

Abstract
Neurotrophin-3 (NT-3) negatively modulates nerve growth factor (NGF) receptor expression and associated nociceptive phenotype in intact neurons, suggesting a beneficial role in treating aspects of neuropathic pain mediated by NGF. We report that NT-3 is effective at suppressing thermal hyperalgesia associated with chronic constriction injury (CCI); however, NT-3 does not alter the mechanical hypersensitivity that also develops with CCI. Thermal hyperalgesia is critically linked to expression and activation of the capsaicin receptor, transient receptor potential vanilloid receptor-1 (TRPV1). Thus, its modulation by NT-3 after CCI was examined. CCI results in elevated TRPV1 expression at both the mRNA and protein levels in predominantly small-to-medium neurons, with the percentage of neurons expressing TRPV1 remaining unchanged at ∼56%. Attenuation of thermal hyperalgesia mediated by NT-3 correlates with decreased TRPV1 expression such that only ∼26% of neurons ipsilateral to CCI expressed detectable TRPV1 mRNA. NT-3 effected a decrease in expression of the activated component of the signaling pathway linked to regulation of TRPV1 expression, phospho-p38 MAPK (Ji et al., 2002), in neurons ipsilateral to CCI. Exogenous NT-3 could both prevent the onset of thermal hyperalgesia and reverse established thermal hyperalgesia and elevated TRPV1 expression 1 week after CCI. Continuous infusion is required for suppression of both thermal hyperalgesia and TRPV1 expression, because removal of NT-3 resulted in a prompt reestablishment of the hyperalgesic state and corresponding CCI-associated TRPV1 phenotype. In conclusion, although NGF drives inflammation-associated thermal hyperalgesia via its regulation of TRPV1 expression, NT-3 is now identified as a potent negative modulator of this state.
Keywords: dorsal root ganglion, NT-3, NGF, trkA, pain, thermal hyperalgesia
Introduction
Neuropathic pain is a complex clinical problem that occurs in response to peripheral nerve damage and is characterized, in part, by hypersensitivity to thermal and/or mechanical stimuli. The signals regulating these responses are complex and include the promotion of thermal hyperalgesia and nociceptor phenotype by the neurotrophin nerve growth factor (NGF) through activation of its receptor, tropomyosin-related kinase A (trkA) (Verge et al., 1989b, 1995; Lewin and Mendell, 1993; Jongsma Wallin et al., 2001, 2003). Recently, it has been shown that the upregulation of NGF after peripheral inflammation leads to the activation of the mitogen-activated protein kinase (MAPK) p38 (Ji et al., 2002). Widmann et al. (1999) and Ji and Woolf (2001) characterized p38 MAPK as a protein kinase that is stress-activated and plays a role in both injury responses and cell death. Secondary to p38 MAPK activation by NGF is the upregulation of the transient receptor potential vanilloid receptor-1 (TRPV1) (Ji et al., 2002). TRPV1, in turn, plays a critical role in inflammation and injury-induced thermal hyperalgesia (Caterina et al., 1997, 2000; Davis et al., 2000; Garcia-Martinez et al., 2002). Thus, NGF is linked to TRPV1-mediated thermal hyperalgesia, having a role in both sensitization and positive regulation of TRPV1 expression (Chuang et al., 2001; Winston et al., 2001; Ji et al., 2002).
Evidence emerging from our lab indicates that another neurotrophin, neurotrophin-3 (NT-3), acts in a manner that is antagonistic to NGF. Although trkA is the cognate receptor for NGF, NT-3 is also capable of activating trkA and trkB in addition to its own cognate receptor (trkC) (for review, see Lindsay, 1996). Our findings reveal that in intact neurons, NT-3 effects a notable reduction in trkA, high-affinity NGF binding sites, brain-derived neurotrophic factor (BDNF), substance P, calcitonin gene-related peptide, and pituitary cAMP-activated peptide levels (Verge et al., 1989a,b, 1992, 1995; Jongsma Wallin et al., 2001; Karchewski et al., 2002; Gratto and Verge, 2003). This ability of NT-3 to antagonize nociceptive phenotype in sensory neurons led us to speculate that exogenous NT-3 may prove beneficial in the treatment of neuropathic pain. Thus, in this study, we examined whether NT-3 could functionally antagonize neuropathic pain associated with chronic constriction injury (CCI), an injury that elicits a series of changes in primary sensory neurons believed to contribute to the ensuing neuropathic pain state (Bennett and Xie, 1988; Kajander and Bennett, 1992; Nahin et al., 1994; Miki et al., 1998). The evaluation focuses on the ability of NT-3 to alter activation of p38 MAPK and subsequent elevation of TRPV1, as well as the resultant thermal hyperalgesia associated with CCI.
Portions of this work have been published previously in abstract form (Wilson and Verge, 2001; Wilson-Gerwing and Verge, 2003).
Materials and Methods
Animal surgery. All animal procedures were conducted in accordance with the National Institutes of Health policy on the use of animals in research and the University of Saskatchewan animal care committee guidelines (protocol 19920164). A total of 59 young adult male Wistar rats (Charles River Laboratories, Wilmington, MA) weighing 250-300 g were used. Animals were anesthetized for surgery with sodium pentobarbital (Somnitol, 65 mg/kg; MTC Pharm, Cambridge, Ontario, Canada). Preoperative and postoperative (for 24 h) subcutaneous injections of buprenorphine (Temgesic, 0.1-0.2 mg/kg) were given to alleviate any postoperative discomfort. To examine the influence of NT-3 on development of neuropathic pain responses, 12 rats were used: 5 underwent 7 d unilateral CCI of the right sciatic nerve as described by Bennett and Xie (1988) during which the sciatic nerve was exposed and loosely ligated with four chromic gut sutures; 5 rats received 7 d CCI with intrathecal infusion of NT-3 for the duration of injury (intact plus NT-3, CCI plus NT-3); and 2 received 7 d CCI with infusion of vehicle for the duration of injury (intact plus vehicle, CCI plus vehicle). To examine whether NT-3 altered pain thresholds in naive animals, 14 rats were used: 5 served as naive controls; 6 received 7 d intrathecal infusion of NT-3 (naive plus NT-3); and 3 received 7 d infusion of vehicle (naive plus vehicle). To examine the ability of NT-3 to reverse neuropathic pain responses and whether continuous infusion is required for the effect, 21 rats were used: 7 underwent 14 d unilateral CCI; 7 received intrathecal infusion of NT-3 on day 7 after CCI [intact plus NT-3 (delayed), CCI plus NT-3 (delayed)]; and 7 rats received intrathecal infusion of NT-3 at the time of CCI with pump removal at 7 d after CCI [intact plus NT-3 (immediately), CCI plus NT-3 (immediately)]. To examine the effect of NT-3 on total protein levels using Western blot analysis, 12 rats were used: 6 rats underwent 7 d unilateral CCI, and 6 rats received 7 d CCI with intrathecal infusion of NT-3 for the duration of the injury (intact plus NT-3; CCI plus NT-3).
NT-3 was delivered intrathecally for 7 d via mini-osmotic pumps (model 2001; Alza, Cupertino, CA) inserted at the lumbar sacral junction as per Verge et al. (1989a) at a concentration and rate of 600 ng · μl-1 · h-1 (Karchewski et al., 2002) in a solution of PBS containing rat serum albumin (1 mg/ml), streptomycin (100 U/ml), and penicillin (100 U/ml). This dose of NT-3 was the minimum dose found to selectively reverse injury-associated gene expression in injured trkC-expressing neurons (Verge et al., 1996; Jongsma Wallin et al., 2001; Karchewski et al., 2002). Pump control rats were infused with vehicle. At the conclusion of the experiments, rats were killed, and tissue was dissected and processed for in situ hybridization and/or immunohistochemistry as described below.
In situ hybridization. Deeply anesthetized animals were perfused via the aorta with 0.1 m PBS, pH 7.4, followed by rapid dissection and freezing of right and left L4 and L5 dorsal root ganglia (DRG) in OCT compound (Tissue Tek; Miles Laboratories, Elkhart, IN) in a cryomold (Tissue Tek, Miles Laboratories). Before sectioning, blocks containing pairs of experimental and control DRG were fused to ensure processing under identical conditions. Sections were cut at 6 μm on a Micron cryostat (Zeiss, Edmonton, Alberta, Canada), thaw mounted onto Probe-On+ slides (Fisher Scientific, Edmonton, Alberta, Canada), and stored with desiccant at -20°C until hybridization.
