12 cells after 48 h of incubation with NGF (50 ng/ml) or NAP (10-12 m) in the presence or absence of PARP-1 inhibitor (3-AB; 1 mm). Top, Confocal microscope images are presented, indicating morphological changes and fluorescent immunolabeling of MAP2 in the fixed cells. Bottom, Cells were also visualized by phase contrast in transmitted light (n = 3). Neurite outgrowth induced by NGF (50 ng/ml) or by NAP (10-12 m) was prevented by the PARP-1 inhibitors 3-AB (0.5 mm) or Phen. (25 μm). b, PARP-1 was activated in PC12 cells exposed for 5 min, 1 h, or 24 h to NGF. PolyADP-ribosylated proteins were immunolabeled by anti-PAR. PolyADP-ribosylation was suppressed in the presence of Phen. (25 μm). Confocal images indicate polyADP-ribosylated proteins in the neuronal nuclei labeled with fluorescein-conjugated secondary antibody (n = 3).
PolyADP-ribosylation was also involved in neuroprotection by NAP. NAP exhibits a potent neuroprotective effect (Gozes et al., 2003). Here, the protective activity of NAP was tested in cultured cortical neurons treated with the Alzheimer's disease neurotoxin β-amyloid peptide (Bassan et al., 1999) (see Materials and Methods). Results showed that the treatment with β-amyloid reduced by 48 ± 3% (n = 3) the number of cultured cortical neurons. In accordance with previous findings, NAP at a very low concentration (10-16-10-15 m) protected some of the nerve cells from death (Bassan et al., 1999). However, suppressing the activity of PARP-1 completely prevented the protective effect of NAP; 68 ± 7% (n = 3; p < 0.05) of the β-amyloid treated neurons survived in the presence of 10-16 m NAP, whereas only 47 ± 4% (n = 3) of the neurons survived when polyADP-ribosylation was suppressed by 0.5 mm 3-AB during the treatment. PARP-1 inhibition by itself had no significant effect on cell survival in the β-amyloid-treated neurons under these conditions. Thus, according to these results, polyADP-ribosylation was involved in neuroprotection by NAP in the β-amyloid-treated neurons.
Linker histone H1 was polyADP-ribosylated along with PARP-1 in neurons treated by NGF-related nerve growth factors or by NAP (Fig. 6). PolyADP-ribosylation of histone H1, one of the prominent substrates of PARP-1 (Buki et al., 1995; Rouleau et al., 2004), causes a transient shuttling of H1 in and out of its binding site on the nucleosome (Kraus and Lis, 2003; Rouleau et al., 2004; Kim et al., 2004). Therefore, polyADP-ribosylation of H1 destabilizes the nucleosomal structure (Boulikas, 1990; Boulikas, 1993; Kim et al., 2004), resulting in a transient loosening of the highly condensed structure of chromatin (D'Amours et al., 1999; Rouleau et al., 2004). This renders DNA accessible to repair and transcription factors (Kraus and Lis, 2003), thereby enabling a fast gene expression and DNA repair. Neurotrophin-induced PARP-1 activation (Figs. 1, 3) could therefore promote neurotrophic effects as a result of polyADP-ribosylation of histone H1 (Fig. 6) and chromatin relaxation.
Figure 6.
