

Abstract
In response to glutamatergic synaptic drive, striatal medium spiny neurons in vivo transition to a depolarized “up state” near spike threshold. In the up state, medium spiny neurons either depolarize enough to spike or remain below spike threshold and are silent before returning to the hyperpolarized “down state.” Previous work has suggested that subthreshold K+ channel currents were responsible for this dichotomous behavior, but the channels giving rise to the current and the factors determining its engagement have been a mystery. To move toward resolution of these questions, perforated-patch recordings from medium spiny neurons in tissue slices were performed. K+ channels with pharmacological and kinetic features of KCNQ channels potently regulated spiking at up-state potentials. Single-cell reverse transcriptase-PCR confirmed the expression of KCNQ2, KCNQ3, and KCNQ5 mRNAs in medium spiny neurons. KCNQ channel currents in these cells were potently reduced by M1 muscarinic receptors, because the effects of carbachol were blocked by M1 receptor antagonists and lost in neurons lacking M1 receptors. Reversal of the modulation was blocked by a phosphoinositol 4-kinase inhibitor, indicating a requirement for phosphotidylinositol 4,5-bisphosphate resynthesis for recovery. Inhibition of protein kinase C reduced the efficacy of the muscarinic modulation. Finally, acceleration of cholinergic interneuron spiking with 4-aminopyridine mimicked the effects of exogenous agonist application. Together, these results show that KCNQ channels are potent regulators of the excitability of medium spiny neurons at up-state potentials, and they are modulated by intrastriatal cholinergic interneurons, providing a mechanistic explanation for variability in spiking during up states seen in vivo.
Keywords: linopirdine, XE991, M1 knock-out, cholinergic interneuron, PIP2, PKC
Introduction
Striatal medium spiny neurons undergo shifts in their membrane potentials in response to coordinated glutamatergic synaptic input, moving from a hyperpolarized “down state” to a depolarized “up state” (Wilson and Kawaguchi, 1996; Wilson, 2004). In the up state, medium spiny neurons either depolarize sufficiently to generate spikes or remain below spike threshold and are silent (Wilson and Kawaguchi, 1996; Wickens and Wilson, 1998; Tseng et al., 2001; Wilson, 2004). Understanding the mechanisms underlying this dichotomous behavior is of obvious importance to models of striatal information processing, because a silent up state will be indistinguishable from a maintained down state to targets in the globus pallidus and substantia nigra.
Previous studies of the up state have focused on voltage-dependent K+ channels (Wilson and Kawaguchi, 1996). Blockade of K+ channels has profound effects on up-state potentials. Several voltage-gated K+ channels might contribute to this dependence. Kv4 channels activate in this voltage range, but they inactivate rapidly, making them poor regulators of sustained depolarizing inputs (Tkatch et al., 2000). Kv1.2 channels are also active in this range (Nisenbaum et al., 1994; Shen et al., 2004). However, these channels inactivate as well, albeit more slowly than Kv4 channels. This characteristic makes it unlikely that they are central players in the phenomenon observed in vivo, in which the up state can reside at subthreshold potentials for seconds.
Another K+ channel that is known to regulate subthreshold membrane potential and excitability in several central and peripheral neurons is the KCNQ (Kv7) channel (Brown and Adams, 1980; Marrion, 1997; Wang et al., 1998; Jentsch, 2000; Shapiro et al., 2000). These channels open at subthreshold membrane potentials and do not inactivate. As with other members of the Kv class, these channels are multimeric (Jentsch, 2000; Hadley et al., 2003). Four known subunits contribute to KCNQ channels found in the brain (KCNQ2-5) (Jentsch, 2000). In situ hybridization (Saganich et al., 2001) and immunocytochemical (Cooper et al., 2001) studies have shown that KCNQ subunits are expressed in the striatum. However, it is unclear from these studies whether they are expressed by medium spiny neurons, because the most prominent labeling appears to be of large interneurons.
Another feature of KCNQ channels that makes them attractive candidates for controlling up-state silencing is their susceptibility to neuromodulation. They were originally called “M-channels” because of their suppression by muscarinic receptor signaling (Brown and Adams, 1980; Adams and Brown, 1982; Jones, 1985). One of the most prominent modulators of medium spiny neurons is acetylcholine (Bolam et al., 1984; Kawaguchi, 1993). All medium spiny neurons express high levels of the M1 muscarinic receptor, the receptor known to modulate KCNQ channels in other cell types (Hersch et al., 1994; Yan et al., 2001). Could cholinergic interneurons toggle medium spiny neurons between spiking and silent up states by regulating KCNQ channel opening? The data presented below are consistent with this hypothesis, showing that medium spiny neurons express functional KCNQ channels that regulate spiking at up-state potentials and that these channels are potently modulated by M1 receptor activation.
Materials and Methods
Slice preparation. All experiments were conducted in accord with the guidelines approved by the Northwestern University Animal Care and Use Committee. Standard techniques were used for the preparation of slices for recording (Shen et al., 2004). Briefly, Sprague Dawley rats of either sex, 16-23 d of age, and M1 muscarinic receptor knock-out mice (Hamilton et al., 1997) were anesthetized deeply with ketamine-xylazine and perfused transcardially with 5-10 ml of ice-cold artificial CSF (ACSF) comprising the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2.0 CaCl2, 1.0 MgCl2, 25 NaHCO3, and 14 glucose, bubbled continuously with carbogen (95% O2 and 5% CO2). The brain was quickly removed, blocked in either coronal or parasagittal plane, glued to the stage of a VT1000S slicer (Leica, Nussloch, Germany), and immersed in the ice-cold ACSF. Sections through the striatum were cut at a thickness of 275-300 nm and then transferred to a holding chamber, where they were completely submerged in ACSF and maintained at 35°C for 30-40 min. Slices were then kept in the holding chamber at the room temperature (22°-23°C) for another 20 min before recording.
Electrophysiological recordings. Individual slices were transferred to a recording chamber and were perfused continuously (2-3 ml/min) with carbogenated ACSF for the duration of the experiment. A 40× water immersion objective (Olympus, Melville, NY) was used to examine the slice with standard infrared differential interference contrast video microscopy. Experiments were performed at room temperature unless otherwise specified.
Patch pipettes were pulled from thick-walled borosilicate glass (outer diameter, 1.5 mm) on a Sutter P-97 puller (Sutter Instruments, Novato, CA) and fire polished before recording. Pipette resistance was typically 3-4 MΩ when filled with recording solution. The internal pipette solution contained the following (in mm): 126 KMeSO4, 14 KCl, 3 MgCl2, 0.5 CaCl2, 5 EGTA, 10 HEPES, pH adjusted to 7.25 with NaOH and osmolarity adjusted to 275-280 mOsm/L. Electrical access was achieved through the perforated-patch method using amphotericin B (Rae et al., 1991). The perforated-patch method was used to preserve the intracellular integrity of the neurons, thus maintaining functional KCNQ current and muscarinic responses that would be lost after cell dialysis in these neurons during whole-cell recording. For perforated-patch experiments, a stock solution of amphotericin B (60 mg/ml) in dimethyl sulfoxide was prepared and diluted in the recording solution immediately before use yielding a final concentration of 180 μg/ml. Capacitance current was monitored continuously by applying a 5-10 mV pulse from a holding potential of -70 mV. Series resistance (20-35 MΩ in voltage-clamp recordings and 40-50 MΩ in current-clamp recordings) was monitored periodically during experiments. To minimize voltage error in voltage-clamp recordings, series resistance was typically compensated by 60-70%. Given small amplitude of the KCNQ currents, this should have produced an error of ∼1-2 mV. If series resistance changed >15% during recording, the data were discarded. The membrane potentials were corrected routinely for a -7 mV liquid junction potential (Neher, 1992).
Data analysis. Data acquisition used Clampex 8.2 software in combination with a MultiClamp 700A amplifier (Molecular Devices, Union City, CA). All data were analyzed with Igor Pro 5 (WaveMetrics, Lake Oswego, OR) and Clampfit 8.2. Statistical analyses were performed with SigmaStat 3.0 (SPSS, Chicago, IL). Central tendency of numerical data were estimated by computing means ± SEM for samples ≥10 or median and range for smaller samples. Box plots were used for graphic presentation of the data with the small sample size. Differences between small samples were analyzed with nonparametric tests.
