

Abstract
Analyses of the effect of ryanodine in vertebrate brain slices have led to the conclusion that presynaptic ryanodine receptors (RYRs) may have several functions in synaptic release, including causing large-amplitude miniature postsynaptic currents (mPSCs) by promoting concerted multivesicular release. However, the role of RYRs in synaptic release is controversial. To better understand the role of RYRs in synaptic release, we analyzed the effect of RYR mutation on mPSCs and evoked postsynaptic currents (ePSCs) at the Caenorhabditis elegans neuromuscular junction (NMJ). Amplitudes of mPSCs varied greatly at the C. elegans NMJ. Loss-of-function mutations of the RYR gene unc-68 (uncoordinated 68) essentially abolished large-amplitude mPSCs. The amplitude of ePSCs was also greatly suppressed. These defects were completely rescued by expressing wild-type UNC-68 specifically in neurons but not in muscle cells, suggesting that RYRs acted presynaptically. A combination of removing extracellular Ca2+ and UNC-68 function eliminated mPSCs, suggesting that influx and RYR-mediated release are likely the exclusive sources of Ca2+ for synaptic release. Large-amplitude mPSCs did not appear to be caused by multivesicular release, as has been suggested to occur at vertebrate central synapses, because the rise time of mPSCs was constant regardless of the amplitude but distinctive from that of ePSCs, and because large-amplitude mPSCs persisted under conditions that inhibit synchronized synaptic release, including elimination of extracellular Ca2+, and mutations of syntaxin and SNAP25 (soluble N-ethylmaleimide-sensitive factor attachment protein 25). These observations suggest that RYRs are essential to normal quantal size and are potential regulators of quantal size.
Keywords: ryanodine receptor, quantal size, C. elegans, neuromuscular junction, calcium, presynaptic
Introduction
It is generally thought that synaptic exocytosis may occur in a Ca2+-independent manner (Knight et al., 1989; Gerber and Sudhof, 2002), and extracellular Ca2+ is the only ionic requirement for depolarization to evoke transmitter release (Katz and Miledi, 1969a,b, 1970). However, emerging evidence suggests that ryanodine receptor (RYR)-mediated Ca2+ release from the endoplasmic reticulum (ER) may also play a role in synaptic exocytosis. Blockade of RYRs with ryanodine in vertebrate brain slices or other preparations inhibits the frequency and amplitude of miniature postsynaptic currents (mPSCs) (Llano et al., 2000; Sharma and Vijayaraghavan, 2003), the amplitude of evoked postsynaptic currents (ePSCs) (Narita et al., 2000; Galante and Marty, 2003), paired pulse facilitation (Emptage et al., 2001), and presynaptic Ca2+ transients (Llano et al., 2000; Narita et al., 2000; Emptage et al., 2001; De Crescenzo et al., 2004).
One major conclusion of the previous studies was that presynaptic RYRs mediate large-amplitude mPSCs by promoting concerted multivesicular release (Llano et al., 2000; Sharma and Vijayaraghavan, 2003). However, the evidence for multivesicular release is not compelling. In one study, the primary evidence for the multivesicular release was a positive correlation between the rise time and amplitude of mPSCs (Sharma and Vijayaraghavan, 2003). This relationship, however, was completely reversed in a different study despite a similar conclusion (Llano et al., 2000). The major evidence of the latter study was a decrease in the mean amplitude of mPSCs after long-term removal of extracellular Ca2+. However, long-term removal of extracellular Ca2+ might reduce the amplitude of mPSCs via other potential mechanisms such as depletion of intracellular Ca2+ stores or interference with endocytosis (Heuser and Reese, 1973; Ceccarelli and Hurlbut, 1980; Thomas et al., 1990; Smith and Neher, 1997; Gad et al., 1998; Neale et al., 1999; Kuromi et al., 2004). Most importantly, several other studies have been unable to show an effect of RYRs on synaptic release (Bardo et al., 2002; Carter et al., 2002; Lim et al., 2003). Thus, further studies are needed to completely understand the role of presynaptic RYRs.
All previous studies have relied on ryanodine as a pharmacological tool. However, ryanodine may either activate or block RYRs depending on its concentration (Xu et al., 1998; Llano et al., 2000; Narita et al., 2000). The lack of a clear boundary between concentrations required for the two different actions may have contributed to the controversy. For example, ryanodine in the concentration range of 10-20 μm has been used either to activate (Llano et al., 2000; Sharma and Vijayaraghavan, 2003) or to block (Narita et al., 2000; Emptage et al., 2001; Bardo et al., 2002; Carter et al., 2002; Simkus and Stricker, 2002) RYRs. A more straight-forward approach would be to analyze animals in which RYRs have been genetically removed.
There are three forms of RYRs encoded by three different genes in human and mouse: RYR1, RYR2, and RYR3 (Verkhratsky and Shmigol, 1996). All three forms of RYR are expressed in the nervous system (Furuichi et al., 1994; Giannini et al., 1995), which complicates the analysis. To overcome these limitations, we used Caenorhabditis elegans as a model system to analyze the function of the RYR in spontaneous and evoked neurotransmitter release. C. elegans has a single RYR gene, which is known as unc-68 (uncoordinated 68) or ryr-1 (Maryon et al., 1996). Null mutants of unc-68 have severe locomotion defects but are viable and fertile (Maryon et al., 1996; Sakube et al., 1997). We show here that RYRs play critical roles in regulating both spontaneous and evoked neurotransmitter release at the neuromuscular junction (NMJ). RYR-mediated large-amplitude mPSCs at the C. elegans NMJ did not appear to be caused by synchronized multivesicular release, suggesting that RYRs are required for normal quantal size and are potential regulators of quantal size.
Materials and Methods
Electrophysiology. C. elegans were grown at room temperature (22-24°C) on agar plates with a layer of OP50 Escherichia coli. (Sulston and Hodgkin, 1988). Adult animals were used for physiological analysis. Postsynaptic currents at the NMJ were recorded as previously described (Wang et al., 2001) using a technique originally developed by Richmond et al. (1999). Briefly, an animal was immobilized on a glass coverslip by applying a cyanoacrylate adhesive along the dorsal side. A longitudinal incision was made in the dorsolateral region. After clearing the viscera, the cuticle flap was folded back and glued to the coverslip, exposing the ventral nerve cord and the two adjacent muscle quadrants. A Nikon (Melville, NY) Eclipse E600FN microscope equipped with a 40× water immersion lens and 15× eyepieces was used for viewing the preparation. Borosilicate glass pipettes with a tip resistance of 3∼5 MΩ were used as electrodes for voltage clamping. The classical whole-cell configuration was obtained by rupturing the patch membrane of a gigaohm seal formed between the recording electrode and a body wall muscle cell. The cell was voltage-clamped at -60 mV to record mPSCs and ePSCs. ePSCs were evoked by applying a 0.5 ms square wave current pulse at a supramaximal voltage through a stimulation electrode placed in close apposition to the ventral nerve cord. Postsynaptic currents were amplified with a Multi-clamp 700A amplifier (Axon Instruments, Foster City, CA), and acquired with Clampex software (Axon Instruments). Data were sampled at a rate of 10 kHz after filtering at 2 kHz.
The recording pipette solution contained the following (in mm): 120 KCl, 20 KOH, 5 Tris, 0.25 CaCl2, 4 MgCl2, 36 sucrose, 5 EGTA, and 4 Na2ATP, pH adjusted to 7.2 with HCl. The standard external solution included the following (in mm): 140 NaCl, 5 KCl, 5 CaCl2, 5 MgCl2, 11 dextrose, and 5 HEPES, pH adjusted to 7.2 with NaOH. In experiments examining the effect of eliminating extracellular Ca2+, the standard extracellular solution was modified by omitting CaCl2 and adding 5 mm EGTA and 5 mm NaCl.