Oligonucleotide probes complementary to and selective for TRPV1 mRNA [complementary to bases 509-542 and bases 2601-2634 (Caterina et al., 1997; Michael and Priestley, 1999)] were synthesized (University of Calgary DNA services, Calgary, Alberta, Canada). All probes were checked against the GenBank database (National Institutes of Health, Bethesda, MD) to ensure that no more than 60% homology was found to sequences other than the cognate transcript. The probes were labeled at the 3′-end with α-[35S]dATP (New England Nuclear, Boston, MA) using terminal deoxynucleotidyl-transferase (Amersham Biosciences, Piscataway, NJ) in a buffer containing 10 mm CoCl2, 1 mm dithiothreitol (DTT), 300 mm Tris base, and 1.4 m potassium cacodylate, pH 7.2, and purified through Bio-Spin Disposable Chromatograph Columns (Bio-Rad Laboratories, Hercules, CA) containing 200 mg of NENSORB PREP Nucleic Acid Purification Resin (DuPont NEN, Boston, MA). Dithiothreitol was added to a final concentration of 10 nm. The specific activities ranged from 4.5 to 5.0 × 106 cpm/ng oligonucleotide.
Hybridization was performed according to published procedures (Dagerlind et al., 1992). Briefly, the sections were hybridized at 43°C for 14-18 h in a buffer containing 50% formamide (Sigma Aldrich, Oakville, Ontario, Canada), 4× SSC (1× SSC = 0.15 m NaCl, 0.015 m sodium citrate), 1× Denhardt's solution (0.02% bovine serum albumin and 0.02% Ficoll), 1% sarcosyl (N-laurylsarcosine), 0.02 m phosphate buffer, pH 7.0, 10% dextran sulfate, 500 μg/ml heat-denatured sheared salmon sperm DNA, 200 mm dithiothreitol, and 107 cpm/ml of probe. After hybridization, the slides were washed four times for 15 min in 1× SSC at 55°C, dehydrated in ascending alcohols, processed for radioautography as per Karchewski et al. (2002), and exposed for 7-10 d before developing in D-19 (Kodak, Rochester, NY).
The specificity of hybridization signal for the TRPV1 probes used in the study was confirmed using serial sections hybridized with labeled probe, labeled probe with a 1000-fold excess of cold probe (which effectively competed all specific binding of labeled probe), or labeled probe with a 1000-fold excess of another, dissimilar cold probe of the same length and similar “G-C” content (which did not alter the hybridization signal pattern).
Quantification and analysis. All slides were analyzed qualitatively, and relative changes in hybridization signal from one experimental group to another were noted for sections mounted on the same slide to avoid bias attributable to the variance in signal observed from slide to slide. Slides selected for quantitative analysis had similar numbers of neurons in all DRG sections. Relative changes in hybridization signal over individual neurons with a visible nucleus were determined in DRG from different experimental treatments mounted on the same slide using computer-assisted image analysis as described previously (Karchewski et al., 1999). Analysis was performed as follows: for TRPV1 mRNA (7 d injury) on 12 DRG sections or 2823 neuronal profiles [intact: n = 3 animals; CCI: n = 3 animals; intact plus NT-3: n = 3 animals; CCI plus NT-3: n = 3 animals); for TRPV1 mRNA (14 d injury) on 12 DRG sections or 1994 neuronal profiles [intact: n = 2 animals; CCI: n = 2 animals; intact plus NT-3 (immediately): n = 2 animals; CCI plus NT-3 (immediately): n = 2 animals; intact plus NT-3 (delayed): n = 2 animals; CCI plus NT-3 (delayed): n = 2 animals].
Cells were considered labeled if they had more than five times background levels of silver grains, as determined by averaging grain densities over defined areas of the neuropil devoid of positively labeled cell bodies. This criterion of determining labeled neuronal profiles correlates well with the identification of labeled versus unlabeled neurons as determined manually using a 63× oil immersion objective.
Immunohistochemistry. Deeply anesthetized animals were perfused via the aorta with 0.1 m PBS followed by 4% paraformaldehyde. Right and left L4 and L5 DRG were dissected, postfixed for 1-1.5 h, and cryoprotected in 20% sucrose overnight at 4°C. Paired experimental and control tissues were frozen in the same cryomold to ensure processing under identical conditions. Transverse 10 μm sections were cut on the cryostat, thaw-mounted onto Probe-ON+ slides (Fisher Scientific), and processed for immunohistochemistry.
For TRPV1, sections were washed three times for 10 min in 0.1 m PBS, blocked in 1.5% BSA, 4% horse serum, and 0.1% Triton X-100 in 0.1 m PBS for 1 h at 4°C, incubated overnight with rabbit anti-VR-1 (TRPV1) (10 μg/ml; Alpha Diagnostic International, San Antonio, TX), diluted in 0.5% BSA, 2% horse serum, and 0.1% Triton X-100 in PBS (0.1 m) at 4°C, and visualized with Alexa Fluor 488 goat anti-rabbit IgG (1:200; Molecular Probes, Eugene, OR) in PBS (0.1 m) with 0.5% BSA and 2% horse serum for 50 min at room temperature. Slides were washed and coverslipped with 50% glycerol/50% PBS. For phosphorylated p38 MAPK (p-p38), sections were washed in 0.1 m PBS, blocked with 10% horse serum and 0.1% Triton X-100 in 0.1 m PBS for 1 h at room temperature, incubated overnight with rabbit anti-phospho p38 MAPK (Cell Signaling Technology, Beverly, MA), diluted 1:50 in 1% BSA and 0.1% Triton X-100 in PBS (0.1 m) at 4°C, and visualized using donkey anti-rabbit F(ab′)2 Cy3 conjugate (1:400; Jackson ImmunoResearch Laboratories, West Grove, PA) in 0.1 m PBS for 1 h at room temperature. The slides were washed and coverslipped with Citifluor (Marivac, Halifax, Nova Scotia, Canada). Control sections were processed in the same manner, but without the primary antibody. Results were viewed using a Zeiss Axioscope 50 microscope equipped with incident-light fluorescence optics and a CCD camera.
Western blots. The L4 and L5 DRG were dissected out and immediately placed in ice-chilled RIPA buffer (50 μl per three DRG) containing an anti-protease mixture (Sigma Aldrich, catalog #P-8340) and 1 m DTT with 10 mm Na acetate, pH 5.3. The DRG were flash freeze/thawed three times and homogenized (Tissue Tearor; Biospec Products, Bartlesville, OK), and protein was extracted using pulse sonication for 3 s. The samples were ultracentrifuged at 14.5 K rpm at 4°C for 30 min. A Bradford assay was conducted on the lysates so an equivalent quantity of protein could be loaded onto each gel. Equivalent loading was verified by Coomassie staining after transfer of the SDS-PAGE.
Protein samples were separated by SDS-PAGE and transferred onto a nitrocellulose paper. The blots were blocked with 7.5% milk powder and 0.5% Tween 20 overnight at 4°C. The blots were then incubated with the p-p38 MAPK (Thr180/Tyr182) (1:1000) (Cell Signaling Technology) or VR-1 (TRPV1) (1:1000) (Alpha Diagnostic International, San Antonio, TX) primary antibody in PBS containing 5% milk powder and 0.5% Tween 20 overnight at 4°C, followed by incubation in the HRP-linked secondary antibody (1:2000) (anti-rabbit IgG; Cell Signaling Technology) for 45 min at room temperature. All washes were done using PBS containing 0.1% Tween 20. A Western Lightening Chemiluminescence kit was used to visualize the bound HRP-conjugated secondary (PerkinElmer Life Sciences, Boston, MA). The membrane was exposed to Kodak X-Omat AR film and developed.
Assessment of behavioral responses. Male Wistar rats (250 g) (Charles River Laboratories) were acclimatized to the facility before the start of testing. Animals were housed individually in clear plastic cages and maintained in a 12 h light/dark cycle. Food and water were available ad libitum. All behavioral testing was performed at the same time of day to avoid potential fluctuations caused by daily hormonal cycling of the male rats.
Thermal hyperalgesia was assessed using the Hargreaves Plantar Test (Hargreaves et al., 1988) (Ugo Basile, Comerio, Italy). The animal was placed in a clear Plexiglas chamber atop a clear glass surface that housed a radiant heat source and acclimatized (∼5 min). The heat source was placed directly under the plantar surface of the hindpaw and activated, starting a digital timer. The timer stopped when the paw was withdrawn from the heat source, indicating the paw withdrawal latency. A maximum time of 30 s was in place to prevent tissue damage. Five readings for each paw were taken (allowing 5 min intervals between paws to prevent sensitization). Baseline levels were established for 5 d before CCI and/or pump implantation. Behavioral testing resumed 2 d after surgery and continued for 6 consecutive days (7 d injury). Alternatively, for 14 d injury, behavioral testing continued on days 2, 4, 6, 9, 11, and 13 after surgery.
