

Abstract
Recent findings have uncovered a role for the Bcl-x gene in the survival of dopaminergic neurons. The exact nature of this role has been difficult to examine because of the embryonic lethality of Bcl-x gene disruption in mouse models. Here we report the generation catecholaminergic cell-specific conditional Bcl-x gene knock-out mice using Cre-lox recombination technology. First we produced transgenic mice that express Cre recombinase from an exogenous rat tyrosine hydroxylase promoter (TH-Cre mice). These mice were crossed to Z/AP and Z/EG reporter mouse strains to verify catecholaminergic (TH-positive) cell-specific Cre expression. The TH-Cre mice then were mated to mice possessing the Bcl-x gene flanked by loxP sites, thereby producing offspring with Bcl-x deletion limited to catecholaminergic cells. The resulting mice are viable but have one-third fewer catecholaminergic neurons than do control animals. They demonstrate a deficiency in striatal dopamine and also tend to be smaller and have decreased brain mass when compared with controls. Surprisingly, surviving neurons were found that lacked Bcl-x immunoreactivity, thereby demonstrating that this gene is dispensable for the ongoing survival of a subpopulation of catecholaminergic cells.
Keywords: Parkinson, Bcl-x, catecholamine, substantia nigra, dopamine, tyrosine hydroxylase
Introduction
Parkinson's disease (PD) is a relatively common neurodegenerative disorder with ∼70,000 new cases diagnosed in the United States annually (Mayeux, 2003). Underlying the symptoms of PD is the degeneration of several neuronal subtypes, including the conspicuous loss of dopamine-containing neurons in the substantia nigra pars compacta (SNPc) (Lang and Lozano, 1998). Loss of 60-80% of the these cells and the resulting decrease in striatal dopamine correlate with symptom onset (Pakkenberg et al., 1991; Subramanian, 2001; Isacson, 2002). In addition to the SNPc, other catecholaminergic regions, including the locus ceruleus (LC), olfactory nuclei, sympathetic ganglia, and adrenal medulla show pathology in PD (Pierce and Bari, 2001; Braak et al., 2003; Micieli et al., 2003). Many of the cells in these regions express tyrosine hydroxylase (TH), the rate-limiting enzyme in catecholamine biosynthesis.
The factors responsible for PD pathology are mostly unknown. Recent data have implicated mitochondrial dysfunction, oxidative stress, and proteasomal dysfunction in the death of nigral neurons (Dawson and Dawson, 2003). Attempts by the cell to counteract these stressors may involve the survival factor Bcl-xL (Hartmann et al., 2002). Specifically, this protein is upregulated in the surviving dopaminergic neurons of PD patients (Hartmann et al., 2002), in neuroblastoma cells exposed to the dopaminergic neurotoxin 1-methyl-4-phenylpyridinium (MPP+) (Veech et al., 2000), and in cybrid cell cultures containing mitochondrial complex I deficiency (Veech et al., 2000). In addition, Bcl-xL overexpression in embryonic stem cells promotes dopaminergic differentiation and enhances MPP+ resistance (Shim et al., 2004). Bcl-xL is a product of the Bcl-2 family gene Bcl-x. The gene is expressed in many regions throughout the developing and adult animal, including the CNS, where it prevents neuronal cell death (Gonzalez-Garcia et al., 1995; Mizuguchi et al., 1996). Indeed, Bcl-x gene inactivation leads to extensive cell death in the hematopoietic system and CNS, resulting in embryonic lethality (Motoyama et al., 1995).
To better study the role of Bcl-x in dopaminergic cell function, we set out to interrupt this gene specifically in catecholaminergic neurons. Our hypothesis was that this would avoid the hematopoietic failure and embryonic lethality seen in the genome-wide knock-out and therefore provide an opportunity to study the role of Bcl-x in catecholaminergic cells beyond the embryonic period. Furthermore, in light of increasing evidence that dopaminergic neurons rely heavily on Bcl-x function for survival, we postulated that dopaminergic cells lacking this gene would not survive. Such a finding would verify that Bcl-x is required for dopaminergic cell survival in vivo and in an environment of normal Bcl-x expression.
To accomplish the targeted deletion of Bcl-x, we generated mice that express Cre recombinase in TH-containing cells and crossed them to mice homozygous for the loxP-flanked Bcl-x gene (Rucker et al., 2000). The resulting animals demonstrate deficiencies in the catecholaminergic system and also show that Bcl-x is dispensable for continued survival of this cell population even at advanced ages. These mice provide the first opportunity to examine the consequences of Bcl-x deletion on postnatal neuronal survival in vivo.
Materials and Methods
Animals. All mice were housed and treated in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. They were kept in a pathogen-free facility and exposed to a 12 h light/dark cycle with food and water provided ad libitum.
Transgenic TH-Cre mice were generated by microinjection of fertilized B6/SJLF2 oocytes at the pronuclear stage with a linearized fragment of the pTH-Cre vector. This vector was created using a pSP73 (Promega, Madison, WI) backbone. The TH promoter consisting of the SalI-EcoRI fragment of the pTH9000 plasmid (Min et al., 1994) was placed upstream from a cassette containing the Cre-coding sequence flanked upstream by a synthetic intron initially derived from the 5′-untranslated sequence of the adenovirus-major late region (Huang and Gorman, 1990) and 3′ by the simian virus 40 (SV40) late region polyadenylation site [all derived from the pOG231 vector (O'Gorman et al., 1997)]. An 11 kb SalI-NotI fragment was isolated using the GELase preparation reagent (Epicenter, Madison, WI), resuspended in 10 mm Tris, pH 7.5, and 0.1 mm EDTA, and provided to the Johns Hopkins Transgenic Core Facility for microinjection. Five lines of mice containing the TH-Cre transgene (TH-Cre 1-TH-Cre 5) were identified by PCR amplification of tail DNA using the primers 5′-AAA TGT TGC TGG ATA GTT TTT ACT GC-3′ and 5′-GGA AGG TGT CCA ATT TAC TGA CCG TA-3′. The expected 300 bp fragment was visualized by standard agarose gel electrophoresis.
