

Abstract
Astrocytes promote the formation and function of excitatory synapses in the CNS. However, whether and how astrocytes modulate inhibitory synaptogenesis are essentially unknown. We asked whether astrocytes regulate the formation of inhibitory synapses between hippocampal neurons during maturation in vitro. Neuronal coculture with astrocytes or treatment with astrocyte-conditioned medium (ACM) increased the number of inhibitory presynaptic terminals, the frequency of miniature IPSCs, and the number and synaptic localization of GABAA receptor (GABAAR) clusters during the first 10 d in vitro. We asked whether neurotrophins, which are potent modulators of inhibitory synaptic structure and function, mediate the effects of astrocytes on inhibitory synapses. ACM from BDNF- or tyrosine receptor kinase B (TrkB)-deficient astrocytes increased inhibitory presynaptic terminals and postsynaptic GABAAR clusters in wild-type neurons, suggesting that BDNF and TrkB expression in astrocytes is not required for these effects. In contrast, although the increase in the number of inhibitory presynaptic terminals persisted, no increase was observed in postsynaptic GABAAR clusters after ACM treatment of hippocampal neurons lacking BDNF or TrkB. These results suggest that neurons, not astrocytes, are the relevant source of BDNF and are the site of TrkB activation required for postsynaptic GABAAR modulation. These data also suggest that astrocytes may modulate postsynaptic development indirectly by stimulating Trk signaling between neurons. Together, these data show that astrocytes modulate inhibitory synapse formation via distinct presynaptic and postsynaptic mechanisms.
Keywords: neurotrophin, BDNF, TrkB, GABAA receptor, synapse formation, astrocyte
Introduction
Astrocytes play important roles in the development and function of neuronal circuitry. Astrocytes upregulate the formation of functional glutamatergic synapses in cultures of retinal ganglion cells, spinal motor neurons, and hippocampal neurons (Pfrieger and Barres, 1997; Ullian et al., 2001, 2004; Zhang et al., 2003; Hama et al., 2004; Christopherson et al., 2005). Although astrocytes appear to signal to neurons by local contact as well as by the release of soluble factors, the cellular and molecular mechanisms by which astrocytes regulate synaptogenesis are not understood.
Several lines of evidence suggest that astrocytes modulate excitatory synapses presynaptically as well as postsynaptically. In purified retinal ganglion cell cultures, astrocyte-conditioned medium (ACM) dramatically increases the number of presynaptic contacts made between neurons, the quantal size and efficacy of neurotransmitter release, and the number of postsynaptic AMPA receptor clusters (Nagler et al., 2001; Ullian et al., 2001, 2004). Astrocyte-derived cholesterol complexed to apolipoprotein E has been shown to be necessary and sufficient to induce functional presynaptic terminals (Mauch et al., 2001; Ullian et al., 2001, 2004). In hippocampal neurons grown in vitro, integrin-mediated contact between astrocytes and pyramidal neurons induces neuron-wide activation of PKC signaling that promotes the maturation of excitatory presynaptic terminals but has no effect on postsynaptic AMPA receptor clusters (Hama et al., 2004). As synapses mature, astrocytes continue to modulate synaptic function by potentiating or suppressing activity at presynaptic glutamatergic terminals via the release of glutamate or ATP, respectively (Zhang et al., 2003; Fiacco and McCarthy, 2004). These studies provide some insights into the signaling mechanisms by which astrocytes modulate glutamatergic presynaptic maturation and function.
Currently, the role of astrocytes in modulating inhibitory synapse formation and function is less well understood. Examination of current density in hippocampal cultures before synaptogenesis suggests that astrocytes contribute to the maintenance of GABAA receptors (GABAARs) in the neuronal membrane (Liu et al., 1996, 1997). Embryonic hippocampal neurons grown on cortical astrocyte monolayers had larger GABA-induced Cl- currents relative to neurons grown only on poly-d-lysine in vitro (Liu et al., 1996). Although the upregulation of GABA current density required Ca2+ elevation in astrocytes, this effect was not dependent on direct contact with astrocytes, because ACM treatment mimicked the effects observed in cocultures (Liu et al., 1996, 1997). Thus, soluble factors released by astrocytes modulate the distribution of GABAA receptors as synapses are formed. Astrocytes continue to provide soluble factors that regulate ongoing inhibitory synaptic transmission. In hippocampal slices, perisynaptic astrocytes release glutamate in response to GABAB receptor activation, which potentiates interneuron GABA release and inhibitory synaptic transmission onto pyramidal neurons (Kang et al., 1998). These observations suggest that astrocytes modulate inhibitory synapse formation and function presynaptically and postsynaptically, prompting us to further examine the signaling mechanisms underlying these effects. Together, our data demonstrate that astrocytes modulate inhibitory synaptogenesis via distinct presynaptic and postsynaptic mechanisms and suggest that astrocytes enhance BDNF and tyrosine receptor kinase B (TrkB) signaling in neurons, thereby promoting the formation and postsynaptic localization of GABAAR clusters.
Materials and Methods
Cell cultures. Primary hippocampal neuronal cultures were prepared as described previously (Goslin et al., 1988) with minor modifications. Briefly, hippocampi were dissected from embryonic day 18 (E18) Sprague Dawley rats, dissociated for 20 min in Ca2+- and Mg2+-free HBSS containing 0.03% trypsin, triturated in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and plated at 100,000 cells/ml in DMEM supplemented with 10% FBS, 10% Ham's F-12 (Invitrogen), and 1% penicillin and streptomycin (Invitrogen) on poly-l-lysine-coated coverslips in 12-well plates. For pure neuronal cultures, cytosine arabinoside (AraC) (10 μm) was added to cultures 18-20 h after plating to prevent glial proliferation. Culture media was changed to Neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen) at 4 d in vitro (div). Cells were maintained at 37°C, 5% CO2, and 95% humidity in defined medium that was changed weekly.
The effects of acutely isolated and mature astrocytes (14-21 div) were examined in neuron-astrocyte cocultures. For acute astrocyte cocultures, neurons were prepared in DMEM supplemented with 10% FBS, 10% Ham's F-12, and 1% penicillin and streptomycin as described above, without the addition of AraC, allowing astrocyte proliferation. At 4 div, when ∼75-80% of cells were astrocytes (n = 35 coverslips), culture media was changed to Neurobasal medium with B27, and cocultures were maintained for up to 21 div. For mature astrocyte cocultures, neurons were plated onto confluent monolayers of astrocytes grown on coverslips and maintained for up to 21 div in Neurobasal medium. No differences were observed in the effects of acutely isolated and mature astrocytes on GABAAR clustering [fold increase in GABAAR cluster number per 20 μm dendrite compared with neuron-only controls, 2.35 ± 0.57 (25) and 2.34 ± 0.61 (25) in acute and mature cocultures at 4 div, respectively; p = 0.40; Student's t test]. Thus, acutely isolated astrocyte cocultures were used for experiments unless otherwise specified.
Primary astrocyte cultures were prepared as described previously (Duan et al., 2003; Zhang et al., 2003). Briefly, hippocampi were dissected and rinsed in cold HEPES-buffered Earle's balanced salt solution (EBSS), dissociated in 0.125% trypsin for 20 min, and plated in T25 flasks in modified minimum essential medium (MMEM) supplemented with 10% heat-inactivated FBS, 2 mm l-glutamine, 14 mm sodium bicarbonate, 40 mm d-glucose, 1% sodium pyruvate, and 1% penicillin and streptomycin. Astrocytes were allowed to proliferate for 14-21 d and, after reaching confluency, were rinsed in cold EBSS and shaken at 260 rpm for 18-20 h in MMEM to remove neurons and other cell types. Purified astrocytes were then plated onto poly-d-lysine-coated coverslips at 400,000 cells/ml in MMEM. Culture medium was changed to Neurobasal medium, and coverslips were used for direct cocultures or ACM treatments within 1-3 d. Primary fibroblast cultures were prepared from meninges of E18 Sprague Dawley rats following a similar protocol. Coverslips were immunostained with an antibody against glial fibrillary acidic protein (GFAP) (1:500; rabbit polyclonal; Chemicon, Temecula, CA) to determine the purity of astrocytes in these cultures. Only coverslips with >90% purity were used for conditioned medium experiments.
For pure neuronal cultures treated with conditioned medium, neurons were plated in Neurobasal medium that had been conditioned by astrocytes or fibroblasts (14-21 d of age) during the previous 24-72 h. Sterile inserts with 3 μm high-pore-density polyethylene terephthalate membranes (BD Biosciences, Franklin Lakes, NJ) were placed into each well, and coverslips with astrocyte or fibroblast monolayers were inverted 0.9 mm above neurons. Inserts remained in place throughout the culture duration.