Because the thermal sensitivity of the uninjured paw contralateral to CCI (intact) was not altered throughout the experiments (T. D. Wilson-Gerwing, unpublished observations), the withdrawal latency of the CCI paw was divided by the withdrawal latency of the uninjured contralateral paw for each day of testing. This ratio was calculated for all five trials on each day of testing. The highest and lowest ratios were excluded. The remaining ratios were transferred into Prism graphing software (GraphPad, version 4.0; GraphPad Software, San Diego, CA), and graphs were generated using the mean value for each day of testing ± SEM. Assessment of thermal hyperalgesia in naive animals was based on the withdrawal time (in seconds) of the right paw alone. ANOVA was used to determine significant differences between experimental and control groups (significance p < 0.05).
Sensitivity of the hindpaw to mechanical stimulation was measured using calibrated von Frey filaments (Stoelting, Kiel, WI). Animals were placed in a clear Plexiglas chamber atop a modified semi-opaque Plexiglas platform [built according to the specifications of Pitcher et al. (1999) with slight modifications] and acclimatized (∼5 min). Briefly, a 3-mm-thick piece of semi-opaque Plexiglas was cut to 30 × 30 cm. Small holes (1.5 mm diameter) were drilled in a grid pattern 5 mm apart to cover the entire surface. This testing surface was then placed on top of a metal frame. Each von Frey filament was applied perpendicular to the plantar surface of the hindpaw five times, and the number of times that the paw was withdrawn from the hair was recorded. Increasing sizes of hairs were applied until the hindpaw was withdrawn four times out of five applications. This was defined as the threshold for withdrawal. The interval between application of increasing size filaments and between paws was 5 min (to prevent sensitization). Baseline levels were established for 5 d before CCI and/or pump implantation. Behavioral testing resumed 2 d after surgery and continued for 6 consecutive days (7 d injury). Alternatively, for 14 d injury, behavioral testing resumed on days 2, 4, 6, 9, 11, and 13 after surgery.
The data obtained from the assessment of mechanical sensitivity, the withdrawal threshold (in grams), for the CCI paw for each day of testing were normalized such that the mean withdrawal thresholds for baseline were given a value of 1. All data collected were graphed in Prism (GraphPad version 4.0). Graphs were generated using the mean values of all normalized withdrawal thresholds for each day of testing ±SEM. Significant differences between experimental groups were determined using ANOVA (p < 0.05).
Results
The effect of NT-3 on neuropathic pain indices associated with CCI: NT-3 significantly attenuates thermal hyperalgesia
The chronic constriction injury model was used to examine whether NT-3 (in its ability to downregulate trkA and associated phenotypes) alters behavioral indices of neuropathic pain. Animals having undergone unilateral CCI of the sciatic nerve for 7 d developed a significant increase in sensitivity to thermal stimulation in the injured hindpaw (p < 0.0001) compared with baseline levels (Fig. 1). Post-CCI sensitivity to thermal stimulation was not altered by infusion of vehicle control, and therefore these data were pooled with those of animals receiving CCI alone; however, intrathecal infusion of NT-3 at the time of unilateral CCI significantly attenuated the development of thermal hyperalgesia compared with those animals having undergone CCI alone (p < 0.0001) (Fig. 1). The ability of NT-3 to alter thermal sensitivity does not extend to normal thermal thresholds, because no significant alteration in thermal sensitivity was observed when NT-3 was infused into naive animals (Fig. 2). A similar lack of effect on thermal thresholds was observed in naive animals receiving vehicle control infusions (Fig. 2).
Figure 1.

NT-3 prevents the development of thermal hyperalgesia but not mechanical sensitivity associated with CCI. Hyperalgesic index plots represent alterations in thermal (A, B) and mechanical (C, D) sensitivities in response to 7 d CCI without (A, C) or with (B, D) 7 d NT-3 intrathecal infusion. Dashed line indicates the time of unilateral CCI. A, Thermal hyperalgesia is significantly increased compared with baseline levels after CCI and is maintained for the duration of the experiment (n = 7). B, Infusion of NT-3 significantly attenuates the development of thermal hyperalgesia after CCI and is maintained for the duration of the infusion (***p < 0.0001, relative to CCI alone; n = 5). The solid arrow indicates the start of NT-3 infusion. C, Mechanical hypersensitivity is significantly increased compared with baseline levels after unilateral CCI and is maintained for the duration of the experiment (7 d; n = 7). D, Infusion of NT-3 does not alter the mechanical hypersensitivity that develops after unilateral CCI (7 d; n = 5). The solid arrow indicates the start of NT-3 infusion.
Figure 2.

NT-3 does not significantly alter thermal or mechanical thresholds in naive animals. Hyperalgesic index plots represent alterations in thermal (A-C) and mechanical (D-F) sensitivities in naive animals in response to vehicle (B, E) or NT-3 (C, F) intrathecal infusion for 7 d. A, Naive, uninjured animals demonstrate little fluctuation in their responses to thermal stimuli compared with baseline (n = 5). B, Infusion of control (1× vehicle) did not alter responses to thermal stimuli in naive animals compared with baseline (n = 3). The arrow indicates the time of infusion of control. C, Infusion of NT-3 did not alter responses to thermal stimuli in naive animals compared with baseline levels (n = 6). The solid arrow indicates the time of infusion of NT-3. D, Naive, uninjured animals do not demonstrate significant fluctuation in their responses to mechanical stimuli compared with baseline (n = 5). E, Infusion of control (1× vehicle) did not significantly alter responses to mechanical stimuli in naive animals compared with baseline levels (n = 3). The arrow indicates the time of infusion of vehicle control. F, Infusion of NT-3 did not significantly alter responses to mechanical stimuli in naive animals compared with baseline levels; however, a nonsignificant increased mechanical sensitivity was observed (n = 6). The solid arrow indicates the start of NT-3 infusion.
To determine whether NT-3 might be effective at reversing thermal hyperalgesia once established and if continuous infusion of NT-3 is required for this effect, a longer injury period was used whereby animals underwent 14 d unilateral CCI of the sciatic nerve. These animals also developed a significant increase in sensitivity to thermal stimulation in the injured hindpaw (p < 0.0001) when compared with baseline levels (Fig. 3). Delayed infusion of NT-3 (days 7-14 after CCI) resulted in a significant reversal of the thermal hyperalgesia compared with those animals having undergone CCI alone (p < 0.0001) (Fig. 3). In contrast, although immediate infusion of NT-3 significantly attenuated the development of thermal hyperalgesia (p < 0.0001), removal of the NT-3 pumps at 7 d after CCI resulted in a quick reestablishment of the thermal hyperalgesic state (Fig. 3). Thus, the NT-3-mediated attenuation of thermal hyperalgesia associated with CCI requires continuous infusion of NT-3.
Figure 3.

Delayed infusion of NT-3 can reverse established thermal hyperalgesia but not mechanical sensitivity after CCI. Hyperalgesic index plots represent alterations in thermal (A-C) and mechanical(D-F) sensitivities in 14 dCCI animals in response to delayed NT-3 [7 d post-CCI, (B, E)] or immediate NT-3 [followed by removal for the last 7 d (C, F)] intrathecal infusion. Dashed line indicates the time of CCI; solid arrow indicates the start of NT-3 infusion; open arrow indicates the time of pump removal. A, Thermal hyperalgesia is significantly increased compared with baseline levels after unilateral CCI and is maintained for the duration of the experiment (n = 7). B, Delayed infusion of NT-3 significantly reverses thermal hyperalgesia compared with CCI alone (***p < 0.0001) and is maintained for the duration of the infusion (7 d) (n = 7). Asterisks indicate significant difference from CCI alone. C, Infusion of NT-3 is required for continuous prevention of thermal hyperalgesia after CCI. Infusion of NT-3 significantly attenuates the development of thermal hyperalgesia after unilateral CCI and is maintained for the duration of the 7 d infusion (***p < 0.0001). After removal of the NT-3 pump, thermal hyperalgesia was reestablished and did not significantly differ from CCI alone (n = 7). The solid arrow indicates the start of NT-3 infusion; open arrow indicates the time of pump removal. Asterisks indicate significant difference from CCI alone. D, Mechanical sensitivity is significantly increased compared with baseline levels after unilateral CCI and is maintained for the duration of the experiment (n = 7). E, Delayed infusion of NT-3 does not alter mechanical sensitivity compared with CCI alone (n = 7). F, Stopping NT-3 infusion does not alter mechanical sensitivity compared with CCI alone (n = 7).