Rescue experiments. The rab-3 (Nonet et al., 1997) and myo-3 (Okkema et al., 1993) promoters were used independently for neuron- and muscle-specific expression of a myc-tagged wild-type UNC-68. Because the unc-68 gene was rather large (∼27 kb from the first to the last exon), a plasmid containing either the rab-3 or myo-3 promoter fused to the initial 1059 bp sequence of unc-68 was coinjected with cosmid M04C11. Cosmid M04C11 contains all the exons, introns, and 3′ untranslated region (UTR) of the unc-68 gene. However, it lacks essential 5′ upstream sequences for muscle cell expression and is unable to rescue unc-68 mutant phenotypes when injected alone (Maryon et al., 1996; Sakube et al., 1997). Homologous recombination was expected to occur in vivo between the plasmid and the cosmid, resulting in neuron- or muscle-specific expression of the transgene (Mello and Fire, 1995; Maryon et al., 1996; Yuan et al., 2000). The 1059 bp fragment of unc-68 was amplified from M04C11 by PCR using the following primer pair: ATA CCG GTC GAA CAA AAA TTA ATT TCT GAA GAA GAT TTA ATG GCC GAC AAG GAG GAG CA (sense) and CGA GAC CGC GGC TGA CCA TCT TCT ACG CCA TAC A (antisense). The sense primer included an AgeI site near the 5′ end and a myc coding sequence fused in-frame to the unc-68 initiation site. The antisense primer had an SacII site near its 5′ end. The PCR fragment was subcloned into two plasmids that contained either the rab-3 promoter plus rim 3′-UTR or myo-3 promoter plus the unc-54 3′ UTR to form two new plasmids: wp137 for neuron-specific expression and wp235 for muscle-specific expression. Before injection, the cosmid and the plasmid were linearized with AgeI and SacII, respectively. AgeI does not cut the unc-68 gene. SacII cuts wp137 and wp235 only at the 3′ end of the 1059 bp insert. M04C11 (13.2 ng/μl), wp137 or wp235 (6.6 ng/μl), and pPD118.20 (10 ng/μl) were coinjected into unc-68(r1162) mutant animals for germ line transformation. pPD118.20 drives the expression of green fluorescent protein in body wall muscle cells and thus served as a marker of transformation. Successful recombinations between the plasmids and the cosmid were confirmed by PCR using a sense primer that is complementary to either the rab-3 or myo-3 promoter and an antisense primer that is complementary to the unc-68 gene downstream of the 1059 bp PCR fragment. The transgenes were integrated by γ irradiation. Animals carrying the integrated transgenes were analyzed.
Immunohistochemistry. Animals were fixed in Bouin's fixative and permeabilized as previously described (Nonet et al., 1997). Rabbit anti-UNC-10 Rim (Rab3a-interacting molecule) sera (Koushika et al., 2001) and mouse anti-Myc 9B11 monoclonal antibody (Cell Signaling Technology, Beverly, MA) were incubated overnight at 4°C at 1:12,500 and 1:5000, respectively. Primary antibodies were detected with Alexa Fluor 488- and 594-conjugated secondary antibodies (Molecular Probes, Eugene, OR). Animals were mounted and photographed using a Regita Exi charge-coupled device camera mounted on an Olympus Optical (Tokyo, Japan) B-60 microscope or an F-view II digital camera (Soft Imaging System GmbH) mounted on a Nikon TE2000-U microscope. Overlays were assembled using Photoshop (Adobe Systems Inc., San Jose, CA).
Measurement of locomotion. Locomotion assays were performed at room temperature on healthy adult animals. A piece of paper with a circular hole (7 mm in diameter) was laid on top of the bacterial lawn in the culture plate to restrict animal movements within the field of observation. A single animal was placed in the center of the field to start the assay. The plate was left undisturbed for 10 min while images of the plate were taken at 1 s intervals with an F-view II digital camera (Soft Imaging System GmbH) mounted on a Nikon SMZ800 stereomicroscope. The camera was operated through analySIS Imager software (Soft Imaging System GmbH). The distance traveled by the animal from the beginning of the third minute to the end of the seventh minute was determined by measuring the length of locomotion tracks left behind by the animal using the “Polygon Length” measurement function of analySIS. Locomotion speed was calculated by dividing the distance with the time.
Electron microscopy. Adult animals were fixed and embedded as described previously (Maryon et al., 1998). Serial sections of ∼60 nm thickness were cut with a Reichert Ultracut E ultramicrotome, collected on Formvar-coated slot grids, and examined at 60 kV in a Philips (Eindhoven, The Netherlands) CM10 transmission electron microscope. Images were recorded on Eastman Kodak Co. (Rochester, NY) 4489 film at an initial magnification of 21,000×; the developed negatives were printed on Kodak photographic paper to a final magnification of 80,850×. NMJs near the dorsal and ventral nerve cords were analyzed. For each synapse, all the sections containing the presynaptic density plus two neighboring sections that sandwich them were included for analysis. The presynaptic density and synaptic vesicles were traced with an ultra-fine marker onto a piece of transparency paper that was overlaid on the electron micrograph. The sections were traced in random sequence by an individual without the knowledge of the genotypes. The tracings were scanned and saved as tagged image file format files, which were subsequently analyzed with the Optimas program version 6.5 (Media Cybernetics) to determine the area of synaptic vesicles (Munirathinam et al., 2004). The diameter of a synaptic vesicle was calculated from its area assuming that the vesicle was a perfect sphere. Vesicles were considered docked if they were located within a single-vesicle radius (∼30 nm) of the presynaptic plasma membrane and within a distance of ∼60 nm on either side of the presynaptic density. Docked vesicles on the two sections without the presynaptic density were estimated by assuming that the presynaptic density was the same as in their neighboring sections. The criteria for docked synaptic vesicles were based on the methods used by two previous publications (Richmond et al., 1999; Koushika et al., 2001).
Data analysis. Amplitude and frequency of mPSCs were analyzed using MiniAnalysis (Synaptosoft, Decatur, GA). A detection threshold of 10 pA was used in initial automatic analysis, followed by visual inspections to include missed smaller events (≥5 pA) and to exclude false events resulting from baseline fluctuations. Ten to 90% rise time and rise slope of mPSCs and ePSCs were determined with MiniAnalysis. Amplitudes of ePSCs were measured with Clampfit (Axon Instruments). The averaged amplitude of the two largest peaks of ePSCs from each experiment was used for statistical analysis. The ePSC decay time constants were determined from a 200 ms trace starting from the ePSC peak by fitting to one or two standard exponentials using Clampfit.
Data were imported into Origin version 7.5 (OriginLab Corp., Northampton, MA) for graphing and statistical analysis. Either the t test (paired or unpaired) or one-way ANOVA was used, depending on whether data were from independent treatment groups and whether more than two groups were compared; p < 0.05 was considered statistically significant. All values are expressed as mean ± SE; n is the number of cells that were voltage-clamped.
Preparation and application of chemicals. Stock solutions of ryanodine (Molecular Probes), dihydro-β-erythroidine (DHβE; Sigma, St. Louis, MO), and d-tubocurarine (d-TBC; Sigma) were made by dissolving them either in DMSO (ryanodine and DHβE) or water (d-TBC) to 100× of their final concentrations, which was pipetted into the recording chamber (volume, 1 ml), followed by mixing with several pipetting actions. Acetylcholine, dissolved in extracellular solution to its final concentration, was applied by puffing directly onto the body wall muscle cell through a glass pipette. The puffing pressure pulse (0.2 psi, 0.1 s) was generated with a FemtoJet injector (Eppendorf).
Results
To determine the role of RYRs in neurotransmitter release, we analyzed mPSCs and ePSCs at the NMJ of wild-type C. elegans and unc-68 mutants. The unc-68 alleles used in the study were r1162 and r1161, both of which completely disrupt RYR function (Maryon et al., 1996). The coding regions of all the putative membrane-spanning domains are deleted in r1162. A substantial portion (7.2 kb) of the unc-68 gene involving many exons is deleted in r1161 (Sakube et al., 1993; Maryon et al., 1996). mPSCs and ePSCs were recorded from body wall muscle cells of C. elegans under the voltage-clamp condition (-60 mV).
Mutations of RYRs inhibited mPSCs and ePSCs
In wild-type preparations, the frequency of mPSCs was 53.4 ± 4.8 Hz (n = 25). A striking feature of mPSCs at the C. elegans NMJ was that their amplitudes varied greatly (Fig. 1A, a). Although mPSCs averaged 28.6 ± 1.5 pA in amplitude, a significant number (14.1%) were >50 pA, with some >100-200 pA. The presence of large-amplitude events resulted in a positive skew in the amplitude distribution (Fig. 1A, d).
Figure 1.