Mice. To evaluate the role of BDNF and TrkB signaling, cultures were prepared from postnatal day 0 mice mutant for TrkB (Klein et al., 1993) (The Jackson Laboratory, Bar Harbor, ME) or BDNF (Ernfors et al., 1994) (The Jackson Laboratory) or from wild-type littermate controls. Mice were genotyped by PCR.
Growth factor or cholesterol treatments. Neurons were treated with 50 ng/ml BDNF or neurotrophin 3 (NT3) (Upstate Biotechnology, Lake Placid, NY) for 48 h at 8-11 div. Neurotrophin scavenging was performed by the addition of 2 μg/ml TrkB-IgG (binds BDNF and NT4/NT5), TrkC-IgG (binds NT3), or control IgGs (a gift from Regeneron Pharmaceuticals, Tarrytown, NY) at 1 div until neurons were immunostained at 4, 7, or 10 div. In a subset of experiments, cultures were treated with IgG constructs for 24-48 h at 8-11 div. All treatments were replenished after 24 h.
To examine the effects of cholesterol on inhibitory synapse formation, pure neuronal cultures were treated with Neurobasal medium with B27 alone, medium plus 10 μg/ml cholesterol (Mauch et al., 2001), or ACM for 48 h starting at 8 div or for 7 d starting at 3 div. In these experiments, neurons were treated with ACM collected from purified confluent astrocyte cultures on the day of treatment, and astrocyte monolayer inserts were not included in the culture wells. All treatments were replenished after 48 h. Neuronal cultures were immunostained at 10 div.
Immunostaining and confocal microscopy. Neurons were fixed in 4% paraformaldehyde and 4% sucrose for 15 min, permeabilized with cold 0.25% Triton X-100 for 5 min, and blocked in 5% normal goat serum for 1 h at room temperature. Double and triple labeling were performed with combinations of primary antibodies: anti-GABAAR-β2/β3 (1:100; monoclonal; Chemicon), GAD-64 (1:10; monoclonal; Developmental Studies Hybridoma Bank, Iowa City, IA), GFAP (1:500; rabbit polyclonal; Chemicon), microtubule-associated protein 2 (MAP2) (1:1000; polyclonal; a gift from Dr. Virginia Lee, University of Pennsylvania School of Medicine), synaptophysin (SP) (1:500; monoclonal; Sigma, St. Louis, MO), synaptophysin (1:200; rabbit polyclonal; NeoMarkers, Fremont, CA), vesicular GABA transporter (VGAT) (1:1000; guinea pig polyclonal; Chemicon) or VGAT (1:200; mouse monoclonal; Synaptic Systems, Göttingen, Germany), and vesicular glutamate transporter 1 (VGLUT) (1:1000; guinea pig polyclonal; Chemicon). Antibodies were visualized after staining with the appropriate FITC-, rhodamine isothiocyanate-, or cyanine 5-conjugated secondary antibodies (all used at 1:200; Jackson ImmunoResearch, West Grove, PA). Cell viability was assessed at 10 div using a terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay to label apoptotic nuclei (ApopTag fluorescein in situ apoptosis detection kit; Chemicon). Images were obtained using a laser-scanning confocal microscope (TCS 4D; Leica, Nussloch, Germany). In each image, laser light levels and detector gain and offset were adjusted so that no pixel values were saturated in regions analyzed.
Cell-surface biotinylation. Biotinylation assays were performed as described previously (Mammen et al., 1997; Jovanovic et al., 2004). Briefly, cultures were incubated with 1 mg/ml sulfo-biotin-N-hydroxysuccinimide ester (Pierce, Rockford, IL) for 30 min at 4°C at 10 div and washed twice with PBS supplemented with 1 mg/ml bovine serum albumin to remove excess biotin. Cell lysates were collected, and biotinylated cell-surface proteins were precipitated using UltraLink Immobilized NeutrAvidin biotin-binding protein (Pierce) and resolved by SDS-PAGE.
Western blot analysis. Total cell and surface protein extracts were harvested into Laemli's buffer from pure neuronal and neuron-astrocyte cocultures at 10 div. After SDS-PAGE, samples were transferred to nitrocellulose membranes and probed for antibodies to GABAAR-β3 (1:1000; rabbit polyclonal; a gift from Dr. S. J. Moss, University of Pennsylvania School of Medicine), neurofilament H (1:1000; monoclonal; Sternberger Monoclonals, Lutherville, MD), or Kv2.1 (1:1000; monoclonal; Chemicon) as loading controls for total and surface fractions, respectively. Alkaline phosphatase-conjugated goat anti-rabbit or anti-mouse antisera (1:5000; Applied Biosystems, Foster City, CA) were used, and signals were visualized using chemiluminescence (WesternStar detection system; Applied Biosystems). Films were digitally scanned, and signals were quantified using Universal Imaging (Downingtown, PA) MetaMorph software.
Electrophysiology. Whole-cell patch-clamp recording was performed at room temperature (20-25°C) on pyramidal neurons at 9-11 and 17-18 div using an Axopatch 200A amplifier and pClamp9 software (Axon Instruments, Union City, CA). Pyramidal neurons were distinguished from interneurons by cellular morphology, including a large pyramidal-shaped soma and the presence of a prominent apical dendrite. Patch pipettes with a resistance of 2-3 MΩ were filled with a solution containing the following (in mm): 140 KCl, 5 NaCl, 2 MgCl2, 1 CaCl2, 10 HEPES, 5 EGTA, 2 Mg-ATP, and 0.2 Li-GTP, pH 7.3, adjusted with KOH (319 mOsm). During recording, coverslips were continuously superfused with HEPES-buffered physiological salt solution composed of the following (in mm): 145 NaCl, 3 KCl, 1 MgCl2, 2 CaCl2, 10 dextrose, and 10 HEPES, pH 7.35, adjusted with NaOH (325 Osm). For recording spontaneous IPSCs, 50 μm APV and 10 μm CNQX were added to block NMDA and AMPA receptor-mediated currents, respectively. To record miniature IPSCs (mIPSCs), 1 μm TTX was also added to block action potentials. Pipette voltage offset was neutralized before the formation of a gigaohm seal. Membrane resistance (Rm), series resistance (Rs), and membrane capacitance (Cm) were determined from current transients elicited by a 5 mV depolarizing step from a holding potential of -60 mV, using the “membrane test” application of pClamp9. Criteria for cell inclusion in the data set included a Rs ≤ 15 MΩ and stability throughout the recording period, typically 2-3 min. Each recording was performed for a minimum of 2 min, IPSCs were amplified and then low-pass filtered at 2.5 kHz, and the sampling rate, performed using pClamp9, was 5 Hz.
The cumulative probability distributions of mIPSC interevent intervals were compared using the Kolmolgorov-Smirnov nonparametric test. Differences in mIPSC amplitude were compared using Student's t test.
Quantification and statistical analysis. For each condition, a minimum of 6-10 randomly selected neurons were examined on each of three coverslips in three to six independent experiments. Pyramidal neurons were distinguished from interneurons by pyramidal morphology and lack of anti-GAD immunoreactivity. In all experiments, the number and colocalization of SP+ and VGAT+ terminals and the number and synaptic localization of GABAARs were determined from confocal images using interactive software (MetaMorph; Universal Imaging). Images were thresholded, and the number of individual clusters along every dendrite of analyzed neurons was determined. Values are presented as mean ± SEM (number of cells). Values for cluster number were compared using the Kruskal-Wallis nonparametric ANOVA test, followed by Dunn's pairwise multiple-comparison test.
Pixel overlap of SP+ clusters with VGAT+ or VGLUT+ clusters was used to distinguish between inhibitory and excitatory terminals, respectively. To quantify synaptic localization of GABAARs, receptor clusters with pixel overlap with SP+ clusters were considered synaptic. For each parameter, the percentage of colocalization between cultures was compared using Student's t test.
Results
Astrocytes increase inhibitory presynaptic terminals and postsynaptic GABAAR clusters
To study the effects of astrocytes on inhibitory synapse formation, we first compared the development of presynaptic and postsynaptic specializations in low-density embryonic rat hippocampal neurons cultured in the presence and absence of astrocytes during the first 10 d in vitro. Immunostaining and confocal microscopy were performed at 4, 7, and 10 div using antibodies against SP to label presynaptic terminals, VGAT to distinguish inhibitory terminals, and the GABAAR-β2/β3 subunits to visualize postsynaptic receptors.