Mice possessing the loxP-flanked Bcl-x gene were generously provided by Dr. Edmund Rucker (University of Missouri, Columbia, MO) (Rucker et al., 2000). The presence of the floxed Bcl-x gene was determined by PCR around the 5′ loxP site using the primers 5′-CGG TTG CCT AGC AAC GGG GC-3′ and 5′-CTC CCA CAG TGG AGA CCT CG-3′, giving a wild-type band of 200 bp and a floxed gene product of 300 bp, or around the 3′ site using primers 5′-TCA GAA GCC GCA ATA TCC CC-3′ and 5′-GCC ACC TCA TCA GTC GGG-3′, yielding a wild-type product of 150 bp and a floxed gene product of ∼200 bp. These fl/fl Bcl-x mice have been used successfully to examine the role of Bcl-x in a variety of cell types, including those in the liver, ovary, and mammary gland, as well as in erythroid and dendritic cells (Rucker et al., 2000; Wagner et al., 2000; Walton et al., 2001; Riedlinger et al., 2002; Hon et al., 2004; Takehara et al., 2004).
The fl/fl Bcl-x mice were crossed to TH-Cre mice (lines 1 and 3), and the offspring were bred to homozygosity at the loxP-flanked Bcl-x locus. This enabled the Cre recombinase to inactivate the Bcl-x gene specifically in cells in which the TH promoter is active (i.e., catecholaminergic cells). Cre-mediated excision of Bcl-x genomic sequences was detected by PCR on DNA isolated from ventral midbrain tissue of adult animals using the forward primers 5′-CGG TTG CCT AGC AAC GGG GC-3′ and 5′-AAT GGC CAG TAC TAG TGA ACC-3′ and the reverse primer 5′-TCA GAA GCC GCA ATA TCC CC-3′ as described previously (Wagner et al., 2000).
The Z/AP mouse line (Lobe et al., 1999) was provided by the Johns Hopkins Transgenic Core Facility, and the Z/EG mouse was purchased from The Jackson Laboratory (Bar Harbor, ME). These strains were genotyped by staining tail tissue for β-galactosidase activity. Tail samples were immersed in lacZ staining solution containing 2 mm magnesium chloride, 0.01% sodium deoxycholate, 0.02% NP-40, 100 mm sodium phosphate, pH 7.3, 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside, 6 mm potassium ferrocyanide, and 5 mm potassium ferricyanide and were incubated for 5 min at 37°C. Positive samples were identified by a blue reaction product.
For the isolation of tissues for immunohistochemistry and enzyme histochemistry, animals were anesthetized with a lethal dose of pentobarbital (100 mg/kg) and perfused through the left ventricle with 50 ml of ice-cold PBS followed by 75 ml of ice-cold 4% paraformaldehyde in PBS. Relevant tissues were dissected and postfixed in 4% paraformaldehyde and PBS overnight at 4°C and cryoprotected for 36-48 h in 30% sucrose and PBS at 4°C.
Enzyme histochemistry. Staining for human placental alkaline phosphatase activity was performed as described previously, with minor modifications (Lobe et al., 1999). Tissue was processed as 30 μm cryostat sections (retina, adrenal gland, and 1 d pup tissue) or 40 μm microtome sections (brain) cut from frozen paraformaldehyde-fixed tissue. Endogenous alkaline phosphatase activity was quenched by incubation at 70°C for 30 min in PBS. The samples were allowed to cool for 2 min and washed with PBS at room temperature. Sections were transferred to a solution containing 100 mm Tris-HCl, pH 9.5, 100 mm NaCl, and 10 mm MgCl2 for 10 min and then transferred to staining solution consisting of 100 mm Tris-HCl, pH 9.5, 100 mm NaCl, 50 mm MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40, 340 μg/ml nitroblue tetrazolium, and 175 μg/ml 5-bromo-4-chloro-3-indolyl-phosphate (Roche Molecular Biochemicals, Indianapolis, IN) for 15-30 min at room temperature in the dark. Sections were washed in PBS and allowed to air dry before dehydration and mounting in distrene plasticizer xylene (Sigma, St. Louis, MO).
Immunohistochemistry. Forty micrometer sections were cut on a sliding microtome (Microm, Kalamazoo, MI) and collected free-floating in PBS. Sections were permeabilized and blocked in PBS containing 0.2% Triton X-100 and 10% normal goat serum at room temperature for 1 h. Sections then were incubated in 1:2000 rabbit anti-TH antibody (Novus Biologicals, Littleton, CO) overnight at 4°C and washed with PBS containing 0.1% Triton X-100. Next, sections were incubated for 2 h in 1:500 biotin-labeled F(ab′)2 fragment goat anti-rabbit antibody (Jackson ImmunoResearch, West Grove, PA) in PBS, 0.2% Triton X-100, and 2% goat serum, washed, and visualized by incubation in biotin-streptavidin-HRP (Vector Laboratories, Burlingame, CA), followed by incubation with 3,3′-diaminobenzidine per the manufacturer's instructions (Sigma). Sections were mounted on glass slides, dehydrated, and cover-slipped for visualization. Sections for double-immunofluorescent labeling were processed similarly using 1:500 rabbit anti-Bcl-x (PharMingen, San Diego, CA) and 1:2000 mouse anti-TH (Immunostar, Hudson, WI) as primary antibodies and cyanine 3 (Cy3)-conjugated goat anti-mouse IgG F(ab′)2-specific and Cy2-conjugated goat anti-rabbit IgG F(ab′)2-specific as secondary antibodies at a 1:500 dilution (Jackson ImmunoResearch). Sections were mounted in Vectashield (Vector Laboratories) and kept in the dark until visualized by confocal microscopy using a Zeiss (Thornwood, NY) LSM-510 microscope. Slides processed for stereology were prepared as above, and TH-positive SNPc and locus ceruleus neurons were counted using nonbiased stereologic methods with the optical fractionator as described previously (West, 1993; Mandir et al., 1999) and using software provided by MicroBrightField (Williston, VT). Selected sections were Nissl-counterstained and counted as described above.
