

Abstract
The fast inhibitory transmitter GABA is robustly expressed in the arcuate nucleus (ARC) and appears to play a major role in hypothalamic regulation of endocrine function and energy homeostasis. Previously, it has not been possible to record selectively from GABA cells, because they have no defining morphological or physiological characteristics. Using transgenic mice that selectively express GFP (green fluorescent protein) in GAD67 (glutamic acid decarboxylase 67)-synthesizing cells, we identified ARC GABA neurons (n > 300) and used whole-cell recording to study their physiological response to neuropeptide Y (NPY), the related peptide YY3-36 (PYY3-36), and pancreatic polypeptide (PP), important modulators of ARC function. In contrast to other identified ARC cells in which NPY receptor agonists were reported to generate excitatory actions, we found that NPY consistently reduced the firing rate and hyperpolarized GABA neurons including neuroendocrine GABA neurons identified by antidromic median eminence stimulation. The inhibitory NPY actions were mediated by postsynaptic activation of G-protein-linked inwardly rectifying potassium (GIRK) and depression of voltage-gated calcium currents via Y1 and Y2 receptor subtypes. Additionally, NPY reduced spontaneous and evoked synaptic glutamate release onto GABA neurons by activation of Y1 and Y5 receptors. The peptide PYY3-36, a peripheral endocrine signal that can act in the brain, also inhibited GABA neurons, including identified neuroendocrine cells, by activating GIRK conductances and depressing calcium currents. The endogenous Y4 agonist PP depressed the activity of GABA-expressing neurons mainly by presynaptic attenuation of glutamate release. Together, these results show that the family of neuropeptide Y modulators reduces the activity of inhibitory GABA neurons in the ARC by multiple presynaptic and postsynaptic mechanisms.
Keywords: neuroendocrine, glutamate, GABA, mediobasal hypothalamus, feeding, neuropeptide Y
Introduction
The arcuate nucleus (ARC) integrates synaptic information and endocrine signals from the blood facilitated by the local weak blood-brain barrier. Axons from the ARC terminate in many regions of the brain, including locally and in the median eminence (ME). ARC neuroendocrine neurons represent the final common neuronal output that regulates the secretions of the anterior pituitary. Many different neurotransmitters and neuromodulators are found in different ARC neurons (Horvath et al., 1997; Kalra, 1997; Elias et al., 1998; Hahn et al., 1998). GABA is found in one-half of the synaptic terminals in ARC (Decavel and van den Pol, 1990) and is synthesized by a subset of ARC neurons (Horvath et al., 1997). GABA neurons play a key role in many of the functions of the ARC, including regulation of the endocrine system and energy homeostasis (Kelly and Grossman, 1979; Kalra and Horvath, 1998; van den Pol et al., 1998; Cone et al., 2001).
ARC GABAergic cells express the functional leptin receptor, suggesting they monitor fat deposition (Ovesjo et al., 2001) and also respond to ghrelin released by the stomach (Wang et al., 2002). Fasting increases the level of the GABA-synthesizing enzyme glutamate decarboxylase (Leonhardt et al., 1999) and GABA agonists increase food intake (Grandison and Guidotti, 1977; Kelly et al., 1977; Morley et al., 1981). GABA axons terminate on ARC cells that play key roles in food intake (Bai et al., 1985; Cowley et al., 2001) and terminate in the ME to modulate anterior pituitary secretions.
One of the highest concentrations of neuropeptide Y (NPY) cell bodies and terminals is in the ARC (McDonald and Koenig, 1993). NPY axons terminate on GABA cells (Cowley et al., 2001). NPY neurons express estrogen and glucocorticoid receptors (Sar et al., 1990; Bonavera et al., 1994; Diano et al., 1998; Fehm et al., 2004). NPY modulates neuroendocrine regulation (luteinizing hormone, luteinizing hormone-releasing hormone, corticotrophin releasing hormone, adrenocorticotropic hormone, vasopressin, growth hormone, thyroid-stimulating hormone) (McDonald and Koenig, 1993) and energy homeostasis (Stanley et al., 1986). G-protein-coupled NPY receptors (Y1-Y5) in the ARC (Inui, 1999; Sun and Miller, 1999) show different affinities for members of the NPY family (Bard et al., 1995; Keire et al., 2000).
Previous work suggested that NPY released by local axons, and peptide YY (PYY) acting as a long-distance signal from the gut, can exert excitatory actions on some ARC cell types, particularly the neurons producing pro-opiomelanocortin (POMC), by reducing tonic inhibitory synaptic transmission (Cowley et al., 2001; Batterham et al., 2002). Other data indicate that NPY inhibits unidentified ARC neurons by activation of Gi/Go-coupled receptors (Rhim et al., 1997; Sun and Miller, 1999). Previous studies used selective reporter gene expression to study ARC neurons that synthesize specific neuropeptides (Cowley et al., 2001, 2003). Because ARC GABA neurons have no defining morphological characteristics, it has not previously been possible to record from them selectively. In the present study using whole-cell recording in hypothalamic slices, we studied the responses of ARC GABA neurons, identified by selective green fluorescent protein (GFP) expression in a novel glutamic acid decarboxylase (GAD)-GFP transgenic mouse (Tamamaki et al., 2003), to NPY, PYY, and pancreatic polypeptide (PP).
Materials and Methods
Animals. GABAergic neurons in the hypothalamic ARC were identified by using GAD67-GFP (Δneo) transgenic mice in which GFP is selectively expressed under the control of the endogenous GAD67 gene promoter as described previously (Tamamaki et al., 2003). In this study, these transgenic mice are called GAD67-GFP mice for simplicity. All of the experimental procedures involving animals were approved by the Yale University Committee on Animal Care and Use.
In situ hybridization and immunohistochemistry for GAD67. In situ hybridization was performed to identify GAD67 mRNA-producing neurons in the hypothalamus. RNA probes were synthesized with a digoxigenin labeling kit, using as a template a plasmid in which a DNA sequence corresponding to a segment of the mouse GAD67 cDNA was cloned as described previously (Tamamaki et al., 2003). Blocks of hypothalamic tissue from adult mice were obtained and treated as described in detail previously (Tamamaki et al., 2003).
Colocalization of GAD67 immunoreactivity and GFP expression was determined in hypothalamic sections (20 μm) prepared after perfusion of GAD67-GFP transgenic mice (n = 5) with 4% paraformaldehyde. Cryostat-cut hypothalamic sections were incubated overnight with mouse primary antibodies selective for GAD67 (Chemicon, Temecula, CA). The tissue was next incubated in goat anti-mouse Texas Red-conjugated secondary antibodies (Molecular Probes, Eugene, OR) for 60 min, and analyzed under an Olympus (Tokyo, Japan) IX70 microscope equipped with fluorescence capabilities. A Spot digital camera (Diagnostic Instruments, Sterling Heights, MI) was used to produce photomicrographs. Contrast and brightness were adjusted with Photoshop 7.0 (Adobe Systems, Mountain View, CA).
Hypothalamic slices. Two- to 3-week-old animals were deeply anesthetized (sodium pentobarbital; 100 mg/kg), and then the brains were rapidly removed and placed in ice-cold oxygenated (95% O2, 5% CO2) high-sucrose solution that contained the following (in mm): 220 sucrose, 2.5 KCl, 6 MgCl2, 1 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose, pH 7.4 with NaOH. A block of tissue containing the ARC was excised and transferred to a vibratome where 250-350 μm transversal sections were obtained.
The hypothalamic slices were gently moved to an equilibrium chamber filled with oxygenated artificial CSF (ACSF) solutions that contained the following (in mm): 124 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 1.23 NaH2PO4, 26 NaHCO3, 10 glucose, pH 7.4 with NaOH. After 2 h of recovery from sectioning, the slices were transferred to a recording chamber mounted on an Olympus BX51WI upright microscope. Tissue was continuously superfused with ACSF solution. The microscope was equipped with video-enhanced infrared-differential interference contrast (DIC) and fluorescence capabilities. The temperature in the recording chamber was kept near 36°C using a dual-channel heater controller (Warner Instruments, Hamden, CT).
Electrophysiological recordings. Whole-cell voltage- and current-clamp recording were performed by using low-resistance (3-6 MΩ) borosilicate pipettes. Recording pipettes were pulled with a PP-83 vertical puller (Narishige, Tokyo, Japan) and filled with an intracellular solution that contained the following (in mm): 130 KMeSO4 (or KCl for IPSCs), 1 MgCl2, 10 HEPES, 1.1 EGTA, 2 Mg-ATP, 0.5 Na2-GTP, and 10 Na2-phosphocreatin, pH 7.3 with KOH. In the experiments in which voltage-activated calcium currents were studied, a Cs-based pipette solution was used and contained the following (in mm): 130 CsCH3SO3, 1 MgCl2, 10 HEPES, 11 EGTA-Cs, 2 Mg-ATP, and 0.5 Na2-GTP, pH 7.3 with CsOH. In these experiments, we used a modified external solution that contained the following (in mm): 78.5 NaCl, 40 tetraethylammonium chloride (TEA-Cl), 3 KCl, 2 MgCl2, 5 BaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4 with HCl.
GABAergic GFP-expressing neurons in the ARC were identified under fluorescence, and then DIC was used to achieve the cell-attached configuration in these cells. Whole-cell mode was obtained after gentle application of negative pressure through the recording pipette by using a glass syringe. Slow and fast capacitive components were automatically compensated using Pulse software (HEKA Elektronik, Lambrecht/Pfalz, Germany). Access resistance was monitored throughout the experiments, and only those cells with stable access resistance (changes <10%) were used for analysis. The recordings were made with an EPC9 amplifier controlled with a Pulse 8.54 software (HEKA Elektronik). Unless otherwise specified, all drugs were directly applied to the recorded cell by using a low-resistance flow pipe aimed at the recorded cell.
In the evoked potential experiments, we used a bipolar electrode (World Precision Instruments, Sarasota, FL) to deliver electrical stimulation (50-100 μA; 0.2-0.5 ms; 0.1-0.2 Hz) as described previously (Acuna-Goycolea et al., 2004). Excitatory potentials were obtained after stimulation with an electrode placed within the ARC, usually lateral to the recorded cell. The experiments on excitatory evoked potentials were done with 30 μm bicuculline (BIC) in the bath to block inhibitory GABAA receptor-mediated synaptic transmission. In some experiments, GABA neurons that sent axons to the median eminence were studied. To identify these neuroendocrine cells, we used antidromic axonal stimulation with a bipolar electrode (50-100 μA; 0.2-0.5 ms; 0.5 Hz) placed in the median eminence (van den Pol et al., 1998). At the end of each experiment, median eminence stimulation was performed in the presence of CdCl2 (200 μm) in the external solution to block all orthodromic axonal synaptic release. TTX was also applied by flow pipe to the median eminence to block voltage-dependent sodium-mediated antidromic spikes.
Data analysis and statistics. PulseFit 8.54 (HEKA Electronik), Axograph 4.7 (Molecular Devices, Palo Alto, CA), and Igor Pro 4.07 (Wave-Metrics, Lake Oswego, OR) software were used for analysis. Spontaneous synaptic currents were detected using Axograph software (Clements and Bekkers, 1997; Gao and van den Pol, 2001). The detection was performed by comparison of the spontaneous events with a simulated template based on a double exponential function in which the time course (rise and decay) as well as the width of a typical synaptic current was used. The template function was moved along the recorded trace, and individual events were compared with the template. All of the events with amplitude >5 pA were included in the analysis. Details of this procedure have been described previously (Bekkers and Stevens, 1995; Gao and van den Pol, 2001).
Data in Results are expressed as mean ± SEM. For synaptic-activity population graphs, the average number of events in control (5 min), drug application (3 min), and recovery (5 min) conditions were obtained and then normalized as the spontaneous synaptic current frequency varies within the population. Data for control conditions did not include 2 min of stabilization that followed whole-cell configuration achievement. The peak peptidergic effects on synaptic activity were typically observed 1 min after peptide application and persisted for at least two more minutes, and therefore this time window was used for data analysis. Statistical analyses were performed using one-way ANOVA followed by a Bonferroni post hoc procedure for pairwise between-group comparison (i.e., control vs treatment). The nonparametric Kolmogorov-Smirnov test was used for comparison of the cumulative fractions before and during drug applications. A value of p < 0.05 was considered statistically significant.