NT-3 has no significant influence on mechanical hypersensitivity associated with CCI
Unilateral CCI of the sciatic nerve for 7 d also resulted in the development of significant mechanical hypersensitivity (p < 0.0001) (Fig. 1). The development of post-CCI mechanical hypersensitivity was not altered by infusion of vehicle control, and therefore these data were pooled with those of animals receiving CCI alone. Neither naive control animals nor naive animals receiving vehicle control infusions developed a significant increase in mechanical hypersensitivity, although there was a nonsignificant trend toward slightly increased sensitivity as the testing progressed (Fig. 2). Infusion of NT-3 at the time of CCI did not lead to any significant changes in mechanical hypersensitivity when compared with animals having undergone CCI alone (Fig. 1).
To determine whether delayed infusion of NT-3 or removal of exogenous NT-3 might alter mechanical responses, the 14 d unilateral CCI model was used. As with 7 d CCI, animals having undergone unilateral CCI of the sciatic nerve for 14 d developed a significant increase in mechanical sensitivity (p < 0.0001) (Fig. 3). Delayed infusion of NT-3 after CCI did not result in any perceptible changes in mechanical sensitivity compared with those animals having undergone CCI alone (Fig. 3). Furthermore, removal of NT-3 pumps at 7 d after injury did not impact the mechanical hypersensitivity (Fig. 3).
NT-3 prevents and counteracts CCI-associated increases in TRPV1 expression
Because of the dramatic influence of NT-3 on neuropathic thermal sensation, the regulation of TRPV1 expression was examined in the DRG of animals used for the behavioral studies. Analysis of sections processed for in situ hybridization to detect neuronal expression of TRPV1 mRNA revealed that in the DRG contralateral to CCI (intact), detectable hybridization signal was localized over small and medium neurons (<40 μm) representing 56.13 ± 0.99% (SEM) of all neurons measured (Fig. 4). Neurons expressing moderate to high levels of hybridization signal (>20× background) accounted for 24.77 ± 1.88% (SEM) of all neurons measured. Seven days after CCI, relative levels of hybridization signal over small and medium neurons were elevated and detectable over a few large neurons, albeit at relatively low levels (Fig. 4). Despite these alterations in expression, the overall percentage of neurons expressing TRPV1 mRNA remained similar to that seen in the DRG contralateral to CCI [55.90 ± 2.95% (SEM)], with no significant change in the percentage of neurons with moderate to high levels of hybridization signal [25.57 ± 5.19% (SEM)].
Figure 4.
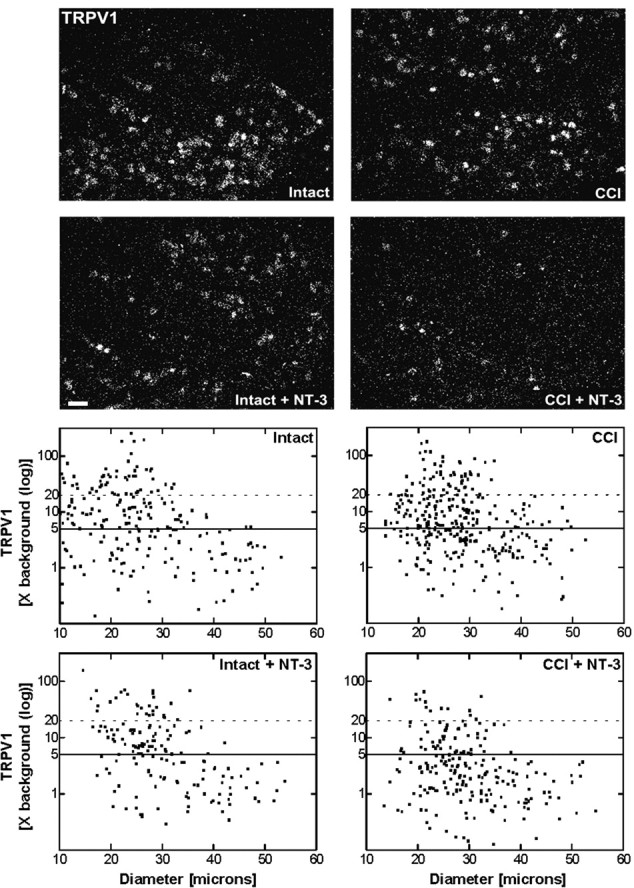
Message levels for TRPV1 are reduced after NT-3 treatment. Top, Dark-field photomicrographs of 6-μm-thick adult rat L5 DRG sections processed for in situ hybridization to detect TRPV1 transcripts contralateral (intact) or ipsilateral to 7 d CCI (CCI) and after a 7 d unilateral CCI plus intrathecal infusion of 600 ng · μl-1 · h-1 NT-3 (Intact + NT-3; CCI + NT-3). Scale bar, 100 μm. Note: NT-3 infusion results in the reduction in relative levels of hybridization signal for TRPV1 over individual neurons, with the influence most apparent after CCI. Bottom, Representative scatterplots whereby each point represents the labeling index of an individual neuron identified in 6-μm-thick sections of L5 DRG processed to detect TRPV1 mRNA. The relationship between TRPV1 mRNA labeling intensity (y-axis, log scale) and perikaryal diameter (x-axis) is depicted. Experimental states are indicated at the top right of each graph and are as described above. Labeling refers to the ratio of silver grain density over the neuronal cytoplasm to grain density over areas of the neuropil devoid of positive hybridization signal. Solid lines divide the plots into labeled and unlabeled populations; dotted lines separate lightly labeled from moderate to heavily labeled populations of TRPV1-expressing neurons. Note: In DRG contralateral to CCI, TRPV1 is expressed predominantly in small-to-medium neurons. CCI results in elevated TRPV1 expression in small-to-medium neurons and a novel but low level of expression in large neurons. NT-3 infusion produced a reduction in the levels and percentage of neurons expressing detectable TRPV1 mRNA DRG both ipsilateral and contralateral to CCI, with a more pronounced effect ipsilateral to injury.
NT-3 infusion effected a reduction in the levels of neuronal TRPV1 mRNA expression both ipsilateral (CCI plus NT-3) and contralateral (intact plus NT-3) to CCI (Fig. 4). This effect was most profound for neurons ipsilateral to CCI. In DRG contralateral to CCI, levels of TRPV1 mRNA hybridization signal were modestly attenuated after infusion of NT-3 (Fig. 4), with the overall percentage of neurons expressing detectable levels reduced from 56.13 ± 0.99% (SEM) to 45.50 ± 6.98% (SEM). In contrast, a far more dramatic attenuation of TRPV1 expression was observed in 7 d CCI animals that received NT-3 infusion for the duration of the injury [from 55.90 ± 2.95% (SEM) to 26.13 ± 0.61% (SEM)[(Fig. 4). The decrease in TRPV1 mRNA was observed in all size ranges of neurons, with those expressing moderate to high levels of hybridization signal now representing only 5.07 ± 1.69% (SEM) of the neuronal population compared with 24.77 ± 1.88% (SEM) in animals with CCI alone. Notably, TRPV1 expression was no longer detectable in large-size neurons after NT-3 infusion (Fig. 4).
Analysis of sections processed to detect neuronal expression of TRPV1 mRNA from animals having undergone 14 d CCI with either immediate (days 0-7) or delayed (days 7-14) infusion of NT-3 revealed that the patterns of TRPV1 expression from CCI plus immediate NT-3 appeared very similar to those of CCI alone. Quantification of hybridization signal over individual neurons from CCI or CCI plus immediate NT-3 infusion revealed that detectable TRPV1 mRNA hybridization signal was observed predominantly over small-to-medium neurons, with some large neurons also expressing at 14 d after CCI (Fig. 5). As with 7 d CCI, neurons expressing moderate to high levels of hybridization signal (>20× background) were observed almost exclusively over the small-to-medium-sized neurons (Fig. 5). This suggests that removal of exogenous NT-3 results in a reinstatement of the CCI phenotype. In contrast, in animals receiving delayed NT-3 infusion, relative levels of TRPV1 mRNA expression were reduced with the effect most prominent for neurons ipsilateral to CCI [Fig. 5, CCI + NT-3(d)].
Figure 5.