In the absence of astrocytes, the number of SP+ boutons and GABAAR clusters gradually increased during the first week in vitro, consistent with previous reports (Table 1) (Brunig et al., 2001; Elmariah et al., 2004). At 4 div, few SP+ terminals were observed, and <2% of these inputs colocalized with VGAT+ clusters, suggesting that inhibitory terminals were rare (Table 1). In a separate experiment, immunostaining with antibodies against SP, VGAT, and VGLUT to label glutamatergic terminals confirmed the presence of VGLUT+ clusters at 4 div and demonstrated that ∼80% of SP+ terminals were excitatory (Table 1). Consistent with the observation of few inhibitory presynaptic terminals, postsynaptic GABAAR immunoreactivity appeared diffuse throughout somata and proximal dendrites of pyramidal neurons with few clusters present (Table 1). The number of presynaptic terminals steadily increased over time in culture to 4.8 SP+ boutons per 20 μm dendritic segment at 7 div and 6.8 boutons per 20 μm at 10 div (Fig. 1A; Table 1). VGLUT+ clusters increased at a similar rate and continued to represent the majority of presynaptic terminals (Table 1). Although VGAT expression increased in the soma and axons of interneurons during the first week, the number of inhibitory synaptic contacts represented only ∼15% of synaptic contacts at 10 div (Fig. 1A; Table 1). By 7-10 div, GABAAR clusters had begun to form, and ∼10% of GABAAR clusters were apposed to presynaptic terminals (Fig. 1A; Table 1). Thus, inhibitory synaptogenesis occurs slowly in the absence of astrocytes, with few inhibitory presynaptic terminals or postsynaptic GABAAR clusters present during the first 2 weeks in vitro.
Table 1.
Astrocyte modulation of presynaptic terminals and GABAAR clusters
|
|
Number of SP boutons (per 20 μm segment) |
Number of VGAT boutons (per 20 μm segment) |
Number of VGLUT boutons (per 20 μm segment) |
Number of GABAAR clusters (per 20 μm segment) |
Percentage of GR colocalization with SP + boutons |
|---|---|---|---|---|---|
| 4 div | |||||
| Pure neuronal cultures | 2.0 ± 0.4 (55) | 0.8 ± 0.1 (88) | 1.7 ± 0.2 (42) | 1.9 ± 0.2 (38) | 0.5 ± 0.2 |
| Astrocyte cocultures | 7.9 ± 0.5 (58)** | 5.4 ± 0.6 (85)** | 4.6 ± 0.5 (46)** | 4.2 ± 0.2 (32)** | 13.3 ± 0.9** |
| ACM-treated cultures | 7.9 ± 0.8 (52)** | 5.1 ± 0.4 (87) | 4.3 ± 0.6 (47)* | 3.9 ± 0.3 (35)* | 13.7 ± 0.7** |
| FCM-treated cultures | 2.2 ± 0.5 (25) | 1.1 ± 0.3 (25) | n.d. | 2.4 ± 0.3 (25) | 0.8 ± 0.6 |
| 7 div | |||||
| Pure neuronal cultures | 4.8 ± 0.3 (55) | 2.0 ± 0.40 (68) | 4.0 ± 0.7 (53) | 2.2 ± 0.2 (35) | 6.0 ± 0.5 |
| Astrocyte cocultures | 9.7 ± 0.6 (55)** | 7.9 ± 0.7 (60)** | 7.2 ± 0.4 (52)* | 4.9 ± 0.3 (35)** | 31.9 ± 2.3** |
| ACM-treated cultures | 9.1 ± 0.4 (55)** | 8.3 ± 0.8 (60)** | 8.5 ± 0.6 (52)** | 4.8 ± 0.2 (38)** | 36.7 ± 1.1** |
| FCM-treated cultures | 4.4 ± 1.0 (25) | 2.1 ± 0.3 (25) | n.d. | 3.2 ± 0.6 (25) | 8.6 ± 2.2 |
| 10 div | |||||
| Pure neuronal cultures | 6.8 ± 0.6 (42) | 3.8 ± 0.4 (50) | 5.8 ± 0.4 (25) | 3.4 ± 0.2 (35) | 10.1 ± 1.0 |
| Astrocyte cocultures | 13.4 ± 0.5 (36)** | 11.2 ± 0.9 (42)** | 8.9 ± 0.6 (25)* | 8.0 ± 0.5 (36)** | 39.1 ± 3.9** |
| ACM-treated cultures | 12.8 ± 0.6 (41)** | 9.3 ± 0.9 (48)** | 10.2 ± 0.5 (25)** | 8.4 ± 0.2 (34)** | 37.1 ± 2.9** |
| FCM-treated cultures | 6.5 ± 0.8 (25) | 2.3 ± 0.6 (25) | n.d. | 4.6 ± 0.8 (25) | 11.0 ± 2.9 |
|
|
Number of SP boutons (per 20 μm segment) |
Number of VGAT boutons (per 20 μm segment) |
Number of VGLUT boutons (per 20 μm segment) |
Number of GABAAR cluster (per 20 μm segment) |
Percentage of GR colocalization with VGAT + boutons |
|---|---|---|---|---|---|
| 10 div | |||||
| Pure neuronal cultures | 1.3 ± 0.2 (15) | 0.7 ± 0.2 (15) | n.d. | 2.4 ± 0.3 (15) | 3.8 ± 1.9 |
| Cholesterol-treated cultures | 1.7 ± 0.2 (18) | 0.9 ± 0.2 (18) | n.d. | 2.2 ± 0.2 (18) | 1.8 ± 0.2 |
| ACM-treated cultures |
2.7 ± 0.4T (12)***
|
1.4 ± 0.3 (12)***
|
n.d. |
3.4 ± 0.4 (12) |
14.7 ± 4.0***
|
Values are shown as mean ± SEM (number of cells from 3-6 separate experiments). In the three experiments at 4, 7, and 10 div, neurons were treated with ACM using a feeder layer of astrocytes on a membrane inverted in the culture well. In the fȯurth experiment at 10 div, ACM collected from purified astrocyte monolayers was added directly to neuronal cultures, and feeder layers were not used (for details, see Materials and Methods). In all experiments, cluster number was compared among conditions using Kruskal-Wallis ANOVA followed by Dunn's pairwise multiple-comparison test. The percentage of presynaptic and postsynaptic colocalization among cultures was compared using Student's t test. *p < 0.001, **p < 0.0001, and ***p < 0.05, significantly different from pure neuronal cultures. n.d., Not determined.
Figure 1.
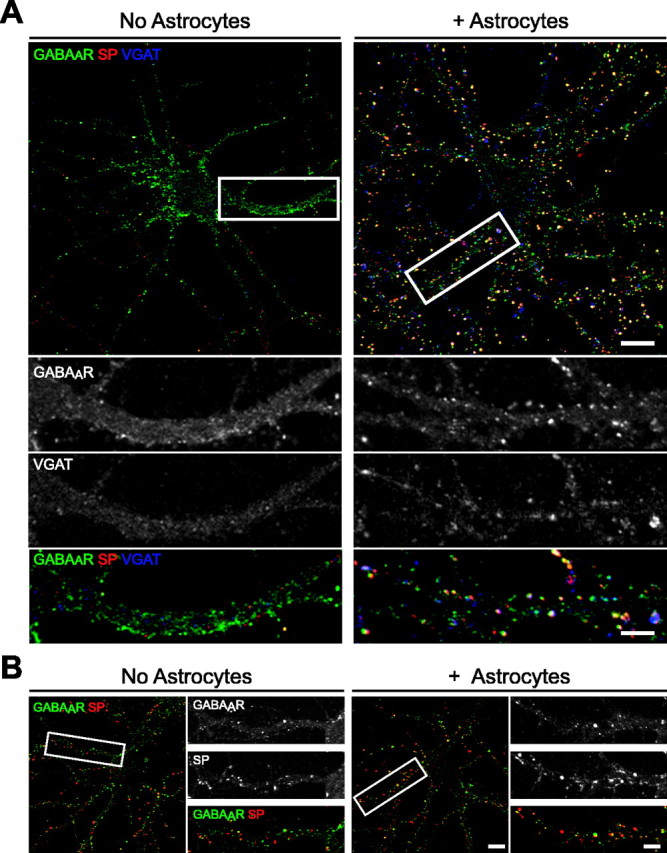
Astrocytes increase the number of inhibitory presynaptic terminals and postsynaptic GABAAR clusters in hippocampal neurons in vitro. Hippocampal neurons were cultured in the presence or absence of astrocytes and immunostained with antibodies against GABAAR-β2/β3 (green), SP (red), and VGAT (blue) to visualize inhibitory presynaptic terminals. A, The number of SP+ and VGAT+ presynaptic terminals and the number of GABAAR clusters increased in neuron-astrocyte cocultures (right) compared with pure neuronal cultures (left) at 10 div. The proportion of GABAAR clusters apposed to presynaptic terminals also increased in neuron-astrocyte cocultures (Table 1). Scale bar, 10 μm. Areas within white boxes are shown in the panels below at a higher magnification. Scale bar (bottom right), 2 μm. B, At 3 weeks in vitro, the proportion of GABAAR clusters localized to synapses (yellow) was approximately threefold greater in neuron-astrocyte cocultures (right) than in pure neuronal cultures (left). No differences were observed in the number of presynaptic terminals (red) or the number of GABAAR clusters (green). Scale bar, 10 μm. Areas within white boxes are shown to the right at a higher magnification. Scale bar (bottom right), 2 μm.