Results
Hypothalamic GFP expression in GAD67 transgenic mice
Previously, GFP expression was shown exclusively in neurons that contained the GABA-synthesizing enzyme GAD67, as detected with in situ hybridization and immunocytochemistry (Tamamaki et al., 2003; Jiang et al., 2004; Li et al., 2005). GFP- and GAD67-double immunocytochemistry on dissociated cells also revealed that the GFP-containing cells were consistently positive for GAD67 and GABA (our unpublished data). We further examined the distribution of GAD67-expressing neurons in the hypothalamus using in situ hybridization histochemistry. In situ hybridization revealed a prominent GAD67 mRNA labeling in ARC as indicated by the black arrows in Figure 1A-C (Ovesjo et al., 2001). This region was the target of the whole-cell recording experiments described below. Robust expression of GAD67 was also evident in the perifornical/lateral hypothalamus (LH) (Rosin et al., 2003) as well as dorsomedial nucleus (DMH) (Herman et al., 2002), but not in the ventromedial hypothalamic nucleus (VMH) (Fig. 1A,B). In the posterior hypothalamus, GAD67 expression was consistently found in the tuberomammillary nucleus (TMN), in accordance with previous findings (Ericson et al., 1991; Airaksinen et al., 1992). Sections obtained from GAD67-GFP transgenic mice, showed a similar distribution of GFP throughout the hypothalamus. GFP-synthesizing neurons were detected in ARC, LH, DMH, and TMN (Fig. 1D-F). Figure 1G-I shows high-power magnifications of the boxed areas in Figure 1D-F, respectively, revealing a large GABAergic cell population throughout the rostrocaudal extent of the ARC.
Figure 1.
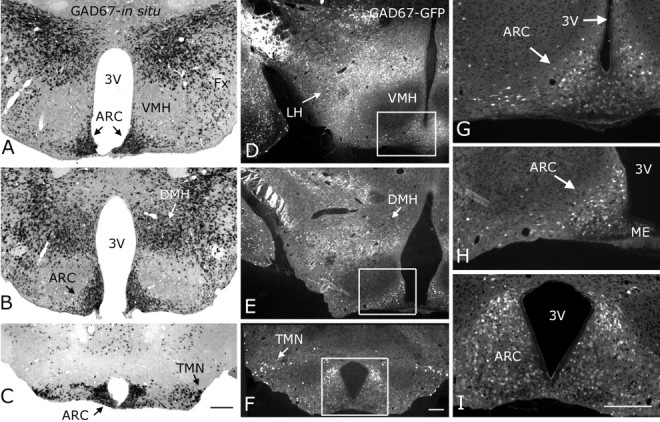
GAD67 mRNA matches GAD67-GFP in transgenic mice. A-C, In situ hybridization showing GAD67 mRNA (small black spots) was strongly expressed in ARC, lateral hypothalamus, dorsomedial nucleus, and tuberomammillary nucleus in the posterior hypothalamus. Very low levels of GAD67 mRNA were found in the ventromedial hypothalamic nucleus. D-F, GFP-producing cells (GFP shown in white) were detected in the same hypothalamic regions where GAD67 mRNA was found. G-I, High-power magnification of the ARC boxed areas in D-E, respectively. Scale bars: C (for A-C), F (for D-F), I (for G-I), 200 μm. 3V, Third ventricle; Fx, fornix.
A GABAergic neuronal population has been reported previously in the suprachiasmatic nucleus (SCN) (van den Pol, 1980; van den Pol and Tsujimoto, 1985; Moore and Speh, 1993). Consistent with this, strong GFP expression was detected in SCN sections from GAD67-GFP transgenic mice (Fig. 2B). No expression of GFP was found in the magnocellular neurons of the paraventricular (PVN) (Fig. 2D) or supraoptic nuclei (SON) (Fig. 2C) of the hypothalamus, consistent with the lack of GABAergic neurons in these hypothalamic magnocellular neurons reported previously (Boudaba et al., 1996; Bowers et al., 1998). GFP was found in the medial parvicellular PVN, in accordance with previous studies (Decavel and van den Pol, 1990). Immunostaining (using secondary antibodies coupled to Texas Red) for GAD67 in hypothalamic sections from GAD67-GFP mice showed fluorescence colocalization, both in the medial (Fig. 2E1,E2) and lateral (Fig. 2F1,F2) hypothalamus (Fig. 2E1-F2, both green GFP and Texas Red are shown in white), supporting the view that the GFP expression in this line of transgenic animals is selectively restricted to GAD67-containing neurons. This is consistent with previous reports on the same transgenic mouse, examining other regions of the brain (Tamamaki et al., 2003; Jiang et al., 2004; Li et al., 2005).
Figure 2.
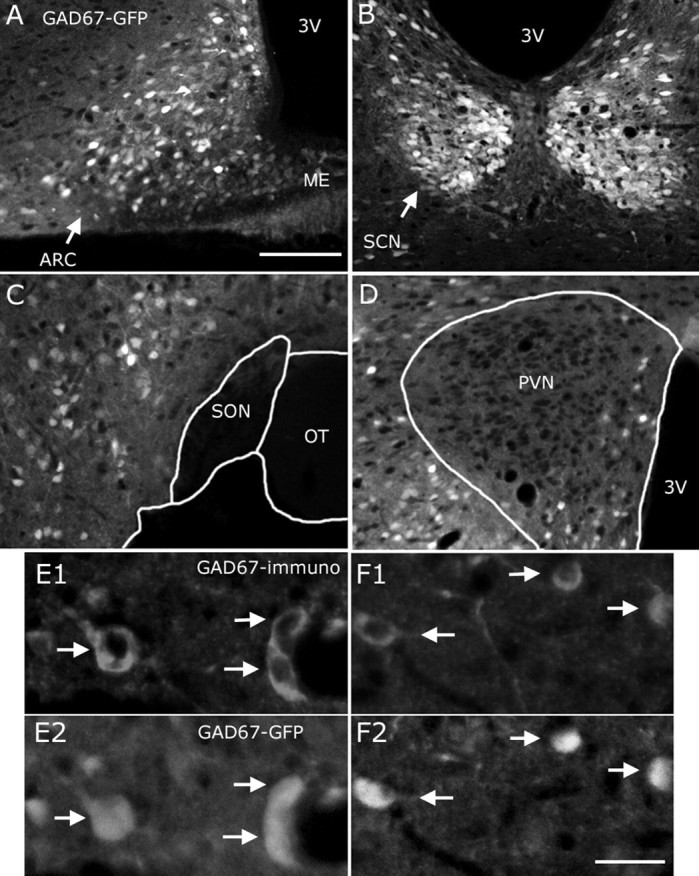
Colocalization of GFP and GAD67 immunoreactivity in the hypothalamus of GAD67-GFP mice. In addition to the ARC (A), GFP was also detected in the suprachiasmatic nucleus (B) in the transgenic mice. GFP was found in neither the supraoptic (C) nor the magnocellular area of the paraventricular hypothalamic nucleus (D). In both the medial (E1-E2, white arrows) and lateral (F1-F2, white arrows) hypothalamus, GFP colocalized with GAD67 immunoreactivity, consistent with the selective GFP expression in GAD67-producing neurons. In this figure, both green (GFP expression) and red (Texas Red from secondary antibody) fluorescence are presented in white. Scale bars: (in A) A-D, 200 μm; (in F2) E1-F2, 20 μm. 3V, Third ventricle; OT, optic tract.
Basic properties of ARC GAD67-GFP cells
ARC neurons identified by GAD67-GFP expression had a mean resting membrane potential of -50.6 ± 3.2 mV (n = 11), showed spontaneous spikes (2.3 ± 0.6 Hz; range, 0-5.51 Hz; n = 9) and had an input resistance of ∼1.1 ± 0.35 GΩ (n = 6). These characteristics are similar to those described after identification of GABA cells by single-cell reverse transcription-PCR (Burdakov et al., 2003). Typically, low-threshold spikes were evoked after the application of a negative current step through the recording pipette (-30 pA during 500 ms). When positive current injection protocols (+60 pA during 500 ms) were delivered to these hypothalamic neurons, spike-frequency adaptation was evident with the last spikes showing a greater interspike interval than the first of the series (the interspike interval increased by 107.5 ± 31.05% when the last and first one-half of a series of spikes were compared; n = 7) (data not shown).
NPY inhibits arcuate GAD67-expressing neurons
We evaluated NPY actions on the frequency of spontaneous action potentials of GAD67-containing neurons in the ARC under current clamp. NPY consistently hyperpolarized and depressed the spontaneous firing by 90.3 ± 5.7% in these hypothalamic cells (n = 18; p < 0.01) (Fig. 3A). In six cells that did not fire under resting conditions, NPY induced a -6.2 ± 3.5 mV shift in membrane potential (p < 0.05). In voltage-clamp conditions, NPY (1 μm) evoked an outward current when the cells were held at -60 mV (Fig. 3B). The mean amplitude of this outward current was 9.56 ± 2.75 pA (n = 8; p < 0.05). Similarly to its actions on spike frequency and membrane potential, the NPY actions on the outward current were long-lasting, and partial recovery was achieved after 10-20 min of peptide washout (Fig. 3B).
Figure 3.
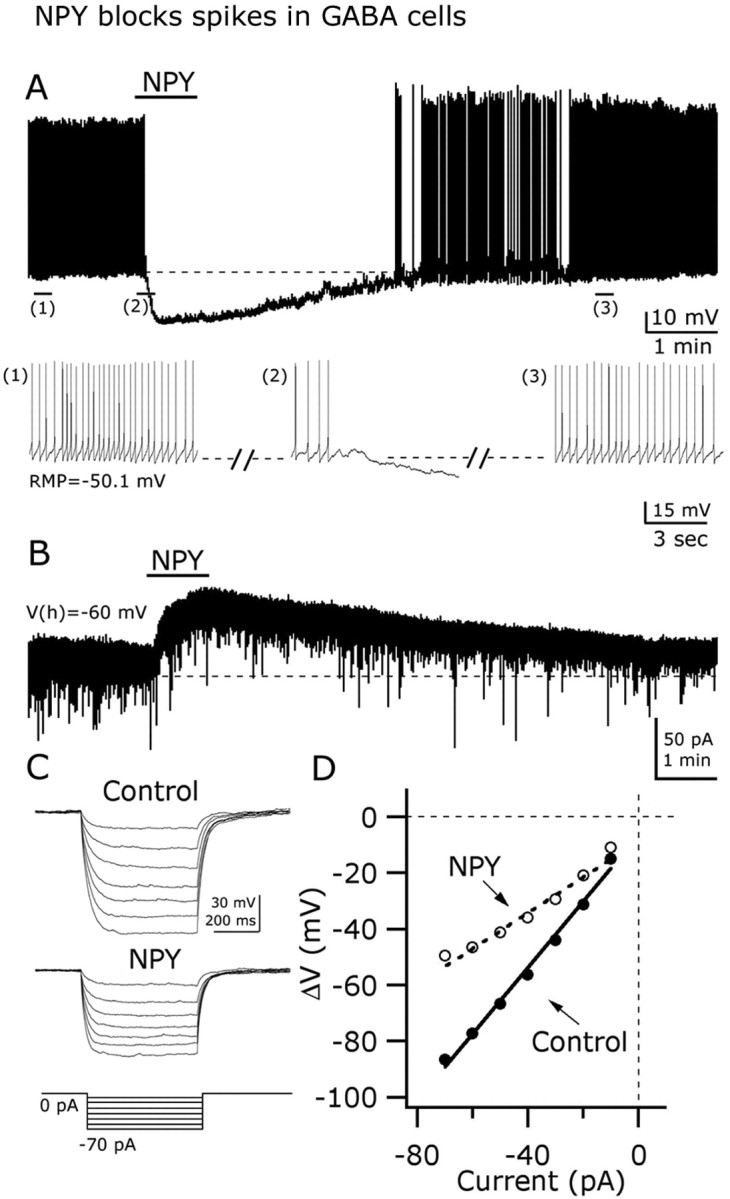
NPY inhibits GAD67-expressing neurons in arcuate nucleus. A, Current-clamp recording from a representative neuron showing the inhibitory effect of 1 μm NPY on the frequency of action potentials. A hyperpolarizing shift in the membrane potential is also evident after NPY application. Extended traces before, during, and after NPY are depicted in the bottom part (1, 2, and 3 indicate selected parts of the trace). B, In voltage-clamp (Vh = -60 mV), NPY evoked a robust and long-lasting outward current. C, NPY decreased the voltage response of GAD67 neurons after hyperpolarizing current steps (left) (see protocol in the bottom part). D, This graph shows the current-voltage relationship in the absence (right, filled circles) and the presence (white circles) of NPY. NPY evoked a decrease in the whole-cell input resistance (slope). RMP, Resting membrane potential.