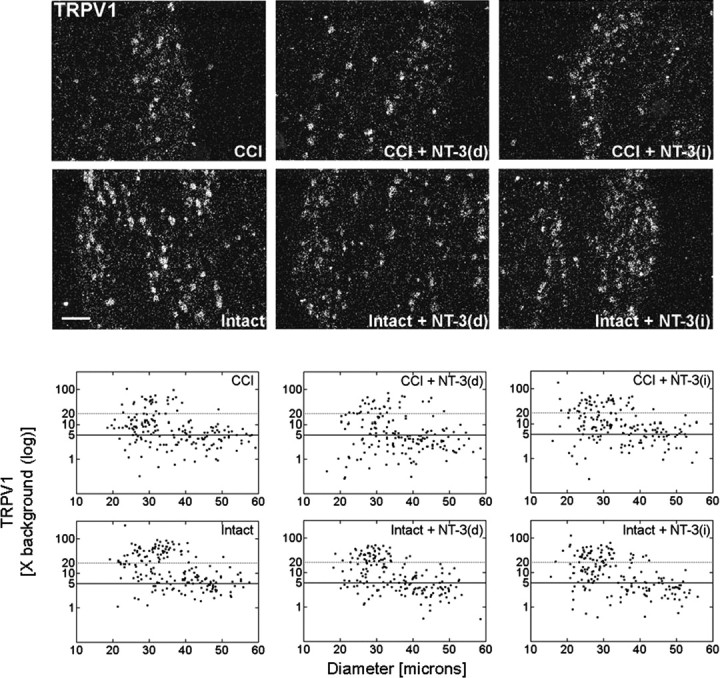
Delayed NT-3 infusion reverses the increase in TRPV1 mRNA detected in DRG neurons subjected to CCI. Top, Dark-field photomicrographs of 6 μm sections of L5 DRG ipsilateral or contralateral to 14 d unilateral CCI, in response to 7 d 600 ng · μl-1 · h-1 NT-3 infusion for the last 7 d of a 14 d unilateral CCI [CCI + NT-3(d); Intact + NT-3(d)], or in response to 7 d 600 ng · μl-1 · h-1 NT-3 infusion for the first 7 d of a 14 d unilateral CCI [CCI + NT-3(i); Intact + NT-3(i)], as indicated. Scale bar, 60 μm. Note: Delayed infusion of NT-3 [CCI + NT-3(d)] results in a decrease in the relative levels of TRPV1 mRNA detected. Bottom, Representative scatterplots whereby each point represents the labeling index of an individual neuron identified in 6-μm-thick sections of L5 DRG processed to detect TRPV1 mRNA. The relationship between TRPV1 mRNA labeling intensity (y-axis, log scale) and perikaryal diameter (x-axis) is depicted. Experimental states as indicated are as described above. Labeling refers to the ratio of silver grain density over the neuronal cytoplasm to grain density over areas of the neuropil devoid of positive hybridization signal. Solid lines divide the plots into labeled and unlabeled populations; dotted lines separate lightly labeled from moderate to heavily labeled populations of TRPV1-expressing neurons. Note: Delayed infusion of NT-3 [CCI + NT-3(d)] results in a decrease in the relative levels of TRPV1 most prominent in medium-to-large neurons.
Tissue sections were also processed for immunohistochemistry to ascertain whether modulation of TRPV1 protein expression by CCI and exogenous NT-3 corresponds to that observed at the mRNA level. Immunohistochemistry revealed that in the DRG contralateral to CCI (intact), staining for TRPV1 protein was intense in a subpopulation of small neurons, whereas a more moderate level of staining was observed in other small and medium neurons. Seven day CCI resulted in a dramatic increase in the relative numbers of neurons that were intensely stained for TRPV1, with predominantly small and medium neurons, but also the occasional large neuron, falling into this category (Fig. 6). Infusion of NT-3 for the duration of CCI (7 d) resulted in decreased intensity of TRPV1 staining that was most evident in the medium and large neurons from DRG ipsilateral to CCI. In DRG contralateral to CCI, NT-3 infusion did not have a dramatic impact on TRPV1 immunostaining; however, more intense staining was observed over some small neurons (intact plus NT-3) (Fig. 6). Consistent with the immunohistochemistry findings, Western blot analysis revealed that TRPV1 protein levels were elevated after 7 d CCI. NT-3 infusion effectively mitigated this increase in DRG ipsilateral to CCI (CCI plus NT-3), whereas slightly increased levels were observed in DRG contralateral to CCI (intact plus NT-3) (Fig. 6).
Figure 6.
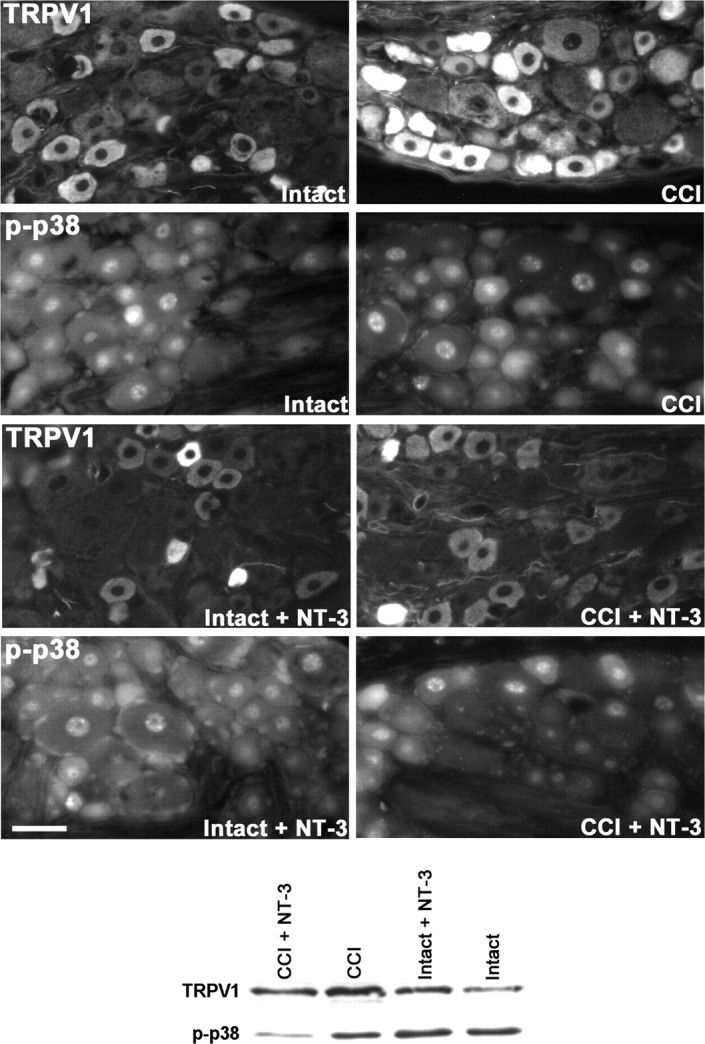
NT-3 infusion results in decreased expression of TRPV1 protein. Top, Fluorescence photomicrographs demonstrate levels of TRPV1 and p-p38 MAPK-like immunoreactivity in 10 μm sections of DRG ipsilateral (CCI) and contralateral to CCI (Intact) L5 DRG with or without immediate intrathecal infusion of NT-3 (CCI + NT-3, Intact + NT-3), as indicated. Scale bar, 60 μm. Note: In the DRG contralateral to CCI (Intact), levels of TRPV1 protein are highest in small neurons, with lower levels of expression observed in several small-to-medium dorsal root ganglion neurons. Seven days after CCI, levels of expression have increased relative to the intact state, with protein now being detected in a few large neurons. Intrathecal infusion of NT-3 at the time of injury results in reduced levels of TRPV1 protein, most notable in medium-to-large CCI neurons. Levels of p-p38 MAPK nuclear expression do not appear to be altered by CCI. Cytoplasmic expression of p-p38 MAPK does decrease slightly with CCI. Infusion of NT-3 results in a dramatic downregulation of p-p38 MAPK after CCI. The DRG contralateral to CCI (Intact) appear to have an increase in perineuronal p-p38 MAPK staining. Bottom, Representative Western blots of TRPV1 (n = 4) and p-p38 MAPK (n = 5) reflect the staining patterns as described above. Experimental states are as indicated.
As with 7 d CCI, expression of TRPV1 protein in DRG ipsilateral to 14 d CCI was increased in small, medium, and some large neurons, relative to contralateral DRG (Fig. 7). Delayed infusion of NT-3 resulted in decreased levels of TRPV1, such that the pattern and level of expression in neurons subjected to CCI [CCI plus NT-3(d)] now approximated that observed in the DRG contralateral to CCI (Intact) (Fig. 7). The effect of delayed NT-3 infusion was less evident for neurons from DRG contralateral to CCI [intact plus NT-3(d)], with only slightly decreased expression observed in some medium neurons. As observed at the mRNA level, the ability of NT-3 to modulate TRPV1 protein expression requires continuous infusion of NT-3, because patterns of protein localization and expression levels resembled those of 14 d CCI animals when NT-3 infusion was halted for the last 7 d of injury (Fig. 7).