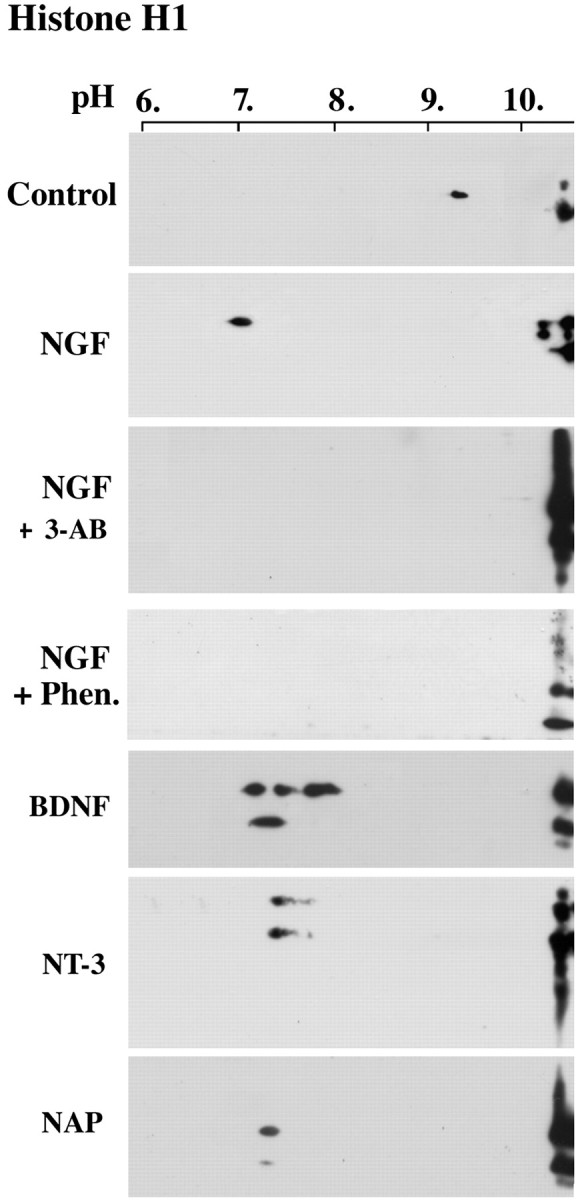
PolyADP-ribosylation of linker histone H1 in neurons treated by nerve growth factors and NAP. PolyADP-ribosylation of histone H1 in nuclei isolated from cortical neurons treated by neurotrophins, as indicated. PolyADP-ribosylation was assayed by the shift in the pI of H1 toward acidic pH values. The shift in pI was prevented by the PARP-1 inhibitors 3-AB (0.5 mm) and Phen. (25 μm). Brain cortical neurons were incubated for 5 min with 100 ng/ml NGF, BDNF, NT-3, or 10-12 m NAP (n=3 for each treatment). Histone H1 was labeled by monoclonal anti-human H1 antibody (Upstate Biotechnology, Lake Placid, NY) on Western blots of nuclear proteins separated by two-dimensional gel electrophoresis (see Materials and Methods).
Discussion
Our findings outline a mechanism for PARP-1 activation by nerve growth factors and by the neuroprotective peptides NAP and ADNF-9 (Figs. 1, 3 and supplemental material, available at www.jneurosci.org). Although PARP-1 activation by damaged DNA has been well documented (D'Amours et al., 1999), these findings present an alternative mode of PARP-1 activation, which is not mediated by a measurable damage in DNA (Fig. 4). This suggested mechanism complies with recent data by Kim et al. (2004), presenting evidence for PARP-1 activation when it is bound to intact polynucleosomes.
The findings, along with previous results indicating IP3-induced PARP-1 activation in isolated nuclei (Homburg et al., 2000), outline an alternative mode of PARP-1 activation downstream to signal transduction cascades initiated by stimulation of Trk receptor tyrosine kinases, activation of PLCγ, PIP2 hydrolysis forming DAG (diacylglycerol) and IP3 (Berridge and Irvine, 1989), IP3-induced Ca2+ mobilization (Mikoshiba, 1997), and activation of Ca2+ dependent kinases (Schlessinger, 2000) (see supplemental material, available at www.jneurosci.org).
PARP-1 auto-polyADP-ribosylation in neurons stimulated by NGF-related nerve growth factors was suppressed by capturing free intracellular Ca2+ (Fig. 1b,c) and by inhibiting the activity of Ca2+-dependent kinases (Fig. 3b). Activation of PARP-1 by NGF-related nerve growth factors is therefore consistent with our previous findings indicating a dose-dependent PARP-1 activation by IP3 and Ca2+ applied to isolated nuclei of cortical neurons (Homburg et al., 2000). This may involve IP3-induced Ca2+ mobilization from perinuclear stores into the nucleoplasm (Malviya and Rogue, 1998). Ca2+-induced PARP-1 activation has been observed recently in a cell-free system (Kun et al., 2004). Other members of the PARP family (Amé et al., 2004) might be similarly activated by Ca2+ and therefore may take part in Ca2+-dependent PARP-1 activation and in polyADP-ribosylation of nuclear proteins.