Single-cell reverse transcriptase-PCR analysis. Dissociated, individual medium spiny neurons were patched in the cell-attached mode and then aspirated into an electrode. Electrodes contained 1-2 μl of diethyl pyrocarbonate (DEPC)-treated water and 1.5 U/ml SUPERase-In (Ambion, Austin, TX). The glass used for making electrodes was heated to 200°C for 2-3 h before use. Sterile gloves were worn at all times during the procedure to minimize RNase contamination. After aspiration of the neuron, the electrode tip was broken in a 0.6 ml presiliconized tube (Midwest Scientific, Valley Park, MO) containing 1.9 μl of DEPC-treated water, 0.7 μl of SUPERase-In (20 U/ml), 0.7 μl of oligo-dT (50 mm), 0.7 μl of BSA (143 mg/ml), 1.0 ml of dNTPs (10 mm), and the contents were ejected. The mixture was heated to 65°C for 5 min to denature the nucleic acids and then placed on ice for at least 1 min. Single-strand cDNA was synthesized from the cellular mRNA by adding 1.2 μl of SuperScript II reverse transcriptase (RT) (200 U/ml), 1 μl of 10× RT buffer, 2 μl of MgCl2 (25 mm), 1 μl of DTT (0.1 m), and 0.5 ml of RNase Out (40 U/ml) and then incubating the mixture at 42°C for 90 min. The reaction was terminated by heating the mixture to 70°C for 15 min. The RNA strand in the RNA-DNA hybrid was then removed by adding 0.5 μl of RNase H (2 U/ml) and incubating at 37°C for 20 min. All reagents except SUPERase-In were obtained from Invitrogen (Carlsbad, CA).
The single-cell cDNA generated from the reverse transcription step was subjected to conventional PCR using a programmable thermal cycler (P-200; MJ Research, Watertown, MA). PCR primers were developed from GenBank sequences with commercially available OLIGO software (version 6.7; Molecular Biology Insights, Cascade, CO). Primers for substance P (SP) and enkephalin (ENK) have been described previously (Surmeier et al., 1996). KCNQ2 (Kv7.2) mRNA (GenBank accession number AF087453) was detected with a pair of primers, 5′-GCAACAGAGCCAGTACCGAGTTC (position 2335) and 5′-CACATT GACCCTGAGAGCGA (position 2811). The predicted PCR product length was 496 bp. KCNQ3 (Kv7.3) mRNA (GenBank accession number NM031597) was detected with a pair of primers, 5′-ACCAGGTGCCCATCGAC (position 2529) and 5′-GCCAGTGA CCCCTTCTAAGTG (position 3043). The predicted PCR product length was 535 bp. KCNQ4 (Kv7.4) mRNA (GenBank accession number AF249748) was detected with a pair of primers, 5′-GCTTCC GGGCCTCTCTAAGACT (position 159) and 5′-TGGACCCTTAT CGCCCTTC (position 484). The predicted PCR product length was 344 bp. KCNQ5 (Kv7.5) mRNA (GenBank accession number AF263836) was detected with a pair of primers, 5′-GTTGCCTCCAAGGAAAGTGTTC (position 2192) and 5′-ACCGTGACCTTCCAGTCCTTAGA (position 2545). The predicted PCR product length was 376 bp. A two-round protocol was used for more efficient amplification of single-cell cDNA. This was done by implementing 35 cycles of denaturation at 94°C for 45 s, annealing at 55°C for 45 s, and elongation at 72°C for 70 s, followed by elongation for 7 min at 72°C.
PCR procedures were performed using procedures designed to minimize the chance of cross-contamination (Cimino et al., 1990). Negative controls for contamination from extraneous and genomic DNA were run for every batch of neurons. To verify that genomic DNA did not contribute to the PCR products, neurons were aspirated and processed in the normal manner, except that the reverse transcriptase was omitted. Contamination from extraneous sources was checked by replacing the cellular template with buffer solution. Both controls were consistently negative in these experiments.
Chemicals and drugs. Reagents were obtained as follow: linopirdine and 10,10-bis(4-pyridinylmethyl)-9(10H)anthracenone dihydrochloride (XE991; Sigma, St. Louis, MO, or Tocris Cookson, Ellisville, MO); (RS)-3,5-dihydroxyphenylglycine(DHPG),7-(hydroxy-imino)cyclopro pa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt), and 2-methyl-6-(phenylethynyl)-pyridine (MPEP) (Tocris Cookson); muscarinic toxin 7 (MT 7; Peptides International, Louisville, KY); and calphostin C, thapsigargin, and wortmannin (Calbiochem, La Jolla, CA). Tetraethylammonium (TEA), 4-aminopyridine (4-AP), mecamylamine, and all other chemicals were obtained from Sigma. When TEA was applied at concentrations ≥1 mm, the osmolarity was adjusted by reducing the concentration of NaCl in ACSF.
Results
Blockade of KCNQ channels increases excitability at up-state potentials
Perforated-patch recordings were obtained from medium-sized neurons (somal diameter, 10-15 μm). These neurons were distinguished readily from large cholinergic interneurons by their size, as illustrated in Figure 1A. To distinguish medium spiny neurons from other interneurons (Kawaguchi, 1993), electrophysiological criteria were used (Fig. 1B). Recordings were made only from neurons having (1) a relatively hyperpolarized resting potential (-88.4 ± 1.1 mV; n = 27); (2) strong inward rectification; and (3) a slow voltage ramp to near rheobase current injection. They were classified as medium spiny neurons (Kerr and Plenz, 2002; Shen et al., 2004; Wilson, 2004).
Figure 1.
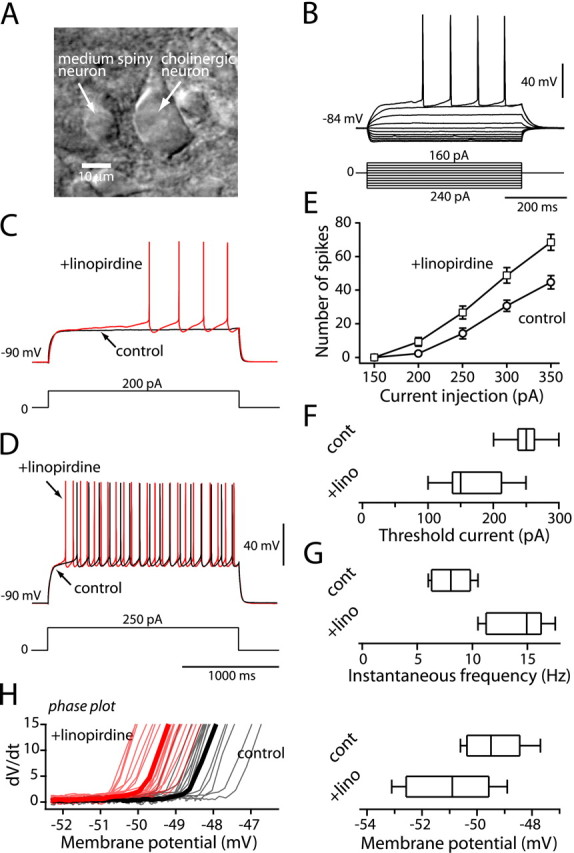
Blockade of KCNQ channels increased evoked activity. A, Photomicrograph of a medium spiny neuron and a large neuron resembling a cholinergic interneuron. B, Typical membrane responses to somatic current injection (shown below). Note the strong inward rectification and delayed firing. C, The response of a medium spiny neuron to a near rheobase current step (200 pA, 2 s) before and after application of linopirdine (10 μm). Linopirdine increased the rate of rise of the membrane potential toward spike threshold (red line). D, The response to a suprathreshold current step (250 pA, 2 s) before and after application of linopirdine. Linopirdine increased firing frequency (red line). E, Average number of spikes evoked to the values of the current injected. Linopirdine reduced the amount of current required to generate a given number of spikes. Error bars represent SEM. Box plots illustrate the difference in rheobase current required for action potential firing (F) and the difference in instantaneous frequency change to a current step (250 pA) (G). H, The phase plane analysis in D shows that spike threshold is lowered in linopirdine (red lines). A box plot summary showing reduction in spike threshold in linopirdine is displayed. cont, Control; lino, linopirdine.