To determine whether the NPY inhibitory actions on GAD67-expressing neurons were accompanied by changes in the whole-cell input resistance, we delivered negative current steps (from -10 to -70 pA during 500 ms; increments of 10 pA) (Fig. 3C, bottom) through the recording pipette and evaluated the changes in the membrane potential before and after 1 μm NPY application. With NPY, the hyperpolarizing shifts in the membrane potential in response to the injection of negative current steps were drastically reduced (Fig. 3C), and current-voltage relationship showed a consistent alteration compared with control pre-NPY conditions (Fig. 3D). A linear function was fitted to the current-voltage relationship, and a significant decrease in the slope was observed after NPY application, consistent with a reduction in the whole-cell input resistance from 1.17 ± 0.16 to 0.64 ± 0.25 GΩ (n = 4; p < 0.05) (Fig. 3D). Together, these results show that NPY actions in ARC GAD67-GFP neurons involve hyperpolarization, an outward current at -60 mV holding potentials, a decrease in the membrane input resistance, and a robust attenuation of spike frequency.
NPY inhibits neuroendocrine GABA cells
It has been reported that a subset of ARC neurons, including some that synthesize GABA, send their axons to the ME where they can release trophic factors that control anterior pituitary hormone secretion (Sawaki and Yagi, 1973; Tappaz et al., 1983; Merchenthaler, 1991). We evaluated whether NPY would affect the activity of these neuroendocrine GABAergic cells in hypothalamic slices in vitro. To identify GFP-expressing cells that innervate the ME, we used antidromic electrical stimulation protocols. A bipolar electrode was placed in the ME (Fig. 4A), and 50-100 μA current steps for 0.2-0.5 ms were delivered at 0.5 Hz. All experiments were done in 300-μm-thick slices to preserve arcuate GABA neurons axonal endings in ME. Typically, fixed-latency (near 2 ms) action potentials were evoked in ARC neurons after ME stimulation (Fig. 4B, inset). These action potentials were completely abolished after focal 0.5 μm TTX flow-pipe application to the median eminence, suggesting that the spikes were dependent on the activation of axonal voltage-gated sodium channels (n = 6) (data not shown). In addition, evoked spikes persisted in the presence of 200 μm external CdCl2, suggesting that they were not attributable to transmitter release after orthodromic activation of recurrent collaterals (Fig. 4B). Not all recorded GAD67-GFP showed antidromic responses to median eminence stimulation, and the number of responding cells was reduced in thinner brain slices.
Figure 4.

NPY inhibits ARC neuroendocrine GABAergic neurons. A, Schematic representation of hypothalamic slice preparation used to study neuroendocrine cells. 3V, Third ventricle. B, Antidromic responses (action potentials) were evoked in GFP-expressing cells after median eminence electrical stimulation with a bipolar electrode. The action potentials induced by ME antidromic stimulation persisted in 200 μm CdCl2 in the bath, indicating that these responses were not attributable to transmitter release induced by orthodromic activation of recurrent axon collaterals. Inset shows the spike latency and initial rise of an action potential evoked by ME stimulation. C, In this typical cell, negative current steps (see protocol in the bottom part) were delivered before (control) and during NPY application. A robust reduction in the hyperpolarizing responses after negative current injection was observed in the presence of NPY, consistent with a decrease in whole-cell input resistance. D, In current-clamp conditions, NPY blocked spikes and hyperpolarized ARC neuroendocrine neurons. E, F, NPY-induced reduction in spike frequency and hyperpolarization in neuroendocrine neurons were not significantly different from that of other ARC GABA cells. Error bars indicate SE. The number of cells is shown in parentheses.
To determine the effect of NPY on the input resistance in ARC neuroendocrine cells, negative current steps (from -10 to -60 pA during 100 ms at 2 s intervals) were delivered through the recording pipette; the membrane potential responses before and during NPY application were evaluated. NPY (1 μm) reduced the hyperpolarization evoked by negative current steps in comparison to control pre-NPY conditions (n = 4) (Fig. 4C), reducing membrane input resistance from 1.03 ± 0.24 to 0.81 ± 0.36 GΩ. In current clamp, 1 μm NPY almost completely blocked neuroendocrine cell spikes (reduction in spike frequency after NPY, 87.8 ± 4.8%; n = 5; p < 0.05) (Fig. 4D). In addition, ARC neuroendocrine GABA cells were hyperpolarized by 8.3 ± 1.2 mV in the presence of NPY (n = 9; p < 0.05). The NPY actions on input resistance, spike frequency, and membrane potential in neuroendocrine GABA cells were not significantly different from NPY effects on the other ARC GABA cells in which the neuroendocrine nature was not tested (Fig. 4E,F).
Direct postsynaptic NPY actions via Y1 and Y2 receptor subtype activation
The hyperpolarization induced by NPY might be explained by direct postsynaptic activation of its receptors in GABA neurons. To evaluate this, we performed current-clamp experiments in the presence of 0.5 μm TTX in the bath. These experiments were also done with AP-5 (50 μm), CNQX (10 μm), and BIC (30 μm) in the ACSF to prevent any modulation of synaptic glutamate or GABA release onto recorded neurons. NPY induced a robust and long-lasting hyperpolarizing shift in the membrane potential as shown in Figure 5A,B, trace 1. NPY (0.1 or 1 μm) dose-dependently hyperpolarized GABA neurons by 6.0 ± 1.2 (n = 6) and 8.7 ± 3.5 mV (n = 5), respectively (p < 0.05) (Fig. 5B). This indicates that NPY-mediated hyperpolarization is caused by direct activation of postsynaptic receptors. Doses of 0.01 μm did not significantly change the membrane potential (n = 5; p > 0.05).
Figure 5.
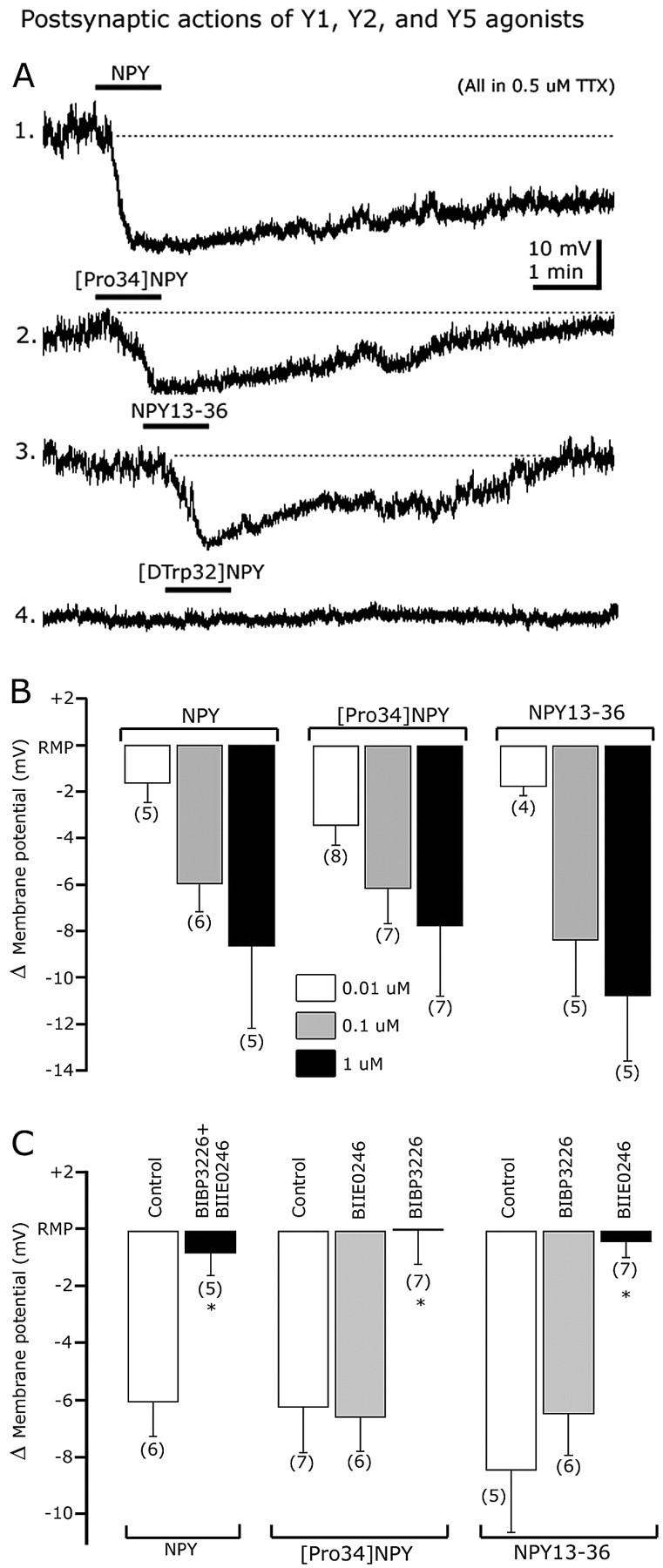
Y1 and Y2, but not Y5, receptor subtypes are involved in the direct dose-dependent postsynaptic actions of NPY. A, Representative traces showing the inhibitory effect of NPY (trace 1), [Pro34]NPY (a selective Y1 agonist) (trace 2), and NPY13-36 (a Y2 agonist) (trace 3) on the membrane potential of identified GAD67 neurons. The Y5 receptor agonist [d-Trp32]NPY1-36 (trace 4) did not alter the membrane potential. B, Dose-dependent effects of NPY (left), [Pro34]NPY1-36 (middle), and NPY13-36 (right) on membrane potential. C, Bar graph summarizing Y1 and Y2 agonist and antagonist actions on hyperpolarization induced by NPY (left), [Pro34]NPY1-36 (middle), and NPY13-36 (right). Error bars indicate SE. *Statistically significant. The number of cells is shown in parentheses.
It has been reported that central NPY effects are mediated by different Y receptors, including Y1, Y2, and Y5 (Chen and van den Pol, 1996; Inui, 1999; Sun and Miller, 1999; Schober et al., 2000; Pronchuk et al., 2002; Fu et al., 2004). To determine the receptor subtypes involved in postsynaptic NPY actions, we first tried the selective Y1 agonist [Pro34]NPY1-36 (Potter et al., 1991). [Pro34]NPY1-36 hyperpolarized GABA cells in a dose-dependent manner (Fig. 5B); 0.01, 0.1, and 1 μm [Pro34]NPY1-36 shifted the membrane potential by -3.5 ± 0.8 (n = 8), -6.2 ± 1.5 (n = 7), and -7.8 ± 2.9 mV (n = 7) (Fig. 5A, trace 1). These [Pro34]NPY1-36 effects were likely attributable to activation of postsynaptic Y1 receptors, because they were eliminated by pretreatment with the selective Y1 antagonist (R)-N2-(diphenylacetyl)-N-[(4-hydroxyphenyl)methyl]-d-arginine-amide (BIBP3226) (Doods et al., 1996; Sun et al., 2003). A selective Y2 antagonist, (S)-N2-[[1-[2-[4-[(R,S)-5,11-dihydro-6(6H)-oxodibenz [b,e]azepin-11-yl]-1-piperazinyl]-2-oxoethyl]cyclopentyl]acetyl]-N-[2-[1,2-dihydro-3,5(4 H)-dioxo-1,2-diphenyl-3H-1,2,4-triazol-4-yl]ethyl]argininamide (BIIE0246) (Doods et al., 1999), did not block the hyperpolarization induced by 100 nm [Pro34]NPY1-36 (n = 6) (Fig. 5C), further substantiating a Y1 selectivity of [Pro34]NPY1-36. Together, these results suggest that postsynaptic NPY effects are mediated in part by somatodendritic Y1 receptors.