Ca2+ release from internal stores could also activate PARP-1 as a result of activation of Ca2+-dependent kinases. Agents modulating the activity of PKC and CAMK affected neurotrophin-induced PARP-1 activation (Fig. 3b), thereby suggesting a possible involvement of these Ca2+-dependent kinases in neurotrophin-induced PARP-1 activation (supplemental material, available at www.jneurosci.org).
PARP-1 activation by neurotrophins may be necessary for their activity. PolyADP-ribosylation was involved in neuroprotection by NAP as well as in NGF-induced and NAP-induced differentiation of PC12 cells. Cole and Perez-Polo (2004) recently showed that PARP-1 inhibitors delay cell death of differentiated PC12 cells under oxidative stress. Interestingly, here, in the absence of stress conditions, inhibition of PARP-1 prevented neurite outgrowth in PC12 cells treated by NGF or NAP (Fig. 5a).
The involvement of polyADP-ribosylation in the neurotrophic effects may be attributed to polyADP-ribosylation of linker histone H1 (Fig. 6). The role of polyADP-ribosylation induced chromatin relaxation in DNA transcription has been demonstrated recently in the giant chromosomes of Drosophila larva salivary glands (Tulin and Spradling, 2003). PolyADP-ribosylation of linker histone H1 may underlie this effect (Kraus and Lis, 2003; Rouleau et al., 2004). PolyADP-ribosylation of histone H1 by activated PARP-1 has been demonstrated in a variety of cell types and in cell-free system (Buki et al., 1995; D'Amours et al., 1999; Rouleau et al., 2004). Although recent data demonstrated different binding sites of PARP-1 and histone H1 on the giant chromosome of Drosophila (Kim et al., 2004), an interaction between these two proteins may still be possible because of the dynamic structure of chromatin (Jenuwein and Allis, 2001; Harrer et al., 2004).
Neurotrophin-induced polyADP-ribosylation of H1, rapidly rendering the DNA accessible to transcription factors and repair enzymes needed for differentiation and cell survival (Kraus and Lis, 2003; Tulin and Spradling, 2003; Rouleau et al., 2004), may take part in neurite outgrowth in PC12 cells treated by NGF or NAP (Fig. 5) as well as in neuroprotection by NGF-related nerve growth factors and NAP (Nakajima and Kohasaka, 1998; Riccio et al., 1999; Sofroniew et al., 2001; Pinhasov et al., 2003; Gozes et al., 2004).
Neurotrophin-induced PARP-1 activation may also polyADP-ribosylate other DNA-binding proteins affecting chromatin structure and DNA transcription. This may involve polyADP-ribosylation of high-mobility group proteins (Tanuma et al., 1986), core histones, histone acetyl transferases, and deacetylases (D'Amours et al., 1999; Kraus and Lis, 2003; Rouleau et al., 2004).
Recently, we associated long-term facilitation in sensory-to-motor synapses of the marine mollusk Aplysia with PARP-1 activation in sensory ganglia of trained Aplysia (Cohen-Armon et al., 2004). Moreover, polyADP-ribosylation was essential during training for the formation of long-term memory in the animal (Cohen-Armon et al., 2004). This may be attributed to a fast and transient chromatin relaxation (Cohen-Armon et al., 2004), enabling a fast gene expression, needed for long-term memory formation (Kandel, 2001). A possible PARP-1 activation by secretion of nerve growth factors during neuronal activity (Chao, 1992; Klintsova and Greenough, 1999; Blondel et al., 2000; Huang and Reichardt, 2001; Keyvani and Schallert, 2002) may take part similarly in long-term memory formation promoted by NGF and BDNF (Schinder and Poo, 2000; Mizuno et al., 2003; Koponen et al., 2004).
Footnotes
This work was supported by the Israel Science Foundation, the Israeli National Institute for Psychobiology, Israel Ministry of Health, The United States-Israel Binational Science Foundation, the Lily and Avraham Gildor Chair for the Investigation of Growth Factors-Tel-Aviv University, the Dr. Diana and Zelman Elton Laboratory for Molecular Neuroendocrinology, and Allon Therapeutics
Correspondence should be addressed to Dr. Malka Cohen-Armon, The Neufeld Cardiac Research Institute, Sackler School of Medicine, Tel-Aviv University, Tel-Aviv 69978, Israel. E-mail: marmon@post.tau.ac.il.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257420-09$15.00/0
References
- Amé J-C, Jacobson EL, Jacobson MK (2000) ADP-ribose polymers metabolism. In: From DNA damage and stress signaling to cell death: polyADP-ribosylation reactions (de Murcia G, Shall S, eds), pp 3-8. Oxford: Oxford UP.