As a first step toward testing the hypothesis that KCNQ channels modulated the subthreshold excitability of medium spiny neurons, the effect of linopirdine, a selective KCNQ channel blocker, on the response to 2 s current steps was examined (Fig. 1C,D, bottom). Linopirdine (10 μm) had no effect on the resting membrane potential (-89.5 ± 1.0 mV in control; -88.5 ± 0.8 mV in linopirdine; n = 6; p > 0.05; Wilcoxon signed rank test). However, near rheobase, linopirdine accelerated the rate of rise of the characteristic slow voltage ramp (Fig. 1C). The median rheobase current was reduced by nearly 100 pA by linopirdine (n = 6; p < 0.05; Wilcoxon) (Fig. 1F). With larger current steps in which repetitive spiking was evoked, linopirdine increased the number of spikes evoked (Fig. 1D), shifting the frequency-current relationship to the left (Fig. 1E). Using a 250 pA step, the median discharge frequency increased from ∼8 to ∼15 Hz in the presence of linopirdine (p < 0.05; Wilcoxon) (Fig. 1G). Phase plane analysis of spiking revealed that linopirdine lowered spike threshold, defined by the discontinuity in the relationship between dV/dt and V (Fig. 1H) without significantly affecting the maximum dV/dt. The median change in spike threshold was 1.4 mV (median, -49.5 mV in control, -50.9 mV in linopirdine; p < 0.05; Wilcoxon) (Fig. 1H). XE991, another selective KCNQ channel blocker, had similar effects on the excitability of medium spiny neurons (n = 2; data not shown).
In whole-cell recordings (as opposed to perforated-patch recordings), linopirdine-sensitive currents were lost in the first few minutes (data not shown). As a consequence, all of our subsequent experiments used perforated-patch recording.
Medium spiny neurons express a slow, noninactivating KCNQ channel currents
Somatic point clamp experiments were performed to generate a biophysical description of the gating properties of the putative KCNQ channel currents. In an attempt to isolate the KCNQ channel currents from other voltage-gated K+ currents, the membrane potential was held at a relatively depolarized potential (VH, -30 mV) to activate KCNQ channels (Brown and Adams, 1980; Adams and Brown, 1982) and to inactivate many of the other K+ channels that activate in this membrane potential region (Nisenbaum et al., 1996; Tkatch et al., 2000; Shen et al., 2004). The membrane potential was then stepped down to more hyperpolarized potentials for 4 s to deactivate the KCNQ channels (Fig. 2A, top). The inward tail currents decayed biexponentially (Fig. 2A,D). Bath application of XE991 (20 μm) reduced the holding current and blocked the deactivating current (Fig. 2A). The calculation of difference currents by subtracting the XE991-resistant currents from control currents yielded records expected of KCNQ channels (Fig. 2A, bottom). Stepping back to -30 mV led to the reactivation of the KCNQ channels and the generation of an outward current.
Figure 2.

Striatal medium spiny neurons express a slow, noninactivating K+ channel current. A, Representative deactivation currents recorded from a medium spiny neuron before and after application of XE991. The cell was held at -30 mV to generate sustained outward currents. The channels were then deactivated by 4 s hyperpolarizing steps in increments of -10 mV (shown above). Subtraction of current traces obtained in the presence of XE991 (20 μm) from in control gives rise to the difference currents in the bottom panel of A is shown. B, Representative activation currents recorded from another medium spiny neuron before and after application of XE991. The cell was held at -90 mV and then activated by 4 s depolarizing steps in increments of -10 mV (shown above). Subtraction of current traces obtained in the presence of XE991 (20 μm) from control yielded the difference currents in the bottom panel of B. C, Conductance-voltage relationship. GM was determined from the amplitude of the current relaxations during the hyperpolarizing and depolarizing steps. Data points were fit with the following equation: GM = Gmax/(1 + exp (V - V1/2)/k), where V1/2 = -43.3 mV, k = -8.1 mV for deactivation and V1/2 = -41.6 mV, k = -7.9 mV for activation. D, Plots of the voltage dependence of fast and slow time constants. Data points were fit with a Boltzmann equation. E, The percentage of the current that was fast, plotted as a function of voltage. Data were fit to a simple linear equation. Error bars represent SEM.
To measure the activation threshold and activation time constant of the KCNQ current, standard activation protocol was applied by stepping to depolarized potentials from a holding potential of -90 mV (Fig. 2B). Again, subtraction of XE991-resistant currents from control currents yielded KCNQ difference currents. The voltage dependence of activation and deactivation was assessed by converting the current amplitudes to chord conductances (Fig. 2C). Data points were then fitted with a first-order Boltzmann function of the following form: GM = Gmax/(1 + exp ((V - V1/2)/k)), where V1/2 is the half-deactivation or activation voltage, and k is the slope factor. KCNQ channel gating measured using the deactivation or the activation protocol yielded the same results: average half-deactivation voltage was -43.3 ± 1.0 mV (n = 7), and the slope factor was -8.1 ± 0.4 mV; the average half-activation voltage was -41.6 ± 0.8 mV (n = 7), and the slope factor was -7.9 ± 0.6 mV (V1/2, p > 0.05, Mann-Whitney rank-sum test; k, p > 0.05, Mann-Whitney).
Both deactivation and activation kinetics were best fit with two exponentials over a range of potentials (Fig. 2D,E). The time constants in medium spiny neurons were close to those found in rat sympathetic neurons and those reported for heterologously expressed KCNQ2/KCNQ3 heteromeric channels (Hadley et al., 2000; Shapiro et al., 2000; Pan et al., 2001). These data were pooled and then fit with a two-state kinetic model plotted as a solid line in Figure 2D. Deactivation accelerated with increasing hyperpolarization; both τfast and τslow shortened, with τfast falling e-fold for a 52.6 mV hyperpolarization (Fig. 2D).
KCNQ channels of medium spiny neurons are comprised of KCNQ2/3 subunits
The biophysical and pharmacological data presented thus far suggest that medium spiny neurons express KCNQ channels. Previous studies have shown KCNQ transcripts are present in the striatum (Saganich et al., 2001), an observation confirmed by our tissue level RT-PCR analysis (data not shown). However, localization of KCNQ transcripts to medium spiny neurons is less clear. To examine directly cellular localization of KCNQ mRNAs, single-cell RT-PCR (scRT-PCR) profiling was performed on 16 medium spiny neurons identified by their expression of ENK or SP mRNA. Neurons were profiled for their expression of KCNQ2, KCNQ3, KCNQ4, and KCNQ5 mRNAs. KCNQ2, KCNQ3, and KCNQ5 mRNAs were consistently detected in both SP- and ENK-expressing medium spiny neurons (Fig. 3A,B). KCNQ4 mRNA was not detected in any cell (n = 16). There was no correlation between SP or ENK detection and the detection of KCNQ mRNAs.
Figure 3.

Medium spiny neurons express functional KCNQ2/KCNQ3 channel subunits. A, scRT-PCR from an ENK-expressing neuron (top) and an SP-expressing neuron (bottom). M, Marker. B, In a sample of medium spiny neurons (n = 16), 94% had detectable levels of KCNQ2 mRNA, 100% had detectable levels of KCNQ3 mRNA, and 88% had KCNQ5 mRNA, with none of the cells having detectable levels of KCNQ4 mRNA. C, KCNQ currents recorded by stepping membrane voltage from -30 to -60 mV (shown above) for 4 s in the absence and presence of increasing concentrations of TEA (0.3-30 mm; different-colored lines). D, The amplitude of the KCNQ channel current was measured as the difference between the instantaneous current at the onset of hyperpolarization and the steady-state current at the end of voltage command, as shown in the inset. The percentage of inhibition was plotted as a function of TEA concentration. The data were fit with a logistic function: IC50 = 4.1 mm and slope factor = 1.07. Error bars represent SEM.