In other hypothalamic neurons, postsynaptic NPY actions have been shown to depend mainly on the activation of Y1 receptor (Rhim et al., 1997; Fu et al., 2004; Roseberry et al., 2004). In hypothalamic slices incubated with 1 μm BIBP3226, NPY still hyperpolarized ARC GABA neurons by 5.2 ± 1.4 mV (n = 5; p < 0.05) (data not shown), suggesting that another NPY receptor, in addition to Y1, is involved in the postsynaptic actions of this peptide. Coapplication of BIBP3226 (1 μm) and BIIE0246 (100 nm) prevented the NPY-induced hyperpolarization (n = 5) (Fig. 5C), further suggesting a role for Y2 receptor subtype in postsynaptic NPY effects. Consistent with this, the specific Y2 agonist NPY13-36 (1 μm) (Guo et al., 2002) also induced a consistent hyperpolarization (Fig. 5A, trace 3) of 10.8 ± 3.8 mV (1 μm NPY13-36), returning toward control levels after 5-10 min of peptide washout (n = 5) (Fig. 5B). Concentrations of 0.01 and 0.1 μm NPY13-36 hyperpolarized GABA neurons by 1.8 ± 0.4 (n = 4) and 8.4 ± 2.4 (n = 5) mV, respectively, consistent with a dose-dependent effect (Fig. 5B). In addition, 100 nm NPY13-36-evoked hyperpolarization was abolished when the slices were pretreated with the Y2 antagonist BIIE0246 (n = 7; p < 0.05) (Fig. 5C). Preincubation with BIBP3226 (1 μm) did not modify 100 nm NPY13-36-mediated changes in membrane potential (p > 0.05), consistent with activation of a Y2 receptor (Guo et al., 2002).
In contrast, the application of the Y5 agonist [d-Trp32] NPY1-36 (1 μm) (Inui, 1999; Guo et al., 2002) had a nonsignificant effect on the membrane potential (0.56 ± 1.09 mV compared with control pretreatment levels; n = 5; p = 0.88) (Fig. 5A, trace 4). Together, these experiments suggest that Y1 and Y2, but not Y5, receptors contribute to the direct inhibitory actions of NPY.
NPY activates inwardly rectifying potassium currents
The experiments above (Fig. 3) showed that inhibitory NPY actions in GAD67 neurons involved a decrease in whole-cell input resistance consistent with channel opening. If these inhibitory effects were mediated by the activation of NPY-sensitive inwardly rectifying potassium channels (Sun et al., 2001a; Fu et al., 2004; Roseberry et al., 2004), a suppression of its actions in the presence of external barium would be expected (Sodickson and Bean, 1996). To evaluate this, we performed experiments in the presence of 0.8 mm barium in the external solution and found that NPY (1 μm) had little hyperpolarizing effect under these conditions (change in the membrane potential, 1.22 ± 0.66 mV; n = 6; p > 0.05). To confirm that NPY activated a potassium current, a voltage ramp (from -140 to -10 mV during 800 ms) (Fig. 6A, top) was used with 0.5 μm TTX and 200 μm CdCl2 in the ASCF to prevent the activation of voltage-dependent sodium and calcium channels, respectively.
Figure 6.

NPY activates a GIRK-like current in arcuate GABA neurons. A, Typical current responses of an identified GAD67 cell before (black trace) and during (gray trace) the application of NPY (1 μm) to the slice. The voltage-ramp protocol used in this experiment is shown in the top part. The net NPY-induced current (inset) was obtained by subtracting control from NPY components. B, NPY-induced current was altered by changing extracellular K+ from 3 to 15 mm. A positive shift in the reversal potential was evident for 15 mm potassium in the bath. C, The current evoked by NPY was G-protein activation dependent, and sensitive to 800 μm barium in the external solution, consistent with a GIRK-type conductance. D, Similarly, both the selective Y1 and Y2 receptor agonists evoked inwardly rectifying currents in arcuate GAD67 neurons. A Y5 receptor agonist, [d-Trp32]NPY1-36, had no effect on the GIRK-like current in these hypothalamic neurons.
Figure 6A shows representative current responses recorded during voltage-ramp commands in control (black trace) and 1 μm NPY (gray trace) in a typical cell (with 15 mm extracellular K+). The net NPY-induced current was obtained by subtracting control from NPY components (Fig. 6A, inset). The reversal potential of the current induced by NPY was dependent on the extracellular K+ concentration (Fig. 6B). Under normal 3 mm extracellular K+ (intracellular potassium concentration, 145 mm), the reversal potential of the NPY-induced current was -96.9 ± 2.8 mV (n = 5). With 15 mm external K+ (KCl was substituted for an equimolar amount of NaCl), the current induced by NPY showed a reversal potential of -58.1 ± 3.85 mV (n = 6). This 38.7 mV shift in the reversal potential after increasing the external K+ concentration from 3 to 15 mm is similar to the value predicted by the Nernst equation for a pure potassium current (positive shift in reversal potential predicted by Nernst under our experimental conditions, 42.9 mV). The NPY-induced current showed inward rectification at potentials near -20 mV (Fig. 6A,B, inset) and was abolished by barium (0.8 mm) in the bath (Fig. 6C, thinner black trace), consistent with an inwardly rectifying potassium conductance (Sodickson and Bean, 1996). We also used the nonhydrolyzable GDP analog GDP-β-S instead of GTP in the recording pipette. After 5-12 min of dialysis under these conditions, we did not detect any NPY effect on current responses induced by voltage-ramp protocols compared with the control pretreatment response (Fig. 6C, gray trace), suggesting that NPY hyperpolarizing actions in GABA neurons may be attributable to activation of G-protein-linked inwardly rectifying potassium (GIRK)-like channels in postsynaptic compartments (Sodickson and Bean, 1996; Fu et al., 2004).
We then tested whether agonists of Y1 and Y2 receptor subtypes might lead to the activation of GIRK-like current in arcuate GABAergic neurons. [Pro34]NPY1-36 (1 μm) induced a current that reversed at -58.8 ± 3.6 mV with 15 mm extracellular K+ concentration (n = 8) (Fig. 6D, thicker black trace). Similarly, the Y2 agonist NPY13-36 (1 μm) evoked an inwardly rectifying current that reversed at -56.4 ± 2.5 mV under the same experimental conditions (15 mm external K+) (Fig. 6D, gray trace). In contrast, application of [d-Trp32]NPY1-36 (1 μm) (a Y5 agonist) did not alter the current response induced by voltage ramps (Fig. 6D, thin black trace), implying that Y1 and Y2, but not Y5, receptor subtypes can activate GIRK-like current in GAD67 neurons in the arcuate.
Y1 and Y2 agonists depress whole-cell calcium currents
The modulation of calcium current represents another important mechanism by which neuromodulators might alter neuronal excitability (Dolphin, 2003). We evaluated whether NPY may influence whole-cell calcium currents in arcuate GAD67-containing neurons. Calcium currents were activated by a voltage step from -80 to 0 mV for 200 ms (Acuna-Goycolea and van den Pol, 2004) (Fig. 7A, top part). CsMeSO3 was used in the recording pipette and 40 mm TEA-Cl in the external solution to prevent the activation of potassium conductances (NaCl was replaced by an equimolar concentration of TEA-Cl). TTX (0.5 μm) was also added to the ACSF to block voltage-dependent sodium channels. To increase the conductance of calcium channels, 5 mm BaCl2 was used in the bath solution instead of CaCl2. Application of NPY (1 μm) to the bath resulted in a robust decrease in the whole-cell barium current as shown in Figure 7A. At the end of this experiment, 200 μm CdCl2 was applied to the ACSF and the current was abolished, confirming that it was attributable to the opening of voltage-dependent calcium channels (Fig. 7A). In eight cells tested, NPY (1 μm) reduced barium current by 25.4 ± 5.1% (p < 0.05) (Fig. 7E). In Figure 7B, the dose dependence of the effect of 0.1 and 1 μm NPY on whole-cell barium currents is presented.
Figure 7.
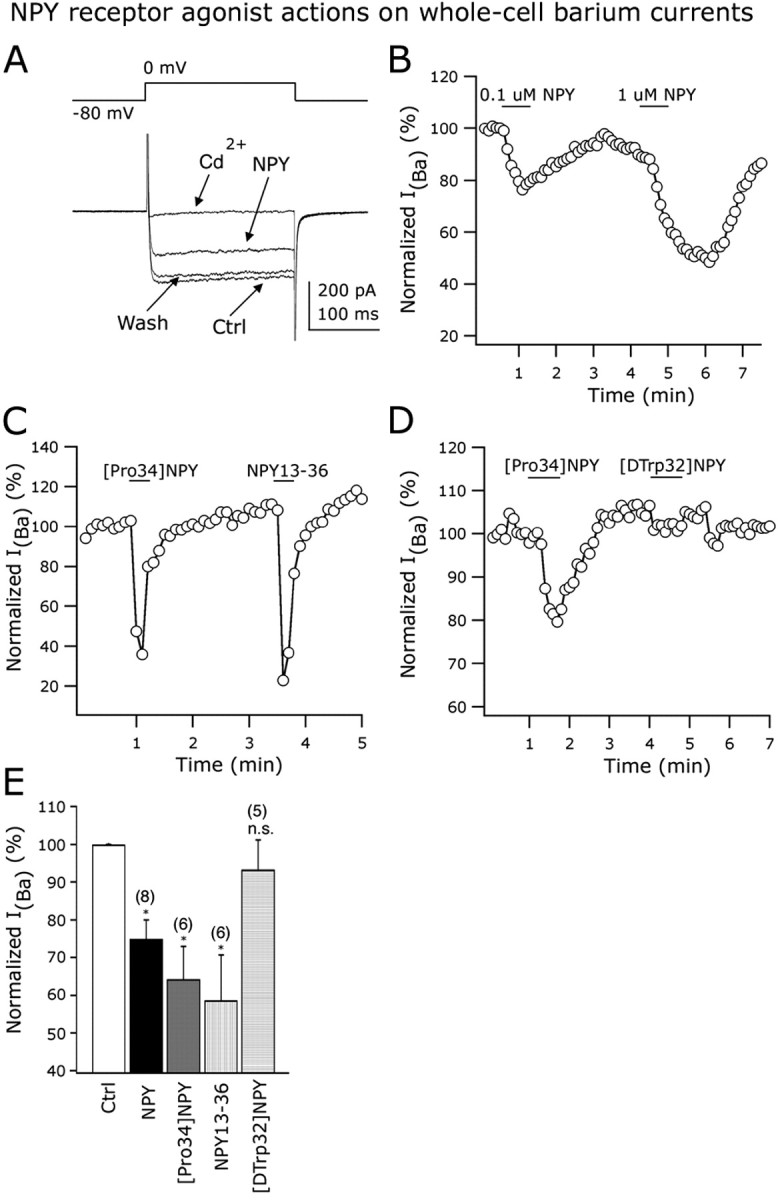
Y1 and Y2 receptors inhibit voltage-dependent calcium channels. External calcium was replaced by barium in all of these experiments. A, A representative neuron showing the inhibitory effect of 1 μm NPY on the whole-cell IBa. Bath application of 200 μm CdCl2 abolished the IBa, consistent with the idea that IBa was mediated by voltage-dependent calcium channels. B, Inhibitory actions of 0.1 and 1 μm NPY on the normalized amplitude of IBa. C, The Y1 ([Pro34]NPY) and Y2 (NPY13-36) agonists inhibited whole-cell IBa in a single GAD67 neuron from the ARC. D, The Y5 receptor agonist [d-Trp32]NPY1-36 did not evoke a reduction in the IBa amplitude. In the same cell, [Pro34]NPY inhibited IBa. E, Bar graph summarizing the NPY receptor agonist actions on whole-cell IBa in identified arcuate GAD67-containing neurons. Error bars indicate SE. Ctrl, Control; n.s., nonsignificant. *Statistically significant. The number of cells is shown in parentheses.