- Amé J-C, Spenlehauer C, de Murcia G (2004) The PARP superfamily. BioEssays 26: 882-893. [DOI] [PubMed] [Google Scholar]
- Barbacid M (1995) Neurotrophic factors and their receptors. Curr Opin Cell Biol 7: 148-155. [DOI] [PubMed] [Google Scholar]
- Barker PA (2004) p75NTR is positively promiscuous. Neuron 42: 529-533. [DOI] [PubMed] [Google Scholar]
- Bassan M, Zamostiano R, Davidson A, Pinhasov A, Giladi E, Perl O, Bassan H, Blatt C, Gibney G, Glazner G, Brenneman DE, Gozes I (1999) Complete cDNA sequence of a novel protein containing a femtomolar-activity-dependent neuroprotective peptide. J Neurochem 72: 1283-1293. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Irvine RF (1989) Inositol phosphates and cell signaling. Nature 341: 197-204. [DOI] [PubMed] [Google Scholar]
- Blondel O, Collin O, McCarran B, Zhu S, Zamostiano R, Gozes I, Brenneman DE, McKay RD (2000) A glia-derived signal regulating neuronal differentiation. J Neurosci 20: 8012-8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhoeffer T (1996) Neurotrophins and activity-dependent development of the neocortex. Curr Opin Neurobiol 6: 119-126. [DOI] [PubMed] [Google Scholar]
- Boulikas T (1990) Poly(ADP-ribosylated) histones in chromatin replication. J Biol Chem 24: 14638-14647. [PubMed] [Google Scholar]
- Boulikas T (1993) Poly(ADP-ribosyl)ation, DNA strand breaks, chromatin and cancer. Toxicol Lett 67: 129-150. [DOI] [PubMed] [Google Scholar]
- Brenneman DE, Gozes I (1996) A femtomolar-acting neuroprotective peptide. J Clin Invest 97: 2299-2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneman DE, Neale EA, Foster GA, D'autremont SW, Westbrook GL (1987) Nonneuronal cells mediate neurotrophic action of vasoactive intestinal peptide. J Cell Biol 104: 1603-1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneman DE, Hauser J, Neale E, Rubinraut S, Fridkin M, Davidson A, Gozes I (1998) Activity-dependent neurotrophic factor: structure-activity relationships of femtomolar-acting peptides. J Pharmacol Exp Ther 285: 619-627. [PubMed] [Google Scholar]
- Brenneman DE, Spong CY, Hauser JM, Abebe D, Pinhasov A, Golian T, Gozes I (2004) Protective peptides that are orally active and mechanistically nonchiral. J Pharmacol Exp Ther 309: 1190-1197. [DOI] [PubMed] [Google Scholar]
- Buki KG, Bauer PI, Hakam A, Kun E (1995) Identification of domains of poly(ADP-ribose)polymerase for protein binding and self association. J Biol Chem 270: 3370-3377. [DOI] [PubMed] [Google Scholar]
- Cameron AM, Steiner JP, Roskams AJ, Ali SM, Ronnett GV, Snyder SH (1995) Calcineurin associated with the inositol 1,4,5-trisphosphate receptor: FKBP12 complex modulates Ca+2 flux. Cell 83: 463-472. [DOI] [PubMed] [Google Scholar]
- Chao MV (1992) Neurotrophin receptors: a window into neuronal differentiation. Neuron 9: 583-593. [DOI] [PubMed] [Google Scholar]
- Chiarugi A (2002) Poly(ADP-ribose) polymerase: killer or conspirator? The `suicide hypothesis' revisited. Trends Pharmacol Sci 23: 122-129. [DOI] [PubMed] [Google Scholar]
- Chiba T, Hashimoto Y, Tajima H, Yamada M, Kato R, Niikura T, Terashita K, Schulman H, Aiso S, Kita Y, Matsuoka M, Nishimoto I (2004) Neuroprotective effect of activity-dependent neurotrophic factor against toxicity from familial amyotrophic lateral sclerosis-linked mutant SOD1 in vitro and in vivo. J Neurosci Res 78: 542-552. [DOI] [PubMed] [Google Scholar]
- Cohen-Armon M, Hammel I, Anis Y, Homburg S, Dekel N (1996) Evidence for endogenous ADP-ribosylation of GTP-binding proteins in neuronal cell nucleus: possible induction by membrane depolarization. J Biol Chem 271: 26200-26208. [DOI] [PubMed] [Google Scholar]