Although KCNQ subunits are capable of forming homomeric channels in heterologous expression systems, in native systems, KCNQ3 subunits are thought to form heteromeric channels with KCNQ2, 4, or 5 subunits (Jentsch, 2000). For example, KCNQ channels in sympathetic neurons are thought to be heteromers of KCNQ2 and KCNQ3 subunits (Wang et al., 1998; Shapiro et al., 2000; Selyanko et al., 2001). Immunoprecipitation studies have shown that in many regions of the brain, KCNQ3 subunits associate with either KCNQ2 (Cooper et al., 2000; Yus-Najera et al., 2003; Devaux et al., 2004) or KCNQ5 (Yus-Najera et al., 2003) subunits. KCNQ2 and KCNQ5 subunits do not appear to associate (Yus-Najera et al., 2003). One way of distinguishing KCNQ channels with differing subunit composition is to determine their sensitivity to TEA (Hadley et al., 2000; Hadley et al., 2003). KCNQ2 homomeric channels have a very high affinity for TEA, whereas the affinity of KCNQ2/KCNQ3 heteromeric channels is ∼10-fold lower because KCNQ3 channels are insensitive to TEA. KCNQ3/KCNQ5 heteromeric channels are blocked poorly by TEA (Schroeder et al., 2000). In medium spiny neurons, KCNQ channel tail currents evoked by stepping to -60 mV from a holding potential of -30 mV had an intermediate sensitivity to TEA (Fig. 3C,D). The dose-response relationship could be well fit by a logistic equation of the following form: Y = ((Y0 - Y∝)/(1 + (C × IC50-1)B)) + Y∝, where Y0 is the amplitude in the absence of TEA, Y∝ is the maximal response to TEA, B is the slope factor, and C is the concentration of TEA. The IC50 was 4.1 ± 0.5 mm, and the slope factor was 0.95 ± 0.09 (n = 7). This value is close to that reported for KCNQ2/KCNQ3 heteromeric channels (Wang et al., 1998; Hadley et al., 2000, 2003; Shah et al., 2002).
M1 muscarinic receptor mediates suppression of KCNQ channel current in medium spiny neurons
KCNQ channel opening is reduced by activation of M1-class receptors (Brown and Adams, 1980; Marrion, 1997; Wang et al., 1998; Shapiro et al., 2000; Robbins, 2001). Both striatonigral and striatopallidal medium spiny neurons express M1-class receptors (Yan et al., 2001). As expected, application of the cholinergic agonist carbamylcholine chloride (carbachol) in the presence of the nicotinic receptor antagonist mecamylamine (10 μm) reduced KCNQ channel currents (Fig. 4A). The carbachol dose-response relationship could be fitted by a logistic equation with IC50 of 0.52 ± 0.14 μm and slope factor of 1.06 ± 0.12 (n = 5). In agreement with the attribution of the modulation to M1-class receptors, pirenzipine (100 nm) blocked the effects of carbachol (10 μm; n = 4) (Fig. 4B,C).
Figure 4.

M1 muscarinic receptor mediates the modulation of KCNQ channel currents. A, Dose-response relationship of the carbachol modulation of KCNQ channel current. The fitting parameters were IC50 = 0.51 μm and slope factor = 1.07. Error bars represent SEM. B, The box plot summary illustrates the M1 receptor-mediated modulation. Carbachol (10 μm) blocked 86% of the KCNQ current (n = 6). The blockade was reversed in the presence of pirenzepine (100 nm; n = 4) and MT 7 (100 nm; n = 5) and was lost in M1 muscarinic receptor knock-out mice (M1 KO; n = 5). The KCNQ current recorded in M1 KO mice was reduced by the group 1 metabotropic glutamate agonist DHPG (50 μm; n = 5). C, Typical KCNQ current traces and their responsiveness to M1 agonist and antagonists and DHPG. CCh, Carbachol; PZ, pirenzepine.
There are three M1-class receptors (M1, M3, M5) that are expressed by medium spiny neurons, with the M1 receptor being the predominant subtype (Hersch et al., 1994; Yan et al., 2001; Zhang et al., 2002). All three receptors are coupled to phospholipase Cβ (PLCβ) through Gq-proteins, and all three have been implicated in the modulation of KCNQ currents (Robbins et al., 1991; Guo and Schofield, 2003). In sympathetic ganglion neurons, M1 receptors have been firmly established as mediating the cholinergic modulation of KCNQ channels (Hamilton et al., 1997; Robbins, 2001). However, in central neurons, the situation is less clear. Several recent studies showed that muscarinic modulation of KCNQ channel currents is not altered in neurons from M1 receptor knock-out mice (Rouse et al., 2000; Fisahn et al., 2002) or by an M1-specific toxin (Rouse et al., 2000).
To determine whether M1 muscarinic receptors mediate the effects of carbachol on KCNQ channels in striatal medium spiny neurons, two approaches were taken. First, MT 7, a highly selective, irreversible antagonist of the M1 receptor, was used (Max et al., 1993). Bath perfusion of the toxin (100 nm) for 40-50 min completely abolished the effects of carbachol on KCNQ channel currents (Fig. 4B,C). Second, medium spiny neurons from M1 receptor knock-out mice were examined for their responsiveness to carbachol. Consistent with the result from the toxin treatment, KCNQ channel currents in medium spiny neurons from M1 knock-out mice (n = 5) were completely insensitive to carbachol (n = 6) (Fig. 4B,C). The failure of carbachol to alter currents was not caused by a defect in the KCNQ channel itself or PLCβ signaling, because the group 1 metabotropic glutamate receptor agonist DHPG (50 μm) potently reduced KCNQ channel currents in neurons from the M1 receptor knock-out (n = 5) (Fig. 4B,C), as it did in wild-type neurons (n = 3; data not shown). The absence of an effect of carbachol was not species dependent either because carbachol potently reduced KCNQ channel currents in wild-type mice (median reduction, 81%; n = 5; data not shown). These results argue that muscarinic receptor mediating the reduction in KCNQ channel currents in medium spiny neurons is the M1 receptor.
Phosphotidylinositol 4,5-bisphosphate is required for KCNQ channel gating
The signaling cascade mediating muscarinic receptor modulation of KCNQ channels is controversial (Marrion, 1997; Brown and Yu, 2000; Ikeda and Kammermeier, 2002; Shapiro, 2004). Recent studies have suggested that PLC-dependent depletion of a membrane lipid, phosphotidylinositol 4,5-bisphosphate (PIP2), results in decreased KCNQ channel opening with membrane depolarization (Suh and Hille, 2002, 2005; Ford et al., 2003; Zhang et al., 2003; Suh et al., 2004; Winks et al., 2005). One of the strongest pieces of evidence for the hypothesis that PIP2 is an allosteric regulator of KCNQ channel gating is that inhibition of the enzyme that maintains membrane PIP2 levels [phosphotidylinositol 4 kinase (PI4K)] dramatically slows recovery from G-protein-coupled receptor modulation (Suh and Hille, 2002; Zhang et al., 2003; Winks et al., 2005). To determine whether the M1 receptor modulation of KCNQ channels in medium spiny neurons had the same PIP2 dependence, the PI4K inhibitor wortmannin was applied after establishment of the KCNQ modulation. Normally, KCNQ channel currents recovered nearly completely after carbachol application (recovery to 81% ± 3.2% of control amplitudes; n = 5) (Fig. 5A,B). However, application of wortmannin (50 μm) significantly reduced recovery of the KCNQ channel currents after termination of carbachol exposure (recovery to 10% ± 1.8% of control amplitudes; n = 5; p < 0.05; Mann-Whitney) (Fig. 5C,D).
Figure 5.

Recovery from M1 muscarinic inhibition requires PIP2 synthesis. A, C, Plots of inhibition and recovery of KCNQ currents elicited by steps from a holding potential of -30 mV. A, Carbachol (3 μm) modulation and recovery in the standard external ACSF solution. B, Representative current traces from A. C, Application of wortmannin (50 μm) prevented reversal of the CCh (3 μm) modulation. D, Current traces used to construct C. E, Box plot summary of the percentage of recovery from muscarinic inhibition (control, 81%, n = 5; wortmannin, 10.2%, n = 5; p < 0.05; Mann-Whitney). CCh, Carbachol; cont, control; wort, wortmannin.