We then studied the Y receptor subtypes involved in the modulation of calcium currents. We first applied the Y1 agonist [Pro34]NPY1-36 (1 μm), which significantly depressed whole-cell barium currents by 35.7 ± 8.6% (n = 6; p < 0.05) (Fig. 7C,E). Similarly, the Y2 agonist NPY13-36 (1 μm) reduced the amplitude of IBa by 41.4 ± 12.1% (n = 6; p < 0.05) (Fig. 7C,E). A single neuron in Figure 7C responded to both Y1 and Y2 receptor subtype agonists, suggesting coexpression of these Y receptor subtypes in individual GAD67-positive cells. The same experiment was repeated in five additional cells with similar results (data not shown). In striking contrast to Y1 and Y2 agonists, the selective Y5 agonist [d-Trp32]NPY1-36 (1 μm) had no significant effect on the amplitude of IBa as shown in Figure 7, D and E (n = 5; p < 0.05), whereas, in the same cells, [Pro34]NPY1-36 did attenuate the current. The same cell in Figure 7D showed a consistent reduction in IBa after the application of 1 μm [Pro34]NPY1-36 (the same result was obtained in three additional experiments in which both Y1 and Y5 agonists were tested in individual arcuate GABA cells). Together, these results show that activation of Y1 and Y2 receptors inhibit calcium currents in GABA neurons from ARC.
NPY depresses synaptic transmission
The actions of NPY on synaptic transmission were explored in arcuate GAD67-expressing neurons. At -60 mV holding potentials and using KMeSO4 as the intracellular solution, NPY (1 μm) induced a robust and long-lasting decrease in the frequency of postsynaptic currents in all of the nine cells tested (Fig. 8A,B, traces; C, bar graph). Representative traces in Figure 8A show the inhibitory effect of NPY on synaptic activity. NPY (1 μm) reduced the frequency of postsynaptic currents by 38.9.9 ± 7.5% (n = 9; p < 0.05). Whereas GIRK and calcium currents recovered rapidly, synaptic release was depressed for an extended period by NPY. Little recovery was detected after 10-15 min of peptide washout (Fig. 8A,B).
Figure 8.

NPY modulates spontaneous excitatory transmission in arcuate nucleus. A, Representative cells showing the NPY-evoked reduction in postsynaptic current frequency. B, This graph shows the time course of the inhibitory NPY effect on the frequency of postsynaptic currents (PSCs) in a representative GAD67 neuron from the ARC. C, D, Bar graphs presenting the mean reduction in the PSC (C) and EPSC (D) frequency with NPY application. Error bars indicate SE. Ctrl, Control. *Statistically significant. The number of cells is shown in parentheses.
EPSCs were then selectively studied in the presence of 30 μm BIC in the bath. The application of NPY (1 μm) to the slice resulted in a 43.2 ± 6.2% decrease in the spontaneous EPSC frequency (n = 8; p < 0.05) (Fig. 8D, bar graph). EPSCs were completely eliminated when AP-5 (50 μm) and CNQX (10 μm) were added to the recorded cells, confirming that they were glutamatergic in nature (n = 10) (data not shown). We then tested the effect of NPY on electrically evoked EPSPs in arcuate GAD67-synthesizing neurons. EPSPs were evoked by stimulating within the ARC with a bipolar electrode (50-100 μA; 0.5 ms; 0.2 Hz) (Acuna-Goycolea et al., 2004). BIC (30 μm) was applied to the external solution to block the GABAA receptor-mediated evoked activity. These EPSPs were abolished by the glutamate receptor antagonists AP-5 (50 μm) and CNQX (10 μm), suggesting that they are mainly glutamate mediated (n = 6) (data not shown). NPY (1 μm) significantly depressed the evoked EPSPs in GAD67-containing neurons, as shown in Figure 9A. The time integral (area) was depressed by 51.2 ± 9.5% (n = 5; <0.05) (Fig. 9B, top bar graph) and peak amplitude reduced by 35.5 ± 8.8% in the presence of NPY (1 μm) (Fig. 9B, bottom bar graph) (n = 5; p < 0.05) (Fig. 9C,D). Together, these results support a role for NPY in modulating glutamate-dependent synaptic transmission in arcuate GAD67-positive neurons.
Figure 9.

Excitatory evoked potentials are depressed by NPY. A, Representative evoked potentials before (top), during (middle), and after (bottom) NPY application in a typical GFP-expressing neuron from the ARC. B, Bar graphs show the suppressing NPY effects on the time-voltage integral (top) and peak amplitude (bottom) of the electrically evoked responses. Error bars indicate SE. Ctrl, Control. *Statistically significant. The number of cells is shown in parentheses.
To explore the possible mechanism of NPY actions on glutamate transmission, additional experiments in the presence of 0.5 μm TTX were performed with the membrane potential held at -60 mV. Miniature EPSCs (mEPSCs) were detected as fast inward currents under these conditions. NPY (1 μm) significantly reduced the mEPSC frequency by 29.5 ± 8.6% in six of the cells tested (p < 0.05) (Fig. 10A, inset). A graph showing the time course of the NPY actions on the frequency of mEPSCs is presented in the Figure 10A. NPY (1 μm) also left shifted the cumulative distribution of the mEPSC amplitude (n = 6; p > 0.05; Kolmorogov-Smirnov) (Fig. 10B) and reduced mean amplitude of the EPSCs by 22.4 ± 5.5% compared with the control pre-NPY levels (n = 5; p < 0.05) (Fig. 10C), suggesting that NPY might postsynaptically modulate glutamatergic actions in GAD67-positive neurons in ARC.
Figure 10.

NPY actions on miniature glutamatergic activity. A, NPY (1 μm) reduced the frequency of mEPSCs as shown in this time course graph. The inset bar graph shows the mean NPY effect on mEPSC frequency. B, Cumulative distributions of mEPSC amplitude were left shifted in NPY. C, The mean amplitude of the mEPSCs was also reduced by this peptide. The inset shows miniature events in control and NPY conditions. Thicker black traces show the averaged events. Error bars indicate SE. Ctrl, Control. *Statistically significant.
We then studied the individual contribution of the Y1, Y2, and Y5 agonists on the frequency and amplitude of mEPSCs. [Pro34]NPY1-36 (1 μm) reduced both the mEPSC frequency (by 20.2 ± 6.2%; p < 0.05) (Fig. 11A1,A2) and amplitude (Fig. 11A3) in 12 cells studied. The time course of the Y1 agonist effects on the mEPSC frequency is shown in Figure 11A1. The cumulative distribution of the mEPSC amplitude was significantly left shifted (n = 12; p > 0.05; Kolmorogov-Smirnov) (Fig. 11A3) and the mean mEPSC amplitude reduced by 14.3 ± 3.7% (n = 12; p < 0.05) (Fig. 11A3, inset). Because [Pro34]NPY1-36 altered the mEPSC amplitude, Y1 receptor-dependent postsynaptic modulation of glutamatergic transmission may also exist in arcuate GAD67 neurons.
Figure 11.

Y5 and Y1 receptors modulate glutamate transmission by presynaptic mechanisms. A1-A3, Inhibitory actions of 1 μm [Pro34]NPY (Y1 agonist) on the frequency and amplitude of mEPSCs. B1-B3, The Y2 selective agonist NPY13-36 (1 μm) did not affect either the frequency or amplitude of mEPSCs. C1-C3, The Y5 agonist [d-Trp32]NPY1-36 (1 μm) reduced the frequency, but not the amplitude, of the mEPSCs in GABA neurons from ARC. Ctrl, Control; n.s., nonsignificant. *Statistically significant.
In contrast, the Y2 receptor agonist NPY13-36 (1 μm) did not affect the mEPSC frequency (n = 5; p = 0.67) (Fig. 11B1,B2) or the cumulative fraction of the mEPSC amplitude (n = 5; p > 0.05; Kolmorogov-Smirnov) (Fig. 11B3) in GAD67-expressing neurons in the ARC. NPY13-36 (1 μm) had no effect on the mean amplitude of mEPSCs (n = 5; p = 0.88) (Figure 11B3). Finally, the application of the Y5 agonist [d-Trp32]NPY1-36 (1 μm) reduced the frequency of mEPSCs by 21.9 ± 2.7% (n = 6; p < 0.05) (Fig. 11C1,C2). A representative cell showing the time course of Y5 agonist actions on mEPSC frequency is presented in Figure 11C1.[d-Trp32]NPY1-36 (1 μm) had no effect on the cumulative fraction of mEPSC amplitude (n = 6; p > 0.05; Kolmorogov-Smirnov) (Fig. 11C3) or on the mean amplitude of the mEPSCs (control, 100%; Y5 agonist, 102.4 ± 5.4%; wash, 98.6 ± 3.9%; n = 8; p = 0.36), consistent with a presynaptic localization of Y5 receptors in glutamate axons innervating GAD67-expressing neurons in the ARC. Together, our results suggest that some of the inhibitory NPY effects on excitatory synaptic transmission can be explained by presynaptic, Y5-mediated, depression of glutamate release.
IPSCs were then also studied using KCl in the recording pipette and AP-5 (50 μm)/CNQX (10 μm) in the bath. Application of 1 μm NPY under these experimental conditions resulted in a 65.4 ± 7.7% decrease in the frequency of IPSCs compared with control pre-NPY values (n = 8; p < 0.05) (data not shown). This result is consistent with recent reports suggesting that Y receptor agonists can depress inhibitory synaptic inputs to ARC neurons (Cowley et al., 2001; Batterham et al., 2002), and it was not explored further in the present paper.
PYY3-36 inhibits GAD67 neurons via postsynaptic mechanisms
In addition to NPY, the pancreatic polypeptide hormone family includes PYY3-36 and PP, which may selectively bind Y2 (Keire et al., 2000; Batterham et al., 2002) and Y4 (Bard et al., 1995) receptors, respectively. Application of PYY3-36 (1 μm) resulted in a hyperpolarization and reduction in spike frequency (reduction in firing, 75.05 ± 6.84%; n = 10; p < 0.05). The typical inhibitory effect of PYY3-36 on spike frequency is shown in Figure 12A. With TTX (0.5 μm) in the bath, 0.01, 0.1, and 1 μm PYY shifted membrane potential by 0.15 ± 0.9 (n = 5), 4.6 ± 0.6 (n = 4), and 5.6 ± 1.7 mV (n = 6), respectively (data not shown), consistent with a direct postsynaptic activation of its receptor in arcuate GAD67-expressing neurons. The effect of PYY3-36 on neuroendocrine GABA cells was also evaluated. These cells were identified by antidromic electrical stimulation in the median eminence as described before (Fig. 4A). PYY3-36 (1 μm) reduced the frequency of action potentials by 83.2 ± 9.6% (p < 0.05). The neuroendocrine cells were hyperpolarized by 8.4 ± 0.8 mV after PYY3-36 application. PYY3-36 effect on spike frequency or membrane potential on physiologically identified neuroendocrine cells was not significantly different from its effect on other ARC GABA neurons in which the neuroendocrine nature was not tested.
Figure 12.

