

Abstract
Ca2+-binding proteins are ubiquitously expressed throughout the CNS and serve as valuable immunohistochemical markers for certain types of neurons. However, the functional role of most Ca2+-binding proteins has to date remained obscure because their concentration in central neurons is not known. In this study, we investigate the intracellular concentration of the widely expressed Ca2+-binding protein calbindin-D28k in adult hippocampal slices using patch-clamp recordings and immunohistochemistry. First, we show that calbindin-D28k freely exchanges between patch pipette and cytoplasm during whole cell patch-clamp recordings with a time constant of ∼10 min. Substituting known concentrations of recombinant calbindin-D28k in patch pipettes enabled us to determine the endogenous calbindin-D28k concentration by postrecording immunohistochemistry. Using this calibration procedure, we find that mature granule cells (doublecortin-) contain ∼40 μm, and newborn granule cells (doublecortin+) contain 0-20 μm calbindin-D28k. CA3 stratum radiatum interneurons and CA1 pyramidal cells enclose ∼47 and ∼45 μm calbindin-D28k, respectively. Numerical simulations showed that 40 μm calbindin-D28k is capable of tuning Ca2+ microdomains associated with action potentials at the mouth of single or clustered Ca2+ channels: calbindin-D28k reduces the increment in free Ca2+ at a distance of 100 and 200 nm by 20 and 35%, respectively, and strongly accelerates the collapse of the Ca2+ gradient after cessation of Ca2+ influx. These data suggest that calbindin-D28k equips hippocampal neurons with ∼160 μm mobile, high-affinity Ca2+-binding sites (κS ∼200) that slow and reduce global Ca2+ signals while they enhance the spatiotemporal fidelity of submicroscopic Ca2+ signals.
Keywords: calcium, calcium binding ratio, adult neurogenesis, confocal laser scanning microscopy, buffered calcium diffusion, single compartment model
Introduction
Ca2+ is a highly versatile intracellular signal known to be important in a wide range of cellular functions such as excitability, transmitter release, gene transcription, or neuronal cell death. How the spatial and temporal extent of a rise in intracellular Ca2+ is shaped is therefore a fundamental question. A key factor in determining the spatiotemporal dynamics of Ca2+ signals is the presence of mobile endogenous Ca2+ buffers in the cytosol. Intracellular Ca2+ signals may be differentially affected by the presence of mobile Ca2+ buffers, depending on the spatial dimension of the signal (for review, see Neher, 1998). Global Ca2+ signals that encompass a large portion of neuronal compartments, such as dendritic Ca2+ signals associated with back-propagating action potentials, will be slowed and their initial amplitude will be decreased. More restricted Ca2+ signals, such as those occurring in spines or dendritic subsegments, can have an enlarged range of action: by competing with fixed cytosolic Ca2+-binding sites, diffusible buffers are capable of enhancing Ca2+ diffusion and thus counteract the compartmentalization of Ca2+ signals. At the scale of the highly localized Ca2+ build-up in the vicinity of single Ca2+-permeable channels (so-called Ca2+ microdomains), Ca2+ transients will be spatially and temporally restricted by the presence of mobile buffers. How the different types of Ca2+ signals are modified depends critically on the Ca2+-binding properties and the intracellular concentration of the buffer. For example, whereas low micromolar concentrations of high-affinity Ca2+ buffers strongly affect global Ca2+ transients (Helmchen et al., 1996) as well as the diffusional spread of Ca2+ (Gabso et al., 1997), high concentrations and fast binding rates of mobile buffers are needed to affect microdomain Ca2+ signaling (Roberts, 1994).
A major endogenous Ca2+ buffer is the Ca2+-binding protein calbindin-D28k, which is prominently expressed in specific types of neurons throughout the CNS, including hippocampal granule and pyramidal cells as well as cerebellar Purkinje cells (Baimbridge et al., 1992). Calbindin-D28k is the primary Ca2+-binding protein of hippocampal principal cells, because they do not contain other Ca2+-binding proteins such as parvalbumin or calretinin. Calbindin-D28k belongs to the EF-hand family of Ca2+-binding proteins and contains four high-affinity Ca2+-binding sites (Veenstra et al., 1997; Berggard et al., 2002) that bind Ca2+ with a fast association rate (Nägerl et al., 2000). Attempts have been made to estimate the endogenous buffer concentration in calbindin-D28k-containing neurons using microfluorometric imaging techniques and quantitative analysis of Ca2+ signaling (Maeda et al., 1999; Jackson and Redman, 2003). However, these studies used models that depend on several parameters that are not known and only speculate about the molecular identity of the underlying buffers. Therefore, the intracellular concentration of calbindin-D28k has remained unclear to date, and the role of this Ca2+-binding protein in shaping the spatiotemporal dynamics of intracellular Ca2+ signals is still difficult to judge.
In this study, we use a novel approach combining patch-clamp recordings with intracellular dialysis of purified calbindin-D28k and postrecording immunohistochemistry to quantitatively determine the intraneuronal concentration of calbindin-D28k in hippocampal neurons.
Materials and Methods
Preparation of slices and recording configuration. Wistar rats or adult mice (20- to 40-d-old) (see below) were decapitated under ether anesthesia. The brain was rapidly dissected and chilled in 4°C cold cutting solution consisting of (in mm): 140 NaCl, 3 KCl, 2 MgSO4, 2 CaCl2, 1 Na pyruvate, 10 HEPES, and 10 glucose, pH 7.4 (NaOH, 100% O2). Four hundred micrometer horizontal hippocampal slices were prepared with a vibratome (Leica, Nussloch, Germany). For recording, slices were transferred to an interface chamber perfused with ACSF (35 ± 0.5°C) that contained (in mm): 125 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 20.3 NaHCO3, and 12 d-glucose, pH 7.4 (95% O2 and 5% CO2). Patch pipettes had resistances between 4 and 5 MΩ. The standard pipette solution was free of any Ca2+ chelators and contained (in mm): 140 K+-gluconate, 0.5 MgCl2, 4 NaCl, 10 HEPES, 5 KCl, and 0.1% Lucifer yellow (LY). A small amount of EGTA (20 μm) was added to the solution to balance Ca2+ contamination of salts.
A 100 μm calbindin-D28k stock solution was prepared by dissolving the lyophilized protein (100 μg in 0.1 mm CaCl2; Swant, Bellinzona, Switzerland) in the standard pipette solution. This stock solution was then diluted with the standard pipette solution to obtain the desired concentration of calbindin-D28k (10, 25, 33, and 66 μm) that was used for filling the patch pipettes.
Whole-cell recordings were obtained from granule cells in hippocampal slices using the “blind technique” and an SEC-05LX amplifier (standard headstage; NPI Electronics, Tamm, Germany). Series resistance of the cells ranged between 20 and 30 MΩ and was estimated by bridge balancing after careful compensation of the pipette capacitance.
