

Abstract
Accumulation of β-amyloid (Aβ) peptides in the cerebral cortex is considered a key event in the pathogenesis of Alzheimer's disease (AD). Presenilin 1 (PS1) plays an essential role in the γ-secretase cleavage of the amyloid precursor protein (APP) and the generation of Aβ peptides. Reduction of Aβ generation via the inhibition of γ-secretase activity, therefore, has been proposed as a therapeutic approach for AD. In this study, we examined whether genetic inactivation of PS1 in postnatal forebrain-restricted conditional knock-out (PS1 cKO) mice can prevent the accumulation of Aβ peptides and ameliorate cognitive deficits exhibited by an amyloid mouse model that overexpresses human mutant APP. We found that conditional inactivation of PS1 in APP transgenic mice (PS1 cKO;APP Tg) effectively prevented the accumulation of Aβ peptides and formation of amyloid plaques and inflammatory responses, although it also caused an age-related accumulation of C-terminal fragments of APP. Short-term PS1 inactivation in young PS1 cKO;APP Tg mice rescued deficits in contextual fear conditioning and serial spatial reversal learning in a water maze, which were associated with APP Tg mice. Longer-term PS1 inactivation in older PS1 cKO;APP Tg mice, however, failed to rescue the contextual memory and hippocampal synaptic deficits and had a decreasing ameliorative effect on the spatial memory impairment. These results reveal that in vivo reduction of Aβ via the inactivation of PS1 effectively prevents amyloid-associated neuropathological changes and can, but only temporarily, improve cognitive impairments in APP transgenic mice.
Keywords: Alzheimer's disease, β-amyloid, γ-secretase, mouse, behavior, synaptic plasticity
Introduction
Alzheimer's disease (AD) is an age-related neurodegenerative disorder in which progressive memory loss is accompanied by cognitive decline. Neuropathologically, AD is characterized by a progressive loss of synapses and neurons as well as the presence of amyloid plaques and neurofibrillary tangles. Mutations in the genes encoding β-amyloid precursor protein (APP) and presenilins are linked to familial AD (FAD) (Hutton and Hardy, 1997). According to the “amyloid cascade” hypothesis, accumulation of β-amyloid (Aβ) peptides, which are the main constituents of amyloid plaques, is the key event initiating AD pathogenesis (Hardy and Selkoe, 2002).
Transgenic (Tg) mice overexpressing mutant forms of APP reproduce several key neuropathological features of AD, such as amyloid plaques and inflammatory responses (Games et al., 1995; Borchelt et al., 1996; Hsiao et al., 1996; Masliah et al., 1996; Sturchler-Pierrat et al., 1997; Mucke et al., 2000). In addition, APP transgenic mice can exhibit age-related impairments of hippocampal synaptic plasticity and learning and memory (Hsiao et al., 1996; Chapman et al., 1999; G. Chen et al., 2000; Morgan et al., 2000; Koistinaho et al., 2001; Corcoran et al., 2002). The synaptic and cognitive deficits often appear before amyloid plaque deposition and synaptic loss (Hsia et al., 1999; Larson et al., 1999; Koistinaho et al., 2001).
If Aβ accumulation is causal with respect to progression of the disease, limiting its accumulation should be beneficial therapeutically. An experimental test of this idea would be to limit the accumulation of Aβ at the point of synthesis or degradation. Aβ peptides are generated by two sequential proteolytic cleavages of APP mediated by β- and γ-secretases. β-Secretase [β-site APP-cleaving enzyme (BACE)] is an aspartyl protease that cleaves APP to generate the β-C-terminal fragment (β-C-TF) of APP (Vassar et al., 1999). Thus one route to limit the accumulation of Aβ would be to block or inhibit BACE (Ohno et al., 2004). An alternative route is via γ-secretase, or presenilin, an essential component of the γ-secretase complex, which further cleaves β-C-TF to generate Aβ. This has led to the hypothesis that targeting presenilin function by γ-secretase inhibitors also may be a suitable therapeutic strategy for AD (Hardy and Selkoe, 2002).
A major problem with this strategy is that presenilin 1 (PS1) plays essential roles in vivo (Wong et al., 1997; Xia et al., 2001; Pan et al., 2004), including neuronal differentiation and migration during embryonic development (Shen et al., 1997; Handler et al., 2000; Wines-Samuelson et al., 2005). However, PS1 conditional knock-out (PS1 cKO) mice, in which PS1 inactivation is restricted to the postnatal forebrain, show reduced Aβ generation and only subtle spatial memory impairment (Yu et al., 2001). Nevertheless, complete inactivation of both presenilins in the adult cerebral cortex results in hippocampal memory and synaptic plasticity impairments, followed by progressive neurodegeneration (Saura et al., 2004). Based on these results, we hypothesized that partial, but not complete, γ-secretase inactivation may prevent amyloid-related neuropathological changes and ameliorate the hippocampal-dependent learning deficits exhibited by APP transgenic mice. To test this hypothesis, we used here a multidisciplinary approach to examine the effects of partial inhibition of γ-secretase activity on amyloid-related neuropathology and cognitive impairments associated with APP transgenic mice via PS1 inactivation.
Materials and Methods
Mice. Generation of PS1 cKO and APP transgenic mice (APP Tg; J20 line) has been described previously (Mucke et al., 2000; Yu et al., 2001). To generate PS1 cKO;APP Tg mice, we first crossed APP transgenic mice, which were maintained in the hybrid background of C57BL/6 and DBA, with homozygous floxed PS1 (fPS1/fPS1) mice, which expressed normal levels of PS1 and were maintained in the hybrid background of C57BL/6 and 129, to obtain the fPS1/+;APP Tg mice, which then were bred to fPS1/fPS1 to obtain fPS1/fPS1;APP Tg mice. fPS1/fPS1;APP Tg mice then were bred to fPS1/fPS1;CaM-Cre (PS1 cKO) to obtain fPS1/fPS1;CaM-Cre;APP (PS1 cKO;APP Tg) mice. The mice used in the study, PS1 cKO; APP Tg, PS1 cKO, APP Tg, and control mice, were littermates obtained from the cross between PS1 cKO;APP Tg and control (fPS1/fPS1) mice. The genetic background of all mice used in the study was in the same hybrid background of C57BL/6, 129, and DBA. Experimenters of the behavioral tests and the electrophysiological analysis were blind to the genotypes of the mice.
Western blotting and ELISA analyses. For biochemical analysis, the mice cortices were dissected and homogenized in cold lysis buffer [consisting of (in mm) 50 Tris-HCl, pH 7.4, 150 NaCl, 1 EDTA plus 1% NP-40, and 0.5% Triton X-100] containing protease and phosphatase inhibitors (Sigma, St. Louis, MO). Lysates were cleared by centrifugation (12,000 rpm for 15 min), and the same amount of total protein was resolved on SDS-PAGE. Polyvinylidene difluoride membranes were incubated with antibodies against PS1 (PS1NT, 1:12,000; Calbiochem, La Jolla, CA), APP (Saeko antiserum APP665-695; 1:7500), phosphorylated tau (Ser202; CP13, 1:250), and β-actin (1:20,000; Abcam, Cambridge, UK) (Yu et al., 2001; Saura et al., 2004). For ELISAs, the total (soluble and insoluble) human Aβ40 and Aβ42 peptides from the cortex of APP Tg and PS1 cKO;APP Tg mice (n = 3-8) were measured by the 2G3/3D6 and 21F12/3D6 sandwich ELISA, respectively (Johnson-Wood et al., 1997).
Immunohistochemistry and immunogold electron microscopy. Paraffin-embedded brain sections (10 μm) were deparaffinized, alcohol-dehydrated, and immunostained with an Aβ antiserum (R1282; 1:1000) or monoclonal glial fibrillary acidic protein (GFAP; 1:500; Sigma) or CD45 (1:5000; Serotec, Raleigh, NC) antibodies as described previously (Stoltzner et al., 2000; Saura et al., 2004). Brain sections were incubated with biotinylated secondary antibodies and developed by using the peroxidase avidin-biotin reagent and the Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA). For double immunostaining, the sections were immunostained with C7 antiserum (APP676-695; 1:1000) and monoclonal synaptophysin (1:200; Sigma), NMDA receptor 1 (NMDAR1; 1:200; Chemicon, Temecula, CA), microtubule-activated protein 2 (MAP2; 1:250; Sigma), or R1282 and phosphorylated tau (Ser202/Thr205) (AT-8, 1:50; Innogenetics, Gent, Belgium) antibodies and were incubated with Alexa Fluor 488 or 594 secondary antibodies (Molecular Probes, Eugene, OR) (Saura et al., 2004). Images were analyzed with a Zeiss (Oberkochen, Germany) 510 confocal laser-scanning microscope.