When neurons were cultured in the presence of astrocytes, the rate of overall synaptogenesis and, in particular, inhibitory synapse formation, was dramatically increased at all ages. At 4 div, the total number of SP+ presynaptic terminals increased by severalfold in the presence of astrocytes and appeared similar to levels observed in pure neuronal cultures at 10 div (Table 1). The astrocyte-induced increase in presynaptic terminals was most robust at younger ages in vitro; however, levels remained at least twofold greater compared with controls at 7 and 10 div. Consistent with the increase in SP+ terminals, more VGAT+ and VGLUT+ clusters were observed in neuron-astrocyte cocultures compared with neuron-only cultures (Table 1). VGAT+ clusters increased by >6.5-fold at 4 div, and by 7 and 10 div, inhibitory terminals were widely observed contacting somata and proximal dendrites of pyramidal neurons in astrocyte cocultures (Fig. 1A; Table 1). In contrast, VGLUT+ clusters increased by only approximately twofold in the presence of astrocytes, such that the proportion of inhibitory and excitatory SP+ terminals appeared similar in astrocyte cocultures (Table 1). That is, in contrast to pure neuronal cultures, in which inhibitory synaptic contacts were rare, VGAT+ terminals consistently represented ∼40-50% of all SP+ presynaptic contacts in neuron-astrocyte cocultures (Fig. 1A). Thus, astrocytes promote synaptogenesis by enhancing the formation of presynaptic terminals during maturation in vitro, robustly upregulating the number and proportion of inhibitory presynaptic contacts in developing hippocampal networks.
Astrocytes also increased the number of GABAAR clusters that had formed at 4, 7, and 10 div by over twofold relative to controls (Fig. 1A; Table 1). Moreover, the proportion of GABAAR clusters apposed to presynaptic terminals was significantly increased, such that postsynaptic GABAAR clusters were present in cultures as early as 4 div, and >30% were synaptically localized at 7 and 10 div. As cultures matured, the effects of astrocytes on cluster number appeared less robust, because approximately the same number of GABAAR clusters had formed in pure neuronal and neuron-astrocyte cocultures by 3 weeks in vitro (Fig. 1B). In contrast, the proportion of GABAAR clusters localized to presynaptic sites remained greater in the presence than in the absence of astrocytes in older cultures (Fig. 1B). These observations suggest that astrocytes accelerate the rate of GABAAR cluster formation during early maturation in vitro and increase the number of inhibitory synapses, in part by increasing the localization of GABAAR clusters to synaptic sites.
We next asked whether the increase in inhibitory synapses resulted from a change in cell viability or interneuron density in the presence of astrocytes. Cell viability was assessed at 10 div using a TUNEL assay to label apoptotic nuclei. Few, if any, TUNEL+ nuclei were observed in either culture condition, and no differences were observed in the frequency of cell death in neuron-only and neuron-astrocyte cocultures [percentage of TUNEL+ nuclei per 16× field (62,500 μm2), 0.2 ± 0.2 (n = 1682 cells) and 0.2 ± 0.1 (n = 1263 cells), p = 0.7, Student's t test; in pure neuronal and neuron-astrocyte cocultures, respectively, not statistically different, Student's t test]. Moreover, analysis of cultures immunostained with antibodies against MAP2 to visualize neuronal processes, immunostained with GAD to distinguish inhibitory interneurons, and stained with 4′,6′-diamidino-2-phenylindole dihydrochloride to label nuclei revealed no differences in neuronal density or the proportion of interneurons in the presence or absence of astrocytes [number of neurons per 16× field, 109.4 ± 14.1 (n = 10 fields) and 103.3 ± 11 (n = 10 fields) in pure neuronal cultures and neuron-astrocyte cocultures, respectively, p = 0.7, Student's t test; percentage of interneuron population: 15.4 ± 1.9 (n = 17 fields) and 12.2 ± 2.8 (n = 18 fields) in pure neuronal cultures and neuron-astrocyte cocultures, respectively, p = 0.3, Student's t test]. Finally, no significant differences were observed in the number or length of primary dendrites or in soma size of interneurons in the presence of astrocytes relative to controls [number of primary dendrites, 5.1 ± 0.2 (n = 65 cells) and 5.1 ± 0.3 (n = 62 cells); length of dendrites (in micrometers), 48.0 ± 1.6 and 47.1 ± 2.1; soma size (in square micrometers): 290.0 ± 12.6 and 273.2 ± 12.0 in pure neuronal and neuron-astrocyte cocultures, respectively; not statistically different; Student's t test). Similarly, pyramidal neuron morphology was unaffected in neuron astrocyte cocultures relative to controls [number of primary dendrites, 5. 9 ± 0.4 (n = 40 cells) and 5.6 ± 0.5 (n = 38 cells); length of dendrites (in micrometers), 44.3 ± 4.9 and 41.7 ± 5.1; soma size (in square micrometers), 347.9 ± 26.4 and 387.5 ± 26.2 in pure neuronal and neuron-astrocyte cocultures, respectively; not statistically different; Student's t test]. These results suggest that the increased number of inhibitory synapses observed in neuron-astrocyte cocultures is not attributable to an increase in the number or density of interneurons or the result of nonspecific trophic effects on cell health.
To determine whether glial modulation of inhibitory presynaptic terminals and/or GABAAR clusters required local contact or the release of soluble factors by astrocytes, pure neuronal cultures were treated with ACM during maturation in vitro. Neurons were plated onto coverslips in ACM, and astrocyte monolayers were plated onto separate coverslips that were inverted above neurons until immunostaining was performed at 4, 7, or 10 div. ACM treatment mimicked the effects of astrocyte coculture on both presynaptic terminal and GABAAR cluster number as well as their colocalization. In the presence of ACM, inhibitory presynaptic terminals increased by approximately fourfold, and the number of GABAAR clusters was increased by at least twofold throughout the first week in vitro, consistent with observations in cocultures (Fig. 2A,B; Table 1). Moreover, ACM treatment also increased the proportion of GABAAR clusters apposed to presynaptic terminals relative to untreated controls at all ages in vitro, to the same extent as in cocultures (Fig. 2B; Table 1). In contrast, treatment with fibroblast-conditioned medium had no effect on presynaptic terminal or GABAAR cluster number and synaptic localization (Table 1). These results show that the increase in presynaptic terminal and GABAAR cluster number, and their colocalization, were specific to ACM.
Figure 2.

Astrocyte-conditioned medium increases the number of inhibitory synapses in hippocampal neurons in vitro. Hippocampal neurons were cultured in the presence of ACM, and inhibitory presynaptic terminals or GABAAR clusters were examined at 4, 7, and 10 div. A, Immunostaining was performed with antibodies against VGAT (green) and SP (red) to visualize inhibitory presynaptic terminals. The number of SP+ boutons and the proportion of VGAT+ inhibitory terminals increased after ACM treatment (bottom) compared with untreated controls (top). Scale bar, 2 μm. B, Immunostaining was performed with antibodies against GABAAR-β2/β3 (green) and SP (red). ACM treatment (bottom) resulted in an increase in the number and synaptic localization of GABAAR clusters (green) compared with pure neuronal cultures (top). Scale bar, 2 μm.
Previous work showed that cholesterol is a component of ACM that was sufficient to increase synaptic number and function (Mauch et al., 2001). To test this possibility, cholesterol was added to purified neuronal cultures grown in Neurobasal medium. No increase in VGAT+ terminal number, GABAAR cluster number, or colocalization with inhibitory presynaptic terminals was observed after 48 h or 7 d of treatment with 10 μg/ml cholesterol compared with untreated controls (Table 1). Thus, the addition of cholesterol was not sufficient to mimic the effects of ACM on inhibitory synapses. Together, these results suggest that soluble factors other than cholesterol, released specifically from astrocytes, regulate the formation of inhibitory presynaptic terminals and postsynaptic GABAAR clusters in hippocampal pyramidal neurons.