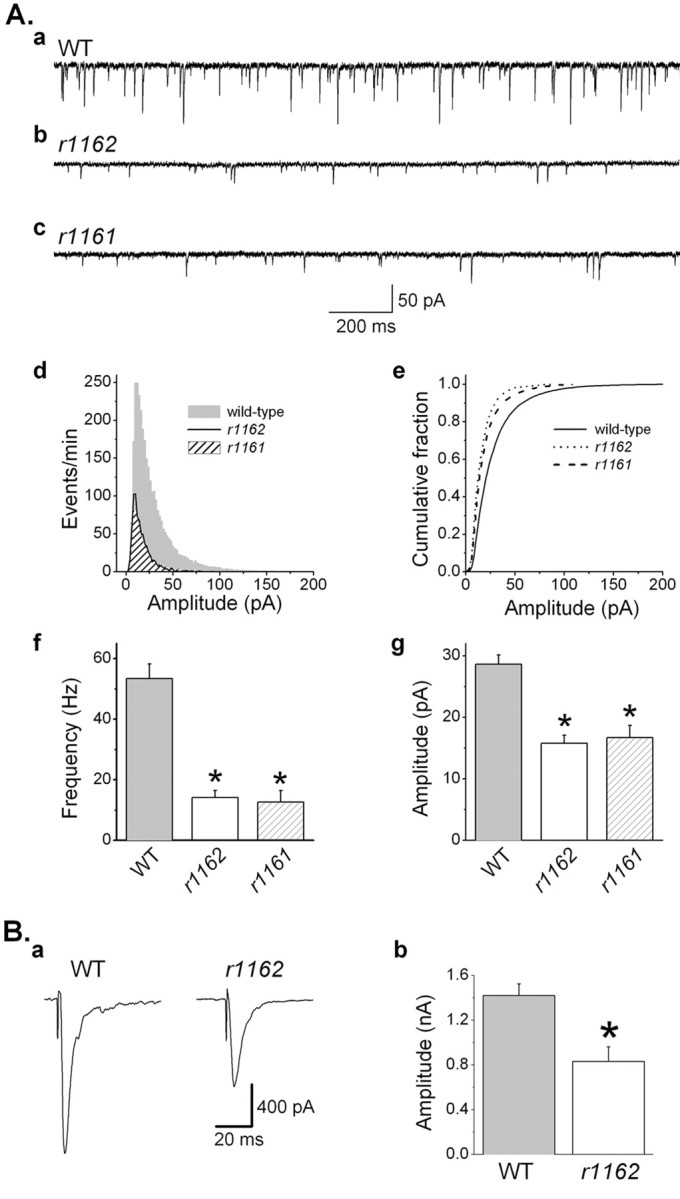
Ryanodine receptor mutations inhibited mPSCs and ePSCs at the C. elegans neuromuscular junction. A, a-c, Representative traces of mPSCs of the wild type (WT) and the unc-68 mutants (r1162, r1162). In WT, mPSCs showed great variability in amplitudes. Many events have amplitudes >50 pA. In r1162 and r1161, the frequency and amplitudes of mPSCs were decreased compared with WT. d, mPSC amplitude histograms showing that large-amplitude mPSCs (operationally defined as >50 pA) were essentially absent in the mutants. e, Cumulative mPSC amplitude distribution showing that mPSCs had smaller amplitudes in the mutants. f, The frequency of mPSCs was significantly decreased in the mutants. g, The mean amplitude of mPSCs was significantly reduced in the mutants (WT, n = 25; r1162, n = 10; r1161, n = 7). B, a, Representative traces of ePSCs in WT and r1162. b, The amplitude of ePSCs was significantly reduced in r1162 (n = 9) compared with WT (n = 14). The asterisk indicates a statistically significant difference (p < 0.05).
The frequency of mPSCs was severely reduced in unc-68 mutants (< 25% of wild-type frequency in both r1161 and r1162) (Fig. 1A). The mean amplitude of mPSCs was also reduced to ∼55% of wild type (Fig. 1A). Overlays of the mPSC amplitude histograms of the wild type and the mutants (Fig. 1A, d,e) revealed that the frequency of large-amplitude mPSCs was reduced more severely than that of the smaller events. Spontaneous events with amplitude ≥50 pA are operationally defined as large-amplitude mPSCs in this report because they were rare in the RYR mutants. The decrease in mPSC frequency in the mutant was not simply because an overall reduction of mPSC amplitudes made small events undetectable. As shown in Figure 1A, RYR mutation caused a 45% drop in the mean amplitude of mPSCs. When all mPSCs in the wild type were scaled down by 45%, only 8% of the mPSCs went below the detection threshold (5 pA), which is much less than the 75% frequency drop observed in the mutant. These results suggest that mPSCs, in particular those of large amplitude, are dependent on the function of UNC-68.
To evaluate the function of UNC-68 in evoked synaptic release, ePSCs were compared between the wild type and the r1162 mutant. The ePSC amplitude was reduced to 58% of wild-type level in the r1162 mutant (Fig. 1B), suggesting that RYRs play a very important role in spontaneous as well as evoked synaptic release.
The changes in mPSCs and ePSCs were not caused by secondary developmental defects
To confirm that the abnormalities of mPSCs and ePSCs in the unc-68 mutants were attributable to the unc-68 mutation rather than a secondary developmental defect, the effects of ryanodine on mPSCs and ePSCs were examined. Ryanodine, which is membrane-permeant, was applied in the extracellular solution at 100 μm during recording. At this concentration, ryanodine is expected to block RYRs (Xu et al., 1998; Llano et al., 2000). In wild-type preparations, ryanodine decreased mPSC frequency to one-third of the starting value. The frequency of larger mPSCs was reduced more than that of the smaller events, resulting in a reduction in the mean amplitude to 70% of the initial value (Fig. 2A). In control experiments with the r1162 mutant, ryanodine showed no effect on the frequency and amplitude of mPSCs (Fig. 2B), confirming that ryanodine acts specifically on UNC-68. Ryanodine also inhibited the amplitude of ePSCs (see the following section). The fact that acute pharmacological blockade of RYRs caused changes of mPSCs and ePSCs similar to those caused by unc-68 mutation suggests that the synaptic transmission defects observed in the unc-68 mutants were indeed caused by RYR loss of function rather than a secondary developmental defect.
Figure 2.

Ryanodine (Rya; 100 μm) inhibited mPSCs in wild-type but not unc-68 mutant preparations. A, In wild-type preparations, ryanodine inhibited the frequency and the mean amplitude of mPSCs (n = 5). B, In the unc-68(r1162) mutant, ryanodine showed no effect on either mPSC frequency or amplitude (n = 5). The preryanodine and postryanodine mPSC amplitude distributions overlap completely (b). The wild-type mPSC amplitude histogram shown in Figure 1 A is replotted here for comparison.
Both cholinergic and GABAergic transmissions were affected by RYR dysfunction
There are two types of cholinergic receptor (nicotinic and levamisole) and one type of GABAergic receptor at the C. elegans NMJ (Richmond and Jorgensen, 1999). We analyzed the effect of ryanodine (100 μm) on synaptic transmission via the three types of receptor independently. To evaluate nicotinic receptor-mediated neuromuscular transmission, the unc-49(e407);unc-29(e1072) mutant was used. In this mutant, only nicotinic receptors are functional at the NMJ because the functions of GABA receptors and levamisole receptors were abolished by the unc-49(e407) and unc-29(e1072) mutations, respectively (Bamber et al., 1999; Richmond and Jorgensen, 1999). To evaluate levamisole receptor-mediated neuromuscular transmission, unc-49(e407) animals were used in the presence 5 μm DHβE, which blocks nicotinic receptors at the C. elegans NMJ (Richmond and Jorgensen, 1999). To evaluate GABA receptor-mediated neuromuscular transmission, wild-type animals were used in the presence of 0.5 mm d-TBC. d-TBC blocks all cholinergic receptors at the C. elegans NMJ (Richmond and Jorgensen, 1999).
When neuromuscular transmissions at the different subsets of synapses were analyzed independently, the amplitudes of mPSCs were still highly variable. The mPSCs mediated by levamisole receptors (11.3 ± 0.9 pA) appeared to be smaller in amplitude than those mediated by nicotinic (24.3 ± 1.5 pA) and GABA (32. 3 ± 5.0 pA) receptors. In response to nerve stimulation, ePSCs were induced when either nicotinic or levamisole receptors were functional. However, the kinetics of ePSCs mediated by the two receptors was very different. The ePSCs mediated by nicotinic receptors returned to the baseline quickly, and the decay phase was best fitted by a single exponential. In contrast, ePSCs mediated by levamisole receptors lasted much longer, and the decay phase was best fitted by two exponentials (Fig. 3B). The existence of two kinetically very different ePSCs at the C. elegans NMJ is reminiscent of the NMDA and non-NMDA receptor-mediated EPSCs at many vertebrate central synapses (Lester et al., 1990; Silver et al., 1992; McBain and Mayer, 1994). No ePSCs could be induced when only the GABA receptors were functional (Fig. 3D), suggesting that GABAergic transmission does not contribute to ePSCs under the current experimental conditions. Consistent with this notion, the amplitude of ePSCs in the unc-49(e407) mutant (Fig. 3D), which lacks the function of GABA receptors, was no less than that in the wild type (Fig. 1B).