Procedures for immunogold electron microscopic analysis have been described previously (Takahashi et al., 2002). For immunogold labeling, the free-floating sections were incubated with 369 antibody (APP649-695) by the immunogold-silver procedure and goat anti-rabbit IgG conjugated to 1 nm gold particles (Amersham Biosciences, Arlington, IL) in 0.01% gelatin and 0.08% bovine serum albumin in PBS. Transmission electron microscopy was performed on a Philips CM10 electron microscope at the Division of Neurobiology of Weill-Cornell Medical College (New York, NY).
Contextual fear conditioning. For contextual fear conditioning, the mice were placed within the conditioning chamber for 3 min before the onset of the unconditioned stimulus (US; footshock; 1 s/1 mA) to allow them to develop a representation of the context via exploration. After the shock, they then were left in the chamber for 2 min (immediate freezing) and returned to the home cages. Conditioning was tested 24 h after training for 4 min in the same conditioning chamber. Freezing response was scored by using the FreezeFrame automated system (Actimetrics, Wilmette, IL).
Water maze. Mice used for all behavioral tests were littermates and were 3 and 15-17 months of age. The serial spatial reversal task was performed in a circular pool (2 m in diameter) with a hidden platform (20 cm in diameter) (G. Chen et al., 2000). For the visible cue task, the pool was surrounded with white curtains to hide the extramaze cues, and the platform was marked with a dark cylinder. Each mouse was given four trials daily for 5 d, with a maximum trial duration of 90 s and an intertrial interval of 10 min. For the serial spatial learning to a hidden platform, each mouse was given a maximum of eight trials daily before reaching the criterion of three trials in a row with an average escape latency of <20 s. After the criterion was reached, the platform was moved to a new position the next day, and the training started again. An initial series of five different hidden platform locations was used, and the mice were allowed to take as many days as they needed to learn each of the platform locations. With the use of additional platform locations, the maximum number that the animals could learn in 10 d was used as a measure of learning capacity. The movement of the mice was monitored by an automated tracking system (Watermaze; Actimetrics).
Electrophysiology. Hippocampal slice preparation was performed as described previously (Yu et al., 2001). Stimulation (200 ms) pulses were delivered with a bipolar concentric metal electrode. Synaptic strength was quantified as the initial slope of field potentials recorded with artificial CSF-filled microelectrodes (1-2 MΩ). Baseline responses were collected at 0.07 Hz with a stimulation intensity that yielded a half-maximal response. Long-term potentiation (LTP) was obtained by five episodes of theta burst stimulation (TBS) delivered at 0.1 Hz. Each episode contained 10 stimulus trains delivered at 5 Hz, and each train consisted of four pulses at 100 Hz. Average responses (mean ± SEM) were expressed as a percentage of pre-TBS baseline response (at least 10 min of stable responses). A repeated-measures ANOVA was used to assess statistical significance.
Results
To evaluate the consequence of partial γ-secretase inactivation on the neuropathological changes and memory impairments in APP transgenic mice, we crossed PS1 cKO mice (Yu et al., 2001) with APP Tg mice, which overexpress a human APP containing the Swedish (K670N/M671L) and Indiana (V717F) mutations (Swe/Ind) (Mucke et al., 2000), to generate double-mutant PS1 cKO;APP Tg mice. The APP transgenic mice have been shown previously to develop amyloid plaques and other AD-like features in an age-dependent manner (Mucke et al., 2000).
Accumulation of APP C-TFs in synaptic terminals
Using Western blot analysis, we first confirmed the marked reduction of PS1 in the cerebral cortex of PS1 cKO;APP Tg mice at 2, 6, and 17 months of age (Fig. 1). The residual level of PS1 in the cortex of PS1 cKO;APP Tg mice likely is attributable to the normal PS1 expression in glia and interneurons, where PS1 expression is not targeted to eliminate in PS1 cKO mice (Yu et al., 2001). We next examined the consequence of PS1 inactivation on the proteolytic processing of APP. Western blot analysis showed an increase (approximately twofold) of full-length APP in APP Tg and PS1 cKO;APP Tg mice compared with nontransgenic controls at 2, 6, and 17 months of age (Fig. 1). The level of APP C-TFs was increased markedly in the cortex of PS1 cKO;APP Tg mice relative to APP Tg mice at 2, 6, and 17 months of age (Fig. 1). Similarly, the level of APP C-TFs was increased in PS1 cKO mice, compared with the control, at all ages. Furthermore, the increase in APP C-TFs was higher at 6 and 17 months of age relative to that at 2 months of age (n = 3-4). These results indicate that there is an age-dependent increase in APP C-TFs in the cortex of PS1 cKO;APP Tg mice.
Figure 1.

Age-dependent accumulation of APP C-TFs in PS1 cKO;APP Tg mice. Levels of APP, APP C-TFs, and PS1 in cortical lysates of control, PS1 cKO, APP Tg, and PS1 cKO;APP Tg mice at 2, 6, and 17 months of age were analyzed by Western blotting with the use of antibodies specific for the C terminal of APP (Saeko) or PS1. An antibody specific for β-actin was used as a loading control. Levels of APP C-TFs in PS1 cKO;APP Tg mice were increased at 6 and 17 months of age compared with 2 months of age.
To determine the subcellular localization of the accumulated APP C-TFs in PS1 cKO;APP Tg mice, we performed immunohistological analysis by using the C7 antibody on PS1 cKO;APP Tg mice at 6 months of age and littermate APP Tg mice as controls. The C7 antibody was raised against the C-terminal region of APP, which can recognize both full-length APP and C-TFs of APP. We found that the C7 immunoreactivity was localized mainly in the cell body of hippocampal neurons in APP Tg mice, whereas in PS1 cKO;APP Tg mice, the C7 immunoreactivity was mostly in neuronal processes, suggesting an ectopic localization of the accumulated APP C-TFs in the absence of PS1 (Fig. 2A-C). To address this issue further, we performed confocal microscopy analysis by using the C7 antibody and antibodies specific for MAP2 (somatodendritic), NMDAR1 (a postsynaptic marker), or synaptophysin (a presynaptic marker). We found that the C7 (green) and MAP2 (red) immunoreactivity were not colocalized in PS1 cKO;APP Tg mice (Fig. 2A). The C7 immunoreactivity, however, did overlap partially (yellow) with the NMDAR1 immunoreactivity in PS1 cKO;APP Tg mice, although there was some C7 immunoreactivity (green) that did not overlap with the NR1 immunoreactivity (red) (Fig. 2B). In contrast, the C7 immunoreactivity overlapped almost entirely with synaptophysin, suggesting that APP C-TFs were localized primarily to the presynaptic terminal (Fig. 2C).
Figure 2.