HPLC. HPLC with electrochemical detection was used to measure striatal levels of dopamine, 3,4-dihydroxyphenylacetic acid, and homovanillic acid. Conditional Bcl-x knock-out mice and age- and gender-matched controls were killed by decapitation, the brains were quickly removed, and striata were dissected. Samples were sonicated in 50 vol of 0.1 m perchloric acid containing 25 μg/ml dihydroxybenzylamine (Sigma) as an internal standard. After centrifugation (15,000 × g, 10 min, 4°C), 20 μl of supernatant was injected onto a Brownlee C-18 reverse phase Spheri 5 RP-18 4.6 mm × 25 cm column. The mobile phase consisted of 0.15 m chloroacetic acid, 0.1 mm EDTA, and 0.86 m sodium octyl sulfate, filtered and adjusted to pH 3, 4% acetonitrile, and 2.5% (v/v) tetrahydrofuran. The flow rate was kept at 1.5 ml/min. Peaks were detected by an Amperometric LC-4C detector (Bioanalytical System, West Lafayette, IN), and the working electrode was kept at 0.7 V. Data were collected and processed on a Star Chromatography 5.52 workstation (Varian, Palo Alto, CA).
Rotorod. A cohort of adult male mice was initially trained on an accelerating rotorod apparatus (Rotamex; Columbus Instruments, Columbus, OH). Animals first were placed on the 3 cm rod and subjected to multiple trials until time spent on the rod reached a plateau (∼16 trials). Trials began with the rod at 4 rpm and accelerated to 50 rpm over 5 min. Age-matched animals were tested during similar times of day by an observer blinded to subject genotype and always in parallel with littermate controls. Once plateau values had been reached, each group of animals was subjected to four independent trials, and their time on the rod was averaged.
Statistics. Throughout the experiments, the investigators were blinded to the genotype of the mice. All values are expressed as mean ± SEM. Differences among means were analyzed by using standard Student's t tests or two-tailed ANOVA followed by the Bonferroni posttest as appropriate. In all analyses, the null hypothesis was rejected at the p = 0.05 level. All statistical analyses were performed using GraphPad (San Diego, CA) InStat and Prism software.
Results
Tissue-specific expression of Cre recombinase in catecholaminergic cells
Microinjection of recombinant DNA containing the Cre recombinase coding sequence downstream of the well characterized 9.0 kb fragment of the rat TH promoter (Fig. 1A) yielded five independent lines of mice containing the Cre transgene (named TH-Cre 1-TH-Cre 5). Cre-expressing mice are fertile, are indistinguishable from their wild-type littermates, and have continued to yield offspring that express the transgene years after their original development. Breeding outcomes suggest that the transgene is present on autosomal chromosomes in all lines except line four, in which the transgene appears to be X-linked (data not shown). Tissue-specific expression of Cre recombinase was demonstrated by crossing each TH-Cre mouse line to Z/AP and Z/EG reporter mice (Lobe et al., 1999; Novak et al., 2000). The Z/AP mouse expresses the lacZ gene during embryonic and adult stages and in nearly all cells except erythrocytes, chondrocytes, and adipocytes. On Cre-mediated rearrangement, the lacZ gene is inactivated and human placental alkaline phosphatase (hPLAP) is activated and expressed at high levels in those cells expressing Cre. The hPLAP enzyme is easily localized by histochemical methods. Z/EG mice function in a similar way, but with activation of the green fluorescent protein (GFP) gene after Cre-mediated recombination. There are no obvious differences between lines 1-5 in the pattern of hPLAP or GFP expression with the exception of minimally altered cell type expression in the retina (data not shown). As expected, Cre recombinase activity is found in those mouse cells containing an active TH promoter, i.e., cathecholaminergic cells (Figs. 2, 3). Specifically, dopaminergic cell populations, including those in the retina (amacrine cells), substantia nigra, retrorubral field, ventral tegmental area, zona incerta, hypothalamus, and olfactory bulb, stain for Cre recombinase activity, as do the noradrenergic cells of the locus ceruleus, sympathetic ganglia, and medulla, as well as the adrenergic cells in the adrenal medulla and lower brainstem. In addition, Cre reporter activity is present in many of the projection areas of these neuronal populations such as the striatum, amygdala, and nucleus accumbens, likely because of filling of axonal processes with hPLAP or GFP (data not shown). Several other areas that are not typically thought to have active TH expression, including the lateral septal nucleus, accessory olfactory bulb, suparafascicular thalamus, and pretectal area, also stain for Cre activity, possibly as a result of TH promoter activity in precursor cell populations or ectopic expression from the exogenous TH promoter. Indeed, ectopic expression driven by the exogenous TH promoter has been described previously (Min et al., 1994; Gelman et al., 2003). Interestingly, some cells closely clustered around and within TH-positive nuclei (locus ceruleus and substantia nigra) demonstrate Cre activity, although they do not stain for TH protein (Fig. 3). Conversely, Cre-mediated recombination is not detected in 100% of the targeted cell population; rather, a mosaic of Cre activity is noted in TH-positive neurons (Fig. 3).
Figure 1.

Targeting the Bcl-x gene in TH+ cells. A, A TH-Cre construct possessing a 9.0 kb fragment of the rat TH promoter, synthetic intron, Cre cDNA, and an SV40 polyadenylation sequence. Numbered arrows represent primers used to screen for the Cre-coding sequence. B, A map of the Bcl-x locus containing a floxed version of the gene and the predicted product of Cre-mediated recombination, including the elimination of a region of the 5′-untranslated sequence, exons 1 and 2, and intervening DNA. Numbered arrows represent PCR primers. C, A montage of ethidium bromide-stained 2.0% agarose gel of PCR products amplified from ventral midbrain DNA of indicated mouse strains. PCR using primers III and V demonstrates selective deletion of DNA between loxP sites in the presence of Cre recombinase in vivo. Each lane represents midbrain DNA samples isolated from an individual mouse of the indicated genotype.
Figure 2.