Astrocytes increase GABAAR clusters expressed at the cell surface
We next asked whether astrocyte modulation of GABAAR clustering and localization was accompanied by changes in total and/or cell-surface GABAAR expression. Protein lysates were collected from cells or after surface biotinylation and avidin precipitation in pure neuronal and neuron-astrocyte cocultures at 10 div, and Western blot analysis was performed.
No significant changes were observed in total GABAAR expression in neurons cultured in the presence or absence of astrocytes, suggesting that astrocytes modulate the distribution of GABAA receptors but do not alter receptor expression or degradation (Fig. 3A). In contrast, levels of GABAAR protein expressed at the cell surface increased by over threefold in neuron-astrocyte cocultures relative to pure neuronal cultures (Fig. 3B). These data suggest that, in the absence of astrocytes, GABAAR proteins are expressed, but the signals necessary for inducing cluster assembly are low or absent. Thus, astrocytes provide cues that promote the assembly of existing GABAARs into clusters and enhance the localization of GABAARs or GABAAR clusters in neuronal membranes.
Figure 3.

Surface GABAAR expression is increased in hippocampal neurons in the presence of astrocytes. Total cell lysates or biotinylated surface protein extracts were harvested from hippocampal neurons cultured in the presence and absence of astrocytes at 10 div. A, Western blot analysis on total cell homogenates was performed using an antibody against the GABAAR-β3 subunit (top) or neurofilament H (bottom) as a loading control. Quantification of relative band intensity compared with loading controls demonstrates no significant difference in the level of GABAAR-β3 expression in the presence of astrocytes. B, Western blot analysis on surface-biotinylated extracts was performed using an antibody against the GABAAR-β3 subunit (top) or Kv2.1 (bottom) as a loading control. Quantification of relative band intensity compared with loading controls shows that GABAAR-β3 expression at the neuronal surface is increased ∼3.5-fold when neurons are cultured in the presence of astrocytes (*p < 0.001). Error bars represent SEM.
Astrocytes increase the frequency of spontaneous inhibitory currents
To determine whether the astrocyte-induced increase in inhibitory terminals and postsynaptic GABAAR clusters resulted in an increase in the number of functional inhibitory connections, whole-cell patch-clamp recordings from pyramidal neurons were performed to examine IPSCs in the presence and absence of astrocytes.
Before the examination of inhibitory transmission, total neuronal activity was assessed at 10 div by recording all PSCs in the absence of glutamate receptor antagonists. In the absence of astrocytes, ∼16% of neurons exhibited spontaneous PSCs at a low frequency (0.09 ± 0.01 Hz; n = 2 of 12 cells). In contrast, spontaneous PSCs were observed in >80% of pyramidal neurons in neuron-astrocyte cocultures and occurred with a mean frequency of 1.5 ± 0.6 Hz (n = 18 of 22 cells). In cocultures in which spontaneous activity was robust, IPSCs were pharmacologically isolated in the presence of APV and CNQX to block excitatory currents. Spontaneous IPSCs were observed at a mean frequency of 0.07 ± 0.01 Hz (range, 0.02-0.1 Hz; n = 10), and currents were 35.0 ± 4.1 pA (n = 17) in amplitude. The low level of spontaneous activity observed in pure neuronal cultures at 10 div before isolation of IPSCs precluded a direct quantitative comparison of the frequency and the amplitude of IPSCs between neurons cultured with or without astrocytes. Nonetheless, these data suggest that astrocytes increase the number of functional inhibitory connections by 10 div.
Whole-cell recordings were also performed at 17-18 div, when spontaneous inhibitory activity could be detected more reliably in pure neuronal cultures and mIPSCs could be isolated. The mean mIPSC frequency in neuron-astrocyte cocultures was twofold greater than in pure neuronal cultures (Fig. 4A) [1.1 ± 0.2 Hz (n = 12) and 2.0 ± 0.3 Hz (n = 13) in pure neuronal cultures and neuron-astrocyte cocultures, respectively; p = 0.03; Student's t test]. Because mIPSCs were not uniformly distributed throughout the recording period, interevent intervals were analyzed, and cumulative histograms of these intervals were compared (Fig. 4B). In the presence of astrocytes, the histogram of interevent intervals is shifted to the left, suggesting that the intervals between mIPSCs are shorter, consistent with an increase in the mean mIPSC frequency (Fig. 4B). In contrast, no difference was observed in the mean amplitude of mIPSCs in the presence or absence of astrocytes (Fig. 4C) [mean amplitude, 46 ± 0.8 pA (n = 12 cells; 1405 events) and 46 ± 0.8 pA (n = 13 cells; 2376 events); p = 0.8; Student's t test]. These data demonstrate that inhibitory transmission is elevated in neuron-astrocyte cocultures relative to controls, because of an increase in the number of inhibitory synapses and/or in the release probability of presynaptic nerve terminals. The astrocyte-induced increase in inhibitory synaptic activity is consistent with structural observations that astrocytes increase the number of inhibitory contacts.
Figure 4.

Spontaneous inhibitory activity is increased in the presence of astrocytes. Whole-cell voltage-clamp recordings were performed on hippocampal pyramidal neurons at 17-18 div to examine mIPSCs in the presence and absence of astrocytes. A, Representative recordings from two neurons in the presence and absence of astrocytes. mIPSCs were recorded in the presence of the following (in μm): 1 TTX, 50 APV, and 10 CNQX. mIPSCs occurred more frequently in neuron-astrocyte cocultures (right) relative to pure neuronal cultures (left). Calibration: vertical, 0.1 nA; horizontal, 0.2 s. B, Cumulative probability distribution of the interevent interval of mIPSCs in pure neuronal and neuron-astrocyte cocultures. *p < 0.0001, significant difference compared with pure neuronal cultures (Kolmogorov-Smirnov test). C, Quantification of mIPSC amplitude in pure neuronal and neuron-astrocyte cocultures (p = 0.77; Student's t test). Error bars represent SEM.
Neurotrophins mediate astrocyte modulation of postsynaptic GABAAR clusters
These results suggest that diffusible factors exchanged between astrocytes and neurons promote the formation of functional inhibitory synapses. Neurotrophins, which can be released by and bind to Trk receptors expressed by neurons and glia, are known to play important roles in the development of inhibitory synapses presynaptically and postsynaptically, thus making them attractive candidates for mediating the effects of astrocytes on inhibitory synaptogenesis. In interneurons, BDNF enhances GAD mRNA and protein expression and facilitates presynaptic GABA release (Vicario-Abejon et al., 1998; Marty et al., 2000; Yamada et al., 2002). In target pyramidal neurons, BDNF and TrkB signaling dynamically modulates the assembly, surface stability, and synaptic localization of GABAAR clusters (Brunig et al., 2001; Elmariah et al., 2004; Jovanovic et al., 2004) and increases postsynaptic GABAA receptor conductance (Rutherford et al., 1997) and mIPSC amplitude (Li et al., 1998; Vicario-Abejon et al., 1998; Marty et al., 2000; Seil and Drake-Baumann, 2000). These studies suggest that neurotrophin and Trk signaling may play a role in mediating the effects of astrocytes on the formation of presynaptic terminals and/or postsynaptic GABAAR clusters.
We tested this hypothesis by examining the effects of astrocytes or ACM on inhibitory terminals and GABAAR clusters when neurotrophin levels were reduced. Scavenging of endogenous BDNF and NT3 was achieved using Trk-IgG fusion proteins, which bind and sequester neurotrophins, preventing their binding to and activation of surface Trk receptors (Binder et al., 1999; Elmariah et al., 2004). Hippocampal cultures were treated with 2.0 μg/ml TrkB-IgG to scavenge BDNF, TrkC-IgG to scavenge NT3, or control IgG. Treatments began at 1 div and were replenished daily until immunostaining was performed at 4, 7, or 10 div.