Figure 3.

RYRs play a similar role in various subsets of neuromuscular junctions. Neuromuscular transmissions through nicotinic (Nic) receptors, levamisole (Lev) receptors, and GABA receptors were analyzed independently using unc-49(e407);unc-29(e1072) mutant, unc-49(e407) mutant in the presence of dihydro-β-erythroidine (5 μm), and wild-type animals in the presence of 0.5 mm d-TBC, respectively. The function of RYRs in synaptic transmission was evaluated by comparing mPSCs and ePSCs during the control period (Ctr) and after the administration of 100 μm ryanodine (Rya). A, Representative current traces and cumulative amplitude plots showing that ryanodine reduced mPSC frequency and shifted mPSCs toward smaller amplitudes. B, Representative traces showing that ryanodine inhibited the amplitudes of ePSCs mediated by either nicotinic or levamisole receptors. The ePSCs mediated by the two receptors were kinetically very different. The ePSCs mediated by nicotinic receptors had a single decay time constant (τ = 7.2 ± 0.9 ms; n = 5), whereas those mediated by levamisole receptors had two decay time constants (τ1 = 5.4 ± 1.2 ms; τ2 = 43.5 ± 7.8 ms; n = 6). C, Bar graphs showing mPSC frequencies (Nic, n = 5; Lev, n = 6; GABA, n = 7) and ePSC amplitudes (Nic, n = 5; Lev, n = 6) before and after ryanodine. D, Sample traces showing the effect of d-TBC on ePSC amplitude in a wild-type preparation and a typical ePSC in unc-49(e407). The ePSCs in wild-type preparations were abolished by d-TBC (Ctr, 1.73 ± 0.36 nA; n = 5; d-TBC, 0.00 ± 0.00 nA; n = 5), which could be partially reversed by washing (wash, 0.69 ± 0.20; n = 3). The amplitude of ePSCs in unc-49(e407) was 2.02 ± 0.16 nA (n = 9).
After recording baseline activities, ryanodine (100 μm) was added to the extracellular solution to determine its effects on mPSCs and ePSCs. After ryanodine application, mPSCs showed a reduction of frequency and a shift toward lower amplitudes at all the subsets of synapses (Fig. 3A,C). Amplitudes of ePSCs mediated by either nicotinic receptors or levamisole receptors were also reduced significantly (Fig. 3B,C). These results suggest that RYRs play a similar role in synaptic transmission at different subsets of synapses at the NMJ.
RYRs acted presynaptically to regulate synaptic transmission
The inhibition of mPSCs and ePSCs by unc-68 mutations could result from either reduced synaptic release or decreased sensitivity of postsynaptic receptors, or both. However, because intracellular concentration of free Ca2+ in the recorded body wall muscle cell was clamped to <100 nm by including EGTA in the pipette solution (see Materials and Methods), postsynaptic RYRs were unlikely to regulate synaptic currents by mobilizing intracellular Ca2+. To further confirm that presynaptic RYR dysfunction caused the synaptic transmission defects, the following two experiments were performed.
First, exogenous acetylcholine (100 μm) was puffed onto body wall muscle cells. The inward currents thus induced were compared between the wild type and the r1162 mutant (Fig. 4). Amplitudes of the inward currents were similar in the two groups, suggesting that the sensitivity of postsynaptic receptors to acetylcholine was not altered in the unc-68 mutant.
Figure 4.

Body wall muscle sensitivity to acetylcholine was not different between the wild-type (WT) and the unc-68(r1162) mutant. A, Representative traces showing acetylcholine-induced inward currents in WT and r1162. B, Comparison of the amplitudes of acetylcholine-induced inward currents between WT and r1162 (WT, n = 8; r1162, n = 6).
Second, an experiment was performed to rescue selectively the neuronal or muscle RYR defects in the r1162 mutant. The rab-3 (Nonet et al., 1997) and myo-3 (Okkema et al., 1993) promoters were used to drive the expression of myc-tagged wild-type UNC-68 in neurons and body wall muscle cells, respectively. Immunostaining with an anti-myc antibody showed that the transgene was expressed specifically in either neurons or body wall muscle cells, depending on the promoter (Fig. 5A). The myc staining in neurons resembled the ER structure observed in C. elegans neuronal soma and processes (Rolls et al., 2002). In the dorsal and ventral nerve cords, myc staining was observed in the vicinity of the active zone marker RIM (Koushika et al., 2001) (Fig. 5A, a-c), suggesting that UNC-68 is present at release sites. The myc staining in body wall muscle cells appeared as regularly spaced puncta (Fig. 5A, e), which resembles the expression pattern of RYRs in wild-type animals (Maryon et al., 1998).
Figure 5.

Expression of the wild-type RYR in neurons but not muscle cells rescued unc-68(r1162) synaptic transmission defects. A myc-tagged wild-type UNC-68 was expressed in neurons and body wall muscle cells independently. A, Animals expressing myc::UNC-68 under the control of the neuron-specific rab-3 promoter show myc immunoreactivity only in neurons and neuronal processes, such as the dorsal cord (a) and neurons in the head (d). The immunoreactivities of myc (a) and the active zone marker RIM (b) in the dorsal cord overlap (c), suggesting that UNC-68 exists at presynaptic release sites. e, Animals expressing myc::UNC-68 under the control of the muscle-specific myo-3 promoter show myc immunoreactivity only in muscle cells. No myc staining was observed in wild-type or r1162 animals that did not express the transgene (data not shown). Scale bars: c, d, 10 μm; e, 20 μm. B, Animals of wild type (WT), r1162, r1162 expressing myc::UNC-68 in neurons (Neuron), and r1162 expressing myc::UNC-68 in muscle cells (Muscle) were singly placed in the center of a circular field (7 mm) in culture plates to track their locomotion. a, Photographs taken 10 min after the animals were placed on the plates showing that the r1162 mutant produced much shorter tracks on the plate than the WT, which was rescued by expressing wild-type UNC-68 in neurons. b, The locomotion velocity of r1162 was significantly slower than that of WT, which was mostly rescued by expressing the wild-type UNC-68 in neurons. Expression of myc::UNC-68 in body wall muscle cells also showed a modest rescuing effect. WT, n = 11; r1162, n = 5; Neuron, n = 5; Muscle, n = 9. C, a, Representative traces of mPSCs from r1162 and rescued animals. b, c, Comparisons of mPSC frequency and amplitudes among WT (n = 25), r1162 (n = 10), and the rescued (Neuron, n = 8; Muscle, n = 7). D, a, Representative traces of ePSCs from r1162 and rescued animals. b, Comparison of ePSC amplitudes among WT (n = 14), r1162 (n = 9), and the rescued (Neuron, n = 5; Muscle, n = 7). The asterisk indicates a statistically significant difference compared with WT (p < 0.05).
The primary behavioral phenotype of RYR loss-of-function mutations in C. elegans is a locomotion defect. Homozygous mutant animals move slowly and exhibit a languid, incomplete flaccid paralysis (Maryon et al., 1996). Mutant animals expressing the wild-type unc-68 transgene in neurons showed greatly improved locomotion, whereas those expressing the transgene in body wall muscle cells were less well rescued (Fig. 5B). Similarly, expression of the transgene in neurons completely rescued abnormalities in mPSCs and ePSCs, whereas that in muscle cells showed no effect (Fig. 5C,D). The neuron- and muscle-specific rescue experiments confirmed that a presynaptic RYR defect was the cause for the changes of mPSCs and ePSCs. In addition, they suggest that the function of presynaptic RYRs is important for animal locomotion.
Thus, several lines of evidence suggest that presynaptic RYRs regulate mPSCs and ePSCs. Although RYR is also expressed in body wall muscle (Maryon et al., 1998; Hamada et al., 2002), its function in these cells does not affect mPSCs and ePSCs.