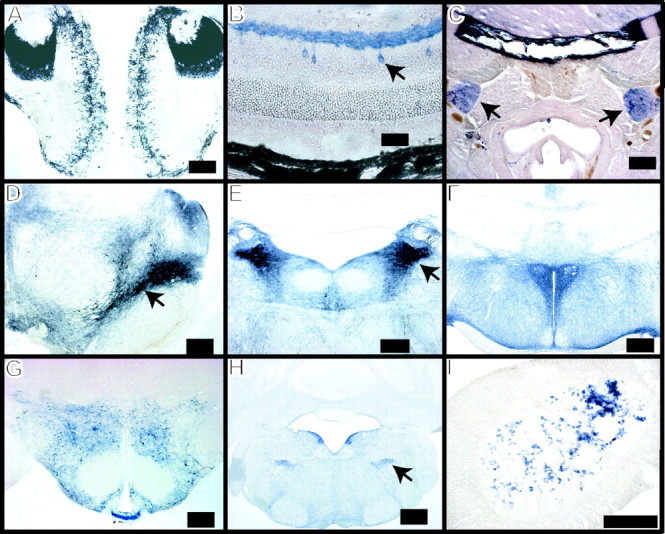
Regional localization of Cre recombinase activity. TH-Cre and Z/AP double transgenic mouse tissue was stained for human placental alkaline phosphatase activity (blue), indicating the presence of Cre recombinase activity. Staining is seen in the periglomerular layer of the olfactory bulb and the accessory olfactory bulb (A), amacrine cells (arrow) and dendritic plexus of the retina (B), superior cervical ganglia (C, arrows), substantia nigra (arrow) and ventral tegmental area of the midbrain (D), locus ceruleus (E, arrow), periventricular and paraventricular nuclei of the hypothalamus (F), hypothalamic area, including the arcuate nucleus, median eminence, and zona incerta (G), dorsal medullary adrenergic cells (H, arrow), and adrenal medulla (I). All tissues are from adult mice, except for C, in which tissue was removed from 24 h paraformaldehyde immersion-fixed 1-d-old pups. Scale bars: B, 25 μm; A, C-I, 200 μm.
Figure 3.

Cellular localization of Cre recombinase activity. Indicated mouse tissue was visualized by confocal microscopy demonstrating the presence of TH immunoreactivity (Cy3; red) and Cre recombinase activity (GFP; green). Colocalization is seen in the glomerular layer of the olfactory bulb, the amacrine cells of the retina (a), the substantia nigra (SN), the ventral tegmental area (VTA), and the locus ceruleus. AOB, Accessory olfactory bulb. Magnification: top four rows, 40×; bottom row, 100×.
Generation of Bcl-x gene deletion in catecholaminergic cells
Conditional Bcl-x gene disruption in catecholaminergic cells was accomplished by crossing TH-Cre mice to mice possessing a floxed Bcl-x gene. Genotyping of the offspring was conducted by PCR using the strategy shown (Fig. 1). Gene disruption is specifically demonstrated by PCR amplification of DNA isolated from the ventral midbrain of mice containing the TH-Cre transgene and two copies (homozygous) of the floxed Bcl-x gene (Fig. 1C). No rearranged product is detected in the absence of the Cre transgene. To verify the loss of Bcl-x expression, mouse brain tissue was double-labeled with antibodies to TH and Bcl-x. Visualization with fluorescent secondary antibodies and subsequent confocal microscopy demonstrates the loss of Bcl-x protein in a subset of TH-positive cells (Fig. 4). Many cells, however, stain for both proteins, suggesting that Bcl-x deletion does not occur in all TH-positive cells. Those TH-positive cells lacking Bcl-x demonstrate TH immunoreactivity and morphology that are indistinguishable from those containing Bcl-x (Fig. 4).
Figure 4.
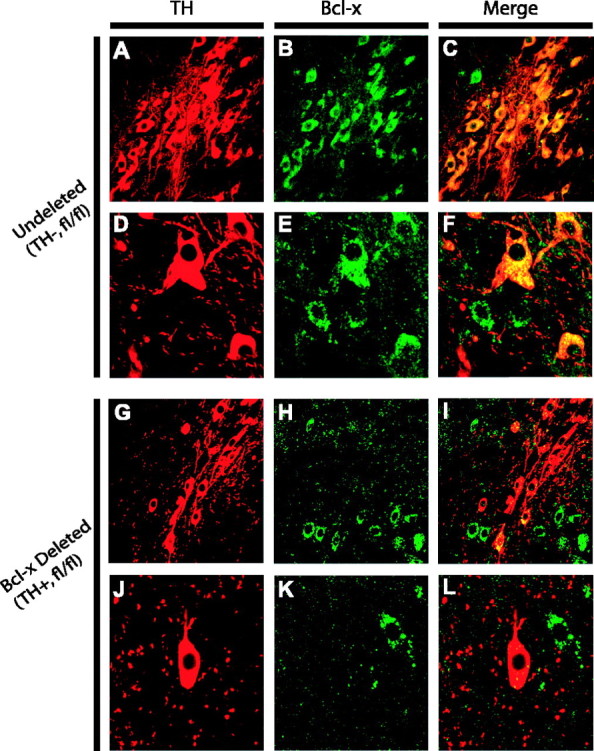
Bcl-x immunoreactivity is lost in a subset of TH-positive neurons of conditional knock-out mice. Tissue sections from the substantia nigra were taken from adult TH-Cre negative, fl/fl Bcl-x (undeleted) (A-F) and TH-Cre positive, fl/fl Bcl-x (Bcl-x-deleted) (G-L) mice, stained for the presence of TH (red) and Bcl-x (green) and visualized by confocal microscopy. Some TH-positive cells in the TH-cre, fl/fl Bcl-x substantia nigra continue to express Bcl-x (I, arrowheads), whereas others clearly do not (L, arrowhead). Magnification: A-C, G-I, 40×; D-F, J-L, 100×.
Animals with the Bcl-x gene disrupted in TH-positive neurons (TH-Cre, fl/fl Bcl-x) appear as healthy and active as TH-Cre-negative littermate controls, and expected genotypic ratios are obtained, suggesting no early survival disadvantage from Bcl-x deletion (data not shown). In individual litters, TH-Cre, fl/fl Bcl-x mice are reduced in weight by 18-30% relative to their control (Cre-negative) littermates (Fig. 5). The overall health, coordination, balance, and activity of the control and Bcl-x conditionally deleted mice were assayed using an accelerating rotorod protocol. Bcl-x-deleted animals are able to stay on the rod significantly longer (127 ± 13 vs 81 ± 9.4 s; p = 0.007) and at higher velocities when compared with control animals, suggesting no major motor system defects in these animals.