Immunohistochemistry. Hippocampal slices from rats and mice were fixed overnight in 8% paraformaldehyde and subsequently resectioned at 40 μm. Those sections were then incubated for 24 h at 4°C with monoclonal mouse anti-calbindin-D28k antibodies (1:500; Swant) in TBS containing 0.3% Triton X-100. Next, biotinylated sheep anti-mouse antibodies (1:200; Vector Laboratories, Linaris, Wertheim-Bettingen, Germany) and streptavidin-conjugated Cy5 (1:250; Jackson ImmunoResearch, West Grove, PA) were each applied overnight. A green fluorescent Nissl stain (“Neurotrace”; Molecular Probes, Eugene, OR) was used in some cases for counterstaining. Sections were analyzed with a confocal laser-scanning microscope (Leica TCS NT, equipped with an argon-krypton laser), and images with different dyes (LY and Cy5) were acquired sequentially. For LY, the following filters and laser lines were used: excitation, 488 nm; dichroic, 510 nm; emission bandpass, 530 ± 30 nm. For Cy5 we used: excitation, 647 nm; dichroic, 660 nm; emission long pass, 665 nm. In separate experiments, we verified (in the absence of Cy5 staining) that LY-filled granule cells do not lead to detectable emission when the Cy5 filter and laser combination was used. All scans were acquired with a 63× objective (oil, numerical aperture 1.4), and the pinhole was set 1 Airy unit. Laser power, detector gain, and offset were adjusted such that in the final scan (two to four averages) only a few pixels had zero or maximal (255) digital units. Typically, detector gain and offset were set to ∼70% and approximately six digital units, respectively.
For calbindin-D28k and doublecortin double-labeling, slices were successively incubated in goat anti-doublecortin antibodies (1:100; Santa Cruz Biotechnology, Santa Cruz, CA), biotinylated anti-goat antibodies (1:200; Vector Laboratories), and streptavidin-Cy5. Then calbindin-D28k staining was performed as above but developed with a FITC-conjugated anti-mouse antibody (1:200; Serotec, Oxford, UK).
Quantification of immunoreactivity of granule cells. Quantitative image analysis was performed with NIH ImageJ software. The immunofluorescence of a given granule cell, referred to as F, was quantified by calculating the mean value over the whole soma and subtracting the background fluorescence. The background fluorescence (the sum of: out of focus light from surrounding granule cells, offset of the photomultiplier, unspecific fluorescence in the tissue, etc.) was determined by measuring the mean fluorescence of small calbindin-D28k-negative presumably glial cells (see Fig. 1 B). The small cells were selected within the granule cell layer either in the same scan or in an immediately adjacent scan that was acquired under identical settings.
Figure 1.
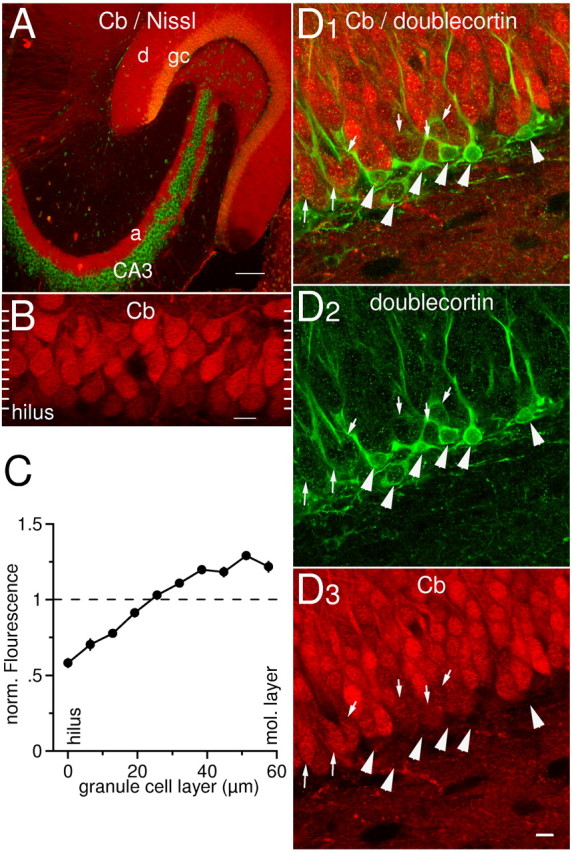
Distribution of calbindin-D28k immunoreactivity in the dentate area. A, Dual channel scan obtained with a confocal laser microscope. Calbindin-D28k (Cb) is present throughout the entire cytoplasm of granule cells: dendrites (d), somata (gc), and axons (a) are stained (red). Note that CA3 pyramidal cells are completely negative, whereas some scattered interneurons are found positive. Green indicates fluorescent counterstain (“Nissl”). Scale bar, 100 μm. 10× objective. B, High-magnification scan of the granule cell layer stained for calbindin-D28k. Note that the calbindin-D28k content of granule cells is heterogeneous: granule cells adjacent to the molecular layer (top) are brighter than hilar dentate granule cells (bottom). The white ticks on the sides of the image indicate how the scan was divided into 10 sections for quantitative analysis in C (see Materials and Methods for details). C, Quantitative analysis of the gradient of calbindin-D28k immunoreactivity across the granule cell layer. The granule cell layer (n = 10) was divided into 10 sections, and the normalized mean fluorescence in each section is plotted versus its position within the layer. D, Double-labeling with anti-calbindin-D28k (red) and anti-doublecortin antibodies (green). Cells that are weakly calbindin-D28k-labeled are doublecortin-positive (small arrows). Strongly calbindin-D28k-positive cells are doublecortin-negative. Note that cells intensely stained for doublecortin appear calbindin-D28k-negative (large arrowheads).
For quantitative analysis of the gradient of calbindin-D28k immunoreactivity (see Fig. 1C), we divided the granule cell layer in each scan into 10 sections of ∼6 μm (see Fig. 1 B). In every section, we calculated the average of the immunofluorescence values F of those parts of the granule cell somata crossing that section. Each of the 10 average values were then normalized on the mean of immunofluorescence values F of all granule cells in that particular scan. This normalization procedure corrects for differences in illumination, focus depth, brightness of staining, and other confounding factors that could be different from slice to slice. The normalized average values were then plotted versus their position within the granule cell layer.
Figure 2 illustrates the time course of the reduction of the immunofluorescence of granule cells during a whole-cell recording. Because granule cells located close to the molecular layer contain more calbindin-D28k than hilar granule cells (compare Fig. 1) we had to compare the immunofluorescence value F of a recorded granule cell with the immunofluorescence of neighboring granule cells in the same section. Therefore, and to correct for the abovementioned confounding factors, F of the recorded cell was normalized on the average of the F values of those granule cells lying in the same section (in analogy to Fig. 1).