Synaptic exocytosis depended on both Ca2+ influx and RYR-mediated Ca2+ release
Ca2+-triggering synaptic exocytosis could come from influx through Ca2+ channels in the plasma membrane and release from intracellular Ca2+ stores. To evaluate the relative contributions of Ca2+ influx and RYR-mediated Ca2+ release to synaptic exocytosis, we analyzed the effects of removing extracellular Ca2+ and unc-68 mutation on mPSC properties.
In the wild type, elimination of extracellular Ca2+ reduced mPSC frequency by 71% (Fig. 6A,E). Nevertheless, the fraction of large-amplitude events remained the same, as indicated by the identical amplitude distributions (Fig. 6B) and the identical shapes of the averaged mPSC traces in the presence and absence of extracellular Ca2+ (Fig. 6C). These results showed that (1) extracellular Ca2+ contributes to the triggering of mPSCs, and (2) large-amplitude mPSCs were not the result of endogenous depolarization-induced responses, which would not occur in the absence of extracellular Ca2+ (Katz and Miledi, 1969a,b, 1970).
Figure 6.

Ca2+ influx and ryanodine receptor-mediated release were the exclusive sources of Ca2+ for synaptic exocytosis, and the amplitude of mPSCs was independent of extracellular Ca2+. A, Representative traces showing the effect of removing extracellular Ca2+ on mPSCs. Removal of Ca2+ greatly reduced the frequency of mPSCs in the wild type but essentially abolished mPSCs in the unc-68(r1162) mutant. B, Amplitude distributions of mPSCs were identical in the presence and absence of extracellular Ca2+ in wild type (n = 5). C, Averaged mPSCs of wild type before and after Ca2+ removal overlap completely. The averages were obtained from the wild-type trace shown in A. D, Removal of extracellular Ca2+ did not change mPSC amplitudes in wild type (n = 5). E, Although removal of extracellular Ca2+ greatly reduced mPSC frequency in wild type (n = 5), it eliminated mPSCs in r1162 (n = 5). The asterisk indicates a statistically significant difference (p < 0.05). F, itr-1(sa73), a hypomorphic mutant of inositol 1,4,5-triphophate receptor, showed normal mPSCs (a, b) and ePSCs (c); mPSC frequency, 50.9 ± 4.8 Hz; mPSC amplitude, 26.7 ± 2.2 pA; ePSC amplitude, 1.43 ± 0.17 nA; n = 5.
In the r1162 mutant, the frequency of mPSCs was low in the presence of standard extracellular solution. On elimination of extracellular Ca2+, mPSCs essentially disappeared (Fig. 6A,E). These experiments with wild-type and unc-68 mutant preparations showed that Ca2+ influx and RYR-mediated Ca2+ release are likely the exclusive sources of Ca2+ for synaptic exocytosis at the C. elegans NMJ. It also suggests that Ca2+ may be absolutely required for synaptic vesicle fusion. Although rare mPSC events (0.19 ± 0.04 Hz) were still observed after removal of extracellular Ca2+ in the unc-68 mutant, the frequency was equivalent to only 0.4% of the wild type in the presence of extracellular Ca2+ (53.4 ± 4.8 Hz). Thus, a combination of RYR mutation and removal of extracellular Ca2+ reduced mPSC frequency by 99.6%. The rare mPSC events might be mediated by residual Ca2+ in motoneurons that was not regulated by RYRs and Ca2+ influx. In contrast to the large effects of RYR mutation on neuromuscular transmission, properties of mPSCs and ePSCs were similar between the loss-of-function inositol 1,4,5-trisphosphate receptor mutant itr-1(sa73) (Dal Santo et al., 1999) (Fig. 6F) and the wild type (Fig. 1), suggesting that RYRs are likely the only meaningful Ca2+-releasing channels in the ER membrane for synaptic exocytosis at the NMJ.
Large-amplitude mPSCs were not caused by multivesicular release
As shown in Figures 1 and 2, RYR dysfunction essentially eliminated large-amplitude mPSCs. RYR-mediated large-amplitude mPSCs at vertebrate central synapses are thought to result from concerted multivesicular release, based on rise time analysis (Sharma and Vijayaraghavan, 2003) or examination of the effect of removing extracellular Ca2+ (Llano et al., 2000; Sharma and Vijayaraghavan, 2003). To determine whether RYR-mediated large-amplitude mPSCs at the C. elegans NMJ were also caused by concerted multivesicular release, we performed similar analyses. Furthermore, we examined mPSCs in mutants that are defective in synchronizing synaptic release. Several lines of evidence suggest that large-amplitude mPSCs at the C. elegans NMJ were not multivesicular events.
First, the fraction of large-amplitude mPSCs remained the same after removal of extracellular Ca2+. Extracellular Ca2+ is required for synchronized synaptic exocytosis, as shown by the desynchronization of ePSCs when extracellular [Ca2+] is reduced from 5 mm to 250 μm (Wang et al., 2001). Large-amplitude mPSCs should be nonexistent in the absence of extracellular Ca2+ if they were indeed caused by concerted multivesicular release. However, the fraction of large-amplitude events remained the same after eliminating extracellular Ca2+ (Fig. 6B,C), despite a severe reduction in mPSC frequency (Fig. 6E).
Second, no correlation was found between the rise time and the amplitude of mPSCs. If large-amplitude events arose from a summation of individual mPSCs, a lack of absolute synchrony among the individual events would cause an increased rise time. However, the rise time remained constant regardless of the amplitude of mPSCs (Fig. 7A, a,c). Consistent with this observation, larger-amplitude events were associated with steeper rise slopes (Fig. 7A, b,d).
Figure 7.
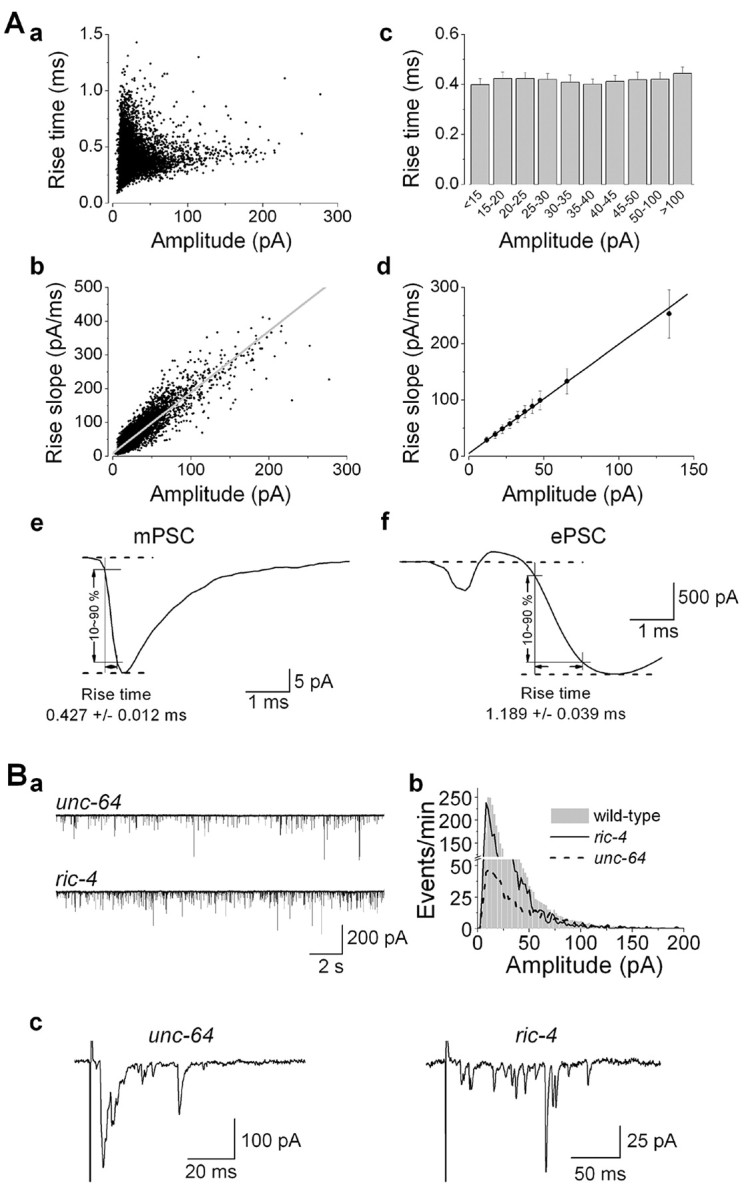
Large-amplitude mPSCs did not result from concerted multivesicular release. A, Scatterplots of mPSCs (a, b) and mPSCs grouped according to amplitudes (c, d) show that the rise time was constant regardless of the amplitude, whereas the rise slope was steeper for events with larger amplitudes; n = 6. e, f, Averaged mPSC and ePSC traces in one representative experiment showing that the rise time of ePSCs is much longer than that of mPSCs. B, Large-amplitude mPSCs still occurred in mutants defective in synchronized synaptic exocytosis. The syntaxin mutant unc-64(e246) and the SNAP25 mutant ric-4(ys7) showed desynchronized ePSCs (c). However, large-amplitude mPSCs still occurred in the mutants (a, b). In ys7, the frequency and amplitude of mPSCs (n = 9) were 40 ± 7 Hz and 25.5 ± 2.5 pA, respectively. ePSC amplitude was 67.6 ± 12.5 pA (n = 5). Statistics on mPSC and ePSC properties of e246 are described in a study by Wang et al. (2001).