Figure 5.

Differences between control and Bcl-x conditional knock-outs. A, Catecholaminergic cell-specific Bcl-x conditional knock-out animals show decreased body mass. B, C, Catecholaminergic cell-specific Bcl-x conditional knock-out animals possess fewer TH-containing neurons in the SNPc and LC. *p < 0.05, statistical significance versus age-matched controls using ANOVA. Values inside bars indicate the numbers of measurements, and cell counts represent total numbers of cells per bilateral nuclei.
Conditional Bcl-x knock-outs have reduced striatal dopamine and fewer TH-positive SNPc and locus ceruleus neurons
TH immunoreactivity is decreased in the midbrain of TH-Cre, fl/fl Bcl-x mice compared with controls (Fig. 6). This effect was quantified by stereological counting of SNPc cells, which shows reduced numbers of TH-immunoreactive cells at all ages tested (Fig. 5). The magnitude of cell loss does not change appreciably with age and ranges from 25 to 33%. This is reflected in fewer Nissl-staining neurons in the SNPc as well, verifying that loss of TH staining is the result of fewer neurons, not merely the down-regulation of TH protein in individual cells (data not shown). A similar finding was seen in the locus ceruleus, where TH-positive cell numbers are reduced by 31% in 1-month-old TH-Cre, fl/fl Bcl-x mice and by 27% in adults (8-12 months of age) relative to age- and gender-matched controls (Fig. 5). Interestingly, confocal double-labeling experiments show occasional surviving TH-positive cells that do not stain for Bcl-x in the brains of TH-Cre, fl/fl Bcl-x mice, whereas no such cells are seen in controls (Fig. 4). These cells are relatively rare, however, and most cells in the TH-Cre, fl/fl Bcl-x mice do show Bcl-x and TH colocalization. HPLC analysis of TH-Cre, fl/fl Bcl-x striatum shows a statistically significant reduction in dopamine and its metabolites when compared with controls (dopamine, 23.3 ± 1.0 vs 18.7 ± 1.2 ng/mg of tissue; 3,4-dihydroxyphenylacetic acid, 1.3 ± 0.07 vs 1.1 ± 0.08 ng/mg of tissue; and homovanillic acid, 0.50 ± 0.08 vs 0.32 ± 0.11 ng/mg of tissue; p < 0.04). Ratios of metabolites to dopamine levels are similar in controls and TH-Cre, fl/fl Bcl-x mice, suggesting no alteration in dopamine metabolism in the surviving cells (data not shown). Overall brain weight also is reduced (508 ± 24 vs 571 ± 8 mg; p < 0.03) in adult TH-Cre, fl/flBcl-x mice compared with age- and gender-matched controls.
Figure 6.

Comparison of TH immunoreactivity in the midbrain of TH-Cre-negative, fl/fl Bcl-x (control) and TH-Cre-positive, fl/fl (Bcl-x-deleted) mice. Every fourth consecutive 40 μm section was stained in the region of the substantia nigra and ventral tegmental area, and similar sections were matched side by side. Staining reactions were performed on adult mouse tissue for identical periods and qualitatively demonstrate fewer TH-positive cells in the Bcl-x-deleted mice. Magnification, 5×.
Discussion
Characterization and utility of TH-Cre mice
In PD, dopaminergic cells of the SNPc and noradrenergic cells of the LC as well as other catecholaminergic cells are among those most affected (Halliday et al., 1990; Zarow et al., 2003). Thus, we hypothesized that targeting catecholaminergic cells via genetic manipulation would provide a valuable tool for the study of PD. We used the well characterized 9.0 kb rat TH promoter to drive expression of Cre recombinase in transgenic mice (Min et al., 1994, 1996). The resulting Cre expression in catecholaminergic cells provides a simple way to selectively alter genes in these cells. Using Z/AP and Z/EG indicator mouse lines, we have shown specific expression of Cre activity in catecholaminergic cells. This along with the generation of multiple mouse lines showing similar Cre localization proves that the TH promoter is driving Cre expression. Also, mice described previously and produced in a similar manner demonstrate Cre localization that parallels that seen here (Gelman et al., 2003).
Despite the presence of TH in most Cre-expressing cells, there are some cells that demonstrated Cre activity but lacked detectable TH. The explanation for this discrepancy likely varies according to cell type. For example, cells in the accessory olfactory nucleus possess an active TH promoter but do not translate appreciable amounts of protein (Min et al., 1994). Such posttranscription control does not exist for Cre mRNA and explains the presence of Cre activity in these cells. Other explanations for the presence of Cre activity in TH-negative cells include ectopic TH promoter activity and the activation of the promoter in less mature cells that either never produced TH protein or subsequently lost this ability. In the latter case, expression of Cre at any time during development would lead to persistent reporter gene (Z/AP or Z/EG) expression. Such cell marking could be useful for tracking cells derived from precursors that possess an active TH promoter. Conversely, Cre activity was not seen in all TH-positive cells, suggesting genetic mosaicism, a common finding in transgenic models (Nagy, 2000; Tronche et al., 2002).