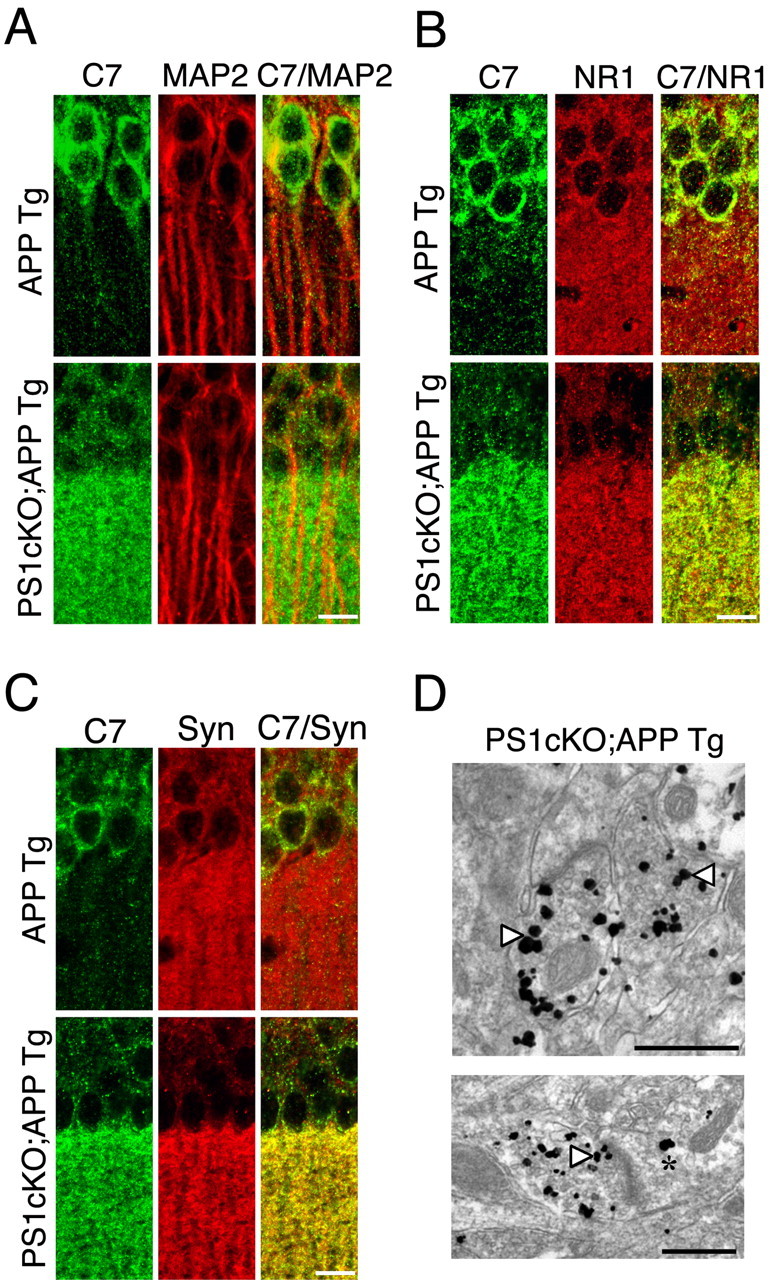
Accumulation of APPC-TFs in presynaptic terminals of PS1 cKO;APP Tg mice. A, Confocal microscopic images showing a marked increase in APP C-TFs recognized by C7 immunoreactivity (green) and a lack of colocalization (yellow) between the C7 immunoreactivity and the somatodendritic marker MAP2 immunoreactivity (red) in CA1 pyramidal neurons of 6-month-old PS1 cKO;APP Tg mice. B, Confocal microscopic images showing an increase in APP C-TFs recognized by C7 immunoreactivity (green) and its partial colocalization (yellow) with the postsynaptic marker NMDAR1 immunoreactivity (NR1; red) in CA1 pyramidal neurons of PS1 cKO;APP Tg mice. C, Confocal microscopic images showing a marked increase in APP C-TFs recognized by C7 immunoreactivity (green) and its abundant colocalization (yellow) with the presynaptic marker synaptophysin immunoreactivity (Syn; red) in CA1 pyramidal neurons of PS1 cKO;APP Tg mice. D, Electron microscopic images showing higher levels of accumulation of APP C-TFs in presynaptic compartments (arrowheads) compared with postsynaptic compartments (asterisk) in CA1 (top) and cortical (bottom) neurons from PS1 cKO;APP Tg mice at 6 months of age. Magnification: top, 19,000×; bottom, 13,500×. Scale bars: A-C, 25 μm; D, 500 nm.
To confirm the synaptic localization of APP C-TFs, we also performed electron microscopy analysis by using the 369 antibody, which is also specific for the C-terminal region of APP. We found that the 369 immunoreactivity was localized predominantly in the presynaptic terminal of cortical and hippocampal neurons in PS1 cKO;APP Tg brains, whereas no immunoreactivity was detected in nontransgenic control or APP Tg mice (Fig. 2D) (data not shown). These results indicate that partial inhibition of γ-secretase activity by PS1 inactivation causes an accumulation of APP C-TFs in the presynaptic terminal.
Prevention of amyloid-related neuropathological changes
To determine whether increased levels of APP C-TFs were accompanied by a decreased production of Aβ peptides, we measured the amount of total (soluble and insoluble) human Aβ peptides in the cerebral cortex of PS1 cKO;APP Tg and APP Tg mice at 2-3, 6, and 17 months of age. Our previous study has shown a reduction of endogenous mouse Aβ peptides in the cortices of PS1 cKO mice (Yu et al., 2001). Sandwich ELISA revealed that Aβ peptides accumulated in an age-dependent manner in the cerebral cortex of APP Tg mice, and the increase in Aβ42 was more marked than Aβ40 at 6 and 17 months of age (Fig. 3). In the cortex of PS1 cKO;APP Tg mice, the cortical Aβ40 levels were reduced significantly at 2-3 months of age (∼58%; p < 0.006; n = 8), 6 months of age (∼78%; p < 0.0001; n = 7), and 17 months of age (∼96%; p < 0.0001; n = 3). Similarly, levels of Aβ42 were reduced at 2-3 months of age (∼55%; p < 0.002; n = 8), 6 months of age (90%; p < 0.0001; n = 7), and 17 months of age (∼99%; p < 0.0001; n = 3). These results indicate that inactivation of PS1 resulted in reductions in the levels of Aβ peptides and prevented age-dependent accumulation of Aβ peptides.
Figure 3.

Reduced accumulation of Aβ peptides in PS1 cKO;APP Tg mice. Levels of Aβ40 (A) and Aβ42 (B) in cortical lysates of APP Tg and PS1 cKO;APP Tg mice were quantified by ELISA. In APP Tg mice, the levels of Aβ40 and Aβ42 were increased from 2 to 17 months of age; levels of Aβ42 were higher than those of Aβ40 at 6 and 17 months of age. In the PS1 cKO;APP Tg mice, Aβ40 and Aβ42 were reduced significantly at all ages. Data represent the mean ± SEM; pmol/g signifies picomoles of Aβ per gram of cortex. *p < 0.01; **p < 0.001; ***p < 0.0001.
Histological analysis with an antibody (R1282) specific for Aβ confirmed the age-dependent deposition of amyloid plaques in the hippocampus and neocortex of APP transgenic mice starting at ∼5 months of age (Fig. 4A), as reported previously (Mucke et al., 2000). In the cortex of PS1 cKO;APP Tg mice, the R1282 immunoreactivity (Fig. 4A) and thioflavine S staining (data not shown), which recognizes fibrillar Aβ, were absent, indicating the lack of amyloid plaque formation in these mice. Similar results were obtained from an analysis of PS1 cKO;APP Tg mice at 12 months of age (data not shown). These results demonstrate that targeting PS1 function in the adult brain effectively prevents amyloid plaque formation in APP transgenic mice.
Figure 4.

Absence of amyloid plaques and inflammation in PS1 cKO;APP Tg mice. A, Sagittal brain sections from APP Tg and PS1 cKO;APP Tg mice were stained with an Aβ antibody (R1282) to reveal the presence of amyloid plaques in the hippocampus of APP Tg mice and their absence in PS1 cKO;APP Tg mice at 6 months of age. Higher-power views of the boxed areas in the top panels are shown at the bottom. CA1 and CA3, CA1 and CA3 fields of the hippocampus, respectively; DG, dentate gyrus. Scale bar, 10 μm. B, Brain sections of APP Tg and PS1 cKO;APP Tg mice at 6 months of age were stained with antibodies specific for CD45 and GFAP. Activated microglial cells labeled by CD45 antibody and reactive astrocytes, which express high levels of GFAP and extend elaborate processes, are found only in the hippocampus of APP Tg mice, but not in PS1 cKO;APP Tg mice. Scale bar, 10 μm. C, Confocal microscopy analysis of APP Tg and PS1 cKO;APP Tg brain sections double-labeled with antibodies for phosphorylated tau (green) and Aβ (red). Dystrophic axons immunoreactive for phosphorylated tau either extend into plaques (arrow) or remain in the surrounding area of the plaque (arrowheads) in the hippocampus of APP Tg mice, whereas the PS1 cKO;APP Tg brain lacks such processes. Scale bar, 10 μm.