Figure 7.

Delayed NT-3 infusion reverses the increase in TRPV1 protein detected in DRG neurons subjected to CCI. Fluorescence photomicrographs demonstrate levels of TRPV1-like immunoreactivity in 10 μm sections of L5 DRG representing DRG ipsilateral (CCI) and contralateral (Intact) to a 14 d, in response to delayed NT-3 infusion for 7 d starting 7 d after CCI [CCI + NT-3(d); Intact + NT-3(d)], or immediate NT-3 infusion for 7 d after CCI, followed by removal for the last 7 d of injury [CCI + NT-3(i); Intact + NT-3(i)], as indicated. Scale bar, 60 μm. Note: Delayed intrathecal infusion of NT-3 results in a dramatic decrease in the levels of TRPV1 protein detected [CCI + NT-3(d)], most notably in medium-to-large neurons. This affect is less pronounced on the DRG contralateral to CCI [Intact + NT-3(d)] (bottom center). Intrathecal infusion of NT-3 for 7 d at the time of CCI, followed by removal for the last 7 d of injury [CCI + NT-3(i)] (top right) results in similar levels of TRPV1 protein detected compared with CCI alone, suggesting that chronic infusion is required for mitigation of CCI-associated elevated TRPV1 expression.
NT-3 reduces the level of activated p38 MAPK after CCI
Tissue sections were processed for immunohistochemistry to ascertain whether CCI and exogenous NT-3 effect a coordinate change in activated p-p38 MAPK protein expression, a component of the signaling pathway shown previously to directly regulate TRPV1 expression in sensory neurons (Ji et al., 2002). In DRG contralateral to CCI (intact), staining for p-p38 MAPK protein was very intense in a subpopulation of small neurons, whereas a more moderate level of staining was observed in other small and medium neurons. Seven day CCI resulted in a slight decrease in the cytoplasmic staining. This does not appear to be reflected in the pattern of nuclear staining that remained relatively unchanged (CCI) (Fig. 6). Infusion of NT-3 effected a marked decrease in the intensity of p-p38 MAPK across all size ranges of neurons ipsilateral to CCI, with the exception of a subpopulation of small neurons that expressed elevated levels of p-p38 MAPK (CCI + NT-3) (Fig. 6). In DRG contralateral to CCI, NT-3 infusion did not appear to alter neuronal nuclear staining patterns; however, there was a noticeable increase in perineuronal staining (intact plus NT-3) (Fig. 6). Consistent with the immunohistochemistry findings, Western blot analysis revealed that a 7 d CCI resulted in a slight decrease in the levels of p-p38 MAPK compared with DRG contralateral to CCI (intact). Infusion of NT-3 dramatically reduced the total p-p38 MAPK protein present in the DRG ipsilateral to CCI (CCI plus NT-3), whereas it slightly increased levels of p-p38 MAPK in the DRG contralateral to CCI (intact plus NT-3) (Fig. 6).
Discussion
Neuropathic pain is a clinical challenge because of the complex mechanisms underlying this pathology. NT-3 can effect a downregulation in trkA expression, NGF high-affinity binding sites, and associated nociceptive phenotype in intact sensory neurons (Jongsma et al., 2001; Karchewski et al., 2002; Gratto and Verge, 2003). Given that enhanced NGF synthesis contributes to various neuropathic pain syndromes, we hypothesized that NT-3 could attenuate these pain syndromes and neuronal expression of molecules complicit in this state through its antagonistic influence on the trkA pathway. Our study supports this hypothesis, identifying NT-3 as a novel potent modulator of thermal hyperalgesia that influences behavioral responses and neuronal expression of TRPV1 and activated p38 MAPK.
NT-3 influences both trkC- and trkA-expressing neurons. There are two lines of evidence suggesting that its influence is selective for these populations. NT-3 reverses injury-associated changes only in the trkC population of sensory neurons (Verge et al., 1996; Jongsma Wallin et al., 2001; Karchewski et al., 2002). Furthermore, NT-3 does not alter somatostatin levels in either intact or injured sensory neurons that are responsive to GDNF and do not colocalize with trkA (Verge et al., 1995; Bennett et al., 1998; K. A. Gratto and V. M. K. Verge, unpublished observations). Finally, the influence of NT-3 on trkA phenotype is most apparent for that subpopulation of trkA neurons that do express trkC (Jongsma Wallin et al., 2001; Karchewski et al., 2002; Gratto and Verge, 2003).
NT-3 influences thermal hyperalgesia but not mechanical hypersensitivity associated with CCI
The ability of NT-3 to prevent and reverse thermal hyperalgesia, believed signaled by C-fibers (Field et al., 1999), is a novel finding with respect to modulation of neuropathic pain. NT-3 is generally thought to influence only large, myelinated proprioceptive neurons, despite evidence that it modulates expression of molecules associated with nociception (Jongsma Wallin et al., 2001; Karchewski et al., 2002; Gratto and Verge, 2003).
Research examining the influence of NT-3 on neuropathic pain-related behaviors is limited. Inflammatory mechanical hyperalgesia was acutely and transiently reversed by local hindpaw injection of NT-3 (Watanabe et al., 2000); however, NT-3 altered neither thermal nor mechanical hypersensitivity resulting from spinal nerve ligation (Boucher et al., 2000). This suggests that the degree of inflammation and/or type of injury may dictate whether NT-3 mitigates C-fiber pathologies. Neuroinflammation resulting from CCI plays a major role in the ensuing hyperalgesic pain state (Maves et al., 1993) and is associated with elevated NGF and cytokine levels (Maves et al., 1993; Sorkin, 2002). Furthermore, CCI preferentially axotomizes large-diameter neurons (Kajander and Bennett, 1992), leaving the remaining intact neurons exposed to a highly inflamed environment in which there is a strong infiltration of immune cells (Wilson-Gerwing, unpublished observations).
The ability of NT-3 to prevent the development of thermal hyperalgesia is not likely caused by the elimination of sensory neurons contributing to this state because removal of NT-3 leads to a prompt reestablishment of the hyperalgesic state and CCI-associated TRPV1 phenotype. The reversion to this state indicates that the influence of NT-3 is transient. NT-3 can also reverse established thermal hyperalgesia. Both the prevention and abolishment of thermal hyperalgesia are likely impacted by the ability of NT-3 to suppress p38 MAPK activation and TRPV1 expression.
In contrast, the mechanical hypersensitivity observed after CCI is not altered by NT-3, despite downregulation of injury-associated peptide expression in larger neurons (Wilson and Verge, 2000). Mechanotransduction is mediated by specialized channels (Garcia-Anoveros et al., 2001; Price et al., 2001; Suzuki et al., 2003a,b), but the manner in which expression or sensitization of these channels is altered by this form of injury or NT-3 is unknown; however, a role for NT-3 in the treatment of chronic mechanical hypersensitivity resulting from intramuscular acid injection indicates a potential role for NT-3 in the treatment of muscular pain (Gandhi et al., 2004).
NT-3 does not significantly alter levels of thermal or mechanical sensitivity in naive animals
NT-3 had no significant influence on thermal thresholds in naive animals. This was surprising because our previous observations that NT-3 modestly influences both levels of trkA expression and associated nociceptive phenotype would have predicted otherwise (Jongsma Wallin et al., 2001; Karchewski et al., 2002; Gratto and Verge, 2003). Past studies are conflicting, differing primarily in the dose injected into the hindpaw, with a higher dose inducing a significant but much delayed thermal sensitivity (Shu et al., 1999; Theodosiou et al., 1999). It remains unknown whether intrathecal delivery of NT-3 used in the present study can directly effect changes in peripheral terminals, as is possible in the former studies.