Requirement for Bcl-x in the SNPc
Bcl-x is a Bcl-2 gene family member and is spliced into five isoforms (Yang et al., 2002), including two major isoforms: the antiapoptotic Bcl-xL and the proapoptotic Bcl-xS (Boise et al., 1993). Most nervous system effects are mediated by Bcl-xL because Bcl-xS is expressed at low or undetectable levels in the brain (Boise et al., 1993; Gonzalez-Garcia et al., 1995; Krajewska et al., 2002). Murine Bcl-x gene interruption results in embryonic lethality likely from hematopoeitic failure (Motoyama et al., 1995; Shindler et al., 1997). These animals exhibit cell death in hematopoetic areas of the liver and in regions of the CNS. The prenatal lethality of Bcl-x deletion prompted the production of several lines of Cre-mediated, conditional Bcl-x knock-out mice (Rucker et al., 2000; Wagner et al., 2000; Hon et al., 2004; Takehara et al., 2004). Similarly, we have generated mice that lack Bcl-x gene products specifically in catecholaminergic cells. These animals have approximately one-third fewer SNPc dopaminergic neurons compared with littermate controls. To determine whether this effect is specific for dopaminergic cells, TH-positive neurons within the noradrenergic LC, another region susceptible to cell loss in PD, also were assayed. The magnitude of cell loss seen here is similar to that in the SNPc, suggesting that Bcl-x is required for the proper development of catecholaminergic cells whether they are dopaminergic or noradrenergic. The majority of the remaining cells contain both Bcl-x and TH. These cells result from failure of Bcl-x deletion, their numbers possibly augmented by a survival advantage conferred on precursor cells lacking Bcl-x recombination. The absence of widespread TH-positive neurons without Bcl-x immunoreactivity in adult animals suggests that in most TH-expressing cells, Bcl-x gene deletion is incompatible with cell survival. This effect likely results from a loss of the antiapoptotic activity of the Bcl-xL isoform. Indeed, Bcl-xL plays an important role in the development and survival of neurons. This is suggested by the expression profile of the protein, which peaks between embryonic day 13 and postnatal day 5 (Krajewska et al., 2002), and by the cell death that occurs in postmitotic but immature neurons when the protein is deficient (Motoyama et al., 1995; Roth et al., 1996, 2000). This neuronal cell death is thought to be cell autonomous because it can be demonstrated in telencephalic cultures of neurons derived from Bcl-x-deficient embryos (Roth et al., 1996) and can be reversed by Bax gene deletion, although Bcl-x/Bax double-knock-out animals still die prenatally (Shindler et al., 1997). Our data lend strong support for the endogenous neuronal requirement of Bcl-x during development.
Our study also supports data demonstrating the antiapoptotic effect of Bcl-xL in dopaminergic neurons. This includes previous studies showing that the dopaminergic cell line MN9D is rescued from staurosporine-mediated cell death by overexpression of Bcl-xL (Kim et al., 1999), and differentiated PC12 cells overexpressing Bcl-xL are more resistant to serum withdrawal (Blomer et al., 1998). Recently, Bcl-xL was used to increase the generation of TH-positive neurons from embryonic stem (ES) cells (Liste et al., 2004; Shim et al., 2004). Indeed, as ES cells are coaxed into a dopaminergic phenotype, levels of endogenous Bcl-xL increase, whereas ES cells expressing exogenous Bcl-xL demonstrate MPP+ resistance and show increased neurite outgrowth and functional recovery in animal PD models (Shim et al., 2004). Last, studies on human neurospheres have demonstrated that TH overexpression is toxic to these cells and that Bcl-xL can partially ameliorate this toxicity (Liste et al., 2004).
Some neurons in our study avoid the lethal effects of Bcl-x deletion. The presence of such cells is demonstrated by their lack of Bcl-x immunoreactivity and by the ability to detect the appropriate genetic rearrangement in tissue from animals containing the TH-Cre transgene and two copies of the floxed Bcl-x allele. This verifies that the appropriate Cre-mediated gene inactivation has taken place in these animals and shows that, despite a loss of Bcl-x protein, catecholaminergic neurons can survive. This suggests a window of vulnerability in TH neuron development. The TH promoter is activated as early as embryonic day 9, and its expression continues into adulthood (Son et al., 1996). Some cells may escape Cre-mediated Bcl-x deletion early in development only to have the gene inactivated at later stages when the requirement for Bcl-x has passed. Similar findings are seen in ovarian and mammary gland tissue, where the requirement for Bcl-x is developmental stage-specific (Walton et al., 2001; Riedlinger et al., 2002). The role of Bcl-x in maintaining postnatal neuronal survival has been inferred by its continued expression into adulthood (Frankowski et al., 1995; Gonzalez-Garcia et al., 1995; Yachnis et al., 1998) and the fact that adult neurons from mice overexpressing Bcl-x are resistant to cell death stimuli such as hypoxia and axotomy (Parsadanian et al., 1998). Our findings suggest that the requirement for Bcl-x is not absolute, and the presence of Bcl-xL in adult brains may serve functions other than maintaining cell survival under baseline conditions. The lack of progressive cell death in our conditional knock-out animals suggests that even advanced age fails to trigger increased loss of dopaminergic neurons.
Curiously the Bcl-x conditional knock-out animals have reduced brain and body mass as well as enhanced rotorod performance. Decreased weight may be the result of effects on the hypothalamus, where Cre expression and thus Bcl-x deletion occur. Also, a change in growth rate may affect brain size, as would the depletion of neurons. Many factors influence rotorod behavior, and further study will be required to determine the reason behind the improved performance. Possible candidate factors include strain differences, body size, and visual impairment, all of which have been correlated to rotorod score (McFadyen et al., 2003). In addition, a recent study has shown a negative correlation between mouse body weight and rotorod fall latency, perhaps making animal size the most likely factor explaining our animals' enhanced performance (Lalonde et al., 2005).
This study has implications regarding our understanding of the pathology of PD. It is known that Bcl-xL is present in human adult substantia nigral cells, and its nigral cell mRNA expression is increased in the brains of PD patients with the caveat that those neurons containing Lewy bodies have reduced levels (Hartmann et al., 2002). In addition, cybrid cell culture models of PD find that cells containing mitochondria derived from PD patients demonstrated increased levels of Bcl-xL compared with controls (Veech et al., 2000). The present study shows that Bcl-x function is dispensable in mature dopaminergic neurons and implies that loss of Bcl-x function is an unlikely primary mechanism for the loss of these cells in PD. Confirmation of the importance of cell-autonomous expression of Bcl-x for the proper development of the SNPc does support the use of this protein in developing methods to generate dopaminergic cells from precursor populations such as embryonic and neural stem cells.
Footnotes
This work was supported by a clinician scientist award from the Passano Foundation (J.M.S.), the Lee Martin Trust, the Sylvia Nachlas Trust, the Morris K. Udall Parkinson's Disease Research Center, and National Institutes of Health-National Institute of Neurological Disorders and Stroke Grant NS 38377. T.M.D. is the Leonard and Madlyn Abramson professor in neurodegenerative diseases. We thank Dr. Jin Son for providing the 9.0 kb rat TH promoter, Dr. S. O'Gorman for providing the Cre construct, Drs. E. Rucker and K. Wagner for providing the Bcl-x floxed mice, and Drs. C. Lobe and A. Nagy for providing the valuable Z/AP and Z/EG reporter mice to the research community.