In addition to amyloid deposition, inflammatory responses such as astrogliosis, microgliosis, and dystrophic neurites are prominent neuropathological features in APP transgenic mice (Masliah et al., 1996; Frautschy et al., 1998; Matsuoka et al., 2001). Reactive astrocytes and activated microglial cells labeled by GFAP and CD45 immunoreactivity, respectively, were present in the hippocampus of APP Tg mice but absent in that of PS1 cKO;APP Tg mice at 6 months of age (Fig. 4B). Similarly, dystrophic neurites labeled with antibodies against phosphorylated tau (AT-8) were present in the cortices of APP Tg mice but absent in PS1 cKO;APP Tg mice (Fig. 4C). Western blot analysis also indicated a modest but statistically insignificant decrease in the level of phosphorylated tau (Ser202) in cortical lysates of PS1 cKO;APP Tg mice compared with that in APP Tg mice at 6 months of age (APP Tg, 100 ± 28%; PS1 cKO;APP Tg, 70.3 ± 2.7%; p > 0.05; n = 3). This likely is attributable to the fact that dystrophic neurites immunoreactive for phosphorylated tau were restricted to areas in or surrounding compact amyloid plaques in APP Tg mice and that the number of compact amyloid plaques was low and varied among individual mice at this age. Overall, these results demonstrate that inactivation of PS1 in APP transgenic mice prevented amyloid-associated inflammatory responses and neuritic degeneration.
Age-dependent rescue of contextual memory deficits
To determine whether reduced levels of Aβ peptides and the absence of amyloid plaques in PS1 cKO;APP Tg mice were associated with the rescue of cognitive deficits, we examined littermate PS1 cKO;APP Tg, APP Tg, PS1 cKO, and control mice by using two behavioral tasks assessing their learning and memory. Although levels of Aβ peptides are similar in PS1 cKO;APP Tg mice between 2-3 and 6 months of age, levels of APP C-TFs accumulate with age. This accumulation may limit the extent of cognitive rescue. We therefore tested these mice at different ages (3 and 6 months).
We first tested all four genotypic groups in contextual fear conditioning, in which memory can be acquired in a single training session. In contextual fear conditioning, the mice learn to associate a conditioned stimulus (CS; test chamber) with a US (footshock) (Phillips and LeDoux, 1992). After the pairing of CS and US, a robust associative memory of the CS-US is formed such that the CS alone can elicit a fear response (e.g., freezing). At 3 months of age, PS1 cKO;APP Tg, APP Tg, PS1 cKO, and control mice displayed similar levels of freezing immediately after training (Fig. 5A). However, APP Tg mice showed significantly reduced levels of freezing to the context 24 h after training compared with the other three genotypic groups (F(3,31) = 3.39; p < 0.03). Post hoc analysis revealed a significant reduction in freezing responses in APP Tg mice (29.3 ± 7.4%; n = 7) compared with control (50.1 ± 3.0%; n = 10), PS1 cKO (49.0 ± 5.2%; n = 9), and PS1 cKO;APP Tg (48.1 ± 6.2%; n = 9) mice (p < 0.032). The fact that PS1 cKO;APP Tg mice performed as well as control mice (p = 0.42) suggests that inactivation of PS1 had prevented the impairment of contextual fear memory normally displayed by APP transgenic mice at this age.
Figure 5.

Age-dependent rescue of contextual memory in PS1 cKO;APP Tg mice. A, Control (n = 10), PS1 cKO (n = 9), APP Tg (n = 7), and PS1 cKO;APP Tg (n = 9) mice at 3 months of age were tested in the one-shock contextual fear conditioning task. All four groups showed similar levels of freezing immediately after the footshock. Control, PS1 cKO, and PS1 cKO;APP Tg mice showed similar levels of freezing (∼50%) at 24 h, whereas APP Tg mice exhibited significantly reduced levels of freezing (∼30%) (F(3,31) = 3.39; *p < 0.03). B, Control (n = 16), PS1 cKO (n = 6), APP Tg (n = 12), and PS1 cKO;APP Tg (n = 13) mice at 6 months of age were tested in the one-shock contextual fear conditioning task. All four groups showed similar levels of freezing immediately after the footshock. However, APP Tg and PS1 cKO;APP Tg mice showed reduced levels of freezing when compared with control and PS1 cKO mice at 24 h (F(3,43) = 9.0; **p < 0.0001). Data represent the mean ± SEM.
A different pattern was observed at 6 months of age. All four genotypic groups again showed similar levels of freezing responses immediately after the footshock (Fig. 5B). However, in the memory retention test conducted 24 h after the initial training, there was no longer any rescue associated with the conditional PS1 inactivation. The overall analysis showed a significant main effect of genotype in freezing responses 24 h after the initial training session (F(3,43) = 9.0; p < 0.0001). Post hoc analysis revealed that the freezing responses shown by APP Tg (28.1 ± 3.4%; n = 12) and PS1 cKO;APP Tg (22.8 ± 4.4%; n = 13) mice were reduced significantly compared with control (50.0 ± 5.04%; n = 16) and PS1 cKO (48.8 ± 5.1%; n = 6) mice (p < 0.02) (Fig. 5B). These results indicate that the ability of PS1 cKO;APP Tg mice to display a rescue of the memory deficit shown by APP transgenic mice deteriorated with age.
Age-dependent rescue of serial spatial learning
Previously, platelet-derived growth factor promoter APP (PDAPP) mice have been shown to exhibit both age-independent and age-related and amyloid plaque-related deficits in serial spatial learning in the water maze (G. Chen et al., 2000). The age-related component of these changes in learning ability occurred gradually over the range of 8-16 months in the PDAPP mice. To determine whether the APP transgenic mice used in our study exhibit similar age-related changes in learning and whether these could be rescued by PS1 inactivation, we used the identical training protocol. The only change was to start testing at a younger age (3 months) at which the APP transgenic mice used in this study previously exhibited an impairment in contextual fear conditioning. The full battery of tests (see Materials and Methods) consisted of cued navigation (5 d), followed by training-to-criterion in each of five different successively learned spatial locations and then continued training for 10 d to obtain a measure of learning capacity.
In the cue task, all genotypic groups at 3 and 15-17 months of age showed excellent learning to locate a visible platform, with rapidly declining swim latencies across days (Greenhouse-Geisser correction for multiple comparisons; F = 99.42; p < 0.001) (Fig. 6A,B). Thus there were no gross sensorimotor abnormalities that might have limited their capacity to learn the more complex hidden platform task that was trained next. In the serial spatial learning task, the escape latency is measured on each swim trial, but the primary measures for analysis are trials-to-criterion and learning capacity. Trials-to-criterion is the number of trials taken to learn each of the five successive spatial locations to the criterion of three trials averaging <20 s. Learning capacity is the total number of spatial locations learned in 10 d. Although distinct, these measures are related and thus not strictly independent.
Figure 6.

Age-dependent rescue of serial spatial learning in PS1 cKO;APP Tg mice. A, B, Control, PS1 cKO, APP Tg, and PS1 cKO;APP Tg mice at 3 months of age (A) and 15-17 months of age (B) were trained in the visible platform version of the water maze for 5 d consecutively (4 trials/d). APP Tg and PS1 cKO;APP Tg mice showed normal performance in the cue task. The number of mice used in each group is indicated in parentheses. C, Mice were trained in the serial spatial reversal task in a water maze for 10 d. Analysis of trials to reach criterion revealed no difference among control, PS1 cKO, and PS1 cKO;APP Tg groups (p > 0.10), but these three groups took fewer trials to learn the five platform locations than the APP Tg group (p < 0.001). These data indicate that PS1 cKO;APP Tg mice at 3 months of age exhibit a rescue of spatial working memory. D, Analysis of trials-to-criterion of mice at 15-17 months of age indicated that performances of the PS1 cKO, APP Tg, and PS1 cKO;APP Tg mice did not differ from each other (p > 0.10), and their performances were poorer than that shown by the control group (p < 0.05). E, Learning capacity was measured as the total number of spatial reversal locations learned in 10 d. Analysis of learning capacity showed similar learning capacity for the control, PS1 cKO, and PS1 cKO;APP Tg groups and reduced learning capacity in the APP Tg group at 3 months of age (p < 0.001). These data indicate a rescue of learning capacity in PS1 cKO;APP Tg mice at 3 months of age. F, The learning capacity of APP Tg, PS1 cKO;APP Tg, and PS1 cKO groups at 15-17 months of age did not differ significantly, but the average performance of these three groups was significantly lower than that of the control group (p < 0.01). Data represent the mean ± SEM.