- Cohen-Armon M, Visochek L, Katzoff A, Levitan D, Susswein AJ, Klein R, Valbrun M, Schwartz JH (2004) Long-term memory requires polyADP-ribosylation. Science 304: 1820-1822. [DOI] [PubMed] [Google Scholar]
- Cole K, Perez-Polo R (2004) Neuronal trauma model: in search of Thanatos. Int J Dev Neurosci 22: 485-496. [DOI] [PubMed] [Google Scholar]
- Corcoran EE, Means AR (2000) Defining Ca2+/calmodulin-dependent protein kinase cascades in transcriptional regulation. J Biol Chem 276: 2975-2978. [DOI] [PubMed] [Google Scholar]
- D'Amours D, Desnoyers S, D'Silva I, Poirier GG (1999) Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 342: 249-268. [PMC free article] [PubMed] [Google Scholar]
- Das KP, Freudenrich TM, Mundy WR (2004) Assessment of PC12 cell differentiation and neurite growth: a comparison of morphological and neurochemical measures. Neurotoxicol Teratol 26: 397-406. [DOI] [PubMed] [Google Scholar]
- de Murcia G, Schreiber V, Molinete M, Saulier B, Poch O, Masson M, Niedergang C, de Murcia JM (1994) Structure and function of poly(ADP-ribose)polymerase. Mol Cell Biochem 138: 15-24. [DOI] [PubMed] [Google Scholar]
- DeSouza N, Reiken S, Ondrias K, Yang YM, Matkovich S, Marks AR (2002) Protein kinase A and two phosphatases are components of the inositol 1,4,5-trisphosphate receptor macromolecular signaling complex. J Biol Chem 277: 39397-39400. [DOI] [PubMed] [Google Scholar]
- Divinski I, Mittelman L, Gozes I (2004) A femtomolar acting octapeptide interacts with tubulin and protects astrocytes against zinc intoxication. J Biol Chem 279: 28531-28538. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S (2000) CREB couples neurotrophin signals to survival messages. Neuron 25: 11-14. [DOI] [PubMed] [Google Scholar]
- Gerwien J, Kaltoft K, Nielsen M, Nielsen MB, Svejgaard A, Geisler C, Ropke C, Odum N (1998) Staphylococcus enterotoxin A modulates interleukin 15-induced signaling and mitogenesis in human T cells. Tissue Antigens 51: 164-173. [DOI] [PubMed] [Google Scholar]
- Gorg A (1999) IPG-dalt of very alkaline proteins. In: 2-D proteome analysis protocols (Link AJ, ed), pp 197-210. Totowa, NJ: Humana. [DOI] [PubMed]
- Gozes I (2001) Neuroprotective peptide drug delivery and development: potential new therapeutics. Trends Neurosci 24: 700-705. [DOI] [PubMed] [Google Scholar]
- Gozes I, Brenneman DE (1996) Activity-dependent neurotrophic factor (ADNF). An extracellular neuroprotective chaperonin. J Mol Neurosci 7: 235-244. [DOI] [PubMed] [Google Scholar]
- Gozes I, Bardea A, Reshef A, Zamostiano R, Zhukovsky S, Rubinraut S, Fridkin M, Brenneman DE (1996) Neuroprotective strategy for Alzheimer disease: intranasal administration of a fatty neuropeptide. Proc Natl Acad Sci USA 93: 427-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozes I, Divinsky I, Pilzer I, Fridkin M, Brenneman DE, Spier AD (2003) From vasoactive intestinal peptide (VIP) through activity-dependent neuroprotective protein (ADNP) to NAP: a view of neuroprotection and cell division. J Mol Neurosci 20: 315-322. [DOI] [PubMed] [Google Scholar]
- Gozes I, Steingart RA, Spier AD (2004) NAP mechanisms of neuroprotection. J Mol Neurosci 24: 67-72. [DOI] [PubMed] [Google Scholar]
- Ha HC, Snyder SH (1999) Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci USA 69: 13978-13982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrer M, Luhrs H, Bustin M, Scheer U, Hock R (2004) Dynamic interaction of HMGA1a proteins with chromatin. J Cell Sci 117: 3459-3471. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Kaneko Y, Tsukamoto E, Frankowski H, Kouyama K, Kita Y, Niikura T, Aiso S, Bredesen DE, Matsuoka M, Nishimoto I (2004) Molecular characterization of neurohybrid cell death induced by Alzheimer's amyloid-beta peptides via p75NTR/PLAIDD. J Neurochem 90: 549-558. [DOI] [PubMed] [Google Scholar]