Correspondence should be addressed to Dr. Ted M. Dawson, Institute for Cell Engineering, Johns Hopkins University School of Medicine, 733 North Broadway, Broadway Research Building, Suite 731, Baltimore, MD 21205. E-mail: tdawson@jhmi.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256721-08$15.00/0
References
- Blomer U, Kafri T, Randolph-Moore L, Verma IM, Gage FH (1998) Bcl-xL protects adult septal cholinergic neurons from axotomized cell death. Proc Natl Acad Sci USA 95: 2603-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB (1993) bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74: 597-608. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24: 197-211. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL (2003) Molecular pathways of neurodegeneration in Parkinson's disease. Science 302: 819-822. [DOI] [PubMed] [Google Scholar]
- Frankowski H, Missotten M, Fernandez PA, Martinou I, Michel P, Sadoul R, Martinou JC (1995) Function and expression of the Bcl-x gene in the developing and adult nervous system. NeuroReport 6: 1917-1921. [DOI] [PubMed] [Google Scholar]
- Gelman DM, Noain D, Avale ME, Otero V, Low MJ, Rubinstein M (2003) Transgenic mice engineered to target Cre/loxP-mediated DNA recombination into catecholaminergic neurons. Genesis 36: 196-202. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Garcia M, Garcia I, Ding L, O'Shea S, Boise LH, Thompson CB, Nunez G (1995) bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc Natl Acad Sci USA 92: 4304-4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday GM, Li YW, Blumbergs PC, Joh TH, Cotton RG, Howe PR, Blessing WW, Geffen LB (1990) Neuropathology of immunohistochemically identified brainstem neurons in Parkinson's disease. Ann Neurol 27: 373-385. [DOI] [PubMed] [Google Scholar]
- Hartmann A, Mouatt-Prigent A, Vila M, Abbas N, Perier C, Faucheux BA, Vyas S, Hirsch EC (2002) Increased expression and redistribution of the antiapoptotic molecule Bcl-xL in Parkinson's disease. Neurobiol Dis 10: 28-32. [DOI] [PubMed] [Google Scholar]
- Hon H, Rucker III EB, Hennighausen L, Jacob J (2004) bcl-xL is critical for dendritic cell survival in vivo. J Immunol 173: 4425-4432. [DOI] [PubMed] [Google Scholar]
- Huang MT, Gorman CM (1990) Intervening sequences increase efficiency of RNA 3′ processing and accumulation of cytoplasmic RNA. Nucleic Acids Res 18: 937-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isacson O (2002) Models of repair mechanisms for future treatment modalities of Parkinson's disease. Brain Res Bull 57: 839-846. [DOI] [PubMed] [Google Scholar]
- Kim JE, Oh JH, Choi WS, Chang II, Sohn S, Krajewski S, Reed JC, O'Malley KL, Oh YJ (1999) Sequential cleavage of poly(ADP-ribose)polymerase and appearance of a small Bax-immunoreactive protein are blocked by Bcl-X(L) and caspase inhibitors during staurosporine-induced dopaminergic neuronal apoptosis. J Neurochem 72: 2456-2463. [DOI] [PubMed] [Google Scholar]
- Krajewska M, Mai JK, Zapata JM, Ashwell KW, Schendel SL, Reed JC, Krajewski S (2002) Dynamics of expression of apoptosis-regulatory proteins Bid, Bcl-2, Bcl-X, Bax and Bak during development of murine nervous system. Cell Death Differ 9: 145-157. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Dumont M, Staufenbiel M, Strazielle C (2005) Neurobehavioral characterization of APP23 transgenic mice with the SHIRPA primary screen. Behav Brain Res 157: 91-98. [DOI] [PubMed] [Google Scholar]
- Lang AE, Lozano AM (1998) Parkinson's disease. First of two parts. N Engl J Med 339: 1044-1053. [DOI] [PubMed] [Google Scholar]
- Liste I, Garcia-Garcia E, Martinez-Serrano A (2004) The generation of dopaminergic neurons by human neural stem cells is enhanced by Bcl-XL, both in vitro and in vivo J Neurosci 24: 10786-10795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobe CG, Koop KE, Kreppner W, Lomeli H, Gertsenstein M, Nagy A (1999) Z/AP, a double reporter for cre-mediated recombination. Dev Biol 208: 281-292. [DOI] [PubMed] [Google Scholar]
- Mandir AS, Przedborski S, Jackson-Lewis V, Wang ZQ, Simbulan-Rosenthal CM, Smulson ME, Hoffman BE, Guastella DB, Dawson VL, Dawson TM (1999) Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci USA 96: 5774-5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeux R (2003) Epidemiology of neurodegeneration. Annu Rev Neurosci 26: 81-104. [DOI] [PubMed] [Google Scholar]
- McFadyen MP, Kusek G, Bolivar VJ, Flaherty L (2003) Differences among eight inbred strains of mice in motor ability and motor learning on a rotorod. Genes Brain Behav 2: 214-219. [DOI] [PubMed] [Google Scholar]
- Micieli G, Tosi P, Marcheselli S, Cavallini A (2003) Autonomic dysfunction in Parkinson's disease. Neurol Sci 24 [Suppl 1]: S32-S34. [DOI] [PubMed] [Google Scholar]
- Min N, Joh TH, Kim KS, Peng C, Son JH (1994) 5′ upstream DNA sequence of the rat tyrosine hydroxylase gene directs high-level and tissue-specific expression to catecholaminergic neurons in the central nervous system of transgenic mice. Brain Res Mol Brain Res 27: 281-289. [DOI] [PubMed] [Google Scholar]
- Min N, Joh TH, Corp ES, Baker H, Cubells JF, Son JH (1996) A transgenic mouse model to study transsynaptic regulation of tyrosine hydroxylase gene expression. J Neurochem 67: 11-18. [DOI] [PubMed] [Google Scholar]
- Mizuguchi M, Sohma O, Takashima S, Ikeda K, Yamada M, Shiraiwa N, Ohta S (1996) Immunochemical and immunohistochemical localization of Bcl-x protein in the rat central nervous system. Brain Res 712: 281-286. [DOI] [PubMed] [Google Scholar]
- Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, Loh DY (1995) Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 267: 1506-1510. [DOI] [PubMed] [Google Scholar]
- Nagy A (2000) Cre recombinase: the universal reagent for genome tailoring. Genesis 26: 99-109. [PubMed] [Google Scholar]
- Novak A, Guo C, Yang W, Nagy A, Lobe CG (2000) Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis 28: 147-155. [PubMed] [Google Scholar]
- O'Gorman S, Dagenais NA, Qian M, Marchuk Y (1997) Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc Natl Acad Sci USA 94: 14602-14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakkenberg B, Moller A, Gundersen HJ, Mouritzen Dam A, Pakkenberg H (1991) The absolute number of nerve cells in substantia nigra in normal subjects and in patients with Parkinson's disease estimated with an unbiased stereological method. J Neurol Neurosurg Psychiatry 54: 30-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsadanian AS, Cheng Y, Keller-Peck CR, Holtzman DM, Snider WD (1998) Bcl-xL is an antiapoptotic regulator for postnatal CNS neurons. J Neurosci 18: 1009-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce RC, Bari AA (2001) The role of neurotrophic factors in psychostimulant-induced behavioral and neuronal plasticity. Rev Neurosci 12: 95-110. [DOI] [PubMed] [Google Scholar]
- Riedlinger G, Okagaki R, Wagner KU, Rucker III EB, Oka T, Miyoshi K, Flaws JA, Hennighausen L (2002) Bcl-x is not required for maintenance of follicles and corpus luteum in the postnatal mouse ovary. Biol Reprod 66: 438-444. [DOI] [PubMed] [Google Scholar]
- Roth KA, Motoyama N, Loh DY (1996) Apoptosis of bcl-x-deficient telencephalic cells in vitro J Neurosci 16: 1753-1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth KA, Kuan C, Haydar TF, D'Sa-Eipper C, Shindler KS, Zheng TS, Kuida K, Flavell RA, Rakic P (2000) Epistatic and independent functions of caspase-3 and Bcl-X(L) in developmental programmed cell death. Proc Natl Acad Sci USA 97: 466-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rucker III EB, Dierisseau P, Wagner KU, Garrett L, Wynshaw-Boris A, Flaws JA, Hennighausen L (2000) Bcl-x and Bax regulate mouse primordial germ cell survival and apoptosis during embryogenesis. Mol Endocrinol 14: 1038-1052. [DOI] [PubMed] [Google Scholar]
- Shim JW, Koh HC, Chang MY, Roh E, Choi CY, Oh YJ, Son H, Lee YS, Studer L, Lee SH (2004) Enhanced in vitro midbrain dopamine neuron differentiation, dopaminergic function, neurite outgrowth, and 1-methyl-4-phenylpyridium resistance in mouse embryonic stem cells overexpressing Bcl-XL. J Neurosci 24: 843-852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindler KS, Latham CB, Roth KA (1997) Bax deficiency prevents the increased cell death of immature neurons in bcl-x-deficient mice. J Neurosci 17: 3112-3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son JH, Min N, Joh TH (1996) Early ontogeny of catecholaminergic cell lineage in brain and peripheral neurons monitored by tyrosine hydroxylase-lacZ transgene. Brain Res Mol Brain Res 36: 300-308. [DOI] [PubMed] [Google Scholar]
- Subramanian T (2001) Cell transplantation for the treatment of Parkinson's disease. Semin Neurol 21: 103-115. [DOI] [PubMed] [Google Scholar]
- Takehara T, Tatsumi T, Suzuki T, Rucker III EB, Hennighausen L, Jinushi M, Miyagi T, Kanazawa Y, Hayashi N (2004) Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology 127: 1189-1197. [DOI] [PubMed] [Google Scholar]
- Tronche F, Casanova E, Turiault M, Sahly I, Kellendonk C (2002) When reverse genetics meets physiology: the use of site-specific recombinases in mice. FEBS Lett 529: 116-121. [DOI] [PubMed] [Google Scholar]
- Veech GA, Dennis J, Keeney PM, Fall CP, Swerdlow RH, Parker Jr WD, Bennett Jr JP (2000) Disrupted mitochondrial electron transport function increases expression of anti-apoptotic bcl-2 and bcl-X(L) proteins in SH-SY5Y neuroblastoma and in Parkinson disease cybrid cells through oxidative stress. J Neurosci Res 61: 693-700. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Claudio E, Rucker III EB, Riedlinger G, Broussard C, Schwartzberg PL, Siebenlist U, Hennighausen L (2000) Conditional deletion of the Bcl-x gene from erythroid cells results in hemolytic anemia and profound splenomegaly. Development 127: 4949-4958. [DOI] [PubMed] [Google Scholar]
- Walton KD, Wagner KU, Rucker III EB, Shillingford JM, Miyoshi K, Hennighausen L (2001) Conditional deletion of the bcl-x gene from mouse mammary epithelium results in accelerated apoptosis during involution but does not compromise cell function during lactation. Mech Dev 109: 281-293. [DOI] [PubMed] [Google Scholar]
- West MJ (1993) New stereological methods for counting neurons. Neurobiol Aging 14: 275-285. [DOI] [PubMed] [Google Scholar]
- Yachnis AT, Giovanini MA, Eskin TA, Reier PJ, Anderson DK (1998) Developmental patterns of BCL-2 and BCL-X polypeptide expression in the human spinal cord. Exp Neurol 150: 82-97. [DOI] [PubMed] [Google Scholar]
- Yang XF, Ye Q, Press B, Han RZ, Bassing CH, Sleckman BP, Alt FW, Cantor H (2002) Analysis of the complex genomic structure of Bcl-x and its relationship to Bcl-x(gamma) expression after CD28-dependent costimulation. Mol Immunol 39: 45-55. [DOI] [PubMed] [Google Scholar]
- Zarow C, Lyness SA, Mortimer JA, Chui HC (2003) Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol 60: 337-341. [DOI] [PubMed] [Google Scholar]