Third, the rise time of ePSCs, which represent synchronized multivesicular release, was much longer than that of mPSCs (Fig. 7A, e,f). Thus, large-amplitude mPSCs are distinct from events of synchronized multivesicular release.
Fourth, the fraction of large-amplitude mPSCs did not decrease in mutants that are defective in synchronized synaptic exocytosis. In unc-64(e246), a hypomorphic syntaxin mutant, mPSC frequency is severely reduced, and ePSCs are desynchronized (Saifee et al., 1998; Wang et al., 2001). Nevertheless, large-amplitude mPSCs were frequent in this mutant (Fig. 7B). In the SNAP25 (soluble N-ethylmaleimide-sensitive factor attachment protein 25) hypomorphic mutant ric-4(ys7), ePSCs were desynchronized and had small amplitudes. However, large-amplitude mPSCs persisted (Fig. 7B). The frequency of mPSCs in the SNAP25 mutant was similar to that in wild type.
Thus, our observations were incompatible with large-amplitude mPSCs being multivesicular events. Because RYRs function presynaptically, regulation of quantal size is the alternative mechanism for RYR to mediate large-amplitude mPSCs.
Synaptic vesicle size and number were not altered in unc-68 mutants
The inhibition of mPSCs and ePSCs observed in unc-68 mutants could potentially result from a decrease in synaptic vesicle size or number. Electron microscopy was used to determine whether synaptic vesicle size and number, including the number of morphologically docked vesicles, were altered in the mutant compared with the wild type. Neither the vesicle size nor the numbers of total and docked vesicles per synapse were different between the wild type and the mutant, suggesting that the inhibitions of mPSCs and ePSCs in the mutant were not attributable to decreased synaptic vesicle size or number (Fig. 8).
Figure 8.
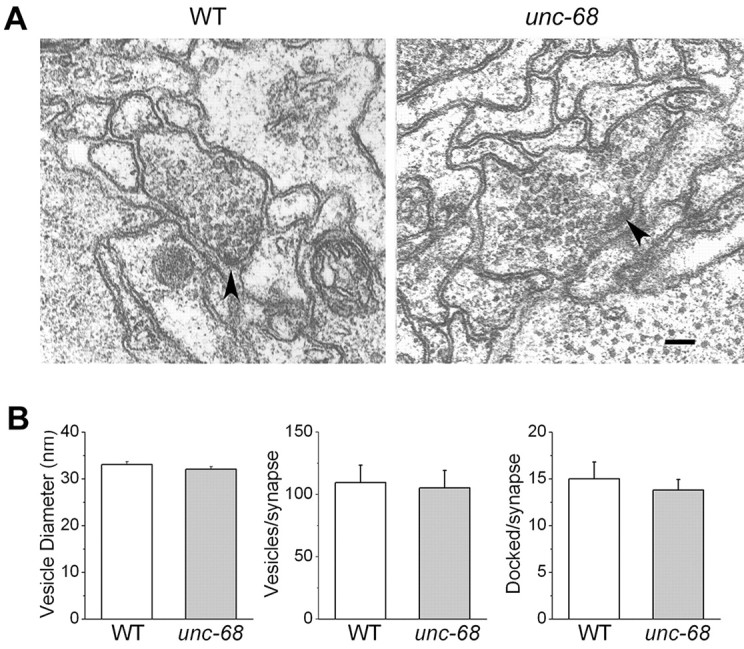
Synaptic vesicle size and number were similar between the wild type (WT) and unc-68 mutant. A, Representative electron micrographs of neuromuscular junctions in the WT and unc-68 mutant. Scale bar, 100 nm. Presynaptic densities are indicated by arrowheads. B, The vesicle diameters were 33.1 ± 0.6 nm in WT and 32.1 ± 0.6 nm in unc-68. The numbers of vesicles per synapse were 109.5 ± 14.1 in WT and 105.4 ± 14.0 in unc-68. The numbers of morphologically docked vesicles per synapse were 15.0 ± 1.8 in WT and 13.8 ± 1.1 in unc-68. No statistically significant differences were found between the WT and unc-68 mutant. The numbers of neuromuscular junctions analyzed were eight for WT and 10 for unc-68(r1161).
Discussion
The present study showed that the RYR plays very important roles in both spontaneous and evoked synaptic release, suggesting that Ca2+ release from the ER may provide an important source of Ca2+ regulating exocytosis at C. elegans NMJs. A striking effect of RYR mutations or blockade was the elimination of large-amplitude mPSCs. An elucidation of the nature of the large-amplitude mPSCs may be a key to understanding the mechanism of RYR action at synapses. At least two previous studies have suggested that RYR-mediated large-amplitude mPSCs resulted from concerted multivesicular release (Llano et al., 2000; Sharma and Vijayaraghavan, 2003). However, our results are inconsistent with this notion. First, the rise time of mPSCs was constant regardless of the amplitude because of the fact that larger mPSCs had steeper rise slopes. Second, the amplitude distribution of mPSCs remained the same after elimination of extracellular Ca2+. Third, the rise time of mPSCs was much shorter than that of ePSCs, which represent concerted multivesicular release. Fourth, the fraction of large-amplitude mPSCs did not decrease in mutants that are defective in synchronized synaptic exocytosis. These lines of evidence suggest that large-amplitude mPSCs at the C. elegans NMJ are probably not caused by concerted multivesicular release.
Because RYRs function presynaptically to cause large-amplitude mPSCs at the C. elegans NMJ, regulation of quantal size is the most plausible explanation. We postulate that reductions in Ca2+ levels in the presynaptic terminal in the absence of RYRs lead to the change in mean quantal size. Mechanistically, this alteration in the manipulation of Ca2+ levels during exocytosis could alter quantal size in a variety of ways. First, RYRs may increase vesicle size. Larger vesicles may contain more neurotransmitters than smaller vesicles do. However, this does not appear to be the case because the size of synaptic vesicles was similar between the wild type and the unc-68 mutant. Second, RYRs may increase the number of docked vesicles, as suggested by Ca2+-dependent translocation of synaptic vesicles at Drosophila NMJs (Koenig et al., 1993), and these newly docked vesicles might contain more transmitters than other docked vesicles. However, the number of morphologically docked vesicles was not changed in the RYR mutant. Third, RYRs may promote vesicle loading with neurotransmitters, resulting in higher concentrations in the wild type compared with unc-68 mutants. Fourth, RYR may stimulate intervesicular fusion to form larger vesicles before exocytosis. The larger vesicles may contain neurotransmitters that were originally packed into several smaller vesicles. Fifth, RYRs may increase either the size or duration of the fusion pore during kiss- and-run synaptic release. Ca2+ entry-dependent regulation of quantal size has been shown to occur for catecholamine release from chromaffin cells, probably through modulation of kiss-and-run exocytosis (Elhamdani et al., 2001). Conceivably, quantal size in neurons might be regulated by RYR-mediated Ca2+ release through a similar mechanism. These possibilities need to be examined in future studies.