Analyses of trials-to-criterion performed separately for young and old mice revealed overall group differences in the young mice, but only a trend in the old mice (for young, F = 5.24, df 3/43, p < 0.005; for old, F = 2.82, df 3/40, 0.10 > p > 0.05). The pattern in the young mice revealed no significant difference among PS1 cKO;APP Tg, PS1 cKO, and control groups (p > 0.10), with all three groups taking fewer trials to learn the five platform locations than the APP Tg group (F = 18.00; df 1/43; p < 0.001) (Fig. 6C). Thus, as in contextual fear conditioning, PS1 cKO;APP Tg mice at 3 months of age showed a rescue of the impaired phenotype displayed by APP Tg mice. However, as shown in Figure 6D, this rescue effect was weaker in the older mice. In these animals, the performance of the APP Tg, PS1 cKO; APP Tg, and PS1 cKO mice did not differ from each other (p > 0.10), but the average performance of these groups was poorer than that shown by the control group (F = 5.25; df 1/40; p < 0.05).
Next we analyzed the learning capacity of mice at 3 and 15-17 months of age (i.e., the number of serial spatial locations they could learn in 10 d). These analyses showed overall differences across groups in both the young and old mice (for young, F = 4.99, df 3/43, p < 0.005; for old, F = 4.19, df 3/40, p < 0.05). In the young mice, orthogonal comparisons showed a rescue effect in the PS1 cKO;APP Tg mice, which performed as well as control and PS1 cKO mice and better than the APP Tg group (p < 0.001) (Fig. 6E). In the older mice, the APP Tg, PS1 cKO;APP Tg, and PS1 cKO groups did not differ significantly, but the average performance of these three groups was significantly poorer than for controls (F = 9.23; df 1/40; p < 0.01) (Fig. 6F). We conclude that, in the serial spatial learning task, which displays an age-related decline in learning by control and APP Tg mice, inactivation of PS1 ameliorates the learning deficit in young APP Tg mice, but this apparent rescue of learning ability deteriorates with age.
Failure to rescue synaptic dysfunction
It has been reported that transgenic mice overexpressing human mutant APP exhibit impaired synaptic transmission and/or plasticity (Chapman et al., 1999; Fitzjohn et al., 2001). To determine whether inactivation of PS1 in PS1 cKO;APP Tg mice can rescue the hippocampal synaptic deficits associated with APP Tg mice, we examined synaptic transmission and plasticity in the Schaffer collateral pathway in control, APP Tg, and PS1 cKO;APP Tg mice at 6 months of age [PS1 cKO mice were reported previously by Yu et al. (2001)], because at this age both APP Tg and PS1 cKO;APP Tg mice exhibited contextual memory impairments (Fig. 5B). To measure basal synaptic transmission, we first determined input/output (I/O) curves in acute hippocampal slices by plotting the amplitude of the fiber volley (FV), which is a measure of the number of recruited axons, versus the initial slope of the evoked field EPSP (fEPSP). ANOVA of the average of the FV-fEPSP slope showed a significant main effect of genotype (F(2,84) = 6.19; p < 0.003). Post hoc analysis revealed reduced basal synaptic transmission in APP Tg and PS1 cKO;APP Tg mice compared with controls (p < 0.02) (Fig. 7A). Paired pulse facilitation (PPF), a presynaptic form of short-term plasticity that correlates inversely with the probability of transmitter release, was similar in all genotypic groups (F(2,12) = 1.99; p = 0.14) (Fig. 7B). These results are consistent with previous reports showing reduced synaptic transmission and unchanged PPF in similar APP Tg mice (Hsia et al., 1999; Jolas et al., 2002). Our previous study has shown that induction of LTP was normal in PS1 cKO mice (Yu et al., 2001); we therefore examined synaptic plasticity in hippocampal slices from APP Tg and PS1 cKO;APP Tg mice and littermate controls by inducing LTP with TBS. The initial induction of LTP was reduced in APP Tg (155.3 ± 11.1) and PS1 cKO;APP Tg (154.3 ± 8.7) mice compared with control mice (180.4 ± 8.1) (Fig. 7C). Repeated-measures ANOVA of the magnitude of LTP showed a significant main effect of genotype (F(2,212) = 4.65; p < 0.05), indicating that LTP was impaired in APP Tg and PS1 cKO;APP Tg mice.
Figure 7.
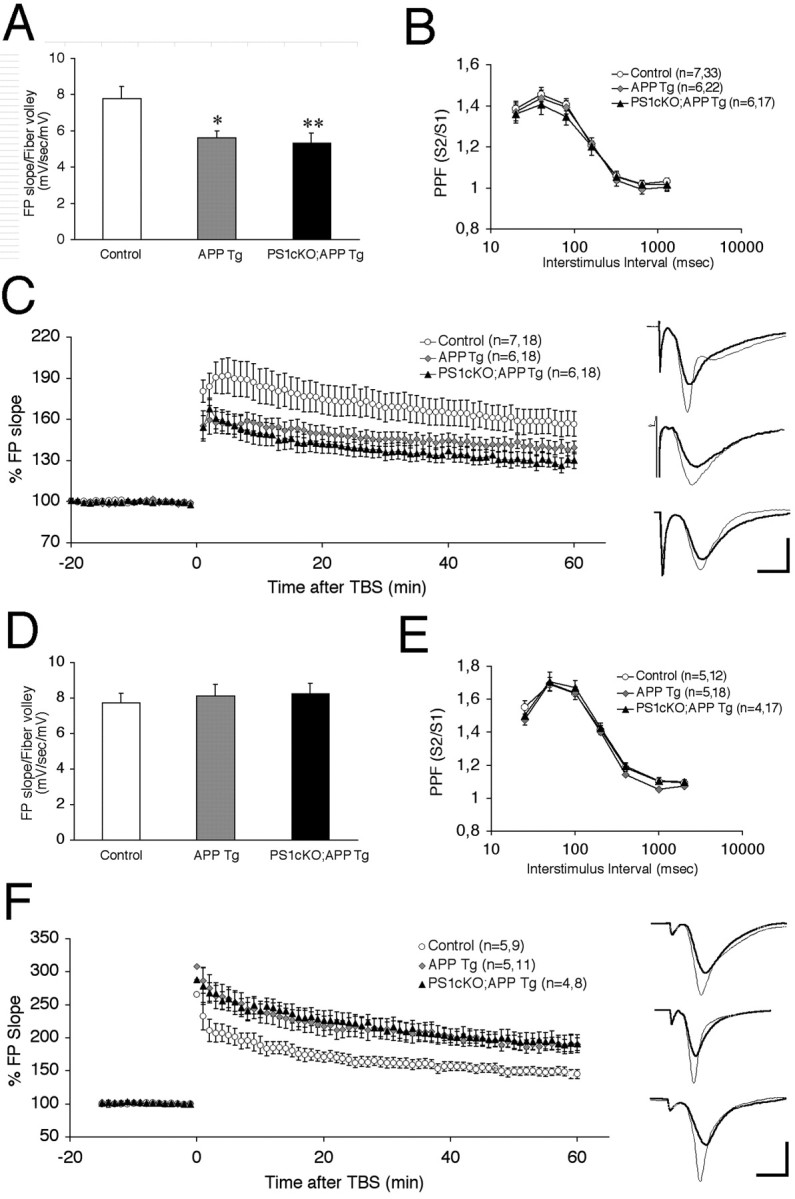
Similar hippocampal synaptic alterations in APP Tg and PS1 cKO;APP Tg mice. A, Basal synaptic transmission is reduced in APP Tg and PS1 cKO;APP Tg mice at 6 months of age. The I/O slope was obtained by plotting the FV amplitude against the initial slope of the evoked fEPSP in acute hippocampal slices of control, APP Tg, and PS1 cKO;APP Tg mice. The graph represents the average slope of the I/O curves. *p < 0.02; **p < 0.005. B, The graph depicts the paired pulse response ratio (second fEPSP/first fEPSP) obtained at different interstimulus intervals in 6-month-old mice. APP Tg and PS1 cKO;APP Tg mice show normal PPF (p = 0.14). C, Left, Time course of the effects of five episodes of TBS on the fEPSP initial slope in 6-month-old mice. Right, Examples of LTP induced in slices from control (top), APP Tg (middle), and PS1 cKO;APP Tg (bottom) mice. Super imposed traces are averages of four consecutive responses recorded 1 min before (thin line) and 60 min after (thick line) TBS. APP Tg and PS1 cKO;APP Tg mice show impaired LTP. D, The graph represents the average slope of the I/O curves in 3-month-old mice. The average of FV-fEPSP slopes in APP Tg and PS1 cKO;APP Tg mice is similar to that of control mice at 3 months of age (p > 0.05). E, The graph depicts the paired pulse response ratio obtained at different interstimulus intervals in 3-month-old mice. APP Tg, PS1 cKO;APP Tg, and control mice show similar PPF (p = 0.13). F, Left, Time course of the effects of five episodes of TBS on the fEPSP initial slope in 3-month-old mice. Right, Examples of LTP induced in slices from control (top), APP Tg (middle), and PS1 cKO;APP Tg (bottom) mice. Super imposed traces are averages of four consecutive responses recorded 1 min before (thin line) and 60 min after TBS (thick line). In B, C, E, F, the numbers of mice (left) and slices (right) are indicated in parentheses. Data represent the mean ± SEM. Calibration: (in C, F) 0.5 mV, 5 ms.