Figure 2.

Endogenous calbindin-D28k is diluted during whole-cell recordings of granule cells. A, A granule cell was recorded and filled with LY for 30s in whole-cell mode. Subsequently, the slice was fixed and stained for calbindin-D28k. Top, Confocal scans of LY fluorescence and of calbindin-D28k (Cy5) fluorescence are merged into one image. The arrow indicates the recorded cell, which is easily identified by the green fluorescence of LY. Bottom, Calbindin-D28k (Cy5) fluorescence only. Note that the recorded cell and neighboring cells show identical levels of fluorescence. Image size in A-D: 116 × 116 μm. B, As in A, but the cell was recorded for 6 min. There is a slight reduction in immunoreactivity of the LY-filled cell. C, As in A, but with a recording time of 12 min. Now, a clear reduction in immunoreactivity of the recorded cell is visible, signifying a dilution of endogenous calbindin-D28k concentration. The cell is less bright than its neighbors but still brighter than background levels when compared with calbindin-D28k-negative cells in the molecular layer or in the hilus (in between positive mossy fibers). D, As in A, but the cell was recorded for a prolonged period, 38 min. Note that the fluorescence of the recorded granule cell is similar to background levels when compared with calbindin-D28k-negative cells in the molecular layer or in the hilus (in between positive mossy fibers), indicating that the cellular calbindin-D28k content is nearly completely lost.E, Quantification of the loss of calbindin-D28k immunoreactivity during whole-cell recordings. The immunofluorescence of each recorded granule cell was normalized on that of neighboring granule cells (fluorescence relationship to neighbors; see Materials and Methods for details) and plotted versus the whole-cell recording time (n = 34). The time-dependent drop in immunofluorescence could be fitted with an exponential function yielding a time constant of ∼10 min.
When we filled granule cells with different concentrations of purified calbindin-D28k (whole-cell recording time, 30-40 min) we intended to estimate the overall mean calbindin-D28k content of granule cells (see Fig. 4). For this reason the immunofluorescence F of a recorded and filled granule cell was normalized on the mean of the immunofluorescence values F of all granule cells in that particular scan.
Figure 4.

Granule cells contain 33 μm calbindin-D28k. Quantitative analysis of 26 experiments as shown in Figure 3. Dentate granule cells were loaded with 0 μm (n = 3), 10 μm (n = 6), 25 μm (n = 7), 33 μm (n = 7), or 66 μm (n = 3) calbindin-D28k. The immunofluorescence of the recorded cell was normalized on the mean fluorescence of all neighboring granule cells, averaged for each concentration, and then plotted versus the concentration. Data points were fitted by a line (superimposed). From this line, the calbindin-D28k concentration that lets the recorded cells appear equifluorescent to surrounding granule cells (normalized fluorescence equals 1; arrow) can be obtained: 32.5 μm (denoted by the arrowhead). The dashed lines originating from the ordinate illustrate translations of immunofluorescence of granule cells (as determined in Fig. 1) into calbindin-D28k concentrations.
To estimate the calbindin-D28k concentration in CA1 pyramidal cells and CA3 stratum radiatum interneurons (see Fig. 6), calbindin-D28k staining was developed with Cy2-conjugated streptavidin (1:250; Jackson ImmunoResearch). Then we proceeded as follows in each slice (n = 5): F was quantified for dendritic subsegments of the three types of neurons. Next, we calculated the average over all granule cells (Fav,gc). Then F of each CA1 pyramidal cell and of each CA3 stratum radiatum interneurons was converted to a calbindin-D28k concentration according to: 33 μm *FCA1/CA3/Fav,gc. Finally, the calbindin-D28k concentrations were averaged over all slices for each type of cell. During all scans within one slice detector settings and laser power were kept constant. Laser power was measured by a photodiode, and variations in light intensity were compensated for manually. The calbindin-D28k concentration for mature granule cells shown in Figure 6 was obtained by multiplying the right-most data point (and the associated error) in Figure 1C with 33 μm calbindin-D28k.
Figure 6.

Different types of hippocampal neurons contain similar calbindin-D28k concentrations. A, High-magnification scans of dendrites of a granule cell (gc), a CA3 interneuron, and a CA1 pyramidal cell as they were used for quantitative analysis. Note that dendritic segments chosen have comparable geometry and are equally bright. Image dimensions are 13 × 34 μm. B, Summary bar graph of intracellular calbindin-D28k concentrations in hippocampal neurons as determined by comparing their calbindin-D28k-related immunofluorescence. All three types of cells investigated contain similar concentrations around 40 μm. For details of calculations, see Materials and Methods.
We tested whether the fluorophore Cy5 accumulates so strongly within granule cells that its emission of fluorescence increases only sublinearly with concentration. We produced a series of streptravidin-Cy5 dilutions that covered the range in which the emission was comparable with that obtain from slices. The streptravidin-Cy5 dilutions contained in microcuvettes were placed under the confocal microscope and were scanned under analogous conditions. We found no signs for saturation of Cy5 emission in this concentration range, and the emission increased proportional to the concentration (data not shown).
Calbindin-D28k knock-out mice. The calbindin-D28k null mutant mice used in this study have been described previously (Airaksinen et al., 1997). We obtained mice from The Jackson Laboratory (Bar Harbor, ME), and homozygous knock-out mice were achieved by heterozygous breeding. All animals were genotyped by PCR on DNA samples extracted from tail tissue. The following primers were used: for wild-type, 5′-GCA AGT AAC TAA TGG CAT CG-3′ and 5′-TGC AGC GGC TAG TTT GAG AGT G-3′; for null mutant, 5′-ACA TCG CAT CGA GCG AGC AC-3′ and 5′-AAG GCG ATG CGC TGC GAA TC-3′.