Our results demonstrated that there are two sources of Ca2+ for synaptic exocytosis: Ca2+ influx and RYR-mediated Ca2+ release from intracellular stores. The two sources of Ca2+ could function both independently and concertedly. The continued occurrence of mPSCs when only one of the two Ca2+ sources was available is an indication of the independent actions. Interactions between the two sources of Ca2+ become obvious when the magnitudes of mPSC frequency inhibition caused by Ca2+ removal and by unc-68 mutation are compared. In wild-type preparations, elimination of extracellular Ca2+ reduced mPSC frequency by ∼70%. If the two Ca2+ sources functioned independently, RYR would be responsible for the remaining 30% of mPSCs. However, unc-68 mutations reduced mPSC frequency by >70%, which is significantly more than what would be expected for independent actions. The most plausible explanation for the discrepancy is that Ca2+ influx is amplified by RYR-mediated Ca2+ release; RYRs are activated by Ca2+ entering the cell [Ca2+-induced Ca2+ release (CICR)] (Verkhratsky and Shmigol, 1996). CICR has been demonstrated in a variety of cell types, including neurons (Narita et al., 1998; Merriam et al., 1999; Sandler and Barbara, 1999). The ER is a continuous network that extends to all parts of the neuron (Berridge, 1998; Rolls et al., 2002). In cardiomyocytes, RYRs in the sarcolemma are closely apposed to Ca2+ channels in the plasma membrane with a gap of only 15 nm. It has been suggested that an analogous mechanism may occur in neurons to allow efficient couplings between Ca2+ influx and RYR activation (Berridge, 1998).
The role of extracellular Ca2+ in mPSCs appears variable in different systems. For example, removal of external Ca2+ has a variable effect at frog NMJs (Fatt and Katz, 1952). It has no effect in cultured rat retinal ganglion cells (Han et al., 2001). Blockade of Ca2+ influx with cadmium does not alter mPSC properties at inhibitory synapses in rat hippocampal slices (Hajos et al., 2000). In contrast, we showed that removal of extracellular Ca2+ reduced mPSC frequency by ∼70%. Our observation is similar to a previous one on the C. elegans NMJ (Richmond et al., 1999). It is unclear why mPSC frequency at the C. elegans NMJ is more susceptible to the removal of extracellular Ca2+ than the other systems.
It has been suggested that exocytotic fusion may occur in a Ca2+-independent manner (Knight et al., 1989; Gerber and Sudhof, 2002). However, there is no compelling evidence to support it. In the present study, a combination of RYR mutation and removal of extracellular Ca2+ reduced mPSC frequency to 0.19 ± 0.04 Hz. Although this value may appear significant compared with many other synapses that have a relatively low mPSC frequency, it represents only 0.4% of the mPSCs frequency observed in wild-type preparations in the presence of extracellular Ca2+. Thus, a combination of zero extracellular Ca2+ and RYR dysfunction eliminated 99.6% of mPSCs. The rare events of mPSCs observed in the absence of both Ca2+ sources could be attributable to residual Ca2+ left inside motoneurons, which would not be surprising given that the preparation had been incubated with zero Ca2+ solution for only a few minutes before the measurement was made. Therefore, this observation is in favor of the notion that synaptic exocytosis has an obligatory requirement for Ca2+, and influx and RYR-mediated release are the exclusive sources of Ca2+ for synaptic exocytosis.
In the present study, we showed that dysfunction of RYR inhibited both spontaneous and evoked synaptic exocytosis. It is well established that evoked synaptic exocytosis is critical to the function of the nervous system because it is required for synaptic transmission at chemical synapses. Miniature postsynaptic currents or potentials were originally thought to result from “spill-over” of neurotransmitters with little physiological significance. However, more recent evidence suggests that they have important physiological functions. For example, mPSCs may play important roles in postsynaptic receptor clustering (Saitoe et al., 2001), shifting of NMDA receptor subunit compositions during development (Barria and Malinow, 2002), dendritic protein synthesis (Sutton et al., 2004), maintenance of dendritic spines (McKinney et al., 1999), and action potential firing (Sharma and Vijayaraghavan, 2003). By regulating both spontaneous and evoked synaptic release, RYR is likely a key player in the function and development of the nervous system.
Footnotes
This work was supported by National Institutes of Health Grants R21 MH070739 (Z.-W.W.) and R01 NS33535 (M.L.N.). We thank Lawrence Salkoff for allowing us to use equipment in preliminary experiments and for helpful suggestions, Erik Jorgensen for providing mutant strains, William Mohler for sharing the fluorescence stereomicroscope, Dingxia Shen, Rose Vincent, Jaya Joshi, and Sijie Wang for technical or graphical assistance, Steve Kempe, Subramani Munirathinam, and Jeff Dutton for assistance with electron microscopic analysis, and Aaron DiAntonio, Dick Mains, Betty Eipper, Douglas Oliver, Matthew Rasband, and Sandhya Koushika for helpful discussions.
Correspondence should be addressed to Zhao-Wen Wang, Department of Neuroscience, University of Connecticut Health Center, 263 Farmington Avenue, Farmington, CT 06030-3401. E-mail: zwwang@uchc.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256745-10$15.00/0
B.C. and M.Y. contributed equally to this work.
References
- Bamber BA, Beg AA, Twyman RE, Jorgensen EM (1999) The Caenorhabditis elegans unc-49 locus encodes multiple subunits of a heteromultimeric GABA receptor. J Neurosci 19: 5348-5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardo S, Robertson B, Stephens GJ (2002) Presynaptic internal Ca2+ stores contribute to inhibitory neurotransmitter release onto mouse cerebellar Purkinje cells. Br J Pharmacol 137: 529-537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Malinow R (2002) Subunit-specific NMDA receptor trafficking to synapses. Neuron 35: 345-353. [DOI] [PubMed] [Google Scholar]
- Berridge MJ (1998) Neuronal calcium signaling. Neuron 21: 13-26. [DOI] [PubMed] [Google Scholar]
- Carter AG, Vogt KE, Foster KA, Regehr WG (2002) Assessing the role of calcium-induced calcium release in short-term presynaptic plasticity at excitatory central synapses. J Neurosci 22: 21-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli B, Hurlbut WP (1980) Ca2+-dependent recycling of synaptic vesicles at the frog neuromuscular junction. J Cell Biol 87: 297-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Santo P, Logan MA, Chisholm AD, Jorgensen EM (1999) The inositol trisphosphate receptor regulates a 50-second behavioral rhythm in C. elegans Cell 98: 757-767. [DOI] [PubMed] [Google Scholar]
- De Crescenzo V, ZhuGe R, Velazquez-Marrero C, Lifshitz LM, Custer E, Carmichael J, Lai FA, Tuft RA, Fogarty KE, Lemos JR, Walsh Jr JV (2004) Ca2+ syntillas, miniature Ca2+ release events in terminals of hypothalamic neurons, are increased in frequency by depolarization in the absence of Ca2+ influx. J Neurosci 24: 1226-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhamdani A, Palfrey HC, Artalejo CR (2001) Quantal size is dependent on stimulation frequency and calcium entry in calf chromaffin cells. Neuron 31: 819-830. [DOI] [PubMed] [Google Scholar]
- Emptage NJ, Reid CA, Fine A (2001) Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron 29: 197-208. [DOI] [PubMed] [Google Scholar]
- Fatt P, Katz B (1952) Spontaneous subthreshold activity at motor nerve endings. J Physiol (Lond) 117: 109-128. [PMC free article] [PubMed] [Google Scholar]
- Furuichi T, Furutama D, Hakamata Y, Nakai J, Takeshima H, Mikoshiba K (1994) Multiple types of ryanodine receptor/Ca2+ release channels are differentially expressed in rabbit brain. J Neurosci 14: 4794-4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gad H, Low P, Zotova E, Brodin L, Shupliakov O (1998) Dissociation between Ca2+-triggered synaptic vesicle exocytosis and clathrin-mediated endocytosis at a central synapse. Neuron 21: 607-616. [DOI] [PubMed] [Google Scholar]
- Galante M, Marty A (2003) Presynaptic ryanodine-sensitive calcium stores contribute to evoked neurotransmitter release at the basket cell→Purkinje cell synapse. J Neurosci 23: 11229-11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber SH, Sudhof TC (2002) Molecular determinants of regulated exocytosis. Diabetes 51 [Suppl 1]: S3-S11. [DOI] [PubMed] [Google Scholar]