To determine whether basal synaptic transmission and LTP changes in APP Tg and PS1 cKO;APP Tg mice were age dependent, we also performed electrophysiological recordings in younger mice at 3 months of age. ANOVA of the FV-fEPSP slope indicated similar responses in younger control (7.74 ± 0.54; n = 5), APP Tg (8.13 ± 0.65; n = 5), and PS1 cKO;APP Tg (8.27 ± 0.58; n = 5) mice (F(2,64) = 0.21; p = 0.81), suggesting normal basal synaptic transmission in mutant mice at this age (Fig. 7D). Similar to the older age, APP Tg and PS1 cKO;APP Tg mice at 3 months of age showed PPF at each interstimulus interval that was similar to that seen for control mice (F(2,12) = 2.1; p = 0.13), indicating intact presynaptic short-term plasticity in these mutant mice (Fig. 7E). Induction of LTP in CA1 hippocampal synapses of younger APP Tg and PS1 cKO;APP Tg mice elicited larger responses than in control mice (Fig. 7F). Repeated-measures ANOVA of the magnitude of LTP in the last 10 min showed a significant main effect of genotype (F(2,253) = 47.9; p < 0.0001), indicating an enhancement of LTP induction in younger APP Tg and PS1 cKO;APP Tg mice at 3 months of age. This enhancement of LTP that occurs before amyloid deposition has been observed previously in the CRND8 transgenic mouse, which also overexpresses the mutant APPSwe/Ind and develops hippocampal-dependent cognitive deficits (Janus et al., 2000; Jolas et al., 2002). Although the exact mechanism or mechanisms underlying the age-dependent synaptic plasticity alterations in these mutants are unclear, varying levels of Aβ and APP C-TFs in APP Tg and PS1 cKO; APP Tg mice at these two ages may account for such a phenotype. Previous in vitro studies have shown both reduction and enhancement of LTP after acute perfusion of Aβ in hippocampal slices (Wu et al., 1995; Q. S. Chen et al., 2000). Secreted forms of APP also have been found to increase LTP (Ishida et al., 1997).
Discussion
Genetic and pharmacological approaches designed to prevent or slow cognitive decline in AD should be evaluated in animal models by a variety of appropriate behavioral tests of learning and memory. The possible causal role of Aβ in AD pathogenesis raises the possibility of therapeutic strategies based on processes that regulate the level of Aβ peptides. Our previous finding that inactivation of PS1 in the postnatal forebrain results in a significant reduction of Aβ peptides without major deleterious consequences in mice supports the view that selective inhibition of PS1 or γ-secretase activity may represent a suitable therapeutic strategy for AD (Yu et al., 2001). Our recent investigations have revealed, however, that disruption of PS1 and PS2 in the mouse postnatal forebrain causes synaptic and memory deficits and age-dependent neurodegeneration (Saura et al., 2004). Because the pharmacological targeting of presenilin function or γ-secretase activity is likely to lead to partial rather than complete inhibition of γ-secretase, a more relevant genetic mouse model of γ-secretase inhibition is a partial γ-secretase inactivation as investigated here, using postnatal forebrain-restricted PS1 cKO mice.
Our findings show that partial inactivation of γ-secretase by conditional inactivation of PS1 ameliorated amyloid-related phenotypes displayed by APP transgenic mice. Specifically, our biochemical and histological analyses indicated that selective inactivation of PS1 in the cerebral cortex reduced the accumulation of Aβ peptides and prevented amyloid plaque formation and inflammatory responses associated with APP transgenic mice (Figs. 3, 4). These results are consistent with a previous report showing reduced Aβ peptides and amyloid deposition in another line of APP transgenic mice with the use of a neuronal-specific PS1 cKO mouse (Dewachter et al., 2002). Furthermore, our behavioral analysis that used the contextual fear conditioning and serial spatial reversal learning revealed that inactivation of PS1 rescued the hippocampal-dependent learning and memory deficits of young APP transgenic mice (Figs. 5, 6). Our observation that Aβ-associated deficits normally observed in two separate hippocampal-dependent memory tasks were rescued by limiting the synthesis of human Aβ in APP transgenic mice via PS1 inactivation is consistent with a previous report stating that germline disruption of the BACE1 gene in another line of APP transgenic mice rescued memory deficits in social recognition and spatial alternation tasks (Ohno et al., 2004).
The second key finding of our study is that the improvement of the cognitive impairment associated with APP Tg mice via PS1 inactivation is age-related and transient. For example, the impairment of contextual memory exhibited by APP Tg mice no longer was rescued by PS1 inactivation in PS1 cKO;APP Tg mice at 6 months of age. Furthermore, alterations in basal synaptic transmission and/or LTP induction observed in APP Tg mice also failed to be rescued by PS1 inactivation in PS1 cKO; APP Tg mice (Fig. 7). Our previous study of PDAPP mice in the serial spatial reversal task showed an age-related decline in performance that could be observed over a period of almost 1 year (G. Chen et al., 2000). Thus older PS1 cKO;APP Tg mice at 15-17 months of age were tested in the serial spatial reversal task, and the amelioration of the learning deficits associated with the APP transgenic mice by PS1 inactivation was no longer statistically significant. This age-related rescue of hippocampal-dependent cognitive function in PS1 cKO;APP Tg mice may explain the apparent discrepancies between our findings and a previous report showing that inactivation of PS1 in neurons of APP[V7171I] transgenic mice rescued LTP impairments but failed to rescue the impairment in spontaneous object recognition at 3-6 months of age (Dewachter et al., 2002).
It may seem surprising that the learning and memory deficits exhibited by old APP transgenic mice are not ameliorated by the prevention of Aβ accumulation in PS1 cKO;APP Tg mice. However, any beneficial effect on learning that results may be more than offset by other biochemical changes taking place in these older mice. Several observations of this study point to the possibility that age-dependent accumulation of APP C-TFs, as a result of γ-secretase inhibition, may underlie the memory deterioration in older PS1 cKO; APP Tg mice. Confocal and electron microscopy analyses showed marked accumulation of APP C-TFs at synaptic terminals in PS1 cKO;APP Tg mice (Fig. 2). The accumulation of APP C-TFs could be caused by deficient trafficking and/or processing of APP and APP C-TFs. Consistent with this interpretation, PS1 has been implicated previously in the trafficking of APP (Naruse et al., 1998; Leem et al., 2002; Cai et al., 2003). PS1 may be regulating the processing of APP and cleavage of APP C-TFs in or near synaptic sites where these precursors containing the entire Aβ domain accumulate (Schubert et al., 1991; Buxbaum et al., 1998). Indeed, we found recently that Aβ42 peptides accumulate in multivesicular bodies at neuronal processes and synaptic compartments of APP transgenic mice and AD brains (Takahashi et al., 2002, 2004). It has been suggested that the Aβ released from presynaptic sites is deposited in extracellular plaques (Buxbaum et al., 1998; Lazarov et al., 2002). Finally, a deleterious effect of abnormally high levels of APP C-TFs on synaptic function is supported by previous findings showing LTP and memory impairments in mice injected with or expressing APP C-TFs (Cullen et al., 1997; Nalbantoglu et al., 1997; Choi et al., 2001).