- Haug LS, Jensen V, Hvalby O, Walaas SI, Ostvold AC (1999) Phosphorylation of the inositol 1,4,5-trisphosphate receptor by cyclic nucleotide-dependent kinases in vitro and in rat cerebellar slices in situ. J Biol Chem 274: 7467-7473. [DOI] [PubMed] [Google Scholar]
- Hirt B (1967) Selective extraction of polyoma DNA from infected mouse cell culture. J Mol Biol 26: 365-369. [DOI] [PubMed] [Google Scholar]
- Homburg S, Visochek L, Moran N, Dantzer F, Priel E, Asculai E, Schwartz D, Rotter V, Dekel N, Cohen-Armon M (2000) A fast signal-induced activation of poly(ADP-ribose)polymerase: a novel downstream target of phospholipase C. J Cell Biol 150: 293-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24: 295-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF (2003) Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 72: 609-642. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074-1080. [DOI] [PubMed] [Google Scholar]
- Ju B-G, Solum D, Song EJ, Lee K-J, Rose DW, Glass CK, Rosenfeld MG (2004) Activating the PARP-1 sensor component of the Groucho/TLE1 corepressor complex mediates a CAMKinase IIδ-dependent neurogenic gene activation pathway. Cell 119: 815-829. [DOI] [PubMed] [Google Scholar]
- Kandel ER (2001) The molecular biology of memory storage: a dialogue between genes and synapses. Science 294: 1030-1038. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD (2000) Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10: 381-391. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Hwang KS, Plummer MR (1997) cAMP-dependent enhancement of dihydropyridine-sensitive calcium channel availability in hippocampal neurons. J Neurosci 17: 5334-5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyvani K, Schallert T (2002) Plasticity-associated molecular and structural events in the injured brain. J Neuropathol Exp Neurol 61: 831-840. [DOI] [PubMed] [Google Scholar]
- Kim M-Y, Mauro S, Gevry N, Lis J-T, Kraus L (2004) NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell 119: 803-814. [DOI] [PubMed] [Google Scholar]
- Klintsova AY, Greenough WT (1999) Synaptic plasticity in cortical systems. Curr Opin Neurobiol 9: 203-208. [DOI] [PubMed] [Google Scholar]
- Koponen E, Vôikar V, Riekki R, Saarelainen T, Rauramaa T, Rauvala H, Taira T, Castrén T (2004) Transgenic mice overexpressing the full-length neurotrophin receptor TrkB exhibit increased activation of the TrkB-PLCγ pathway, reduced anxiety, and facilitated learning. Mol Cell Neurosci 26: 166-181. [DOI] [PubMed] [Google Scholar]
- Kraus LW, Lis TJ (2003) PARP goes transcription. Cell 113: 677-683. [DOI] [PubMed] [Google Scholar]
- Kun E, Kirsten E, Mendeleyev J, Ordahl CP (2004) Regulation of the enzymatic catalysis of poly(ADP-ribose) polymerase by dsDNA, polyamines, Mg+2, Ca+2, histone H1 and H3 and ATP. Biochemistry 43: 210-216. [DOI] [PubMed] [Google Scholar]
- Lautier D, Lagueux J, Thibodeau J, Menard L, Poirier GG (1993) Molecular and biochemical features of poly(ADP-ribose) metabolism. Mol Cell Biochem 122: 171-193. [DOI] [PubMed] [Google Scholar]
- Leker RR, Teichner A, Nussen R, Grigoriadis N, Cohen Y, Ovadia H, Brenemann DE, Fridkin M, Giladi E, Romano J, Gozes I (2002) NAP, a femtomolar-acting peptide, protects the brain against ischemic injury by reducing apoptotic death. Stroke 33: 1085-1092. [DOI] [PubMed] [Google Scholar]