- Giannini G, Conti A, Mammarella S, Scrobogna M, Sorrentino V (1995) The ryanodine receptor/calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. J. Cell Biol 128: 893-904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajos N, Katona I, Naiem SS, MacKie K, Ledent C, Mody I, Freund TF (2000) Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci 12: 3239-3249. [DOI] [PubMed] [Google Scholar]
- Hamada T, Sakube Y, Ahnn J, Kim do H, Kagawa H (2002) Molecular dissection, tissue localization and Ca2+ binding of the ryanodine receptor of Caenorhabditis elegans J Mol Biol 324: 123-135. [DOI] [PubMed] [Google Scholar]
- Han MH, Kawasaki A, Wei JY, Barnstable CJ (2001) Miniature postsynaptic currents depend on Ca2+ released from internal stores via PLC/IP3 pathway. NeuroReport 12: 2203-2207. [DOI] [PubMed] [Google Scholar]
- Heuser JE, Reese TS (1973) Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol 57: 315-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R (1969a) Tetrodotoxin-resistant electric activity in presynaptic terminals. J Physiol (Lond) 203: 459-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R (1969b) Spontaneous and evoked activity of motor nerve endings in calcium Ringer. J Physiol (Lond) 203: 689-706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R (1970) Further study of the role of calcium in synaptic transmission. J Physiol (Lond) 207: 789-801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight DE, von Grafenstein H, Athayde CM (1989) Calcium-dependent and calcium-independent exocytosis. Trends Neurosci 12: 451-458. [DOI] [PubMed] [Google Scholar]
- Koenig JH, Yamaoka K, Ikeda K (1993) Calcium-induced translocation of synaptic vesicles to the active site. J Neurosci 13: 2313-2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML (2001) A post-docking role for active zone protein Rim. Nat Neurosci 4: 997-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuromi H, Honda A, Kidokoro Y (2004) Ca(2+) Influx through distinct routes controls exocytosis and endocytosis at Drosophila presynaptic terminals. Neuron 41: 101-111. [DOI] [PubMed] [Google Scholar]
- Lester RA, Clements JD, Westbrook GL, Jahr CE (1990) Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature 346: 565-567. [DOI] [PubMed] [Google Scholar]
- Lim R, Oleskevich S, Few AP, Leao RN, Walmsley B (2003) Glycinergic mIPSCs in mouse and rat brainstem auditory nuclei: modulation by ruthenium red and the role of calcium stores. J Physiol (Lond) 546: 691-699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Gonzalez J, Caputo C, Lai FA, Blayney LM, Tan YP, Marty A (2000) Presynaptic calcium stores underlie large-amplitude miniature IPSCs and spontaneous calcium transients. Nat Neurosci 3: 1256-1265. [DOI] [PubMed] [Google Scholar]
- Maryon EB, Coronado R, Anderson P (1996) unc-68 encodes a ryanodine receptor involved in regulating C. elegans body-wall muscle contraction. J Cell Biol 134: 885-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryon EB, Saari B, Anderson P (1998) Muscle-specific functions of ryanodine receptor channels in Caenorhabditis elegans J Cell Sci 111: 2885-2895. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Mayer ML (1994) N-methyl-d-aspartic acid receptor structure and function. Physiol Rev 74: 723-760. [DOI] [PubMed] [Google Scholar]
- McKinney RA, Capogna M, Durr R, Gahwiler BH, Thompson SM (1999) Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat Neurosci 2: 44-49. [DOI] [PubMed] [Google Scholar]
- Mello C, Fire A (1995) DNA transformation. Methods Cell Biol 48: 451-482. [PubMed] [Google Scholar]
- Merriam LA, Scornik FS, Parsons RL (1999) Ca(2+)-induced Ca(2+) release activates spontaneous miniature outward currents (SMOCs) in parasympathetic cardiac neurons. J Neurophysiol 82: 540-550. [DOI] [PubMed] [Google Scholar]
- Munirathinam S, Ostapoff EM, Gross J, Kempe GS, Dutton JA, Morest DK (2004) Organization of inhibitory feed-forward synapses from the dorsal to the ventral cochlear nucleus in the cat: a quantitative analysis of endings by vesicle morphology. Hear Res 198: 99-115. [DOI] [PubMed] [Google Scholar]
- Narita K, Akita T, Osanai M, Shirasaki T, Kijima H, Kuba K (1998) A Ca2+-induced Ca2+ release mechanism involved in asynchronous exocytosis at frog motor nerve terminals. J Gen Physiol 112: 593-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita K, Akita T, Hachisuka J, Huang S, Ochi K, Kuba K (2000) Functional coupling of Ca(2+) channels to ryanodine receptors at presynaptic terminals: amplification of exocytosis and plasticity. J Gen Physiol 115: 519-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale EA, Bowers LM, Jia M, Bateman KE, Williamson LC (1999) Botulinum neurotoxin A blocks synaptic vesicle exocytosis but not endocytosis at the nerve terminal. J Cell Biol 147: 1249-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Staunton JE, Kilgard MP, Fergestad T, Hartwieg E, Horvitz HR, Jorgensen EM, Meyer BJ (1997) Caenorhabditis elegans rab-3 mutant synapses exhibit impaired function and are partially depleted of vesicles. J Neurosci 17: 8061-8073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkema PG, Harrison SW, Plunger V, Aryana A, Fire A (1993) Sequence requirements for myosin gene expression and regulation in Caenorhabditis elegans Genetics 135: 385-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Jorgensen EM (1999) One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat Neurosci 2: 791-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Davis WS, Jorgensen EM (1999) UNC-13 is required for synaptic vesicle fusion in C. elegans Nat Neurosci 2: 959-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls MM, Hall DH, Victor M, Stelzer EH, Rapoport TA (2002) Targeting of rough endoplasmic reticulum membrane proteins and ribosomes in invertebrate neurons. Mol Biol Cell 13: 1778-1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saifee O, Wei L, Nonet ML (1998) The Caenorhabditis elegans unc-64 locus encodes a syntaxin that interacts genetically with synaptobrevin. Mol Biol Cell 9: 1235-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoe M, Schwarz TL, Umbach JA, Gundersen CB, Kidokoro Y (2001) Absence of junctional glutamate receptor clusters in Drosophila mutants lacking spontaneous transmitter release. Science 293: 514-517. [DOI] [PubMed] [Google Scholar]
- Sakube Y, Ando H, Kagawa H (1993) Cloning and mapping of a ryanodine receptor homolog gene of Caenorhabditis elegans Ann NY Acad Sci 707: 540-545. [DOI] [PubMed] [Google Scholar]
- Sakube Y, Ando H, Kagawa H (1997) An abnormal ketamine response in mutants defective in the ryanodine receptor gene ryr-1 (unc-68) of Caenorhabditis elegans J Mol Biol 267: 849-864. [DOI] [PubMed] [Google Scholar]
- Sandler VM, Barbara JG (1999) Calcium-induced calcium release contributes to action potential-evoked calcium transients in hippocampal CA1 pyramidal neurons. J Neurosci 19: 4325-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S (2003) Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron 38: 929-939. [DOI] [PubMed] [Google Scholar]
- Silver RA, Traynelis SF, Cull-Candy SG (1992) Rapid-time-course miniature and evoked excitatory currents at cerebellar synapses in situ. Nature 355: 163-166. [DOI] [PubMed] [Google Scholar]
- Simkus CR, Stricker C (2002) The contribution of intracellular calcium stores to mEPSCs recorded in layer II neurones of rat barrel cortex. J Physiol (Lond) 545: 521-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Neher E (1997) Multiple forms of endocytosis in bovine adrenal chromaffin cells. J Cell Biol 139: 885-894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Hodgkin J (1988) Methods. In: The nematode Caenorhabditis elegans (Wood WB, Community of C. elegans Researchers, eds), pp 587-606. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Sutton MA, Wall NR, Aakalu GN, Schuman EM (2004) Regulation of dendritic protein synthesis by miniature synaptic events. Science 304: 1979-1983. [DOI] [PubMed] [Google Scholar]
- Thomas P, Surprenant A, Almers W (1990) Cytosolic Ca2+, exocytosis, and endocytosis in single melanotrophs of the rat pituitary. Neuron 5: 723-733. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Shmigol A (1996) Calcium-induced calcium release in neurones. Cell Calcium 19: 1-14. [DOI] [PubMed] [Google Scholar]
- Wang ZW, Saifee O, Nonet ML, Salkoff L (2001) SLO-1 potassium channels control quantal content of neurotransmitter release at the C. elegans neuromuscular junction. Neuron 32: 867-881. [DOI] [PubMed] [Google Scholar]
- Xu L, Tripathy A, Pasek DA, Meissner G (1998) Potential for pharmacology of ryanodine receptor/calcium release channels. Ann NY Acad Sci 853: 130-148. [DOI] [PubMed] [Google Scholar]
- Yuan A, Dourado M, Butler A, Walton N, Wei A, Salkoff L (2000) SLO-2, a K+ channel with an unusual Cl- dependence. Nat Neurosci 3: 771-779. [DOI] [PubMed] [Google Scholar]