Numerical simulation of buffered Ca2+ diffusion around a single Ca2+ channel. Numerical simulations were performed to study the buffering of calbindin-D28k on Ca2+ microdomains. We consider a single Ca2+ channel that is embedded in a planar membrane that limits a pseudoinfinitely large hemisphere (radius, 30 μm) around the channel. We are interested in the spatial and temporal development of free Ca2+ around the channel after its opening in the presence and absence of various buffers. We make the following standard assumptions: Fickian diffusion of calbindin-D28k and of Ca2+ and second order reaction of calbindin-D28k with Ca2+ according to the law of mass action. We further assume that the diffusion coefficient of calbindin-D28k does not change when it binds Ca2+. These assumptions lead to a well known set of differential equations that can be found previously (Nowycky and Pinter, 1993; Roberts, 1994; Naraghi and Neher, 1997). The diffusion coefficient, KD, and kon of calbindin-D28k were 20 μm2/s, 700 nm, 2.7 × 107 M-1s-1, respectively (Nägerl et al., 2000; Berggard et al., 2002; Schmidt et al., 2003). A 40 μm concentration of calbindin-D28k was modeled as 160 μm Ca2+-binding sites with identical Ca2+ association rates and affinities that were derived as averages of the corresponding values of the four Ca2+-binding sites recently published. The hemisphere (radius 30 μm) was divided into 30,000 shells. Within the first 600 nm from the origin, the shell size was constant at 0.06 nm, and the size of the following shells increased by 5% successively. The maximum single-channel Ca2+ current (at physiological levels of divalent cations) was assumed to be 0.2 pA (Gollasch et al., 1992). Because we are interested in Ca2+ microdomains that develop during action potentials, we took into account the varying driving force of the single-channel current during the repolarizing phase of the action potential. Thus, the single-channel current is not square-like: the channel opens and closes in a step-like manner, but the current steeply increases during the open time and reaches a value of 0.2 pA at the end of the opening. The time course of single-channel current amplitude was empirically approximated with the following function (in picoamperes): 0.2 *(1 + tanh(5 *(t-0.2)))/2, where t (ms) is the time (see Fig. 5A, inset). Simulation starts at t = 0 ms where the channel opens, ends at 1.6 ms, and the channel closes abruptly at t = 0.8 ms. Before the simulation starts, calbindin-D28k and Ca2+ are homogenously distributed and have reached chemical equilibrium at resting Ca2+ concentration (50 nm). The differential equations were integrated using CalC (Calcium Calculator) software [version 4.9.8, freeware by V. Matveev et al. (2004), http://web.njit.edu/~matveev/calc.html]. This script-operated program uses the Crank-Nicholson differences scheme (one-dimensional) and an adaptive time-step method to maintain a given level of accuracy (<0.1%). Because CalC only offers the full spherical geometry, we increased the single-channel current by factor 2, which yields results identical to simulating a hemisphere because of the complete symmetry. The correctness of the program was thoroughly checked against the transient analytical solutions of unbuffered Ca2+ diffusion and of buffered Ca2+ diffusion in case of a very mobile buffer (Crank, 1975; Pape et al., 1998). The output of the program was imported into IgorPro for additional analysis and for graphing.
Figure 5.

Numerical simulation of buffered Ca2+ diffusion around a single Ca2+ channel. A, Left, Time course of free Ca2+ after opening and closing of a single Ca2+ channel in the absence and presence of calbindin-D28k at three distances from the channel (15, 50, and 150 nm). The channel opens at 0 ms and closes at 0.8 ms. The inset shows the time-dependent variation of the simulated single-channel current according to the varying driving force during repolarization of the action potential. For comparison, an action potential is aligned on the same time scale (gray dotted trace). Calbindin-D28k exerts a distance-dependent inhibition on free Ca2+. A notable percentage reduction of free Ca2+ is observed only at a distance of 150 nm. In contrast, calbindin-D28k strongly reduces free Ca2+ after closure of the channel. This is best seen in the right panel, where data are plotted on a semilogarithmic graph. The arrow indicates the prominent reduction in Ca2+ when the channel has closed. The single-channel open time chosen here signifies an upper limit, because most Ca2+ channels would close earlier (Lee and Elmslie, 1999) during the repolarization phase of an action potential. B, Radial gradient of action potential associated free Ca2+ reached around the channel after 0.8 ms (black lines) and after 1.6 ms (when the channel is closed for 0.8 ms, red lines). The continuous lines denote the Ca2+ profile in the absence of calbindin-D28k (Cb); the dashed lines denote the Ca2+ profile in the presence of calbindin-D28k. Black lines, A steep Ca2+ gradient is present that reaches >80 μm close to the channel. It can be seen that close to the channel (<30 nm), the black line and the black dotted line superimpose, indicating that free Ca2+ is hardly affected by calbindin-D28k. Red lines, The Ca2+ gradient has collapsed, and a residual Ca2+ concentration is effectively removed at all distances through shuttling by calbindin-D28k. Ca2+ concentration for red lines is indicated by the right axis, which is scaled up for clarity. Note that the resting Ca2+ concentration (50 nm) of the left and the right axis are on the same level. Distance indicates the distance from the Ca2+ source. C, Radial gradients of the percentage inhibition of the increase in Ca2+ above resting Ca2+ (Δ[Ca2+]) caused by calbindin-D28k at the end of a 0.8-ms-long channel opening. For each condition, we ran the simulation for 0.8 ms and calculated a radial profile of the percentage inhibition of ΔCa2+ at the end of the channel opening as shown by black lines in B. Three different families of curves are plotted (red, black, and blue) that represent three different values of kon (indicated by small numbers, in M-1s-1). Within each of the three families, we varied the single-channel current amplitude as coded by the mode of the lines (0.2, 0.8, 1.6, and 3.2 pA; see key). At 15 nm, the concentration of Ca2+-bound calbindin-D28k increased at 0.8 ms by ∼8 μm (0.2 pA) and by ∼100 μm (3.2 pA). It should be noted that the percentage inhibition plotted here represents an upper estimate, because no other competing buffer is included in the model. If, for example, a 500 μm concentration of a fixed buffer with a relatively fast on-rate (kon = 1 × 108 m/s; KD = 50 μm) is present, then the inhibition caused by addition of calbindin-D28k will be reduced by a factor of 2.
Formulas. Ca2+-free buffer concentration was calculated according to the law of mass action
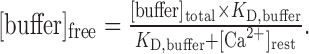 |
1 |
Ca2+-binding ratio was calculated as (Neher, 1995):
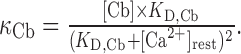 |
2 |
Influence of calbindin-D28k on global Ca2+ signals. Under the assumptions of the single compartment model (Neher, 1998), the buffering effect of calbindin-D28k on the initial amplitude (A) and on the decay time constant (τ) of free Ca2+ after brief Ca2+ influx can be described by the following equations (cf. Helmchen et al., 1996). Let κCb be the Ca2+-binding ratio of calbindin-D28k and κX that of other endogenous Ca2+ buffers. Then, one can write for the amplitude and decay of free Ca2+:
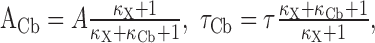 |
3 |
where τ and A denote the values of the amplitude and the decay time constant of free Ca2+ in the absence of calbindin-D28k.
Data are given as mean ± SEM throughout. The level of statistical significance was set to α = 0.05.
Results
The distribution of calbindin-D28k immunoreactivity observed in rat hippocampal slices using confocal laser-scanning microscopy was comparable with that described previously (Baimbridge and Miller, 1982; Sloviter, 1989). All granule cells were intensely labeled throughout their entire cytoplasm including dendrites, somata, and axons (Fig. 1A). In contrast, CA3 pyramidal cells remained completely unstained (Fig. 1A), and only ∼50% of CA1 pyramidal cells were labeled by the antibody (data not shown). An identical staining pattern was observed in slices obtained from mice, but this staining was completely absent in slices from calbindin-D28k(-/-) mice (n = 4; data not shown).