Although we and others have not found a significant influence of NT-3 in altering baseline mechanical sensitivity (Theodosiou et al., 1999; Zhou et al., 2000), we did find a nonsignificant trend toward increased mechanical sensitivity. Significantly decreased mechanical thresholds were observed in a delayed manner in naive animals subjected to 14 d intrathecal NT-3 infusion (White, 1998), and NT-3 sequestration attenuates mechanical hypersensitivity at later stages of nerve transection injury (Deng et al., 2000; Zhou et al., 2000). The delayed nature of this influence implies that the latency to onset may require alterations in circuitry or protein expression. Whether NT-3-induced changes in mechanosensation correlate with alterations in recently identified channels mediating this function is unknown (Garcia-Anoveros et al., 2001; Price et al., 2001; Suzuki et al., 2003a,b)
Attenuation of thermal hyperalgesia by NT-3 correlates with a reduction in TRPV1 and activated p38 MAPK expression
Localization of TRPV1 expression to primarily small and medium neurons in contralateral intact DRG is in agreement with past studies (Caterina et al., 1997; Eglen et al., 1999; Ma, 2002). Although axotomy results in a dramatic reduction in TRPV1 expression (Michael and Priestley, 1999), spared neurons express elevated levels of TRPV1 (Fukuoka et al., 2001; Hudson et al., 2001), the latter presumably via NGF-mediated activation of p38 MAPK (Ji et al., 2002). Although CCI resulted in slightly elevated TRPV1 expression observed in the size range of neurons consistent with C-fibers, it also induced a low level of expression in a novel population of large neurons. It is unknown whether this phenotypic switch factored into the observed thermal hyperalgesia. Alterations in TRPV1 expression effected by NT-3 were most robust for neurons ipsilateral to injury. In accordance with Ji et al. (2002), the observed modulation of TRPV1 expression was most apparent at the protein versus mRNA level. The alterations in TRPV1 expression effected by CCI and exogenous NT-3 were mirrored by a coordinate modulation of activated p38 MAPK, a known regulator of TRPV1 expression (Ji et al., 2002), implying a direct link to outcome; however, when infusion was halted, TRPV1 expression reverted back to that observed with CCI alone, as did the thermal hyperalgesia, suggesting a suppressive and not curative role for NT-3 in modulation of this state.
Potential mechanisms underlying NT-3 antagonism of thermal hyperalgesia
Thermal hyperalgesia associated with inflammation is conditioned by NGF and requires trkA and TRPV1 (Michael and Priestley, 1999; Shu and Mendell, 1999; Davis et al., 2000; Galoyan et al., 2003). NGF promotes the sensitization of TRPV1 via release from PtdIns(4,5)P2-mediated inhibition (Chuang et al., 2001). Furthermore, trkA signaling can be enhanced by transactivation via G-protein-coupled receptors that are activated by inflammatory mediators and the pituitary adenylate cyclase-activating peptide, the latter being expressed at high levels in an NGF-dependent manner in trkA neurons and downregulated by NT-3 (Jongsma Wallin et al., 2001, 2003). The ability of NT-3 to effect a robust downregulation in TRPV1 expression after CCI and a coordinate decrease in trkA (Wilson-Gerwing, unpublished observations) and activated p38 MAPK expression would impact on the degree to which an individual neuron responds to noxious heat. The rapid NT-3-mediated abolition or reestablishment of the response after NT-3 removal implies that it is not solely dependent on alterations in protein expression but may also involve direct influence on signaling pathways that either alter the efficiency of trkA signaling or mediate sensitization of TRPV1. Whether endogenous nerve-derived sources of NT-3 serve to tonically inhibit this pathway is not known; however, constitutive levels of NT-3 in normal skin are higher than that of NGF, and they drop dramatically in the acute phase of the inflammatory response, suggesting a role in tonic inhibition in the uninjured state (Watanabe et al., 2000). Finally, because the NT-3 was delivered intrathecally, one cannot exclude a potential effect of NT-3 on central sensitization events that impact on the development of thermal hyperalgesia (for review, see Melzack et al., 2001; Romanelli and Esposito, 2004). Although not the focus of this study, our previous work has shown that this form of NT-3 delivery mitigates the level of BDNF expressed by primary sensory afferents and linked to central sensitization events (Groth and Aanonsen, 2002; Garraway et al., 2003; Lever et al., 2003). The degree to which other molecules or cell types involved in this process are similarly influenced remains to be elucidated.
In conclusion, past studies reveal a major role for NGF in driving inflammation-associated thermal hyperalgesia via its activation of p38 MAPK and subsequent regulation of TPVR1 expression and other mediators of inflammatory responses. NT-3 is now identified as a potent negative modulator of this state. The extent to which it might ameliorate other aspects of inflammatory neuropathic pain responses remains to be elucidated.
Footnotes
This work was supported by grants from the Canadian Institutes of Health Research, Neurotrauma Partners of Canada, and Saskatchewan Regional Partnership Program. T.D.W.-G. is supported by a University of Saskatchewan Graduate Scholarship. We are indebted to R. W. Wilson for manufacture of the von Frey platform. We thank Regeneron Pharmaceuticals (Tarrytown, NY) for the generous supply of NT-3 used in this study.
Correspondence should be addressed to Valerie M. K. Verge, Cameco Multiple Sclerosis/Neuroscience Research Center, Saskatoon City Hospital, Room 5800, 701 Queen Street, Saskatoon, Saskatchewan, Canada S7N 5E5. E-mail: verge@sask.usask.ca.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250758-10$15.00/0
References
- Bennett DL, Michael GJ, Ramachandran N, Munson JB, Averill S, Yan Q, McMahon SB, Priestley JV (1998) A distinct subgroup of small DRG cells express GDNF receptor components and GDNF is protective for these neurons after injury. J Neurosci 18: 3059-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett GJ, Xie Y-K (1988) A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33: 87-107. [DOI] [PubMed] [Google Scholar]
- Boucher TJ, Okuse K, Bennett DL, Munson JB, Wood JN, McMahon SB (2000) Potent analgesic effects of GDNF in neuropathic pain states. Science 290: 124-127. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389: 816-824. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D (2000) Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288: 306-313. [DOI] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Sheilds S, Jordt S-E, Basbaum AI, Chao MV, Julius D (2001) Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411: 957-962. [DOI] [PubMed] [Google Scholar]
- Dagerlind A, Friberg K, Bean AJ, Hokfelt T (1992) Sensitive mRNA detection using unfixed tissue: combined radioactive and non-radioactive in situ hybridization histochemistry. Histochemistry 98: 39-49. [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA (2000) Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405: 183-187. [DOI] [PubMed] [Google Scholar]
- Deng YS, Zhong JH, Zhou XF (2000) Effects of endogenous neurotrophins on sympathetic sprouting in the dorsal root ganglia and allodynia following spinal nerve injury. Exp Neurol 164: 344-350. [DOI] [PubMed] [Google Scholar]
- Eglen RM, Hunter JC, Dray A (1999) Ions in the fire: recent ion-channel research and approaches to pain therapy. Trends Pharmacol Sci 20: 337-342. [DOI] [PubMed] [Google Scholar]
- Field MJ, Bramwell S, Hughes J, Singh L (1999) Detection of static and dynamic components of mechanical allodynia in rat models of neuropathic pain: are they signaled by distinct primary sensory neurones? Pain 83: 303-311. [DOI] [PubMed] [Google Scholar]
- Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K (2001) Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci 21: 4891-4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galoyan SM, Petruska JC, Mendell LM (2003) Mechanisms of sensitization of the response of single dorsal root ganglion cells from adult rat to noxious heat. Eur J Neurosci 18: 535-541. [DOI] [PubMed] [Google Scholar]
- Gandhi R, Ryals JM, Wright DE (2004) Neurotrophin-3 reverses chronic mechanical hyperalgesia induced by intramuscular acid injection. J Neurosci 24: 9405-9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Anoveros J, Samad TA, Zuvela-Jelaska L, Woolf CJ, Corey DP (2001) Transport and localization of the DEG/EnaC ion channel BNaC1α to peripheral mechanosensory terminals of dorsal root ganglia neurons. J Neurosci 21: 2678-2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Martinez C, Humet M, Planella-Cases R, Gomis A, Caprini M, Viana F, De La Pena E, Sanchez-Baeza F, Carbonell T, De Filipe C, Perez-Paya E, Belmonte C, Messeguer A, Ferrer-Montiel A (2002) Attenuation of thermal nociception and hyperalgesia by VR1 blockers. Proc Natl Acad Sci USA 99: 2374-2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway SM, Petruska JC, Mendell LM (2003) BDNF sensitized the response of lamina II neurons to high threshold primary afferent inputs. Eur J Neurosci 18: 2467-2476. [DOI] [PubMed] [Google Scholar]