- Li Oei S, Griesenbeck J, Scheiger M, Ziegler M (1998) Regulation of RNA polymerase II-dependent transcription by poly(ADP-ribosyl)ation of transcription factors. J Biol Chem 273: 31644-31647. [DOI] [PubMed] [Google Scholar]
- Lindahl T, Satoh MS, Poirier GG, Klungland A (1995) Post-translational modification of poly(ADP-ribose)polymerase induced by DNA strand breaks. Trends Biochem Sci 20: 405-412. [DOI] [PubMed] [Google Scholar]
- Lyall R, Zilberstein A, Gazit A, Gilon C, Levitzki A, Schlessinger J (1989) Tyrphostins inhibit epidermal growth factor (EGF)-receptor tyrosine kinase activity in living cells and EGF-stimulated cell proliferation. J Biol Chem 264: 14503-14509. [PubMed] [Google Scholar]
- Majdan M, Lachance C, Gloster A, Aloyz R, Zeindler C, Bamji S, Bhakar A, Belliveau D, Fawcett J, Miller FD, Baker PA (1997) Transgenic mice expressing the intracellular domain of the p75 neurotrophin receptor undergo neuronal apoptosis. J Neurosci 17: 6988-6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malviya AN, Rogue PJ (1998) “Tell me where the calcium bred”: clarifying the roles of nuclear calcium. Cell 92: 17-23. [DOI] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazaietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C (1993) Selective inhibition of protein kinase C isozymes by the indolocarbazole Gő 6976. J Biol Chem 268: 9194-9197. [PubMed] [Google Scholar]
- Mikoshiba K (1997) The InsP3 receptor and intracellular Ca+2 signaling. Curr Opin Neurobiol 7: 339-345. [DOI] [PubMed] [Google Scholar]
- Mizuno M, Yamada K, He J, Nakajima A, Nabeshima T (2003) Involvement of BDNF receptor TrkB in spatial memory formation. Learn Mem 10: 108-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Kohasaka S (1998) Functional roles of microglia in the central nervous system. Hum Cell 11: 141-155. [PubMed] [Google Scholar]
- Ohmichi M, Decker JS, Saltiel AR (1992) Nerve growth factor stimulates the tyrosine phosphorylation of a 38-kDa protein that specifically associates with the src homology domain of phospholipase C-γ1. J Biol Chem 267: 21601-21606. [PubMed] [Google Scholar]
- Ohmichi M, Pang L, Ribon V, Gazit A, Levitzki A, Saltiel AR (1993) The tyrosine kinase inhibitor tyrphostin blocks the cellular actions of nerve growth factor. Biochemistry 32: 4650-4658. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF (2001) Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol 11: 272-280. [DOI] [PubMed] [Google Scholar]
- Pawson T, Nash P (2000) Protein-protein interactions define specificity in signal transduction. Gene Dev 14: 1027-1047. [PubMed] [Google Scholar]
- Pieper AA, Blackshaw S, Clements EE, Brat DJ, Krug DK, Alison J, White AJ, Pinto-Garcia P, Antonella Favit A, Conover JR, Snyder SH, Verma A (2000) Poly(ADP-ribosyl)ation basally activated by DNA strand breaks reflects glutamate-nitric oxide neurotransmission. Proc Natl Acad Sci USA 97: 1845-1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinhasov A, Mandel S, Torchinsky A, Giladi E, Pittel Z, Goldsweig AM, Servoss SJ, Brenneman DE, Gozes I (2003) Activity-dependent neuroprotective protein: a novel gene essential for brain formation. Brain Res Dev Brain Res 144: 83-90. [DOI] [PubMed] [Google Scholar]
- Poole AW, Pula G, Hers I, Crosby D, Jones ML (2004) PKC-interacting proteins: from function to pharmacology. Trends Pharmacol Sci 25: 528-535. [DOI] [PubMed] [Google Scholar]