Scavenging endogenous BDNF reduced the number of postsynaptic GABAAR clusters in pure neuronal cultures but had no effect on the number of inhibitory presynaptic terminals, consistent with our previous results (Elmariah et al., 2004). In the presence of TrkB-IgG, fewer GABAAR clusters were observed at 4, 7, or 10 div in pure neuronal cultures (Fig. 5A-C). Although no differences were observed in the number of presynaptic terminals between TrkB-IgG-treated and untreated cultures, BDNF scavenging reduced the proportion of GABAAR clusters localized to synapses by one-half during the first week in vitro (Fig. 5A-C). Moreover, scavenging BDNF prevented the increase in postsynaptic GABAAR clusters observed in astrocyte-neuron cocultures or after ACM treatment. The decrease in synaptic GABAAR clusters primarily reflected postsynaptic changes, because the number of inhibitory presynaptic terminals remained elevated in cocultures relative to pure neuronal cultures, despite reduced BDNF levels (Fig. 5A-C). Consistent with this hypothesis, TrkB-IgG treatment led to an approximately sixfold reduction in the number of GABAAR clusters and an approximately eightfold decrease in their synaptic localization relative to untreated cocultures at 10 div (Fig. 5A-C). Scavenging BDNF not only abolished the increase in postsynaptic GABAAR clusters induced by astrocytes but resulted in clustering similar to that observed in pure neuronal cultures after scavenging. These results suggest that astrocytes coordinately modulate the presynaptic and postsynaptic development of inhibitory synapses via Trk-dependent and -independent mechanisms and that BDNF signaling is required for neuronal as well as glial modulation of GABAAR clusters at inhibitory synapses.
Figure 5.

Scavenging endogenous BDNF prevents the astrocyte-induced increase in the number and synaptic localization of GABAAR clusters. Hippocampal cultures with and without astrocytes were treated with 2.0 μg/ml TrkB-IgG to scavenge endogenous BDNF or with TrkC-IgG to scavenge NT3 at different ages in vitro. Immunostaining was performed with antibodies against GABAAR-β2/β3 (green) and SP (red) at 4, 7, or 10 div. A, Treatment with 2.0 μg/ml TrkB-IgG beginning at 1 div resulted in fewer postsynaptic GABAAR clusters in pure neuronal cultures (top) and neuron-astrocyte cocultures (bottom) at 4, 7, and 10 div. No change was observed in the number of SP+ boutons after BDNF scavenging. Scale bar, 2 μm. B, Quantification of TrkB-IgG effects on the number of GABAAR clusters per 20 μm dendrite segment. *p < 0.001, significant difference compared with no treatment in pure neuronal cultures; **p < 0.001, significant decrease compared with no treatment in neuron astrocyte cocultures. C, Quantification of TrkB-IgG effects on the synaptic localization of GABAAR clusters. *p < 0.001, significant difference compared with no treatment in pure neuronal cultures; **p < 0.001, significant decrease compared with no treatment in neuron astrocyte cocultures. D, Treatment with 2.0 μg/ml TrkB-IgG at 8-10 div for 48 h decreased the number of postsynaptic GABAAR clusters in neuron-astrocyte cocultures (middle) (Table 2) compared with untreated controls (left). In contrast, treatment with 2.0 μg/ml TrkC-IgG for 48 h increased the proportion of GABAAR clusters localized to synapses but had no effect on the number of GABAAR clusters (right) (Table 2). No change was observed in the number of SP+ boutons after BDNF or NT3 scavenging. Scale bar, 2 μm. Error bars represent SEM.
We next asked whether TrkB signaling was necessary to maintain the increase in postsynaptic GABAAR clusters induced by astrocytes. Cultures were treated with 2.0 μg/ml TrkB-IgG for 24-48 h at 8-10 div, after which astrocytes or ACM have already induced a robust increase in the number of postsynaptic GABAAR clusters (compare Figs. 1, 2, and Table 1). Scavenging available BDNF levels in cultures containing astrocytes or treated with ACM even transiently resulted in significantly fewer GABAAR clusters and a decrease in the proportion GABAAR clusters localized to synapses (Fig. 5D; Table 2). Together, these findings indicate that BDNF signaling is necessary for and mediates, at least in part, astrocyte promotion of GABAAR cluster formation and synaptic maintenance.
Table 2.
Effects of neurotrophin manipulations on astrocyte modulation of postsynaptic GABAAR clusters
|
|
Number of GABAAR clusters (per 20 μm dendritic segment) |
Percentage of GR colocalization with SP + boutons |
|---|---|---|
| Pure neuronal cultures | 3.4 ± 0.2 (35) | 10.1 ± 1.0 |
| + 50 ng/ml BDNF | 10.2 ± 1.3 (32)* | 32.7 ± 3.4* |
| + 2 μg/ml TrkB-lgG | 0.9 ± 0.3 (31)* | 5.7 ± 0.5* |
| + 50 ng/ml NT3 | 3.5 ± 0.2 (35) | 8.8 ± 1.8 |
| + 2 μg/ml TrkC-lgG | 3.0 ± 0.5 (35) | 9.5 ± 1.2 |
| Astrocyte cocultures | 8.0 ± 0.5 (36)* | 39.1 ± 3.9 |
| + 50 ng/ml BDNF | 11.3 ± 1.1 (30)** | 46.9 ± 2.1 |
| + 2 μg/ml TrkB-lgG | 1.2 ± 0.3 (30)# | 4.8 ± 1.3# |
| + 50 ng/ml NT3 | 7.4 ± 0.3 (35) | 9.2 ± 2.8# |
| + 2 μg/ml TrkC-lgG | 9.0 ± 0.4 (35) | 58.8 ± 4.9** |
| + BDNF + NT3 | 12.0 ± 0.9 (25)# | 25.0 ± 3.1** |
| + TrkB-lgG + TrkC-lgG |
1.7 ± 0.4 (25)#
|
5.9 ± 1.2#
|
Values are shown as mean ± SEM (number of cells from 3-6 separate experiments). Cluster number was compared between conditions using Kruskal-Wallis ANOVA followed by Dunn's pairwise multiple-comparison test. The percentage of synaptic localization between cultures was compared using Student's t test. *p < 0.001, significantly different from pure neuronal cultures (no treatment); **p < 0.01, significantly different from neuron-astrocyte coculture (no treatment); #p < 0.001, significantly different from neuron-astrocyte coculture (no treatment).
We next asked whether the increase in GABAAR clustering and synaptic localization was specific to BDNF. In parallel experiments, the role of NT3 signaling in GABAAR cluster modulation in the presence and absence of astrocytes was examined. At early ages in vitro, scavenging endogenous NT3 with 2.0 μg/ml TrkC-IgG had no effect on the number of GABAAR clusters observed in pure neuronal or neuron-astrocyte cocultures compared with untreated controls (data not shown). Similarly, no change was observed in the number or postsynaptic localization of GABAAR clusters when NT3 levels were reduced at 8-10 div in pure neuronal cultures, consistent with previous results (Table 2) (Elmariah et al., 2004). In the presence of astrocytes, however, scavenging NT3 for 48 h significantly increased the proportion of synaptically localized GABAAR clusters without changing the number of clusters observed at 10 div (Fig. 5E; Table 2). These observations indicate that, in contrast to BDNF signaling, NT3 signaling attenuates the localization of GABAAR clusters to synapses when neurons are cultured with astrocytes. This finding suggests that BDNF and NT3 signaling may be antagonistic with respect to inhibitory synapse formation.
We next examined the interactions of BDNF and NT3 signaling in modulating synaptic localization of GABAAR clusters in the presence of astrocytes. Neuron-astrocyte cocultures were treated with 50 ng/ml recombinant BDNF, NT3, or both, at 8-10 div. Treatment with BDNF increased postsynaptic GABAAR clusters dramatically in pure neuronal cultures and, to a lesser degree, in neuron-astrocyte cocultures (Table 2). In contrast, NT3 treatment had no effect on postsynaptic GABAAR clusters in the absence of astrocytes and decreased the synaptic localization of GABAAR clusters in neuron-astrocyte cocultures (Table 2). When neuron-astrocyte cocultures were treated with BDNF plus NT3, the number of GABAAR clusters appeared similar to BDNF-treated cultures, but fewer synaptically localized GABAAR clusters were observed (Table 2). These results suggest that NT3 reduces the synaptic localization of GABAAR clusters in the presence of astrocytes. In contrast, cultures treated with TrkB-IgG and TrkC-IgG exhibited low levels of postsynaptic GABAAR clusters, similar to TrkB-IgG treatment alone, suggesting that BDNF must be present to increase GABAAR cluster formation and synaptic localization (Table 2). Together, these findings suggest that astrocytes dynamically modulate the number of postsynaptic GABAAR clusters, and thus inhibitory connections, via the regulation of neurotrophin and Trk signaling.