After closer inspection of the immunoreactivity of granule cells, we found a gradient of the fluorescence intensity within the granule cell layer indicating a heterogeneity of the intracellular calbindin-D28k concentration: granule cells at the hilar border appeared significantly less bright than granule cells adjacent to the molecular layer (Figs. 1B, 2A-D). The fluorescence intensity of granule cell somata ranged from 58 ± 3% at the hilar border to 122 ± 4% at the outer edge of the cell layer when compared with the overall average fluorescence of all granule cells in each scan (n = 10) (Fig. 1C) (see Materials and Methods for details).
It is well established that new granule cells are generated throughout postnatal life (van Praag et al., 2002) and that those cells appear first at the border to the hilus and later integrate into outer parts of the granular layer (Cameron et al., 1993). Newborn granule cells do not contain calbindin-D28k (Rami et al., 1987). To test whether the gradient of calbindin-D28k content reflects successive maturation of granule cells after their generation, we performed a double staining for calbindin-D28k and a marker of newly generated granule cells in adult hippocampus, doublecortin (Brown et al., 2003). Indeed, we observed an overlap of calbindin-D28k and doublecortin labeling supporting this hypothesis (Fig. 1D). Numerous weakly calbindin-D28k-positive cells displayed doublecortin-positive filaments in their cytoplasm (small arrows). On the contrary, strongly calbindin-D28k-positive cells were doublecortin-negative. Notably, cells prominently marked with doublecortin antibodies, morphologically resembling the earliest postmitotic neuroblast stage with horizontally oriented processes (large arrowheads), were virtually calbindin-D28k-negative.
Considering that calbindin-D28k is present in all compartments of granule cells, it is likely that this protein is a mobile cytoplasmic Ca2+ buffer. To test the intracellular mobility of calbindin-D28k, we performed whole-cell recordings of granule cells in hippocampal slices. During whole-cell recordings, there is a direct connection between the interior of the patch pipette and the cytoplasm of the recorded cell such that molecules up to a molecular weight of 150,000 show diffusional equilibration between the pipette and the cytoplasm (Pusch and Neher, 1988). In this experiment, the pipette solution contained no calbindin-D28k. Because the volume of the pipette solution (>20 μl) exceeds that of the cytoplasm (<20 pl) by several orders of magnitude, there should be a strong dilution (“washout”) of calbindin-D28k during the course of a whole-cell recording. Indeed, when slices were fixed and stained after prolonged whole-cell recordings, granule cells displayed only very weak calbindin-D28k immunoreactivity (Fig. 2D). In contrast, when the patch pipette was withdrawn from the cell after a short recording period of 30 s, calbindin-D28k immunoreactivity was comparable with that of the neighboring cells (Fig. 2A). To estimate the time course of the washout of calbindin-D28k, we systematically varied the recording time by withdrawing the patch pipette (Fig. 2A-D). Figure 2E summarizes the measured fluorescence obtained from 34 whole-cell recordings of different durations. It is evident that the immunoreactivity of the recorded cells decreases with increasing whole-cell recording periods. Fitting an exponential function to the data suggests that calbindin-D28k washes out of granule cell somata with a time constant of ∼10 min (Fig. 2E).
The experiments so far show that the intracellular calbindin-D28k concentration can be controlled via the solution in the patch pipette provided that there is sufficient time for diffusional equilibration. The idea of the next series of experiments was to supplement purified calbindin-D28k to the pipette solution to find the concentration of calbindin-D28k that is needed to restore the immunoreactivity of the recorded cell. To ensure comparable diffusional equilibration, cells were recorded for a period of 30-40 min. Increasing the calbindin-D28k concentration in the patch pipette to 25, 33, and 66 μm caused a corresponding increase in the fluorescence intensity of the recorded cell (Fig. 3). Cells filled with 66 μm are clearly brighter than surrounding granule cells. A plot of the normalized calbindin-D28k immunofluorescence of filled granule cells (see Materials and Methods) versus the concentration of calbindin-D28k in the patch pipette is depicted for 24 granule cells in Figure 4. From this calibration curve, the mean endogenous concentration of calbindin-D28k over all granule cells was estimated to be ∼33 μm (Fig. 4, arrowhead). Using data from Figure 1, we calculate that mature granule cells close to the molecular layer enclose ∼40 μm and that newborn granule cells at the hilar border contain ∼18 μm calbindin-D28k.
Figure 3.

Purified calbindin-D28k (Cb) in the patch pipettes rescues immunoreactivity after prolonged whole-cell recordings.A, The granule cell shown was recorded with a pipette solution containing LY and 25 μm purified calbindin-D28k. The arrow indicates the recorded granule cell. The dashed line denotes the area that was used to quantify the mean calbindin-D28k-related fluorescence of this cell. Notice that the loss of immunoreactivity as observed in Figure 2 is clearly prevented. The immunoreactivity of this recorded cell is slightly lower than that of neighboring granule cells. For comparison, the brightness of the red color (Cy5) has been decreased in A and B such that the surrounding granule cells appear similarly bright between A-C. A low level of brightness had to be chosen to avoid saturation of the intensity of the granule cell in C. Left, Confocal scans of LY and of calbindin-D28k (Cy5) fluorescence are merged into one image. The arrow indicates the recorded granule cell. Right, Calbindin-D28k (Cy5) fluorescence only. B, As in A, but the cell was filled with 33 μm calbindin-D28k. The recorded cell appears as bright as the neighboring cells, indicating that the patch pipette contains the same concentration of calbindin-D28k as surrounding granule cells. C, As inA, but the recording electrode contained 66 μm calbindin-D28k. This concentration caused an immunofluorescence that is clearly higher than that of surrounding granule cells. Scale bar (applies to A-C), 10 μm.