The products of PLCβ metabolism of membrane PIP2 are diacylglycerol (DAG) and inositol trisphosphate (IP3). DAG is a potent activator of protein kinase C (PKC) isoforms. Attempts to determine whether PKC affects KCNQ channels have yielded seemingly contradictory results. Namely, KCNQ channels appear to be responsive to PKC activators but resistant to PKC inhibitors (Marrion, 1997; Brown and Yu, 2000). Recent work reconciles these discrepant results by showing that inhibitors interacting with the PKC DAG binding site attenuate KCNQ channel modulation, but those that interact with the PKC catalytic domain are less effective because this site may be shielded by scaffolding proteins (Hoshi et al., 2003). In agreement with this view, calphostin C(1 μm), a PKC inhibitor acting on the DAG binding site (Kobayashi et al., 1989), reduced the effect of low concentrations of carbachol on KCNQ channel currents (Fig. 6A,B). However, higher concentrations of carbachol were equally effective in the presence of calphostin C. More complete analysis of this result revealed that calphostin C reduced the efficacy of carbachol in modulating KCNQ channels, approximately tripling the IC50 (Fig. 6C,D). These results suggest that PKC activation acts cooperatively with PIP2 depletion to reduce KCNQ channel opening.
Figure 6.
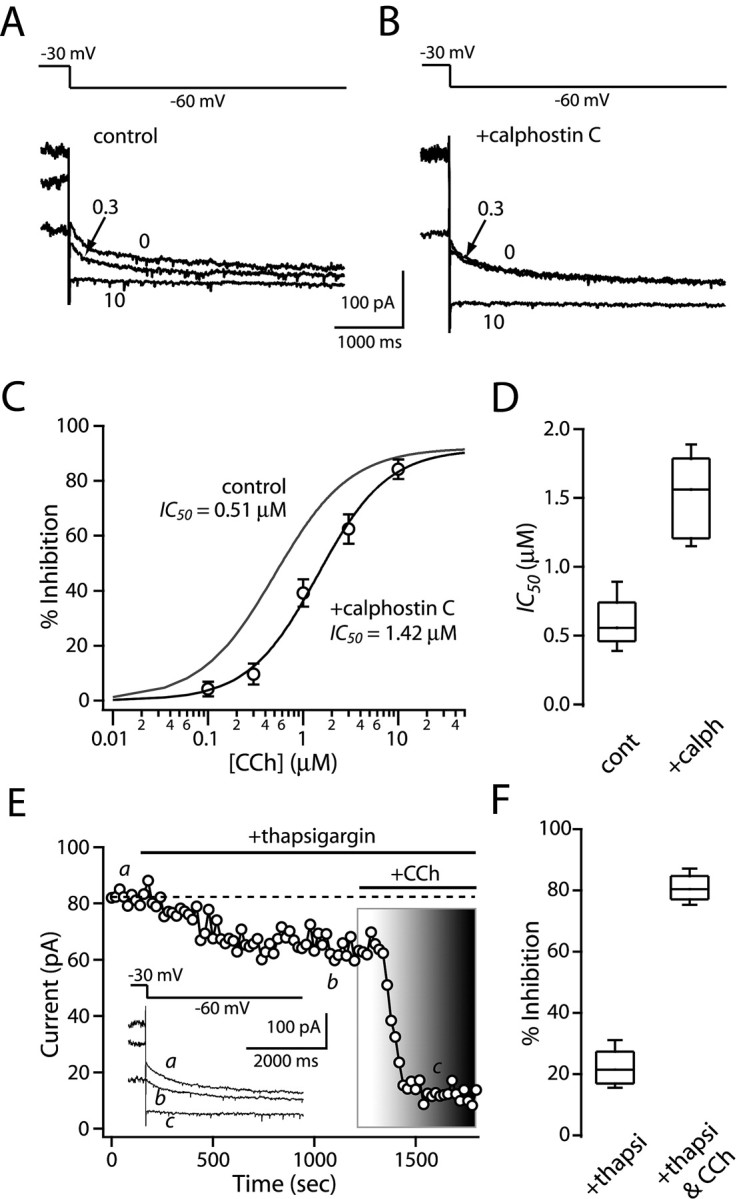
PKC activation increases M1 receptor efficacy. A, B, Representative current traces in the absence (control) and presence of the PKC inhibitor calphostin C (1 μm) during application of carbachol at the indicated concentrations (micromolar). C, Dose-response relationship in control (gray line from Fig. 4 A) and in calphostin C. PKC inhibition shifts the relationship to the right, without changing the maximum modulation. Error bars represent SEM. D, The box plot summary illustrates that PKC inhibitors increases IC50 of carbachol inhibition (n = 5; p < 0.05; Mann-Whitney). E, Bath application of thapsigargin (2 μm) reduced (∼20%) KCNQ currents (inset) but did not disrupt the CCh (3 μm)-mediated modulation (80%). F, Box plot summary from a sample of five neurons. CCh, Carbachol; cont, control; calph, calphostin C; thapsi, thapsigargin.
The other leg of the PLCβ signaling cascade involves IP3 liberation and release of Ca2+ from intracellular stores. Several studies have implicated intracellular Ca2+ in KCNQ regulation (Delmas et al., 2002; Wen and Levitan, 2002; Gamper and Shapiro, 2003). The open probability of KCNQ channel appears to be enhanced by modest elevations in cytosolic Ca2+ concentration (50-150 nm) and inhibited by greater elevations (>200 nm) (Marrion et al., 1991; Yu et al., 1994; Selyanko and Brown, 1996; Gamper and Shapiro, 2003). To determine whether intracellular Ca2+ release was a necessary component of the signaling cascade mediating M1 receptor reductions in KCNQ channel currents, these stores were depleted with Ca2+-ATPase inhibitor thapsigargin (2 μm) before application of carbachol. Bath application of thapsigargin alone reduced KCNQ tail currents by ∼20% (n = 5) (Fig. 6E,F). Although intracellular Ca2+ levels were not directly measured, inhibition of the Ca2+-ATPase has been shown to elevate these levels significantly (Cruzblanca et al., 1998; del Rio et al., 1999), consistent with the proposition that high free-Ca2+ levels reduce KCNQ opening. However, subsequent application of carbachol still resulted in a profound reduction in KCNQ channel currents (Fig. 6E,F), arguing that intracellular Ca2+ release was not necessary for the M1 receptor effects.
Activation of striatal cholinergic interneurons suppresses the KCNQ current
The data presented thus far show that KCNQ channel currents in medium spiny neurons are reduced by M1 muscarinic receptor activation. Activation of these receptors is dependent on acetylcholine release by large aspiny interneurons in the striatum. These interneurons are autonomously active but modulate their spiking rate in response to synaptic input (Bennett and Wilson, 1998). To determine whether modest enhancement of interneuron acetylcholine release would recapitulate the modulation seen with exogenous agonist application, 4-AP (100 μm) was bath applied. At this concentration, 4-AP modestly accelerates interneuron discharge rates (Fig. 7B,E). Perforated-patch recording from a medium spiny neuron in the vicinity of an interneuron showing elevated discharge rates revealed a robust 4-AP associated reduction in KCNQ channel currents (Fig. 7C,D). KCNQ currents were reduced by 4-AP over 40% on average (Fig. 7C,D). These recordings were made in the presence of ionotropic glutamate and GABA receptor antagonists [20 μm CNQX, 20-40 μm d-AP-5, and 10 μm bicuculline methiodide or 10 μm SR95531 [6-imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid] as well as group 1 metabotropic glutamate receptor antagonists (100 μm CPCCOEt and 10 μm MPEP), eliminating potentially confounding changes in these transmitters. To verify that the modulation was mediated by acetylcholine release, scopolamine (10 μm) was coapplied with 4-AP. As expected, the KCNQ channel currents in medium spiny neurons were unchanged by this mixture, despite the fact that interneuron discharge rate increased as a consequence of autoreceptor blockade (Fig. 7C,D).
Figure 7.

Activation of cholinergic interneurons reduces KCNQ channel current in medium spiny neurons. A, Photomicrograph showing the placement of recording electrodes for a cholinergic interneuron and a medium spiny neuron. B, Somatic cell-attached recording of autonomous action potential firing in a cholinergic interneuron before and after application of 4-AP (100 μm)in the continuous presence of ionotropic glutamate receptor antagonists, GABAA receptor antagonist, and group 1 metabotropic glutamate receptor antagonists (see Results). At this concentration of 4-AP, the interneuron discharge rate was increased. Left, Time course plot of the action of 4-AP on the autonomous discharge rate of the cholinergic interneuron shown in the right panel. C, KCNQ channel currents in the medium spiny neuron were reduced by the interneuron activation, and this modulation was blocked by scopolamine (10 μm). The time course of action of 4-AP and 4-AP plus scopolamine is shown in the left panel. D, Box plot summary of the action of 4-AP (4-AP, 56% of control, n = 9; 4-AP and scopolamine, 97% of control, n = 5; p < 0.01; Mann-Whitney). E, The box plot illustrates increased discharge rate in 4-AP (n = 12; control, 0.8 Hz; 4-AP, 1.8 Hz; p < 0.01; Wilcoxon). scop, Scopolamine; cont, control.