PYY3-36 inhibits arcuate GABA neurons by direct and indirect mechanisms. A, PYY3-36 (1 μm) inhibited the frequency of spontaneous action potentials in arcuate neurons. The representative traces on the right show the spontaneous discharge before (1), during (2), and after (3) PYY application to the slice in this neuron. B, PYY3-36 (1 μm) activated an inwardly rectifying current that reversed at -56.8 ± 2.5 mV with 15 mm of external [K+]o (black trace). This current was blocked by barium (0.8 μm) (gray trace). C, These traces show the inhibitory effect of 1 μm PYY3-36 on the amplitude of whole-cell barium currents. These currents were activated by voltage steps from -80 to 0 mV (top part). D, PYY3-36 (1 μm) reduced the frequency of EPSCs. E, Traces showing the inhibitory effect of PYY3-36 on EPSCs. F, G, With TTX (0.5 μm) in the ASCF, PYY3-36 (1 μm) had no effect on the frequency of miniature EPSCs. Ctrl, Control.
PYY3-36 (1 μm) activated an inwardly rectifying current when voltage ramps (from -140 to -10 mV for 800 ms) were delivered to the recorded cells (n = 6) (Fig. 12B, black trace). The reversal potential for the PYY3-36-induced current was -58.8 ± 3.4 mV with 15 mm extracellular potassium concentration (n = 6). This current was sensitive to 800 μm barium in the external solution (Fig. 12B, gray trace), consistent with an inwardly rectifying potassium current. PYY3-36 also affected voltage-dependent calcium channels in GABA neurons (Fig. 12C). PYY3-36 (1 μm) reversibly decreased the amplitude of whole-cell barium (replaced by calcium in ACSF) currents by 29.2 ± 2.2% (n = 5; p < 0.05) (data not shown).
PYY3-36 (1 μm) consistently decreased the frequency of EPSCs (reduction by PYY3-36, 32.5 ± 3.5%; n = 9; p < 0.05) (Fig. 12D,E). With 0.5 μm TTX in the extracellular solution, 1 μm PYY3-36 did not depress the frequency (n = 10; p < 0.05) (Fig. 12F,G) or the cumulative distribution of the miniature EPSC amplitude (n = 10; p > 0.05; Kolmorogov-Smirnov) (data not shown), consistent with an indirect spike-dependent effect on the somatodendritic region of presynaptic glutamate neurons innervating the recorded cells. Local glutamate interneurons have been described in this hypothalamic area (Belousov and van den Pol, 1997) and may contribute to these indirect actions of PYY3-36 on the activity of GAD67-synthetizing neurons in the mediobasal hypothalamus.
PP inhibits arcuate GAD67-expressing neurons by presynaptic attenuation of glutamate release
PP (1 μm) reduced the firing rate of GAD67 neurons as shown in the time course graph in Figure 13A. The spontaneous discharge was reversibly reduced by 47.7 ± 5.1% in PP (n = 6; p < 0.05). With 0.5 μm TTX in the bath, PP (1 μm) had little effect on the membrane potential (-0.56 ± 0.74 mV; n = 5; p = 0.23; not significant). Additionally, PP did not evoke a current at -60 mV holding potential (n = 15) and did not significantly affect current responses after voltage-ramp protocols (from -140 to -10 mV for 800 ms; n = 7) (data not shown). PP (1 μm) had little effect on calcium currents activated by voltage steps from -60 to 0 mV (n = 6; p > 0.05) (data not shown). Although PP had no postsynaptic effect on the group means, in three cells PP evoked a modest outward current, or a slight attenuation of calcium currents, suggesting that a possible heterogeneity in PP response might exist.
Figure 13.
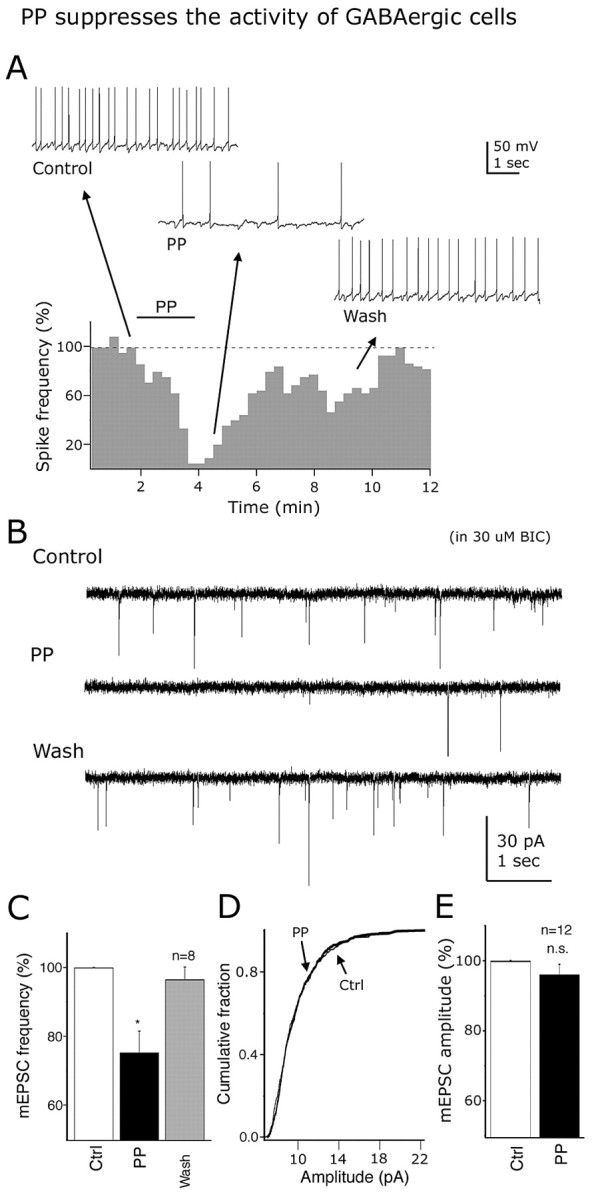
Pancreatic polypeptide inhibits GAD67-expressing neurons. A, Time course and representative recordings showing PP (1 μm) inhibition of spikes in a typical GABA cell. B, These traces show the inhibitory effect of 1 μm PP on the frequency of EPSCs. C-E, With TTX (0.5 μm) in the external solution, 1 μm PP decreased the mESPC frequency (C), but not the cumulative distribution (D) of the mEPSC amplitude, consistent with a presynaptic mechanism of action. E, Bar graph shows the mean mEPSC amplitude before and during PP application to the recorded cells. Error bars indicate SE. Ctrl, Control; n.s., nonsignificant. *Statistically significant.
Spontaneous excitatory glutamate-mediated synaptic activity was reversibly depressed by 25.1 ± 6.8% in the presence of 1 μm PP application (n = 8; p < 0.05) (Fig. 13B). Representative traces before, during, and after PP (1 μm) application are presented in Figure 13B. With 0.5 μm TTX in the bath, PP (1 μm) reduced the frequency of mEPSCs by 24.7 ± 6.3% (n = 8; p < 0.05) (Fig. 13C). The cumulative distribution of the mEPSC amplitude was not significantly altered by PP (1 μm) (n = 12; p > 0.05) (Fig. 13E), and it had no effect on the mean amplitude of the mEPSCs (change in mean amplitude after 1 μm PP, 3.9 ± 3.1%; n = 12; p = 0.29; nonsignificant) (Fig. 13F). These results suggest that the PP may presynaptically inhibit glutamate release onto arcuate GABA cells.
Discussion
In the present paper, we focused on ARC GABA neurons. GABA is found in one-half of all terminals here (Decavel and van den Pol, 1990) and GABAA receptor antagonists block all inhibitory synaptic actions, underlining the critical importance of GABA to ARC intercellular communication. Because live GABA cells in ARC have no defining characteristics, it has not been possible previously to record selectively from them. We evaluated NPY effects on ARC GABA cells by using whole-cell recording in hypothalamic slices from new transgenic mice that selectively express GFP in GAD67-synthesizing neurons. We found that NPY inhibited ARC GABA neurons, including neuroendocrine GABA cells identified by median eminence antidromic stimulation. NPY inhibitory actions included a reduction in spike frequency and a barium-blockable hyperpolarization via Y1 and Y2 receptor modulation. GIRK-like currents were activated and whole-cell calcium currents depressed by NPY through Y1 and Y2 receptors. NPY reduced glutamatergic synaptic activity to GABA neurons by presynaptic inhibition. PYY3-36 and PP depressed GABA cells by activation of a GIRK current and reduction in glutamate release from presynaptic axons, respectively. Importantly, the consistent inhibitory actions of the NPY family of peptides on GABA neurons would tend to reduce GABA-mediated inhibition, resulting in a potential increase in activity in neurons that are the synaptic targets of these GABA cells.
Ionic mechanisms of NPY actions on ARC GABA cells
In some neurons of the hypothalamus, for instance, the predominately GABA neurons of the suprachiasmatic nucleus, NPY exerts little effect on membrane potential (Chen and van den Pol, 1996). In contrast, the inhibitory actions of NPY on the firing rate in ARC GABA neurons were accompanied by a dose-dependent hyperpolarization. NPY decreased input resistance, and evoked an outward current at -60 mV. The NPY-induced hyperpolarization was abolished by barium, suggesting that a potassium current was involved (Sodickson and Bean, 1996; Sun et al., 2001a). Voltage ramps revealed that NPY activated a current that reversed near -100 mV under normal (3.3 mm) extracellular K+, whereas an increase in K+ shifted the reversal potential in a positive direction. This current was blocked by external barium or GDP-β-S in the recording pipette consistent with a GIRK current (Rhim et al., 1997; Sun et al., 2001a; Fu et al., 2004).
Previous studies have suggested that postsynaptic NPY actions may be attributable to Y1 receptor activation (Rhim et al., 1997; Sun et al., 2001b; Sun et al., 2003; Fu et al., 2004; Roseberry et al., 2004). Our results were consistent with this. However, in the presence of the Y1 antagonist BIBP3226 (Doods et al., 1996) in the bath solution, NPY still hyperpolarized the membrane potential, supporting the view that, in addition to Y1, other NPY receptors may be involved. Coapplication of Y1 and Y2 antagonists completely eliminated NPY-evoked hyperpolarization, suggesting a role for postsynaptic Y2 receptors. In some regions of the brain, such as the hippocampus, Y2 receptors are primarily involved in presynaptic inhibition with little postsynaptic effect (Colmers et al., 1991; McQuiston and Colmers, 1996). In contrast, ARC GABA neurons showed a direct robust hyperpolarizing response to Y2 agonists. Previous work on unidentified hypothalamic neurons has suggested that Y2 receptors can postsynaptically alter activity (Sun and Miller, 1999), consistent with histochemical localization of Y2 receptors in the ARC (Broberger et al., 1997).
Both Y1 and Y2 receptor agonists activated GIRK currents, which can account for the hyperpolarization of ARC GABA neurons. Voltage-dependent calcium channels were attenuated by Y1 and Y2, but not Y5 agonists. This would also lead to depression in cellular activity and transmitter release. Single GABA cells showed postsynaptic responses to BIBP-sensitive [Pro34]NPY1-36 and BIIE0246-sensitive NPY13-36, but not to Y4 or Y5 agonists, suggesting that some GABA cells express both Y1 and Y2 receptor types, as previously described in the GABA neurons of the suprachiasmatic nucleus (Chen and van den Pol, 1996).
Presynaptic inhibition
NPY showed long-lasting depression of spontaneous and evoked glutamate-dependent synaptic transmission. Similar long-term effects have been described in other hypothalamic (Obrietan and van den Pol, 1996; Rhim et al., 1997; Fu et al., 2004) and extrahypothalamic regions (Bacci et al., 2002). One explanation for the extended actions of the peptide is that NPY is a large sticky peptide that washes out of slices slowly (Klapstein and Colmers, 1997). Interestingly, both attenuated calcium currents and activated GIRK currents recovered more rapidly, whereas synaptic activity recovered very slowly. This suggests that the NPY-mediated depression of synaptic activity may be a real long-term effect, rather than simply a complication of poor washout, perhaps associated with a peptidergic action on synaptic release apparatus.