Knowing the concentration and the Ca2+ affinity (Veenstra et al., 1997; Berggard et al., 2002), it is possible to predict how calbindin-D28k will affect spatially homogenous Ca2+ elevations as they occur in dendritic segments and in nerve terminals (see Discussion). However, it is less straightforward to estimate the buffering effect of calbindin-D28k on very localized, submicroscopic Ca2+ signals (Ca2+ microdomains). Ca2+ microdomains are steep Ca2+ concentration gradients around a Ca2+ channel (or channels) that build up very rapidly after opening of the channel and reach values in the high micromolar range. Ca2+ microdomains have been implicated in the release of neurotransmitter and in the activation of Ca2+-sensitive potassium channels. Because on this spatial scale Ca2+ ions diffuse some distance before they are captured by the buffer [capture timecalbindin-D28k = ([buffer]free × kon)-1 = 248 μs; Eq. 1, kon = 2.8 × 107 M-1s-1], there is a region around the channel (<200 nm) where chemical equilibrium is not achieved among Ca2+ and buffers. We used numerical simulations to explore how strong 40 μm calbindin-D28k reduces the increment of free Ca2+ around a Ca2+ channel in that nonequilibrium domain (see Materials and Methods for details). We depict a physiologically realistic scenario where a Ca2+ channel opens around the peak of an action potential and remains open for 0.8 ms (Fig. 5A, inset). When calbindin-D28k is added, the maximal free Ca2+ concentration reached during the channel opening is hardly diminished close to the Ca2+ source at 15 nm (Fig. 5A, black dashed line). Only farther away from the channel at a distance of 150 nm (red dashed line) does calbindin-D28k cause a notable reduction in free Ca2+ (Fig. 5A). A more pronounced inhibitory effect of the buffer is observed after closure of the channel. This is best seen in the right panel of Figure 5A where traces are replotted on a semilogarithmic scale (see black arrow). In the absence of buffers, Ca2+ needs ∼5 ms to return to resting levels, whereas in the presence of calbindin-D28k, resting levels are restored within ∼600 μs (Fig. 5A, right). Figure 5B summarizes the distance-dependent inhibition of free Ca2+ at the end of the channel open time (black lines) and the distance-independent acceleration of the decay of free Ca2+ after closure of the channel (red lines). To evaluate the robustness of the results, we have modified different parameters of the simulation. We varied kon (colored groups of lines), from an extreme lower limit of 0.6 × 107 to an upper limit of 5.6 × 107 M-1s-1 (Nägerl et al., 2000), as well as the single-channel current amplitude (indicated by different line modes) to explore clustering of 10-30 Ca2+ channels (Fig. 5C). The buffering effect of calbindin-D28k was consistent even when high single-channel current amplitudes were simulated. As expected, the extent of inhibition was sensitive to alterations of kon: an upper limit of the inhibition of ΔCa2+ by calbindin-D28k at 150-200 nm is ∼50%, and a lower limit at that distance is ∼10%, which is reached when extremely low EGTA-like values for kon are used.
Next, we tested whether other types of hippocampal neurons contain levels of calbindin-D28k similar to that seen for granule cells. For this purpose, we compared the immunofluorescence of CA1 pyramidal cells and CA3 stratum radiatum interneurons with that of granule cells (n = 60) in the same slice (five slices) acquired under identical conditions (see Materials and Methods for details). In high-magnification confocal laser scans, we quantified the intensity of dendritic segments that have approximately the same geometry among the three different cell types (Fig. 6A). In this way, we estimated that CA1 pyramidal cells and CA3 stratum radiatum interneurons contain on average 45 ± 2 μm (n = 59) and 47 ± 6 μm (n = 22) calbindin-D28k, respectively.
Discussion
Calbindin-D28k is mobile and reaches 40 μm in hippocampal neurons
Our experiments indicate that cellular calbindin-D28k freely exchanges with the solution in the patch pipette. The calbindin-D28k content of granule cells decreases during whole-cell recordings with a time constant of ∼10 min (Fig. 2). Taking into account that granule cells possess a large dendritic tree (∼200 μm; compare Fig. 1), from which calbindin-D28k initially can be replenished by diffusion into the soma, this time constant well matches the value expected for free diffusional equilibration of a 28 kDa protein under our conditions (Pusch and Neher, 1988).
The fact that the cellular calbindin-D28k concentration equilibrates with that of the patch pipette enabled us to load single granule cells with defined concentrations of purified calbindin-D28k. We estimated the endogenous calbindin-D28k concentration to be 40 μm in mature granule cells by testing which concentration of purified calbindin-D28k mimics the immunofluorescence of neighboring, untouched granule cells (Figs. 3, 4). This procedure should be readily applicable to quantify the intracellular concentration of other low-abundance soluble proteins in different types of cells.
The concentration of calbindin-D28k in newly generated granule cells was estimated by double labeling with antibodies against doublecortin and calbindin-D28k. Newly generated granule cells transiently express doublecortin for 2-3 weeks after they have entered the postmitotic stage (Brown et al., 2003). We found a gradient of doublecortin immunoreactivity opposite to that of calbindin-D28k: neuroblasts at the hilar border were strongly positive for doublecortin but virtually negative for calbindin-D28k. On average, doublecortin-immunoreactive cells displayed a lower calbindin-D28k content (0-20 μm) compared with mature granule cells. Because strongly calbindin-D28k-labeled granule cells were found to be doublecortin-negative, it appears that granule cells need ∼2-3 weeks to increase their endogenous Ca2+ buffer content to mature levels. It has been reported that newborn granule cells show enhanced Ca2+-dependent synaptic plasticity because of immature membrane properties (Schmidt-Hieber et al., 2004). In light of our findings, it seems possible that the very weak Ca2+ buffer capacity of young granule cells additionally facilitates the induction of synaptic plasticity. Similar considerations also apply to granule cells and CA1 pyramidal cells when investigated within the first 2 postnatal weeks. At these early stages the cellular calbindin-D28k content has not yet fully developed (Tortosa and Ferrer, 1994; Yoon et al., 2000), and Ca2+ signaling might significantly differ from the adult pattern.
The virtually complete exchange of calbindin-D28k between the patch pipette and the cell interior provides strong evidence that most of calbindin-D28k in neurons constitutes a mobile Ca2+ buffer. The diffusional mobility is an important buffer property, as outlined above, and also is crucial for microfluorometric imaging studies investigating cellular Ca2+ signaling using ion-sensitive dyes. Those dyes are frequently loaded into the cell via the patch pipette solution, and this procedure typically requires whole-cell recordings of >30 min. Considering the rapid washout of calbindin-D28k, such imaging studies potentially underestimate the endogenous Ca2+-binding capacity profoundly. Furthermore, Ca2+-dependent processes such as induction of synaptic plasticity or the occurrence and propagation of Ca2+ waves could be strongly altered by washout of cellular Ca2+ buffers. A brief bolus loading with a high indicator concentration could reduce the dilution effect and disclose the role of endogenous buffers in shaping Ca2+ signals. However, when aiming to measure cellular Ca2+ buffering quantitatively, additional efforts have to be made to obtain an approximation of the resulting dye concentration (cf. Helmchen et al., 1996).