Discussion
Our studies show that striatal medium spiny neurons express KCNQ K+ channels that shape the response to depolarizing currents. These widely expressed but little studied channels activate at subthreshold membrane potentials and do not inactivate, making them suitable mediators of variation in up-state potential and spiking observed in vivo. What is more, these channels are modulated potently by acetylcholine released by striatal interneurons. This modulation appears to be mediated primarily by membrane depletion of PIP2 subsequent to activation of an M1 muscarinic receptor pathway coupled to PLCβ and PKC. This modulatory pathway was engaged not only by exogenous agonist application but also by modest acceleration of the autonomous spiking of cholinergic interneurons themselves, demonstrating the functional relevance of the modulation. If cholinergic tone is not uniform in the striatum, then this linkage provides a viable explanation for the variability in up-state spiking seen within the medium spiny population and within individual neurons in vivo. It also establishes a novel mechanism by which event driven modulation in the activity of cholinergic interneurons can shape the spiking of medium spiny neurons in response to cortical and thalamic glutamatergic signals.
Striatal projection neurons express KCNQ channel mRNA and channels
Neuronal KCNQ channels are multimeric transmembrane proteins constructed from a family of at least four subunits (KCNQ2-5) (Wang et al., 1998; Jentsch, 2000; Robbins, 2001). These subunits are expressed widely in the brain and in the striatum in particular (Cooper et al., 2001; Saganich et al., 2001). Our tissue-level RT-PCR analysis has extended this work, showing robust expression of KCNQ2, 3, 4, and 5 mRNA. However, previous attempts to localize KCNQ mRNA and protein in the striatum have focused attention on cholinergic interneurons, leaving it uncertain as to whether KCNQ channels were expressed by the principal medium spiny neurons. Electrophysiological studies of these neurons have not yielded evidence of their expression either. However, KCNQ channels are very sensitive to disruption of the intracellular milieu, as might occur with conventional electrophysiological approaches (Simmons and Schneider, 1998). In our hands, whole-cell dialysis of medium spiny neurons led to the loss of KCNQ-like currents in minutes. With perforated patches, however, linopirdine-sensitive KCNQ channel currents were very stable. The biophysical properties of these pharmacologically isolated currents were very similar to those of KCNQ channel currents found in other neurons (Pan et al., 2001; Shah et al., 2002; Passmore et al., 2003).
The identification of the linopirdine-sensitive channels as KCNQ channels was supported by scRT-PCR. Both striatopallidal and striatonigral medium spiny neurons had readily detectable levels of KCNQ2, 3, and 5 mRNA but not KCNQ4 mRNA (Jentsch, 2000). Based on TEA sensitivity, currents were primarily attributable to heteromeric KCNQ2/KCNQ3 channels (Jentsch, 2000; Schroeder et al., 2000; Hadley et al., 2003).
M1 receptor activation reduces KCNQ K+ channel currents through PIP2-PKC signaling pathway
As in peripheral sympathetic neurons (Marrion, 1997), KCNQ channel currents in medium spiny neurons were suppressed by activation of M1 muscarinic receptors. This conclusion is based on (1) the broad expression of M1 receptors in medium spiny neurons (Yan et al., 2001) and (2) the loss of the muscarinic modulation after genetic deletion of the M1 receptor or pharmacological blockade with muscarinic toxin 7. This signaling configuration differs from that in hippocampal pyramidal neurons in which the muscarinic modulation was not altered by genetic deletion of the M1 receptor or by the M1 receptor-specific toxin (Rouse et al., 2000; Fisahn et al., 2002).
The identity of signaling molecules linking M1 muscarinic receptors to KCNQ channels has been something of a mystery. Recent work has shown that the recovery of KCNQ channels from muscarinic inhibition was blocked by inhibitors of phosphatidylinositol 4-kinase, suggesting that PIP2 resynthesis is a necessary step in the recovery process (Suh and Hille, 2002; Ford et al., 2003; Winks et al., 2005). This dependence suggests that PIP2 binding to the KCNQ subunit is necessary for channel gating (Zhang et al., 2003). Our results show that the M1 receptor modulation of KCNQ channels in medium spiny neurons has a similar PIP2 dependence.
In addition to PIP2 depletion, M1 receptor activation of PLC leads to the production of DAG and PKC stimulation. Several studies have implicated PKC in the modulation of KCNQ channels (Marrion, 1997; Hoshi et al., 2003). Like recent studies (Hoshi et al., 2003), our results show that PKC activation increases the efficacy of muscarinic agonists without changing the maximal modulation.
KCNQ channel currents and up states
In vivo, medium spiny neurons move between hyperpolarized down states to depolarized up states in response to cortical and thalamic glutamatergic synaptic activity. The up-state event is critical to striatal signaling, because medium spiny neurons lack any autonomous activity. These up-state transitions are of variable duration, sometimes lasting seconds (Wilson and Kawaguchi, 1996; Wickens and Wilson, 1998). Previous work has revealed that K+ channels are critical determinants of the up-state membrane potential (Wilson and Kawaguchi, 1996). The ability of KCNQ channels to open and not inactivate in the membrane potential range of the up state perfectly suits them for this role. Our work shows that the linopirdine-sensitive KCNQ channels controlled rheobase and spike threshold. Because of their recruitment by sustained depolarization, these same channels dampened spike frequency over a broad range of somatic currents. Although they may subserve other functions when positioned at other cellular locations (Devaux et al., 2004; Martire et al., 2004), the functional role played by KCNQ channels in medium spiny neurons appears to be similar to that found in peripheral and hippocampal neurons (Wang and McKinnon, 1995; Hu et al., 2002).
KCNQ channels in medium spiny neurons do not act alone in shaping the response to excitatory synaptic input. There are at least three other K+ channels that also participate in regulation of the up state. Inwardly rectifying Kir2 channels are major factors in governing the transition from down state to up state and may contribute a modest sustained outward current during the up state itself (Nisenbaum and Wilson, 1995; Wilson, 2004). Rapidly inactivating Kv4 K+ channels are expressed at modest levels in medium spiny neurons and contribute to the slowing of the membrane potential trajectory to up-state potentials (Nisenbaum and Wilson, 1995; Tkatch et al., 2000). Perhaps the major regulator of this initial phase of the up-state transition is the Kv1.2 K+ channel (Nisenbaum et al., 1994; Shen et al., 2004). But like Kv4 channels, Kv1.2 (or D-type) channels inactivate, albeit more slowly. As these channels inactivate, KCNQ channels open, providing a sustained hyperpolarizing current to modulate the up-state potential for as long as the up state lasts.
Cholinergic modulation of KCNQ channels creates a striatal gating mechanism in learning paradigms
Cholinergic interneurons are thought to act as striatal “teachers.” In associative learning paradigms, presentation of primary, and then secondary, reinforcers induces a pause in interneuronal autonomous activity. This dopamine-dependent pause in activity and lowering of striatal cholinergic tone has been hypothesized to coordinate striatal activity in a way that allows execution of motor tasks (Aosaki et al., 1994; Morris et al., 2004; Yamada et al., 2004). How do fluctuations in cholinergic tone modulate the activity of the principal neurons of the striatum, medium spiny neurons? Voltage-dependent K+ channels regulating state transitions appear to be a major target of cholinergic signaling. Previous studies have suggested that M1 receptor signaling reduces the opening of both Kir2 and Kv4 K+ channels in medium spiny neurons (Akins et al., 1990; Galarraga et al., 1999). Our work complements these studies, showing that KCNQ K+ channel opening is reduced potently by M1 receptor activation. The coordinated modulation of these K+ channels by M1 receptor activation undoubtedly increases the responsiveness of medium spiny neurons to excitatory cortical and thalamic synaptic inputs. Conversely, pauses in the activity of cholinergic interneurons should transiently reduce the excitability of medium spiny neurons.