NPY reduced the frequency and amplitude of glutamatergic mEPSCs. Similarly, [Pro34]NPY1-36, but not the Y2 agonist NPY13-36, depressed both the frequency and amplitude of mEPSCs. Interestingly, a selective Y5 agonist reduced the frequency but not the amplitude of mEPSCs, supporting the view that changes in the frequency of mEPSCs observed after NPY are attributable in part to activation of Y5 receptors in presynaptic glutamate axons terminating onto GABA neurons, similar to Y5 receptors on glutamate axons innervating hypocretin/orexin neurons (Fu et al., 2004). If postsynaptic NPY modulation of glutamate transmission exists in ARC GABA neurons, it can be accounted for by postsynaptic Y1 receptor activation, because [Pro34]NPY1-36 depresses mEPSC amplitude in these hypothalamic cells. The findings that each agonist generated a specific set of responses is consistent with the view that they activated select receptor subtypes.
Functional implications of NPY family inhibition of ARC GABA neurons
GABA is one of many neurotransmitter/neuromodulators that have been described within the ARC. Previous studies have recorded identified ARC peptidergic neurons (Cowley et al., 2001, 2003; Batterham et al., 2002). Our work is the first to use transgenic mice to detect presumptive hypothalamic GABA cells by their expression of GFP, which allowed us to discriminate GABA cells from other types of ARC neurons including those that use glutamate as a neurotransmitter (Belousov and van den Pol, 1997). The importance of GABA neurons is underlined by the fact that all inhibitory synaptic activity in the ARC is blocked by selective GABA receptor antagonists, and that the further addition of glutamate receptor antagonists results in synaptic silence in the ARC (van den Pol et al., 1990; Belousov and van den Pol, 1997).
NPY could be released by local axons within the ARC, or by axons originating outside the ARC (Everitt and Hokfelt, 1989). A previous report showed that NPY attenuated GABA release onto identified POMC cells (Cowley et al., 2001), similar to the findings reported here. Interestingly, little glutamate synaptic input was reported in POMC neurons, and that was not influenced by NPY (Cowley et al., 2001). In striking contrast, in all of the GABA neurons we recorded, a consistent glutamate-mediated excitatory synaptic input was found, and this was presynaptically attenuated by NPY. POMC neurons have been reported to also contain GABA (Hentges et al., 2004) or glutamate (Collin et al., 2003). NPY may disinhibit POMC cells by reducing GABA synaptic tone, and this has become an important feature of a primary model of hypothalamic regulation of food intake (Cowley et al., 2001, 2003; Jobst et al., 2004). Our data showing a direct NPY-mediated action on GABA neurons would be an additional mechanism of disinhibition by NPY. In addition to release of GABA by anorexigenic POMC cells, nearby orexigenic neurons that synthesize NPY might also release GABA (Horvath et al., 1997). In our recordings from large numbers (>300) of GABA cells, NPY agonists were consistently inhibitory, suggesting that little heterogeneity of NPY general actions exists in this subset of neurons.
Inhibitory endocrine modulation by PYY3-36
PYY3-36 is released as a hormone into the circulatory system during food intake (Sundler et al., 1993) and can cross the weak ARC blood-brain barrier. PYY3-36 modulates food intake (Batterham et al., 2002; Tschop et al., 2004), reportedly based on a mechanism of exciting anorexigenic ARC POMC cells by presynaptic attenuation of GABA release (Batterham et al., 2002). In the present work, we find additional evidence for PYY3-36 attenuation of GABA transmission by multiple mechanisms including indirect presynaptic inhibition of excitatory glutamate input, and direct activation of GIRK currents and attenuation of calcium currents. We did not, however, find any evidence that PYY3-36 exerted excitatory actions on any GABA cells.
Both presynaptic and postsynaptic mechanisms would contribute to the robust inhibitory actions of PYY3-36 on GABA release. In contrast, PP had little direct postsynaptic effect and appears to modulate activity mainly by presynaptic mechanisms. We found that PYY3-36 inhibits physiologically identified GABA neuroendocrine neurons and this may represent an inhibitory endocrine circuit, with hormonal PYY3-36 reducing the median eminence release of neuroendocrine peptides colocalized in ARC GABA axons. Similarly, CNS release of NPY would also inhibit GABAergic neuroendocrine cells. Thus, some of the effects of NPY on neuroendocrine regulation (see Introduction) may be mediated by attenuation of neuropeptide tropin release into the median eminence from ARC GABA neurons.
Footnotes
This work was supported by National Institutes of Health Grants NS34887, NS41454, and NS48476. We thank Y. Yang and V. Rougulin for excellent technical assistance and G. Wollmann for suggestions.
Correspondence should be addressed to Anthony N. van den Pol, Department of Neurosurgery, Yale University School of Medicine, 333 Cedar Street, New Haven, CT 06520. E-mail: anthony.vandenpol@yale.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257406-14$15.00/0
References
- Acuna-Goycolea C, van den Pol A (2004) Glucagon-like peptide 1 excites hypocretin/orexin neurons by direct and indirect mechanisms: implications for viscera-mediated arousal. J Neurosci 24: 8141-8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acuna-Goycolea C, Li Y, van den Pol AN (2004) Group III metabotropic glutamate receptors maintain tonic inhibition of excitatory synaptic input to hypocretin/orexin neurons. J Neurosci 24: 3013-3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Airaksinen MS, Alanen S, Szabat E, Visser TJ, Panula P (1992) Multiple neurotransmitters in the tuberomammillary nucleus: comparison of rat, mouse, and guinea pig. J Comp Neurol 323: 103-116. [DOI] [PubMed] [Google Scholar]
- Bacci A, Huguenard JR, Prince DA (2002) Differential modulation of synaptic transmission by neuropeptide Y in rat neocortical neurons. Proc Natl Acad Sci USA 24: 17125-17130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai FL, Yamano M, Shiotani Y, Emson PC, Smith AD, Powell JF, Tohyama M (1985) An arcuato-paraventricular and -dorsomedial hypothalamic neuropeptide Y-containing system which lacks noradrenaline in the rat. Brain Res 331: 172-175. [DOI] [PubMed] [Google Scholar]
- Bard JA, Walker MW, Branchek TA, Weinshank RL (1995) Cloning and functional expression of a human Y4 subtype receptor for pancreatin polypeptide, neuropeptide Y, and PYY. J Biol Chem 270: 26762-26765. [DOI] [PubMed] [Google Scholar]
- Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA, Cone RD, Bloom SR (2002) Gut hormone PYY3-36 physiologically inhibits food intake. Nature 418: 650-654. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF (1995) Quantal analysis of EPSCs recorded from small numbers of synapses in hippocampal cultures. J Neurophysiol 73: 1145-1156. [DOI] [PubMed] [Google Scholar]
- Belousov AB, van den Pol AN (1997) Local synaptic release of glutamate from neurons in the rat hypothalamic arcuate nucleus. J Physiol (Lond) 499: 747-761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonavera JJ, Dube MG, Kalra PS, Kalra SP (1994) Anorectic effects of estrogen may be mediated by decreased neuropeptide-Y release in the hypothalamic paraventricular nucleus. Endocrinology 134: 2367-2370. [DOI] [PubMed] [Google Scholar]
- Boudaba C, Szabo K, Tasker JG (1996) Physiological mapping of local inhibitory inputs to the hypothalamic paraventricular nucleus. J Neurosci 16: 7151-7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers G, Cullinan WE, Herman JP (1998) Region-specific regulation of glutamic acid decarboxylase (GAD) mRNA expression in central stress circuits. J Neurosci 18: 5938-5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broberger C, Landry M, Wong H, Walsh JN, Hokfelt T (1997) Subtypes Y1 and Y2 of the neuropeptide Y receptor are respectively expressed in pro-opiomelanocortin- and neuropeptide-Y-containing neurons of the rat hypothalamic arcuate nucleus. Neuroendocrinology 66: 393-408. [DOI] [PubMed] [Google Scholar]
- Burdakov D, Liss B, Ashcroft FM (2003) Orexin excites GABAergic neurons of the arcuate nucleus by activating the sodium-calcium exchanger. J Neurosci 23: 4951-4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, van den Pol AN (1996) Multiple NPY receptors coexist in pre- and postsynaptic sites: inhibition of GABA release in isolated self-innervating SCN neurons. J Neurosci 16: 7711-7724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM (1997) Detection of spontaneous synaptic events with an optimally scaled template. Biophys J 73: 220-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin M, Backberg M, Ovesjo ML, Fisone G, Edwards RH, Fujiyama F, Meister B (2003) Plasma membrane and vesicular glutamate transporter mRNAs/proteins in hypothalamic neurons that regulate body weight. Eur J Neurosci 18: 1265-1278. [DOI] [PubMed] [Google Scholar]
- Colmers WF, Klapstein GJ, Fournier A, St.-Pierre S, Treherne KA (1991) Presynaptic inhibition by neuropeptide Y in rat hippocampal slice in vitro is mediated by a Y2 receptor. Br J Pharmacol 102: 41-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone RD, Cowley MA, Butler AA, Fan W, Marks DL, Low MJ (2001) The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int J Obes Relat Metab Disord 25: 63-67. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ (2001) Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411: 480-484. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smith RG, Diano S, Tschop M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, Garcia-Segura LM, Nillni EA, Mendez P, Low MJ, Sotonyi P, Friedman JM, Liu H, Pinto S, Colmers WF, Cone RD, et al. (2003) The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37: 649-661. [DOI] [PubMed] [Google Scholar]
- Decavel C, van den Pol AN (1990) GABA: a dominant neurotransmitter in the hypothalamus. J Comp Neurol 302: 1019-1037. [DOI] [PubMed] [Google Scholar]
- Diano S, Kalra SP, Sakamoto H, Horvath TL (1998) Leptin receptors in estrogen receptor-containing neurons of the female rat hypothalamus. Brain Res 812: 256-259. [DOI] [PubMed] [Google Scholar]
- Dolphin AC (2003) G protein modulation of voltage-gated calcium channels. Pharmacol Rev 55: 607-627. [DOI] [PubMed] [Google Scholar]
- Doods H, Gaida W, Wieland HA, Dollinger H, Schnorrenberg G, Esser F, Engel W, Eberlein W, Rudolf K (1999) BIIE0246: a selective and high affinity neuropeptide Y Y2 receptor antagonist. Eur J Pharmacol 384: 3-5. [DOI] [PubMed] [Google Scholar]
- Doods HN, Wieland HA, Engel W, Eberlein W, Willim KD, Entzeroth M, Wienen W, Rudolf K (1996) BIBP 3226, the first selective neuropeptide Y1 receptor antagonist: a review of its pharmacological properties. Regul Pept 65: 71-77. [DOI] [PubMed] [Google Scholar]
- Ekblad E, Sundler F (2002) Distribution of pancreatic polypeptide and peptide YY. Peptides 23: 251-261. [DOI] [PubMed] [Google Scholar]
- Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, Tatro JB, Hoffman GE, Ollmann MM, Barsh GS, Sakurai T, Yanagisawa M, Elmquist JK (1998) Chemical defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J Comp Neurol 402: 442-459. [PubMed] [Google Scholar]
- Ericson H, Kohler C, Blomqvist A (1991) GABA-like immunoreactivity in the tuberomammillary nucleus: an electron microscopic study in the rat. J Comp Neurol 305: 462-469. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Hokfelt T (1989) The coexistence of neuropeptide Y with other peptides and amines in the central nervous system. In: Neuropeptide Y (Mutt V, Fuxe K, Hokfelt T, Lundberg J, eds), pp 61-72. New York: Raven.