- Gratto KA, Verge VMK (2003) Neurotrophin-3 down-regulates trkA mRNA, NGF high-affinity binding sites, and associated phenotype in adult DRG neurons. Eur J Neurosci 18: 1535-1548. [DOI] [PubMed] [Google Scholar]
- Groth R, Aanonsen L (2002) Spinal brain-derived neurotrophic factor (BDNF) produces hyperalgesia in normal mice while antisense directed against either BDNF or trkB prevents inflammation-induced hyperalgesia. Pain 100: 171-181. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32: 77-88. [DOI] [PubMed] [Google Scholar]
- Hudson LJ, Bevan S, Wotherspoon G, Gentry C, Fox A, Winter J (2001) VR1 protein expression increases in undamaged DRG neurons after partial nerve injury. Eur J Neurosci 13: 2105-2114. [DOI] [PubMed] [Google Scholar]
- Ji RR, Woolf CJ (2001) Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis 8: 1-10. [DOI] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ (2002) p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 36: 57-68. [DOI] [PubMed] [Google Scholar]
- Jongsma Wallin H, Danielsen N, Johnston JM, Gratto KA, Karchewski LA, Verge VMK (2001) Exogenous NT-3 and NGF differentially modulate PACAP expression in adult sensory neurons, suggesting distinct roles in injury and inflammation. Eur J Neurosci 14: 267-282. [DOI] [PubMed] [Google Scholar]
- Jongsma Wallin H, Pettersson LM, Verge VM, Danielsen N (2003) Effect of anti-nerve growth factor treatment on pituitary adenylate cyclase activating polypeptide expression in adult sensory neurons exposed to adjuvant induced inflammation. Neuroscience 120: 325-331. [DOI] [PubMed] [Google Scholar]
- Kajander KC, Bennett GJ (1992) Onset of a painful peripheral neuropathy in rat: a partial and differential deafferentation and spontaneous discharge in A beta and A delta primary afferent neurons. J Neurophysiol 68: 734-744. [DOI] [PubMed] [Google Scholar]
- Karchewski LA, Kim FA, Johnston J, McKnight RM, Verge VMK (1999) Anatomical evidence supporting the potential for modulation by multiple neurotrophins in the majority of adult lumbar sensory neurons. J Comp Neurol 413: 327-341. [DOI] [PubMed] [Google Scholar]
- Karchewski LA, Gratto KA, Wetmore C, Verge VMK (2002) Dynamic patterns of BDNF expression in injured sensory neurons: differential modulation by NGF and NT-3. Eur J Neurosci 16:1449-1462. Lever I, Cunningham J, Grist J, Yip PK, Malcangio M (2003) Release of BDNF and GABA in the dorsal horn of neuropathic rats. Eur J Neurosci 18: 1169-1174. [DOI] [PubMed] [Google Scholar]
- Lever I, Cunningham J, Grist J, Yip PK, Malcangio M (2003) Release of BDNF and GABA in the dorsal horn of neuropathic rats. Eur J Neurosci 18: 1169-1174. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Mendell LM (1993) Nerve growth factor and nociception. Trends Neurosci 16: 353-358. [DOI] [PubMed] [Google Scholar]
- Lindsay RM (1996) Role of neurotrophins and trk receptors in the development and maintenance of sensory neurons: an overview. Philos Trans R Soc Lond B Biol Sci 351: 365-373. [DOI] [PubMed] [Google Scholar]
- Ma QP (2002) Expression of capsaicin receptor (VR-1) by myelinated primary afferent neurons in rats. Neurosci Lett 319: 87-90. [DOI] [PubMed] [Google Scholar]
- Melzack R, Coderre TJ, Katz J, Vaccarino AL (2001) Central neuroplasticity and pathological pain. Ann NY Acad Sci 933: 157-174. [DOI] [PubMed] [Google Scholar]
- Maves TJ, Pechman PS, Gebhart GF, Meller ST (1993) Possible chemical contribution from chromic gut sutures produces disorders of pain sensation like those seen in man. Pain 54: 57-69. [DOI] [PubMed] [Google Scholar]
- Michael GJ, Priestley JV (1999) Differential expression of the mRNA for the vanilloid receptor subtype 1 in cells of the adult rat dorsal root and nodose ganglia and its downregulation by axotomy. J Neurosci 19: 1844-1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki K, Fukuoka T, Tokunaga A, Noguchi K (1998) Calcitonin gene-related peptide increase in the rat spinal dorsal horn and dorsal column nucleus following peripheral nerve injury: up-regulation in a subpopulation of primary afferent sensory neurons. Neuroscience 82: 1243-1252. [DOI] [PubMed] [Google Scholar]
- Nahin RL, Ren K, De Leon M, Ruda M (1994) Primary sensory neurons exhibit altered gene expression in a rat model of neuropathic pain. Pain 58: 95-108. [DOI] [PubMed] [Google Scholar]
- Pitcher GM, Ritchie J, Henry JL (1999) Paw withdrawal threshold in the von Frey hair test is influenced by the surface on which the rat stands. J Neurosci Methods 87: 185-193. [DOI] [PubMed] [Google Scholar]
- Price MP, McIlwrath SL, Xie J, Cheng C, Qiao J, Tarr DE, Sluka KA, Brennan TJ, Lewin GR, Welsh MJ (2001) The DRASIC cation channel contributes to the detection of cutaneous touch and acid stimuli in mice. Neuron 32: 1071-1083. [DOI] [PubMed] [Google Scholar]
- Romanelli P, Esposito V (2004) The functional anatomy of neuropathic pain. Neurosurg Clin N Am 15: 257-268. [DOI] [PubMed] [Google Scholar]
- Shu XQ, Llinas A, Mendell LM (1999) Effects of trkB and trkC neurotrophin receptor agonists on thermal nociception: a behavioral and electrophysiological study. Pain 80: 463-470. [DOI] [PubMed] [Google Scholar]
- Sorkin LS (2002) Neuroinflammation, cytokines and neuropathic pain. In: Progress in inflammation research (Malmberg AB, Chaplan SR, eds), pp 67-75. Berlin: Birkhauser Verlag.
- Suzuki M, Mizuno A, Kodaira K, Imai M (2003a) Impaired pressure sensation in mice lacking TRPV4. J Biol Chem 278: 22664-22668. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Watanabe Y, Oyama Y, Mizuno A, Kusano E, Hirao A, Ookawara S (2003b) Localization of mechanosensitive channel TRPV4 in mouse skin. Neurosci Lett 353: 189-192. [DOI] [PubMed] [Google Scholar]
- Theodosiou M, Rush RA, Zhou XF, Hu D, Walker JS, Tracey DJ (1999) Hyperalgesia due to nerve damage: role of nerve growth factor. Pain 81: 245-255. [DOI] [PubMed] [Google Scholar]
- Verge VMK, Riopelle RJ, Richardson PM (1989a) Nerve growth factor receptors on normal and injured sensory neurons. J Neurosci 9: 914-922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verge VMK, Richardson PM, Benoit R, Riopelle RJ (1989b) Histochemical characterization of sensory neurons with high-affinity receptors for nerve growth factor. J Neurocytol 18: 583-591. [DOI] [PubMed] [Google Scholar]
- Verge VMK, Zhang X, Xu X-J, Wiesenfeld-Hallin Z, Hokfelt T (1992) Marked increase in nitric oxide synthase mRNA in rat dorsal root ganglia after peripheral axotomy: in situ hybridization and functional studies. Proc Natl Acad Sci USA 89: 11617-11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verge VMK, Richardson PM, Wiesenfeld-Hallin Z, Hokfelt T (1995) Differential influence of nerve growth factor on neuropeptide expression in vivo: a novel role in peptide suppression in adult sensory neurons. J Neurosci 15: 2081-2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verge VMK, Gratto KA, Karchewski LA, Richardson PM (1996) Neurotrophins and nerve injury in the adult. Philos Trans R Soc Lond B Biol Sci 351: 423-430. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Endo Y, Kimoto K, Katoh-Semba R, Arakawa Y (2000) Inhibition of adjuvant-induced inflammatory hyperalgesia in rats by local injection of neurotrophin-3. Neurosci Lett 282: 61-64. [DOI] [PubMed] [Google Scholar]
- White DM (1998) Contribution of neurotrophin-3 to the neuropeptide Y-induced increase in neurite outgrowth of rat dorsal root ganglion cells. Neuroscience 86: 257-263. [DOI] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL (1999) Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev 79: 143-180. [DOI] [PubMed] [Google Scholar]
- Wilson TD, Verge VMK (2001) NT-3 alters responses to thermal but not to mechanical stimulation following chronic constriction injury (CCI) of the sciatic nerve in rats. Soc Neurosci Abstr 27: 136.15. [Google Scholar]
- Wilson-Gerwing TD, Verge VMK (2003) NT-3 attenuation of thermal hyperalgesia is associated with decreased VR-1 expression. Soc Neurosci Abstr 29: 381.15. [Google Scholar]
- Winston J, Toma H, Shenoy M, Pasricha PJ (2001) Nerve growth factor regulates VR-1 mRNA levels in cultures of adult dorsal root ganglion neurons. Pain 89: 181-186. [DOI] [PubMed] [Google Scholar]
- Zhou XF, Deng YS, Xian CJ, Zhong JH (2000) Neurotrophins from dorsal root ganglia trigger allodynia after spinal nerve injury in rats. Eur J Neurosci 12: 100-105. [DOI] [PubMed] [Google Scholar]