The modulation of KCNQ channels is likely to be a particularly important part of this coordinated cholinergic gating of medium spiny neuron responsiveness. In anesthetized animals, transitions to the up state can either result in spiking or silence, because neurons remain several millivolts below spike threshold for as long as seconds (Wilson and Kawaguchi, 1996; Tseng et al., 2001). Our results show that relatively small changes in the discharge rate of cholinergic interneurons can translate into substantial differences in KCNQ availability in nearby medium spiny neurons, providing a potential mechanism by which up-state potential could be modulated. What is less clear is whether differences between nearby medium spiny neurons in up-state potential can be attributed to local variation in cholinergic tone or to differences in intracellular mechanisms, such as regulators of G-protein signaling proteins, which regulate receptor coupling (Dohlman and Thorner, 1997; Hepler, 1999).
Alterations in cholinergic tone and KCNQ channel are also likely to be important factors in disease states such as Parkinson's disease (PD). In PD, disinhibition of cholinergic interneurons leads to an elevation in striatal cholinergic tone. Our results and those of others shows how elevated cholinergic tone is likely to be translated into a reduction in KCNQ and Kir2/Kv4 channel opening in medium spiny neurons, increasing their excitability. This augmented responsiveness to excitatory inputs could prove to be particularly important to striatopallidal medium spiny neurons in which enhanced cholinergic tone could synergize with diminished D2 dopamine receptor tone (Albin et al., 1989).
Footnotes
This work was supported by National Institutes of Health Grants NS 34696 (D.J.S.) and NS 26920 (N.M.N.). We thank Dr. Savio Chan for his participation in the initial phase of the experiments and Dr. Tatiana Tkatch, Sasha Ulrich, and Yu Chen for their excellent technical support.
Correspondence should be addressed to Dr. D. James Surmeier, Department of Physiology, Feinberg School of Medicine, Northwestern University, 303 East Chicago Avenue, Chicago, IL 60611. E-mail: j-surmeier@northwestern.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257449-10$15.00/0
References
- Adams PR, Brown DA (1982) Synaptic inhibition of the M-current: slow excitatory post-synaptic potential mechanism in bullfrog sympathetic neurones. J Physiol (Lond) 332: 263-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akins PT, Surmeier DJ, Kitai ST (1990) Muscarinic modulation of a transient K+ conductance in rat neostriatal neurons. Nature 344: 240-242. [DOI] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB (1989) The functional anatomy of basal ganglia disorders. Trends Neurosci 12: 366-375. [DOI] [PubMed] [Google Scholar]
- Aosaki T, Graybiel AM, Kimura M (1994) Effect of the nigrostriatal dopamine system on acquired neural responses in the striatum of behaving monkeys. Science 265: 412-415. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Wilson CJ (1998) Synaptic regulation of action potential timing in neostriatal cholinergic interneurons. J Neurosci 18: 8539-8549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolam JP, Wainer BH, Smith AD (1984) Characterization of cholinergic neurons in the rat neostriatum. A combination of choline acetyltransferase immunocytochemistry, Golgi-impregnation and electron microscopy. Neuroscience 12: 711-718. [DOI] [PubMed] [Google Scholar]
- Brown BS, Yu SP (2000) Modulation and genetic identification of the M channel. Prog Biophys Mol Biol 73: 135-166. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR (1980) Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 283: 673-676. [DOI] [PubMed] [Google Scholar]
- Cimino GD, Metchette K, Isaacs ST, Zhu YS (1990) More false-positive problems. Nature 345: 773-774. [DOI] [PubMed] [Google Scholar]
- Cooper EC, Aldape KD, Abosch A, Barbaro NM, Berger MS, Peacock WS, Jan YN, Jan LY (2000) Colocalization and coassembly of two human brain M-type potassium channel subunits that are mutated in epilepsy. Proc Natl Acad Sci USA 97: 4914-4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EC, Harrington E, Jan YN, Jan LY (2001) M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J Neurosci 21: 9529-9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruzblanca H, Koh DS, Hille B (1998) Bradykinin inhibits M current via phospholipase C and Ca2+ release from IP3-sensitive Ca2+ stores in rat sympathetic neurons. Proc Natl Acad Sci USA 95: 7151-7156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Wanaverbecq N, Abogadie FC, Mistry M, Brown DA (2002) Signaling microdomains define the specificity of receptor-mediated InsP3 pathways in neurons. Neuron 34: 209-220. [DOI] [PubMed] [Google Scholar]
- del Rio E, Bevilacqua JA, Marsh SJ, Halley P, Caulfield MP (1999) Muscarinic M1 receptors activate phosphoinositide turnover and Ca2+ mobilisation in rat sympathetic neurones, but this signalling pathway does not mediate M-current inhibition. J Physiol (Lond) 520: 101-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux JJ, Kleopa KA, Cooper EC, Scherer SS (2004) KCNQ2 is a nodal K+ channel. J Neurosci 24: 1236-1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohlman HG, Thorner J (1997) RGS proteins and signaling by heterotrimeric G proteins. J Biol Chem 272: 3871-3874. [DOI] [PubMed] [Google Scholar]
- Fisahn A, Yamada M, Duttaroy A, Gan JW, Deng CX, McBain CJ, Wess J (2002) Muscarinic induction of hippocampal gamma oscillations requires coupling of the M1 receptor to two mixed cation currents. Neuron 33: 615-624. [DOI] [PubMed] [Google Scholar]
- Ford CP, Stemkowski PL, Light PE, Smith PA (2003) Experiments to test the role of phosphatidylinositol 4,5-bisphosphate in neurotransmitter-induced M-channel closure in bullfrog sympathetic neurons. J Neurosci 23: 4931-4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarraga E, Hernandez-Lopez S, Reyes A, Miranda I, Bermudez-Rattoni F, Vilchis C, Bargas J (1999) Cholinergic modulation of neostriatal output: a functional antagonism between different types of muscarinic receptors. J Neurosci 19: 3629-3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Shapiro MS (2003) Calmodulin mediates Ca2+-dependent modulation of M-type K+ channels. J Gen Physiol 122: 17-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Schofield GG (2003) Activation of muscarinic m5 receptors inhibits recombinant KCNQ2/KCNQ3 K+ channels expressed in HEK293T cells. Eur J Pharmacol 462: 25-32. [DOI] [PubMed] [Google Scholar]
- Hadley JK, Noda M, Selyanko AA, Wood IC, Abogadie FC, Brown DA (2000) Differential tetraethylammonium sensitivity of KCNQ1-4 potassium channels. Br J Pharmacol 129: 413-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley JK, Passmore GM, Tatulian L, Al-Qatari M, Ye F, Wickenden AD, Brown DA (2003) Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J Neurosci 23: 5012-5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton SE, Loose MD, Qi M, Levey AI, Hille B, McKnight GS, Idzerda RL, Nathanson NM (1997) Disruption of the m1 receptor gene ablates muscarinic receptor-dependent M current regulation and seizure activity in mice. Proc Natl Acad Sci USA 94: 13311-13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler JR (1999) Emerging roles for RGS proteins in cell signalling. Trends Pharmacol Sci 20: 376-382. [DOI] [PubMed] [Google Scholar]
- Hersch SM, Gutekunst CA, Rees HD, Heilman CJ, Levey AI (1994) Distribution of m1-m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci 14: 3351-3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Zhang JS, Omaki M, Takeuchi T, Yokoyama S, Wanaverbecq N, Langeberg LK, Yoneda Y, Scott JD, Brown DA, Higashida H (2003) AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat Neurosci 6: 564-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Storm JF (2002) Two forms of electrical resonance at theta frequencies, generated by M-current, h-current and persistent Na+ current in rat hippocampal pyramidal cells. J Physiol (Lond) 545: 783-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR, Kammermeier PJ (2002) M current mystery messenger revealed? Neuron 35: 411-412. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ (2000) Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci 1: 21-30. [DOI] [PubMed] [Google Scholar]
- Jones SW (1985) Muscarinic and peptidergic excitation of bull-frog sympathetic neurones. J Physiol (Lond) 366: 63-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y (1993) Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J Neurosci 13: 4908-4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JN, Plenz D (2002) Dendritic calcium encodes striatal neuron output during up-states. J Neurosci 22: 1499-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi E, Ando K, Nakano H, Iida T, Ohno H, Morimoto M, Tamaoki T (1989) Calphostins (UCN-1028), novel and specific inhibitors of protein kinase C. I. Fermentation, isolation, physico-chemical properties and biological activities. J Antibiot (Tokyo) 42: 1470-1474. [DOI] [PubMed] [Google Scholar]