- Fehm HL, Born J, Peters A (2004) Glucocorticoids and melanocortins in the regulation of body weight in humans. Horm Metab Res 36: 360-364. [DOI] [PubMed] [Google Scholar]
- Fu L-Y, Acuna-Goycolea C, van den Pol AN (2004) Neuropeptide Y inhibits hypocretin/orexin by multiple presynaptic and postsynaptic mechanisms: tonic depression of the hypothalamic arousal system. J Neurosci 24: 8741-8751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XB, van den Pol AN (2001) Melanin concentrating hormone depresses synaptic activity of glutamate and GABA neurons from rat lateral hypothalamus. J Physiol (Lond) 533: 237-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandison L, Guidotti A (1977) Stimulation of food intake by muscimol and beta endorphin. Neuropharmacology 16: 533-536. [DOI] [PubMed] [Google Scholar]
- Guo H, Castro PA, Palmiter RD, Baraban SC (2002) Y5 receptor mediate neuropeptide Y actions at excitatory synapses in area CA3 of the mouse hippocampus. J Neurophysiol 87: 558-566. [DOI] [PubMed] [Google Scholar]
- Hahn TM, Breininger JF, Baskin DG, Schwartz MW (1998) Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat Neurosci 1: 271-272. [DOI] [PubMed] [Google Scholar]
- Hentges ST, Nishiyama M, Overstreet LS, Stenzel-Poore M, Williams JT, Low MJ (2004) GABA release from proopiomelanocortin neurons. J Neurosci 24: 1578-1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Tasker JG, Ziegler DR, Cullinan WE (2002) Local circuit regulation of paraventricular nucleus stress integration: glutamate-GABA connections. Pharmacol Biochem Behav 71: 457-468. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Bechmann I, Naftolin F, Kalra SP, Leranth C (1997) Heterogeneity in the neuropeptide Y-containing neurons of the rat arcuate nucleus: GABAergic and non-GABAergic subpopulations. Brain Res 756: 283-286. [DOI] [PubMed] [Google Scholar]
- Inui A (1999) Neuropeptide Y feeding receptors: are multiple subtypes involved? Trends Pharmacol Sci 20: 43-46. [DOI] [PubMed] [Google Scholar]
- Jiang B, Kitamura A, Yasuda H, Sohya K, Maruyama A, Yanagawa Y, Obata K, Tsumoto T (2004) Brain-derived neurotrophic factor acutely depresses excitatory synaptic transmission to GABAergic neurons in visual cortical slices. Eur J Neurosci 20: 709-718. [DOI] [PubMed] [Google Scholar]
- Jobst EE, Enriori PJ, Cowley MA (2004) The electrophysiology of feeding circuits. Trends Endocrinol Metab 15: 488-499. [DOI] [PubMed] [Google Scholar]
- Kalra SP (1997) Appetite and body weight regulation: is it all in the brain? Neuron 19: 227-230. [DOI] [PubMed] [Google Scholar]
- Kalra SP, Horvath TL (1998) Neuroendocrine interactions between galanin, opioids, and neuropeptide Y in the control of reproduction and appetite. Ann NY Acad Sci 21: 236-240. [DOI] [PubMed] [Google Scholar]
- Keire DA, Mannon P, Kobayashi M, Walsh JH, Solomon TE, Reeve Jr JR (2000) Primary structures of PYY, [Pro34]PYY, and PYY-(3-36) confer different conformations and receptor selectivity. Am J Physiol 279: G126-G131. [DOI] [PubMed] [Google Scholar]
- Kelly J, Grossman SP (1979) GABA and hypothalamic feeding systems. II. A comparison of GABA, glycine and actylcholine agonists and their antagonists. Pharmacol Biochem Behav 11: 647-652. [DOI] [PubMed] [Google Scholar]
- Kelly J, Alheid GF, Newberg A, Grossman SP (1977) GABA stimulation and blockade in the hypothalamus and midbrain: effects on feeding and locomotor activity. Pharmacol Biochem Behav 7: 537-541. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, Colmers WF (1997) Neuropeptide Y suppresses epileptiform activity in rat hippocampus in vitro. J Neurophysiol 78: 1651-1661. [DOI] [PubMed] [Google Scholar]
- Leonhardt S, Boning B, Luft H, Wuttke W, Jarry H (1999) Activation of gene expression of the gamma-aminobutyric acid rather than the glutamatergic system in the preoptic area during the preovulatory gonadotropin surge of the rat. Neuroendocrinology 71: 8-15. [DOI] [PubMed] [Google Scholar]
- Li JL, Wu SX, Tomioka R, Okamoto K, Nakamura K, Kaneko T, Mizuno N (2005) Efferent and afferent connections of GABAergic neurons in the supratrigeminal and the intertrigeminal regions. An immunohistochemical tract-tracing study in the GAD67-GFP knock-in mouse. Neurosci Res 51: 81-91. [DOI] [PubMed] [Google Scholar]
- McDonald JK, Koenig JI (1993) Neuropeptide Y actions on reproductive and endocrine functions. In: The biology of neuropeptide Y and related peptides (Colmers WF, Wahlestedt C, eds), pp 419-456. Totowa, NJ: Humana.
- McQuiston AR, Colmers WF (1996) Neuropeptide Y2 receptors inhibit the frequency of spontaneous but not miniature EPSCs in CA3 pyramidal cells of rat hippocampus. J Neurophysiol 76: 3159-3168. [DOI] [PubMed] [Google Scholar]
- Merchenthaler I (1991) Neurons with access to the general circulation in the central nervous system of the rat: a retrograde tracing study with fluorogold. Neuroscience 44: 655-662. [DOI] [PubMed] [Google Scholar]
- Moore RY, Speh JC (1993) GABA is the principal neurotransmitter of the circadian system. Neurosci Lett 150: 112-116. [DOI] [PubMed] [Google Scholar]
- Morley JE, Levine AS, Kneip J (1981) Muscimol induced feeding: a model to study the hypothalamic regulation of appetite. Life Sci 29: 1213-1218. [DOI] [PubMed] [Google Scholar]
- Obrietan K, van den Pol AN (1996) Neuropeptide Y depresses GABA-mediated calcium transients in developing suprachiasmatic nucleus neurons: a novel form of calcium long-term depression. J Neurosci 16: 3521-3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovesjo ML, Gamstedt M, Collin M, Meister B (2001) GABAergic nature of hypothalamic leptin target neurones in the ventromedial arcuate nucleus. J Neuroendocrinol 13: 505-516. [DOI] [PubMed] [Google Scholar]
- Potter EK, Fuhlendorff J, Schwartz TW (1991) [Pro34]neuropeptide Y selectively identifies postjunctional-mediated actions of neuropeptide Y in vivo in rats and dogs. Eur J Pharmacol 193: 15-19. [DOI] [PubMed] [Google Scholar]
- Pronchuk N, Beck-Sickinger AG, Colmers WF (2002) Multiple NPY receptors inhibit GABAA synaptic responses of rat medial parvocellular effector neurons in the hypothalamic paraventricular nucleus. Endocrinology 143: 535-543. [DOI] [PubMed] [Google Scholar]
- Rhim H, Kinney GA, Emmerson PJ, Miller RJ (1997) Regulation of neurotransmission in the arcute nucleus of the rat by different neuropeptide Y receptors. J Neurosci 17: 2980-2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseberry AG, Liu H, Jackson AC, Cai X, Friedman JM (2004) Neuropeptide Y-mediated inhibition of proopiomelanocortin neurons in the arcuate nucleus shows enhanced desensitization in ob/ob mice. Neuron 41: 711-722. [DOI] [PubMed] [Google Scholar]
- Rosin DL, Weston MC, Sevigny CP, Stornetta RL, Guyenet PG (2003) Hypothalamic orexin (hypocretin) neurons express vesicular glutamate transporters VGLUT1 or VGLUT2. J Comp Neurol 465: 593-603. [DOI] [PubMed] [Google Scholar]
- Sar M, Sahu A, Crowley WR, Kalra SP (1990) Localization of neuropeptide-Y immunoreactivity in estradiol-concentrating cells in the hypothalamus. Endocrinology 127: 2752-2756. [DOI] [PubMed] [Google Scholar]
- Sawaki Y, Yagi K (1973) Electrophysiological identification of cell bodies of the tubero-infundibular neurones in the rat. J Physiol (Lond) 230: 75-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober DA, Gackenheimer SL, Heiman ML, Gehlert DR (2000) Pharmacological characterization of 125I-1229U91 binding to Y1 and Y4 neuropeptide Y/peptide YY receptors. J Pharmacol Exp Ther 293: 275-280. [PubMed] [Google Scholar]
- Sodickson DL, Bean BP (1996) GABAB receptor-activated inwardly rectifying potassium current in dissociated hippocampal CA3 neurons. J Neurosci 16: 6374-6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley BG, Kyrkouli SE, Lampert S, Leibowitz SF (1986) Neuropeptide Y chronically injected into the hypothalamus: a powerful neurochemical inducer of hyperphagia and obesity. Peptides 7: 1189-1192. [DOI] [PubMed] [Google Scholar]
- Sun L, Miller R (1999) Multiple neuropeptide Y receptors regulate K+ and Ca2+ channels in acutely isolated neurons from the rat arcuate nucleus. J Neurophysiol 81: 1391-1403. [DOI] [PubMed] [Google Scholar]
- Sun QQ, Huguenard JR, Prince DA (2001a) Neuropeptide Y receptors differentially modulate G-protein-activated inwardly rectifying K+ channels and high-voltage-activated Ca2+ channels in rat thalamic neurons. J Physiol (Lond) 531: 67-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun QQ, Akk G, Huguenard JR, Prince DA (2001b) Differential regulation of GABA release and neuronal excitability mediated by neuropeptide Y1 and Y2 receptors in rat thalamic neurons. J Physiol (Lond) 531: 81-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun QQ, Baraban SC, Prince DA, Huguenard JR (2003) Target-specific neuropeptide Y-ergic synaptic inhibition and its network consequences within the mammalian thalamus. J Neurosci 23: 9639-9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundler F, Bottcher G, Ekblad E, Hakanson R (1993) PP, PYY, and NPY: Occurrence and distribution in the periphery. In: The biology of neuropeptide Y and related peptides (Colmers WF, Wahlestedt C, eds), pp 157-196. Totowa, NJ: Humana.
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T (2003) Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol 467: 60-79. [DOI] [PubMed] [Google Scholar]
- Tappaz ML, Wassef M, Oertel WH, Paut L, Pujol JF (1983) Light- and electron-microscopic immunocytochemistry of glutamic acid decarboxylase (GAD) in the basal hypothalamus: morphological evidence for neuroendocrine γ-aminobutyrate (GABA). Neuroscience 9: 271-287. [DOI] [PubMed] [Google Scholar]
- Tschop M, Castaneda TR, Joost HG, Thone-Reineke C, Ortmann S, Klaus S, Hagan MM, Chandler PC, Oswald KD, Benoit SC, Seeley RJ, Kinzig KP, Moran TH, Beck-sickinger AG, Koglin N, Rodgers RJ, Blundell JE, Ishii Y, Beattie AH, Holch P, et al. (2004) Physiology: does gut hormone PYY3-36 decrease food intake in rodents? Nature 430: 165. [DOI] [PubMed] [Google Scholar]
- van den Pol AN (1980) The hypothalamic suprachiasmatic nucleus of rat: intrinsic anatomy. J Comp Neurol 191: 661-702. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Tsujimoto KL (1985) Neurotransmitters of the hypothalamic suprachiasmatic nucleus: immunocytochemical analysis of 25 neuronal antigens. Neuroscience 15: 1049-1086. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Wuarin JP, Dudek FE (1990) Glutamate, the dominant excitatory transmitter in neuroendocrine regulation. Science 250: 1276-1278. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Gao XB, Obrietan K, Kilduff TS, Belousov AB (1998) Presynaptic and postsynaptic actions and modulation of neuroendocrine neurons by a new hypothalamic peptide, hypocretin/orexin. J Neurosci 18: 7962-7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Saint-Pierre DH, Tache Y (2002) Peripheral ghrelin selectively increases Fos expression in neuropeptide Y-synthesizing neurons in mouse hypothalamic arcuate nucleus. Neurosci Lett 325: 47-51. [DOI] [PubMed] [Google Scholar]