An alternative explanation of the age-dependent deterioration of contextual fear conditioning and serial spatial reversal in PS1 cKO;APP Tg mice is that inactivation of PS1 itself causes cognitive impairments. However, we have demonstrated previously, by using an intensive training protocol, that PS1 cKO mice perform similarly to littermate controls in contextual fear conditioning and spatial reference memory (Saura et al., 2004), although they do exhibit a spatial reference memory deficit under a more difficult training protocol (Yu et al., 2001). Furthermore, PS1 cKO mice exhibit normal synaptic transmission and plasticity in the Schaffer collateral pathway (Yu et al., 2001). These observations suggest that PS1 inactivation alone is unlikely to account for the observed phenotype. Moreover, although it may be argued that residual levels of Aβ in the cortex of PS1 cKO;APP Tg mice contribute to the age-dependent cognitive deterioration, this is also unlikely, because similar residual levels of Aβ40 and Aβ42 were found in these mice at 2-3, 6, and 17 months of age. It is therefore reasonable to suggest that abnormal accumulation of APP C-TFs in neurons of the cerebral cortex, especially in the synaptic terminals, contributes to memory impairments in older PS1 cKO;APP Tg mice. However, we cannot yet rule out the possibility that long-term PS1/γ-secretase inactivation could contribute to the described phenotype by the deficient processing of other known or still unidentified synaptic substrates (Kim et al., 2002; May et al., 2002; Marambaud et al., 2003; Schulz et al., 2003).
Are presenilins potential drug targets for AD therapy? The results presented here showing prevention of Aβ accumulation and amyloid deposition in PS1 cKO;APP Tg mice provide support for the view that targeting PS1 function or γ-secretase activity could be an effective approach for anti-amyloidogenic treatment. In addition, our extensive behavioral analysis demonstrates that inactivation of PS1 can provide temporary benefits to improve cognitive function in APP transgenic mice. Nevertheless, presenilins have many important physiological functions ranging from synaptic plasticity to epidermal proliferation (Doerfler et al., 2001; Xia et al., 2001; Qyang et al., 2004; Saura et al., 2004; Tournoy et al., 2004), and complete loss of presenilin function in the cerebral cortex, which is the most relevant anatomical substrate for AD pathogenesis, leads to progressive memory loss and neurodegeneration (Saura et al., 2004). We recently proposed that loss of presenilin function together with increases in Aβ peptides collectively could lead to memory loss and neurodegeneration in FAD (Saura et al., 2004). The finding that PS1 mutations devoid of γ-secretase activity are associated with frontotemporal dementia (Raux et al., 2000; Amtul et al., 2002; Dermaut et al., 2004) also supports a possible link between loss of PS1 function and pathogenic mechanisms of dementia. The requirement of presenilins for synaptic plasticity, memory, and neuronal survival is mediated partly by the regulation of the cAMP response element-binding protein pathway, likely by γ-secretase-dependent Notch cleavage and signaling (Saura et al., 2004). Therefore, it is unlikely that γ-secretase inhibitors that affect Notch signaling will be useful as potential drugs for AD treatment. Alternatively, the development of specific γ-secretase inhibitors that inhibit Aβ production, and specifically Aβ42, without disturbing other presenilin functions and specifically those involved in synaptic function, still may provide a rational therapeutic approach for the treatment of AD. In conclusion, we have demonstrated that conditional inactivation of PS1 in the postnatal forebrain efficiently prevents amyloid-associated neuropathological changes in APP transgenic mice. We provide evidence for the first time that reduction of Aβ as a result of γ-secretase inhibition in vivo may provide transient cognitive improvements in an amyloid mouse model. These findings raise the possibility that γ-secretase inhibitors that specifically inhibit Aβ generation without altering other essential presenilin functions may provide temporary therapeutic benefits.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant R01NS041783 (J.S.), the Alzheimer's Association (C.A.S., J.S.), the Medical Research Council, and the Alzheimer's Research Trust (R.G.M.M). We thank L. Mucke for the APP transgenic mice, D. Selkoe for the C7 and Aβ antibodies, M. Shoji for the Saeko antiserum, and W. Xia and J. Zheng for ELISA. We are grateful to V. Beglopoulos, W. Cheng, M. Goldberg, and C. Lemere for assistance.
Correspondence should be addressed to Jie Shen, Harvard Medical School, New Research Building 636E, 77 Avenue Louis Pasteur, Boston, MA 02115. E-mail: jshen@rics.bwh.harvard.edu.
C. A. Saura's present address: Institut de Neurociències, Universitat Autònoma de Barcelona, Barcelona 08193, Spain.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256755-10$15.00/0
References
- Amtul Z, Lewis PA, Piper S, Crook R, Baker M, Findlay K, Singleton A, Hogg M, Younkin L, Younkin SG, Hardy J, Hutton M, Boeve BF, Tang-Wai D, Golde TE (2002) A presenilin 1 mutation associated with familial frontotemporal dementia inhibits γ-secretase cleavage of APP and Notch. Neurobiol Dis 9: 269-273. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada C-M, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS (1996) Familial Alzheimer's disease-linked presenilin 1 variants elevate Aβ1-42/1-40 ratio in vitro and in vivo Neuron 17: 1005-1013. [DOI] [PubMed] [Google Scholar]
- Buxbaum J, Thinakaran G, Koliatsos V, O'Callahan J, Slunt H, Price D, Sisodia S (1998) Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci 18: 9629-9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Leem JY, Greenfield JP, Wang P, Kim BS, Wang R, Lopes KO, Kim SH, Zheng H, Greengard P, Sisodia SS, Thinakaran G, Xu H (2003) Presenilin-1 regulates intracellular trafficking and cell surface delivery of β-amyloid precursor protein. J Biol Chem 278: 3446-3454. [DOI] [PubMed] [Google Scholar]
- Chapman P, White G, Jones M, Cooper-Blacketer D, Marshall V, Irizarry M, Younkin L, Good M, Bliss T, Hyman B, Younkin S, Hsiao K (1999) Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 2: 271-276. [DOI] [PubMed] [Google Scholar]
- Chen G, Chen K, Knox J, Inglis J, Bernard A, Martin S, Justice A, McConlogue L, Games D, Freedman S, Morris RGM (2000) A learning deficit related to age and β-amyloid plaques in a mouse model of Alzheimer's disease. Nature 408: 975-979. [DOI] [PubMed] [Google Scholar]
- Chen QS, Kagan BL, Hirakura Y, Xie CW (2000) Impairment of hippocampal long-term potentiation by Alzheimer amyloid beta-peptides. J Neurosci Res 60: 65-72. [DOI] [PubMed] [Google Scholar]
- Choi S, Park C, Koo J, Seo J, Kim H, Jeong S, Lee J, Kim S, Suh Y (2001) Memory impairment and cholinergic dysfunction by centrally administered Aβ and carboxyl-terminal fragment of Alzheimer's APP in mice. FASEB J 15: 1816-1818. [DOI] [PubMed] [Google Scholar]
- Corcoran KA, Lu Y, Turner RS, Maren S (2002) Overexpression of hAPPswe impairs rewarded alternation and contextual fear conditioning in a transgenic mouse model of Alzheimer's disease. Learn Mem 9: 243-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen WK, Suh YH, Anwyl R, Rowan MJ (1997) Block of LTP in rat hippocampus in vivo by β-amyloid precursor protein fragments. NeuroReport 8: 3213-3217. [DOI] [PubMed] [Google Scholar]
- Dermaut B, Kumar-Singh S, Engelborghs S, Theuns J, Rademakers R, Saerens J, Pickut BA, Peeters K, Van Den Broeck M, Vennekens K, Claes S, Cruts M, Cras P, Martin JJ, Van Broeckhoven C, De Deyn PP (2004) A novel presenilin 1 mutation associated with Pick's disease but not β-amyloid plaques. Ann Neurol 55: 617-626. [DOI] [PubMed] [Google Scholar]
- Dewachter I, Reverse D, Caluwaerts N, Ris L, Kuiperi C, Van den Haute C, Spittaels K, Umans L, Serneels L, Thiry E, Moechars D, Mercken M, Godaux E, Van Leuven F (2002) Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice. J Neurosci 22: 3445-3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerfler P, Shearman MS, Perlmutter RM (2001) Presenilin-dependent γ-secretase activity modulates thymocyte development. Proc Natl Acad Sci USA 98: 9312-9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzjohn SM, Morton RA, Kuenzi F, Rosahl TW, Shearman M, Lewis H, Smith D, Reynolds DS, Davies CH, Collingridge GL, Seabrook GR (2001) Age-related impairment of synaptic transmission but normal long-term potentiation in transgenic mice that overexpress the human APP695SWE mutant form of amyloid precursor protein. J Neurosci 21: 4691-4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM (1998) Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 152: 307-317. [PMC free article] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, et al. (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 373: 523-527. [DOI] [PubMed] [Google Scholar]