Astrocyte-mediated upregulation of GABAAR clusters requires neuronal BDNF release and TrkB activation
Previous work from our laboratory showed that BDNF and TrkB signaling in pure neuronal cultures are important for the formation and maintenance of GABAAR clusters (Elmariah et al., 2004). Hippocampal pyramidal neurons produce and release BDNF, which binds and activates TrkB receptors on postsynaptic pyramidal neurons or interneurons (Ernfors et al., 1990; Du et al., 2000; Ivanova and Beyer, 2001; Kohara et al., 2001). Astrocytes may modulate the magnitude or efficacy of this signaling between presynaptic and postsynaptic neurons, thereby enhancing inhibitory synapse formation. However, hippocampal astrocytes in vitro express BDNF mRNA and can release BDNF after stimulation (Condorelli et al., 1994; Alderson et al., 2000; Ivanova and Beyer, 2001; Miklic et al., 2004). Thus, one alternative explanation for astrocyte effects on GABAAR clustering and synaptic localization is that astrocytes condition the culture medium with BDNF. To determine the relative importance of neurotrophin release from neurons and astrocytes on inhibitory synaptogenesis, neurons and astrocytes from postnatal mice deficient in BDNF and wild-type littermates were cultured in several combinations.
We first examined whether astrocyte-dependent modulation of GABAAR clusters required neuronal release of BDNF. Hippocampal neurons were cultured from postnatal BDNF-deficient mice or wild-type littermates in the presence or absence of wild-type ACM, and GABAAR clusters were examined after 10 div. In pure neuronal cultures from mutant mice, the formation and synaptic localization of GABAAR clusters was reduced in the absence of TrkB signaling relative to control wild-type cultures. Although the number of presynaptic terminals was similar, fewer postsynaptic GABAAR clusters were observed at 10 div in BDNF-deficient pyramidal neurons relative to wild-type neurons (Fig. 6A-C). In contrast to wild-type neurons, no changes were observed in GABAAR cluster number or synaptic localization at 10 div when BDNF-deficient neurons were cultured in the presence of wild-type ACM (Fig. 6A-C). These results suggest that astrocyte modulation of postsynaptic GABAAR clusters requires neuronal BDNF.
Figure 6.
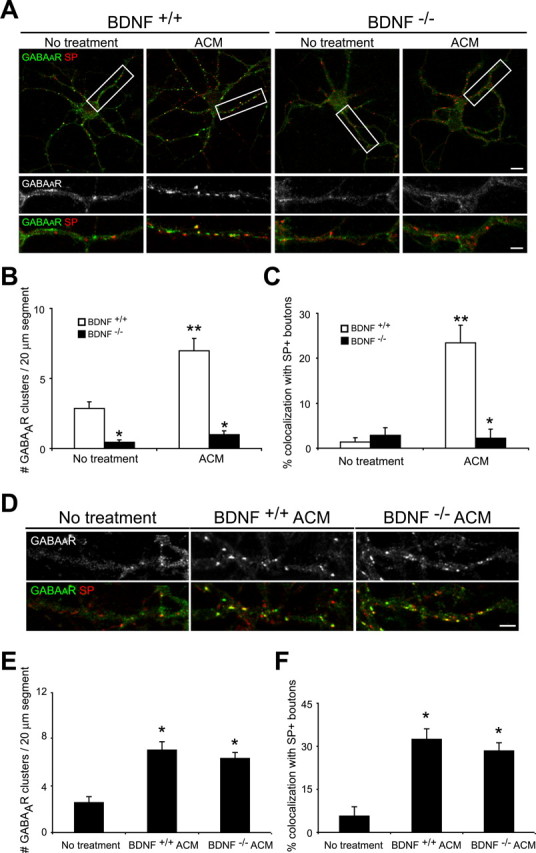
Neuronal BDNF release is required for the astrocyte-induced increase in postsynaptic GABAAR clusters. A-C, Hippocampal neurons were cultured from BDNF-/- mice and wild-type littermates in the presence and absence of ACM. Immunostaining was performed with antibodies against GABAAR-β2/β3 (green) and SP (red) at 10 div. A, GABAAR cluster number and synaptic localization were decreased in BDNF-/- neurons (right) compared with BDNF+/+ neurons (left) at 10 div. ACM treatment increased the number and synaptic localization of GABAAR clusters in BDNF+/+ neurons but had no effect on GABAAR clusters in BDNF-/- neurons. Scale bar (top right), 10 μm. Areas within white boxes are shown below at higher magnification. Scale bar (bottom right), 2 μm. B, Quantification of GABAAR cluster number per 20 μm dendrite segment. *p < 0.001, significant difference compared with BDNF+/+ neurons within a condition; **p < 0.001, significant decrease compared with no treatment in BDNF+/+ neurons. C, Quantification of GABAAR cluster synaptic localization. *p < 0.001, significant difference compared with BDNF+/+ neurons within a condition; **p < 0.001, significant decrease compared with no treatment in BDNF+/+ neurons. D-F, Hippocampal neurons were grown in the presence and absence of ACM from BDNF-/- and wild-type littermate mice, and GABAAR clusters were examined. D, ACM from BDNF+/+ (middle) and BDNF-/- (right) astrocytes increased GABAAR cluster number and synaptic localization compared with untreated controls (left) by a similar magnitude. Scale bar, 2 μm. E, Quantification of GABAAR cluster number per 20 μm dendrite segment. *p < 0.001, significant difference compared with no treatment controls. F, Quantification of GABAAR cluster synaptic localization. *p < 0.001, significant difference compared with no treatment controls. Error bars represent SEM.
We next evaluated the role of astrocyte-derived BDNF in the modulation of inhibitory synapses. Embryonic rat hippocampal neurons were cultured for 10 div in the presence of ACM from astrocytes from BDNF-deficient or wild-type control mice, and GABAAR clusters were examined. After treatment with ACM from astrocytes from BDNF-deficient mice, the number of GABAAR clusters increased by ∼2.5-fold, and synaptic localization increased by approximately fivefold compared with un-treated controls (Fig. 6D-F) and appeared similar to cultures treated with wild-type ACM (Fig. 6E, F). These results show that astrocyte production of BDNF is not required for modulation of postsynaptic GABAAR clusters. Together, our data suggest that astrocytes may modulate the neuronal release of BDNF via additional, as yet unknown, factors.
After its release, BDNF may bind to TrkB receptors expressed by hippocampal neurons or astrocytes. BDNF activation of full-length TrkB receptor signaling in neurons plays numerous roles in neural development, including modulation of synapse formation and plasticity. Recent work has also shown that hippocampal astrocytes in vitro express high levels of truncated TrkB receptors and that BDNF activation of these receptors stimulates inositol-1,4,5-trisphosphate-dependent calcium release (Rose et al., 2003). Thus, TrkB signaling in neurons and/or astrocytes may play a role in modulating inhibitory synaptogenesis in the cultures studied here. To determine whether glial modulation of GABAAR clusters requires the activation of TrkB signaling within neurons or astrocytes, cultures were prepared from postnatal mice lacking all isoforms of the TrkB receptor. First, to determine the importance of TrkB activation within neurons, GABAAR clusters were examined in hippocampal neurons cultured from TrkB-deficient or wild-type littermates in the presence or absence of wild-type ACM. Similar to BDNF-deficient neurons, TrkB-deficient neuronal cultures had fewer synaptic GABAAR clusters compared with control wild-type cultures. Moreover, in contrast to wild-type neurons, no changes were observed in GABAAR cluster number or synaptic localization when TrkB-deficient neurons were cultured in the presence of wild-type ACM (Fig. 7A-C).
Figure 7.