- Marrion NV (1997) Control of M-current. Annu Rev Physiol 59: 483-504. [DOI] [PubMed] [Google Scholar]
- Marrion NV, Zucker RS, Marsh SJ, Adams PR (1991) Modulation of M-current by intracellular Ca2+ Neuron 6: 533-545. [DOI] [PubMed] [Google Scholar]
- Martire M, Castaldo P, D'Amico M, Preziosi P, Annunziato L, Taglialatela M (2004) M channels containing KCNQ2 subunits modulate norepinephrine, aspartate, and GABA release from hippocampal nerve terminals. J Neurosci 24: 592-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Max SI, Liang JS, Potter LT (1993) Purification and properties of m1-toxin, a specific antagonist of m1 muscarinic receptors. J Neurosci 13: 4293-4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G, Arkadir D, Nevet A, Vaadia E, Bergman H (2004) Coincident but distinct messages of midbrain dopamine and striatal tonically active neurons. Neuron 43: 133-143. [DOI] [PubMed] [Google Scholar]
- Neher E (1992) Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol 207: 123-131. [DOI] [PubMed] [Google Scholar]
- Nisenbaum ES, Wilson CJ (1995) Potassium currents responsible for inward and outward rectification in rat neostriatal spiny projection neurons. J Neurosci 15: 4449-4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisenbaum ES, Xu ZC, Wilson CJ (1994) Contribution of a slowly inactivating potassium current to the transition to firing of neostriatal spiny projection neurons. J Neurophysiol 71: 1174-1189. [DOI] [PubMed] [Google Scholar]
- Nisenbaum ES, Wilson CJ, Foehring RC, Surmeier DJ (1996) Isolation and characterization of a persistent potassium current in neostriatal neurons. J Neurophysiol 76: 1180-1194. [DOI] [PubMed] [Google Scholar]
- Pan Z, Selyanko AA, Hadley JK, Brown DA, Dixon JE, McKinnon D (2001) Alternative splicing of KCNQ2 potassium channel transcripts contributes to the functional diversity of M-currents. J Physiol (Lond) 531: 347-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passmore GM, Selyanko AA, Mistry M, Al-Qatari M, Marsh SJ, Matthews EA, Dickenson AH, Brown TA, Burbidge SA, Main M, Brown DA (2003) KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci 23: 7227-7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M (1991) Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods 37: 15-26. [DOI] [PubMed] [Google Scholar]
- Robbins J (2001) KCNQ potassium channels: physiology, pathophysiology, and pharmacology. Pharmacol Ther 90: 1-19. [DOI] [PubMed] [Google Scholar]
- Robbins J, Caulfield MP, Higashida H, Brown DA (1991) Genotypic m3 muscarinic receptors preferentially inhibit M-currents in DNA-transfected NG108-15 neuroblastoma x glioma hybrid cells. Eur J Neurosci 3: 820-824. [DOI] [PubMed] [Google Scholar]
- Rouse ST, Hamilton SE, Potter LT, Nathanson NM, Conn PJ (2000) Muscarinic-induced modulation of potassium conductances is unchanged in mouse hippocampal pyramidal cells that lack functional M1 receptors. Neurosci Lett 278: 61-64. [DOI] [PubMed] [Google Scholar]
- Saganich MJ, Machado E, Rudy B (2001) Differential expression of genes encoding subthreshold-operating voltage-gated K+ channels in brain. J Neurosci 21: 4609-4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Hechenberger M, Weinreich F, Kubisch C, Jentsch TJ (2000) KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J Biol Chem 275: 24089-24095. [DOI] [PubMed] [Google Scholar]
- Selyanko AA, Brown DA (1996) Intracellular calcium directly inhibits potassium M channels in excised membrane patches from rat sympathetic neurons. Neuron 16: 151-162. [DOI] [PubMed] [Google Scholar]
- Selyanko AA, Hadley JK, Brown DA (2001) Properties of single M-type KCNQ2/KCNQ3 potassium channels expressed in mammalian cells. J Physiol (Lond) 534: 15-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Mistry M, Marsh SJ, Brown DA, Delmas P (2002) Molecular correlates of the M-current in cultured rat hippocampal neurons. J Physiol (Lond) 544: 29-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MS (2004) Why biophysicists make models: quantifying modulation of the M current. J Gen Physiol 123: 657-662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B (2000) Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. J Neurosci 20: 1710-1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Hernandez-Lopez S, Tkatch T, Held JE, Surmeier DJ (2004) Kv1.2-containing K+ channels regulate subthreshold excitability of striatal medium spiny neurons. J Neurophysiol 91: 1337-1349. [DOI] [PubMed] [Google Scholar]
- Simmons MA, Schneider CR (1998) Regulation of M-type potassium current by intracellular nucleotide phosphates. J Neurosci 18: 6254-6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Hille B (2002) Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35: 507-520. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B (2005) Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr Opin Neurobiol 15: 370-378. [DOI] [PubMed] [Google Scholar]
- Suh BC, Horowitz LF, Hirdes W, Mackie K, Hille B (2004) Regulation of KCNQ2/KCNQ3 current by G protein cycling: the kinetics of receptor-mediated signaling by Gq J Gen Physiol 123: 663-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Song WJ, Yan Z (1996) Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci 16: 6579-6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkatch T, Baranauskas G, Surmeier DJ (2000) Kv4.2 mRNA abundance and A-type K+ current amplitude are linearly related in basal ganglia and basal forebrain neurons. J Neurosci 20: 579-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Kasanetz F, Kargieman L, Riquelme LA, Murer MG (2001) Cortical slow oscillatory activity is reflected in the membrane potential and spike trains of striatal neurons in rats with chronic nigrostriatal lesions. J Neurosci 21: 6430-6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, McKinnon D (1995) Potassium currents in rat prevertebral and paravertebral sympathetic neurones: control of firing properties. J Physiol (Lond) 485: 319-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D (1998) KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282: 1890-1893. [DOI] [PubMed] [Google Scholar]
- Wen H, Levitan IB (2002) Calmodulin is an auxiliary subunit of KCNQ2/3 potassium channels. J Neurosci 22: 7991-8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickens JR, Wilson CJ (1998) Regulation of action-potential firing in spiny neurons of the rat neostriatum in vivo J Neurophysiol 79: 2358-2364. [DOI] [PubMed] [Google Scholar]
- Wilson CJ (2004) Basal ganglia. In: The synaptic organization of the brain, Ed 5 (Shepherd GM, ed), pp 361-414. Oxford: Oxford UP.
- Wilson CJ, Kawaguchi Y (1996) The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci 16: 2397-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winks JS, Hughes S, Filippov AK, Tatulian L, Abogadie FC, Brown DA, Marsh SJ (2005) Relationship between membrane phosphatidylinositol-4,5-bisphosphate and receptor-mediated inhibition of native neuronal M channels. J Neurosci 25: 3400-3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Matsumoto N, Kimura M (2004) Tonically active neurons in the primate caudate nucleus and putamen differentially encode instructed motivational outcomes of action. J Neurosci 24: 3500-3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Flores-Hernandez J, Surmeier DJ (2001) Coordinated expression of muscarinic receptor messenger RNAs in striatal medium spiny neurons. Neuroscience 103: 1017-1024. [DOI] [PubMed] [Google Scholar]
- Yu SP, O'Malley DM, Adams PR (1994) Regulation of M current by intracellular calcium in bullfrog sympathetic ganglion neurons. J Neurosci 14: 3487-3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yus-Najera E, Munoz A, Salvador N, Jensen BS, Rasmussen HB, Defelipe J, Villarroel A (2003) Localization of KCNQ5 in the normal and epileptic human temporal neocortex and hippocampal formation. Neuroscience 120: 353-364. [DOI] [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohacs T, Lopes CM, Jin T, Logothetis DE (2003) PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37: 963-975. [DOI] [PubMed] [Google Scholar]
- Zhang W, Yamada M, Gomeza J, Basile AS, Wess J (2002) Multiple muscarinic acetylcholine receptor subtypes modulate striatal dopamine release, as studied with M1-M5 muscarinic receptor knock-out mice. J Neurosci 22: 6347-6352. [DOI] [PMC free article] [PubMed] [Google Scholar]