- Handler M, Yang X, Shen J (2000) Presenilin-1 regulates neuronal differentiation during neurogenesis. Development 127: 2593-2606. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe D (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353-356. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L (1999) Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA 96: 3228-3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Ekman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274: 99-102. [DOI] [PubMed] [Google Scholar]
- Hutton M, Hardy J (1997) The presenilins and Alzheimer's disease. Hum Mol Genet 6: 1639-1646. [DOI] [PubMed] [Google Scholar]
- Ishida A, Furukawa K, Keller JN, Mattson MP (1997) Secreted form of β-amyloid precursor protein shifts the frequency dependency for induction of LTD and enhances LTP in hippocampal slices. NeuroReport 8: 2133-2137. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HTJ, Nixon RA, Mercken M, Bergeron C, Fraser PE, St. George-Hyslop P, Westaway D (2000) Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature 408: 979-982. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L (1997) Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA 94: 1550-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolas T, Zhang XS, Zhang Q, Wong G, Del Vecchio R, Gold L, Priestley T (2002) Long-term potentiation is increased in the CA1 area of the hippocampus of APPswe/ind CRND8 mice. Neurobiol Dis 11: 394-409. [DOI] [PubMed] [Google Scholar]
- Kim DY, Ingano LA, Kovacs DM (2002) Nectin-1α, an immunoglobulin-like receptor involved in the formation of synapses, is a substrate for presenilin/γ-secretase-like cleavage. J Biol Chem 277: 49976-49981. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Ort M, Cimadevilla JM, Vondrous R, Cordell B, Koistinaho J, Bures J, Higgins LS (2001) Specific spatial learning deficits become severe with age in β-amyloid precursor protein transgenic mice that harbor diffuse β-amyloid deposits but do not form plaques. Proc Natl Acad Sci USA 98: 14675-14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Lynch G, Games D, Seubert P (1999) Alterations in synaptic transmission and long-term potentiation in hippocampal slices from young and aged PDAPP mice. Brain Res 840: 23-35. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Lee M, Peterson DA, Sisodia SS (2002) Evidence that synaptically released β-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci 22: 9785-9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leem JY, Saura CA, Pietrzik C, Christianson J, Wanamaker C, King LT, Veselits ML, Tomita T, Gasparini L, Iwatsubo T, Xu H, Green WN, Koo EH, Thinakaran G (2002) A role for presenilin 1 in regulating the delivery of amyloid precursor protein to the cell surface. Neurobiol Dis 11: 64-82. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK (2003) A CBP binding transcriptional repressor produced by the PS1/ϵ-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell 114: 635-645. [DOI] [PubMed] [Google Scholar]
- Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D (1996) Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F β-amyloid precursor protein and Alzheimer's disease. J Neurosci 16: 5795-5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y, Picciano M, Malester B, LaFrancois J, Zehr C, Daeschner JM, Olschowka JA, Fonseca MI, O'Banion MK, Tenner AJ, Lemere CA, Duff K (2001) Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer's disease. Am J Pathol 158: 1345-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May P, Reddy YK, Herz J (2002) Proteolytic processing of low density lipoprotein receptor-related protein mediates regulated release of its intracellular domain. J Biol Chem 277: 18736-18743. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW (2000) Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 408: 982-985. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu G-Q, Mallory M, Rockenstein E, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L (2000) High-level neuronal expression of Aβ1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 20: 4050-4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbantoglu J, Tirado-Santiago G, Lahsaini A, Poirier J, Goncalves O, Verge G, Momoli F, Welner SA, Massicotte G, Julien JP, Shapiro ML (1997) Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature 387: 500-505. [DOI] [PubMed] [Google Scholar]
- Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, Wong PC, Sisodia SS (1998) Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron 21: 1213-1221. [DOI] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF (2004) BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron 41: 27-33. [DOI] [PubMed] [Google Scholar]
- Pan Y, Lin MH, Tian X, Cheng HT, Gridley T, Shen J, Kopan R (2004) γ-Secretase functions through Notch signaling to maintain skin appendages but is not required for their patterning or initial morphogenesis. Dev Cell 7: 731-743. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE (1992) Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci 106: 274-285. [DOI] [PubMed] [Google Scholar]
- Qyang Y, Chambers SM, Wang P, Xia X, Chen X, Goodell MA, Zheng H (2004) Myeloproliferative disease in mice with reduced presenilin gene dosage: effect of γ-secretase blockage. Biochemistry 43: 5352-5359. [DOI] [PubMed] [Google Scholar]
- Raux G, Gantier R, Thomas-Anterion C, Boulliat J, Verpillat P, Hannequin D, Brice A, Frebourg T, Campion D (2000) Dementia with prominent frontotemporal features associated with L113P presenilin 1 mutation. Neurology 55: 1577-1578. [DOI] [PubMed] [Google Scholar]
- Saura CA, Choi S-Y, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher III RJ, Kandel ER, Duff K, Kirkwood A, Shen J (2004) Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42: 23-36. [DOI] [PubMed] [Google Scholar]
- Schubert W, Prior R, Weidemann A, Dircksen H, Multhaup G, Masters CL, Beyreuther K (1991) Localization of Alzheimer beta A4 amyloid precursor protein at central and peripheral synaptic sites. Brain Res 563: 184-194. [DOI] [PubMed] [Google Scholar]
- Schulz JG, Annaert W, Vandekerckhove J, Zimmermann P, De Strooper B, David G (2003) Syndecan 3 intramembrane proteolysis is presenilin/γ-secretase-dependent and modulates cytosolic signaling. J Biol Chem 278: 48651-48657. [DOI] [PubMed] [Google Scholar]
- Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S (1997) Skeletal and CNS defects in presenilin-1 deficient mice. Cell 89: 629-639. [DOI] [PubMed] [Google Scholar]
- Stoltzner SE, Grenfell TJ, Mori C, Wisniewski KE, Wisniewski TM, Selkoe DJ, Lemere CA (2000) Temporal accrual of complement proteins in amyloid plaques in Down's syndrome with Alzheimer's disease. Am J Pathol 156: 489-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold K-H, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 94: 13287-13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Milner T, Li F, Nam E, Edgar M, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK (2002) Intraneuronal Alzheimer Aβ42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol 161: 1869-1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK (2004) Oligomerization of Alzheimer's β-amyloid within processes and synapses of cultured neurons and brain. J Neurosci 24: 3592-3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournoy J, Bossuyt X, Snellinx A, Regent M, Garmyn M, Serneels L, Saftig P, Craessaerts K, De Strooper B, Hartmann D (2004) Partial loss of presenilins causes seborrheic keratosis and autoimmune disease in mice. Hum Mol Genet 13: 1321-1331. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, et al. (1999) β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286: 735-741. [DOI] [PubMed] [Google Scholar]
- Wines-Samuelson M, Handler M, Shen J (2005) Role of presenilin-1 in cortical lamination and survival of Cajal-Retzius neurons. Dev Biol 277: 332-346. [DOI] [PubMed] [Google Scholar]
- Wong P, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LHT, Sisodia SS (1997) Presenilin 1 is required for Notch 1 and D111 expression in the paraxial mesoderm. Nature 397: 288-292. [DOI] [PubMed] [Google Scholar]
- Wu J, Anwyl R, Rowan MJ (1995) Beta-amyloid1-40 increases long-term potentiation in rat hippocampus in vitro Eur J Pharmacol 284: R1-R3. [DOI] [PubMed] [Google Scholar]
- Xia X, Qian S, Soriano S, Wu Y, Fletcher A, Wang X, Koo E, Wu X, Zheng H (2001) Loss of presenilin 1 is associated with enhanced β-catenin signaling and skin tumorigenesis. Proc Natl Acad Sci USA 98: 10863-10868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Saura CA, Choi S-Y, Sun LD, Yang X, Handler M, Kawarabayashi T, Younkin L, Fedeles B, Wilson MA, Younkin S, Kandel ER, Kirkwood A, Shen J (2001) APP processing and synaptic plasticity in presenilin-1 conditional knockout mice. Neuron 31: 713-726. [DOI] [PubMed] [Google Scholar]