Calbindin-D28k and global Ca2+ signals
The quantitative determination of the intracellular concentration of calbindin-D28k allows theoretical predictions of how this Ca2+ buffer will affect different types of intracellular Ca2+ signals. The influence of buffers on (small) global Ca2+ signals caused by brief Ca2+ entry as they typically occur in dendritic compartments or in synaptic terminals is well described by the single compartment model (Neher and Augustine, 1992). In this model, the key parameter regarding the action of the buffer is the Ca2+-binding ratio, κ, which is the ratio of the change in buffer-bound Ca2+ over the change in free Ca2+ (δ[Ca Bf]/δ[Ca]). Given that calbindin-D28k displays a Ca2+ affinity of 700 nm (Berggard et al., 2002) and assuming a resting Ca2+ concentration of 50 nm, it follows that 40 μm calbindin-D28k (= 160 μm Ca2+-binding sites) provides a κCb of ∼200 (Eq. 2). Thus, only ∼0.5% of entering Ca2+ ions appear as free ions, whereas the rest will be bound to buffer molecules. As a result, the initial amplitude of a rise in free Ca2+ will be significantly decreased because Ca2+ is bound by calbindin-D28k. Subsequently, the decay of free Ca2+ will be slowed because the bound ions are not available for removal by extrusion mechanisms. If we assume that endogenous Ca2+ buffers other than calbindin-D28k constitute a Ca2+-binding ratio (κX) of ∼50 (Neher, 1995), then calbindin-D28k should decrease the amplitude of Ca2+ transients and slow their decay as much as fivefold (Eq. 3).
A recent imaging study used Ca2+-sensitive dyes to investigate endogenous Ca2+ buffers in axon terminals of granule cells. The total endogenous Ca2+ buffers could be well described mathematically assuming a 130 μm concentration of a single buffer species with a high Ca2+ affinity (∼500 nm) (Jackson and Redman, 2003). No attempts have been made to reveal the number or the molecular identity of the underlying buffers. In light of our findings (160 μm mobile Ca2+-binding sites of calbindin-D28k) and considering that there are probably additional low-affinity Ca2+-binding sites present in granule cells, it appears likely that Jackson and Redman (2003) still underestimated the total Ca2+-binding capacity of granule cells. Using microfluorometric imaging techniques, κS of CA1 pyramidal cell dendrites was determined to be ∼190 (Helmchen et al., 1996). This value is close to our estimate of the κS mediated by calbindin-D28k in CA1 pyramidal cells. However, as argued above, the undisturbed κS of CA1 pyramidal cells could be even higher.
Calbindin-D28k and Ca2+ microdomains
The influence of calbindin-D28k on highly localized Ca2+ signals that are beyond the resolution of current imaging techniques was assessed by numerical simulations. Having identified the intracellular concentration of calbindin-D28k, simulations could be performed with relatively high accuracy because the Ca2+-binding properties and the diffusion coefficient of calbindin-D28k have been determined (Nägerl et al., 2000; Berggard et al., 2002; Schmidt et al., 2003). Calbindin-D28k exerts two main actions on microdomains caused by single or by clustered Ca2+ channels. First there is a distance-dependent inhibition of the increase in free Ca2+. Calbindin-D28k is ineffective close to the Ca2+ source (<30 nm) but inhibits the increase in free Ca2+ at 200 nm by ∼35%. In effect, calbindin-D28k reduces the spatial extent of a Ca2+ microdomain to ∼200-400 nm without affecting its amplitude at the origin. The second result is that calbindin-D28k greatly speeds up the temporal collapse of the microdomain at all distances after closure of the Ca2+ channel or channels. If, for example, ∼30 tightly clustered Ca2+ channels close after an action potential, Ca2+ influx stops abruptly, but a residual increase in free Ca2+ of ∼6 μm persists for several milliseconds in the absence of calbindin-D28k. If mobile calbindin-D28k is added, this Ca2+ is effectively shuttled away, and free Ca2+ drops below ∼200 nm within ∼0.6 ms. Thus, calbindin-D28k sharpens Ca2+ microdomains in space and time and thereby enables a more precise discrimination of the site and the time course of Ca2+ influx by downstream effectors.
Because of their high Ca2+ affinity, the Ca2+-binding sites of calbindin-D28k can be saturated after sufficiently large Ca2+ influx. It has been reasoned that saturation of endogenous Ca2+ buffers can cause a facilitation of neurotransmitter release (Neher, 1998). A requirement for this process is that the unsaturated Ca2+ buffer is able to capture some Ca2+ ions before they reach the Ca2+ sensor of the release machinery. Release will then be facilitated when previous activity depletes some Ca2+-binding sites such that more Ca2+ reaches the Ca2+ sensor. Indeed, it was suggested recently that paired-pulse facilitation (200% at 100 ms intervals) at the mossy fiber-CA3 synapse apparently results from saturation of calbindin-D28k (Blatow et al., 2003; but see Mori-Kawakami et al., 2003). However, given that mossy fiber terminals contain 40 μm calbindin-D28k, it seems unlikely that calbindin-D28k contributes to paired-pulse facilitation by saturation under their conditions (Matveev et al., 2004). First, this would imply that Ca2+ sensors and Ca2+ channels are more loosely associated than previously thought. In fact, calbindin-D28k reduces Ca2+ microdomains by the required extent (∼20%) only at a mean distance of ≥100 nm. Second, saturation of calbindin-D28k can at most be partial (<25%), because during a single action potential, the total Ca2+ concentration does not increase by more than ∼40 μm (Bischofberger et al., 2002; Jackson and Redman, 2003). Third, within the interstimulus interval used (100 ms), Ca2+ nearly completely decays back to baseline levels (Vogt and Regehr, 2001; Dietrich et al., 2003; Jackson and Redman, 2003). Instead of acting via buffer saturation, calbindin-D28k might play a direct regulatory role in paired-pulse facilitation at this synapse.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (SFB TR3, GK 246, and Mu809/6-3), University Clinic Bonn grants (BONFOR), and the Gertrud-Reemtsma-Stiftung of the Max-Planck-Gesellschaft.