- Rankin PW, Jacobson EL, Benjamin RC, Moss J, Jacobson MK (1989) Quantitative studies of inhibitors of ADP-ribosylation in vitro and in vivo. J Biol Chem 264: 4312-4317. [PubMed] [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD (1999) Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 286: 2358-2361. [DOI] [PubMed] [Google Scholar]
- Richardson DS, Allen PD, Kelsey SM, Newland AC (1999) (1999) Effects of PARP inhibition on drug and Fas-induced apoptosis in leukaemic cells. Adv Exp Med Biol 457: 267-279. [DOI] [PubMed] [Google Scholar]
- Rouleau M, Rémy AA, Poirier GG (2004) Poly(ADP-ribosyl)ated chromatin domains: access granted. J Cell Sci 117: 815-825. [DOI] [PubMed] [Google Scholar]
- Sanchez C, Diaz-Nido J, Avila J (2000) Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog Neurobiol 61: 133-168. [DOI] [PubMed] [Google Scholar]
- Satoh MS, Poirier GG, Lindahl T (1994) Dual effect for poly(ADP-ribose) synthesis in response to DNA strand breakage. Biochemistry 33: 7099-7106. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Poo M (2000) The neurotrophin hypothesis for synaptic plasticity. Trend Neurosci 23: 639-645. [DOI] [PubMed] [Google Scholar]
- Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103: 211-225. [DOI] [PubMed] [Google Scholar]
- Segal RA (2003) Selectivity in neurotrophin signaling: theme and variations. Annu Rev Neurosci 26: 299-330. [DOI] [PubMed] [Google Scholar]
- Shah GM, Kaufmann SH, Poirier GG (1995) Detection of polyADP-ribose polymerase and its apoptosis-specific fragment by nonisotopic activity-Western blot technique. Anal Biochem 232: 251-254. [DOI] [PubMed] [Google Scholar]
- Smyth JT, Abbott AL, Lee B, Sienaert I, Nadif Kasri N, De Smedt H, Ducibella T, Missiaen L, Parys JB, Fissore RA (2002) Inhibition of the inositol trisphosphate receptor of mouse eggs and A7r5 cells by KN-93 via a mechanism unrelated to Ca2+2/calmodulin-dependent protein kinase II antagonism. J Biol Chem 277: 35061-35070. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Howe CL, Mobley WC (2001) Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci 24: 1217-1281. [DOI] [PubMed] [Google Scholar]
- Steingart RA, Solomon B, Brenneman DE, Fridkin M, Gozes I (2000) VIP and peptides related to activity-dependent neurotrophic factor protect PC12 cells against oxidative stress. J Mol Neurosci 15: 137-145. [DOI] [PubMed] [Google Scholar]
- Sutherland BM, Bennett PV, Sutherland JC (1999) DNA damage quantitation by alkaline gel electrophoresis. In: DNA repair protocols, eukaryotic systems (Henderson DS, ed), pp 183-193. Totowa, NJ: Humana.
- Tanuma S, Kawashima K, Endo H (1986) Acceptor proteins for (ADP-ribose)n in the HeLa S3 cell cycle. J Biochem (Tokyo) 99: 915-922. [DOI] [PubMed] [Google Scholar]
- Thoenen H (1995) Neurotrophins and neuronal plasticity. Science 270: 593-598. [DOI] [PubMed] [Google Scholar]
- Tischler AS, Greene LA, Kwan PW, Slayton VW (1983) Ultrastructural effects of nerve growth factor on PC 12 pheochromocytoma cells in spinner culture. Cell Tissue Res 228: 641-648. [DOI] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhamel L, Charron D, Kirilovsky DL (1991) The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem 266: 15771-15781. [PubMed] [Google Scholar]
- Tulin A, Spradling A (2003) Chromatin loosening by poly(ADP-ribose polymerase (PARP) at Drosophila puff loci. Science 299: 560-562. [DOI] [PubMed] [Google Scholar]
- Wong DL, Tai TC, Wong-Faull DC, Claycomb R, Kvetnansky R (2004) Genetic mechanisms for adrenergic control during stress. Ann NY Acad Sci 1018: 387-397. [DOI] [PubMed] [Google Scholar]
- Zemlyak I, Furman S, Brenneman DE, Gozes I (2000) A novel peptide prevents death in enriched neuronal cultures. Regul Pept 96: 39-43. [DOI] [PubMed] [Google Scholar]