Correspondence should be addressed to Dr. Dirk Dietrich, Department of Neurosurgery, NCH U1 R035, Experimental Neurophysiology, University Clinic Bonn, Sigmund-Freud Strasse 25, D-53105 Bonn, Germany. E-mail: dirk.dietrich@ukb.uni-bonn.de.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250558-08$15.00/0
References
- Airaksinen MS, Eilers J, Garaschuk O, Thoenen H, Konnerth A, Meyer M (1997) Ataxia and altered dendritic calcium signaling in mice carrying a targeted null mutation of the calbindin D28k gene. Proc Natl Acad Sci USA 94: 1488-1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baimbridge KG, Miller JJ (1982) Immunohistochemical localization of calcium-binding protein in the cerebellum, hippocampal formation and olfactory bulb of the rat. Brain Res 245: 223-229. [DOI] [PubMed] [Google Scholar]
- Baimbridge KG, Miller JJ, Parkes CO (1982) Calcium-binding protein dis-tribution in the rat brain. Brain Res 239: 519-525. [DOI] [PubMed] [Google Scholar]
- Baimbridge KG, Celio MR, Rogers JH (1992) Calcium-binding proteins in the nervous system. Trends Neurosci 15: 303-308. [DOI] [PubMed] [Google Scholar]
- Berggard T, Miron S, Onnerfjord P, Thulin E, Akerfeldt KS, Enghild JJ, Akke M, Linse S (2002) Calbindin D28k exhibits properties characteristic of a Ca2+ sensor. J Biol Chem 277: 16662-16672. [DOI] [PubMed] [Google Scholar]
- Bischofberger J, Geiger JR, Jonas P (2002) Timing and efficacy of Ca2+ channel activation in hippocampal mossy fiber boutons. J Neurosci 22: 10593-10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatow M, Caputi A, Burnashev N, Monyer H, Rozov A (2003) Ca2+ buffer saturation underlies paired pulse facilitation in calbindin-D28k-containing terminals. Neuron 38: 79-88. [DOI] [PubMed] [Google Scholar]
- Brown JP, Couillard-Despres S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG (2003) Transient expression of doublecortin during adult neurogenesis. J Comp Neurol 467: 1-10. [DOI] [PubMed] [Google Scholar]
- Cameron HA, Woolley CS, McEwen BS, Gould E (1993) Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience 56: 337-344. [DOI] [PubMed] [Google Scholar]
- Crank J (1975) The Mathematics of diffusion. Oxford: Oxford UP.
- Dietrich D, Kirschstein T, Kukley M, Pereverzev A, von der Brèlie C, Schneider T, Beck H (2003) Functional specialization of presynaptic Cav2.3 Ca2+ channels. Neuron 39: 483-496. [DOI] [PubMed] [Google Scholar]
- Gabso M, Neher E, Spira ME (1997) Low mobility of the Ca2+ buffers in axons of cultured Aplysia neurons. Neuron 18: 473-481. [DOI] [PubMed] [Google Scholar]
- Gollasch M, Hescheler J, Quayle JM, Patlak JB, Nelson MT (1992) Single calcium channel currents of arterial smooth muscle at physiological calcium concentrations. Am J Physiol 263: C948-C952. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Imoto K, Sakmann B (1996) Ca2+ buffering and action potential-evoked Ca2+ signaling in dendrites of pyramidal neurons. Biophys J 70: 1069-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MB, Redman SJ (2003) Calcium dynamics, buffering, and buffer saturation in the boutons of dentate granule-cell axons in the hilus. J Neurosci 23: 1612-1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Elmslie KS (1999) Gating of single N-type calcium channels recorded from bullfrog sympathetic neurons. J Gen Physiol 113: 111-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda H, Ellis-Davies GC, Ito K, Miyashita Y, Kasai H (1999) Supralinear Ca2+ signaling by cooperative and mobile Ca2+ buffering in Purkinje neurons. Neuron 24: 989-1002. [DOI] [PubMed] [Google Scholar]
- Matveev V, Zucker RS, Sherman A (2004) Facilitation through buffer saturation: constraints on endogenous buffering properties. Biophys J 86: 2691-2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori-Kawakami F, Kobayashi K, Takahashi T (2003) Developmental decrease in synaptic facilitation at the mouse hippocampal mossy fiber synapse. J Physiol (Lond) 553: 37-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägerl UV, Novo D, Mody I, Vergara JL (2000) Binding kinetics of calbindin-D28k determined by flash photolysis of caged Ca(2+) Biophys J 79: 3009-3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naraghi M, Neher E (1997) Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci 17: 6961-6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E (1995) The use of fura-2 for estimating Ca buffers and Ca fluxes. Neuropharmacology 34: 1423-1442. [DOI] [PubMed] [Google Scholar]
- Neher E (1998) Usefulness and limitations of linear approximations to the understanding of Ca2+ signals. Cell Calcium 24: 345-357. [DOI] [PubMed] [Google Scholar]
- Neher E, Augustine GJ (1992) Calcium gradients and buffers in bovine chromaffin cells. J Physiol (Lond) 450: 273-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowycky MC, Pinter MJ (1993) Time courses of calcium and calcium-bound buffers following calcium influx in a model cell. Biophys J 64: 77-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape PC, Jong DS, Chandler WK (1998) Effects of partial sarcoplasmic reticulum calcium depletion on calcium release in frog cut muscle fibers equilibrated with 20 mm EGTA. J Gen Physiol 112: 263-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Neher E (1988) Rates of diffusional exchange between small cells and a measuring patch pipette. Pflügers Arch 411: 204-211. [DOI] [PubMed] [Google Scholar]
- Rami A, Brehier A, Thomasset M, Rabie A (1987) Cholecalcin (28-kDa calcium-binding protein) in the rat hippocampus: development in normal animals and in altered thyroid states. An immunocytochemical study. Dev Biol 124: 228-238. [DOI] [PubMed] [Google Scholar]
- Roberts WM (1994) Localization of calcium signals by a mobile calcium buffer in frog saccular hair cells. J Neurosci 14: 3246-3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Brown EB, Schwaller B, Eilers J (2003) Diffusional mobility of parvalbumin in spiny dendrites of cerebellar purkinje neurons quantified by fluorescence recovery after photobleaching. Biophys J 84: 2599-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Hieber C, Jonas P, Bischofberger J (2004) Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature 429: 184-187. [DOI] [PubMed] [Google Scholar]
- Sloviter RS (1989) Calcium-binding protein (calbindin-D28k) and parvalbumin immunocytochemistry: localization in the rat hippocampus with specific reference to the selective vulnerability of hippocampal neurons to seizure activity. J Comp Neurol 280: 183-196. [DOI] [PubMed] [Google Scholar]
- Tortosa A, Ferrer I (1994) Poor correlation between delayed neuronal death induced by transient forebrain ischemia, and immunoreactivity for parvalbumin and calbindin D28k in developing gerbil hippocampus. Acta Neuropathol (Berl) 88: 67-74. [DOI] [PubMed] [Google Scholar]
- van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH (2002) Functional neurogenesis in the adult hippocampus. Nature 415: 1030-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenstra TD, Johnson KL, Tomlinson AJ, Naylor S, Kumar R (1997) Determination of calcium-binding sites in rat brain calbindin D28k by electrospray ionization mass spectrometry. Biochemistry 36: 3535-3542. [DOI] [PubMed] [Google Scholar]
- Vogt KE, Regehr WG (2001) Cholinergic modulation of excitatory synaptic transmission in the CA3 area of the hippocampus. J Neurosci 21: 75-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SP, Chung YY, Chang IY, Kim JJ, Moon JS, Kim HS (2000) Postnatal development of parvalbumin and calbindin D28k immunoreactivities in the canine hippocampus. J Chem Neuroanat 19: 143-154. [DOI] [PubMed] [Google Scholar]