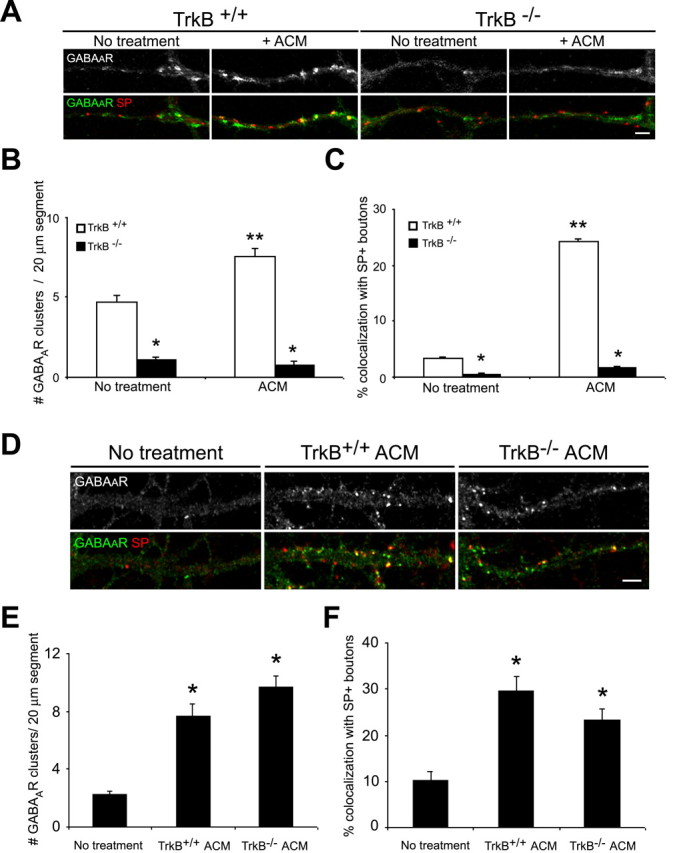
TrkB signaling in astrocytes is not required for GABAAR cluster modulation. A-C, Hippocampal neurons were cultured from TrkB-/- mice and wild-type littermates in the presence and absence of ACM. Immunostaining was performed with antibodies against GABAAR-β2/β3 (green) and SP (red) at 10 div. A, GABAAR cluster number and synaptic localization were decreased in TrkB-/- neurons (right) compared with TrkB+/+ neurons (left) at 10 div. ACM treatment increased the number and synaptic localization of GABAAR clusters in TrkB+/+ neurons but had no effect on GABAAR clusters in TrkB-/- neurons. Scale bar, 2 μm. B, Quantification of GABAAR cluster number per 20 μm dendrite segment. *p < 0.001, significant difference compared with TrkB+/+ neurons within a condition; **p < 0.001, significant decrease compared with no treatment in TrkB+/+ neurons. C, Quantification of GABAAR cluster synaptic localization. *p < 0.001, significant difference compared with TrkB+/+ neurons within a condition; **p < 0.001, significant decrease compared with no treatment in TrkB+/+ neurons. D-F, Hippocampal neurons were grown in the presence and absence of ACM from TrkB-/- and wild-type littermate mice, and GABAAR clusters were examined. D, ACM from TrkB+/+ (middle) and TrkB-/- (right) astrocytes increased GABAAR cluster number and synaptic localization compared with untreated controls (left) by a similar magnitude. Scale bar, 2 μm. E, Quantification of GABAAR cluster number per 20 μm dendrite segment after treatment with TrkB+/+ and TrkB-/- ACM. *p < 0.001, significant difference compared with no treatment controls. F, Quantification of GABAAR cluster synaptic localization after treatment with TrkB+/+ and TrkB-/- ACM. *p < 0.001, significant difference compared with no treatment controls. Error bars represent SEM.
We next asked whether TrkB activation in astrocytes was also necessary for glial modulation of GABAAR cluster formation and synaptic localization. Hippocampal neurons were treated with ACM from TrkB-deficient or wild-type astrocytes, and GABAAR clusters were examined at 10 div. No differences were observed in the ability of ACM from TrkB-deficient or wild-type mouse astrocytes to increase GABAAR cluster formation and synaptic localization in wild-type neurons. Both TrkB-deficient and wild-type astrocytes induced an approximately fourfold increase in the number of GABAAR clusters and an approximately fivefold increase in the synaptic localization of these clusters (Fig. 7D-F). Thus, TrkB-mediated signaling in astrocytes is not required for glial modulation of inhibitory synaptogenesis. Together, these results demonstrate that neuronal release of BDNF is necessary, and furthermore, that neuronal TrkB activation is required for astrocyte modulation of GABAAR clusters. These data are consistent with our previous findings in pure neuronal cultures (Elmariah et al., 2004) and further suggest that astrocytes modulate neurotrophin signaling between neurons.
Discussion
We demonstrate that astrocytes promote the formation of presynaptic and postsynaptic specializations at inhibitory synapses in vitro via Trk-dependent and -independent pathways. Although astrocytes promote the formation of inhibitory presynaptic terminals via TrkB-independent signals, neurotrophin signaling between neurons modulates the formation and localization of postsynaptic GABAAR clusters. BDNF and TrkB signaling between neurons is necessary for the formation and maintenance of synaptic GABAAR clusters regardless of whether astrocytes are present. Astrocytes increase TrkB-mediated modulation of postsynaptic GABAAR clusters by enhancing neuronal BDNF release and/or TrkB activation in neurons. In contrast, NT3 and TrkC signaling, which have no significant effect on GABAAR clusters in the absence of astrocytes, limit the number of postsynaptic GABAAR clusters when neurons and astrocytes are cultured together. Overall, these data suggest that astrocytes enhance the formation of functional inhibitory synapses and tightly regulate postsynaptic GABAAR clusters via the opposing actions of BDNF and NT3 signaling.
Astrocytes promote the development of inhibitory synapses
The present study demonstrates that astrocytes play a key role in the establishment of functional inhibitory synapses between interneurons and target pyramidal neurons in hippocampal cultures. Astrocytes significantly increased the number of inhibitory presynaptic terminals and postsynaptic GABAAR clusters relative to pure neuronal cultures. At early ages in vitro, the robust increase in synaptic GABAAR clusters observed in the presence of astrocytes reflects an increase in the number of inhibitory presynaptic terminals and GABAAR clusters. However, the synaptic localization of GABAAR clusters is elevated in the presence of astrocytes at later ages when the number of GABAAR clusters and presynaptic terminals remains constant. This observation suggests that the astrocyte-induced increase in synaptic localization of GABAAR clusters at younger ages does not simply reflect an increase in SP+ and VGAT+ terminals. Rather, astrocytes actively promote the localization of newly formed GABAAR clusters to synaptic sites and increase the prevalence of spontaneous IPSCs at 10 div. Thus, astrocytes promote and coordinate the development of presynaptic and postsynaptic specializations and modulate ongoing function at inhibitory synapses during maturation in vitro.
Important questions remain about the signals and mechanisms by which astrocytes regulate synaptogenesis. Astrocytes provide cholesterol, a key component of cell membranes that has been shown to modulate excitatory synaptogenesis (Mauch et al., 2001). This observation is consistent with the fact that the bulk of synaptogenesis in vivo occurs after the generation of astrocytes (Pfrieger and Barres, 1996; Ullian et al., 2001). Although cholesterol increases the number and function of excitatory presynaptic terminals in cultured retinal ganglion cells (Mauch et al., 2001), our data suggest that cholesterol alone does not mimic the effects of astrocytes on inhibitory synapse formation in hippocampal neurons. Cholesterol treatment had no effect on inhibitory terminal number, postsynaptic GABAAR cluster number, or colocalization with VGAT+ presynaptic terminals in our cultures, consistent with previous reports that cholesterol alone is insufficient to modulate excitatory hippocampal synapses (Hama et al., 2004). Thus, although cholesterol may play a permissive role, other factors provided by astrocytes probably play instructive roles in presynaptic and postsynaptic development of excitatory and inhibitory synapses. Christopherson et al. (2005) showed recently that thrombospondin-1 and thrombospondin-2, abundant high-molecular-weight extracellular matrix proteins that are present in ACM, enhance synapse formation and presynaptic terminal function in purified retinal ganglion-cell cultures. Hama et al. (2004) showed that integrin-mediated contact with individual astrocytes increased the formation of glutamatergic autapses via PKC signaling. Together, these studies suggest that soluble-mediated as well as contact-mediated signaling from astrocytes is required for the formation of presynaptic and postsynaptic specializations.
Here, we show that astrocytes also contribute to the formation of inhibitory synapses and that astrocyte-dependent increases in postsynaptic GABAAR clusters require neurotrophin signaling in neurons. When endogenous BDNF levels are reduced or TrkB activation is abolished in neurons, ACM treatment no longer increases postsynaptic GABAAR clusters. Glial modulation of GABAAR clusters requires that neurons produce and release BDNF, whereas astrocyte production of BDNF is not necessary. Moreover, although both neurons and astrocytes express isoforms of the TrkB receptor, only the activation of TrkB signaling within neurons is required for astrocyte-mediated modulation of postsynaptic GABAAR clusters. Although BDNF and TrkB signaling is necessary for glial regulation of postsynaptic GABAAR clusters, whether NT4/5 signaling also plays a role in modulation of inhibitory synaptogenesis requires additional examination.
BDNF and TrkB signaling have been reported to modulate inhibitory synaptic activity via presynaptic and postsynaptic effects, including the regulation of GAD expression, GABA release, and GABAAR expression and clustering (Marty et al., 2000; Brunig et al., 2001; Yamada et al., 2002; Elmariah et al., 2004; Palizvan et al., 2004). Astrocytes significantly increased the number of inhibitory presynaptic terminals and postsynaptic GABAAR clusters relative to pure neuronal cultures. In contrast to its role in postsynaptic GABAAR regulation, abolishing neurotrophin signaling had no effect on the astrocyte-mediated increase in inhibitory presynaptic terminals, demonstrating that BDNF is not required for the glial upregulation of inhibitory terminals. That BDNF cannot account for all of the synapse-promoting effects of ACM suggests that astrocytes coordinately regulate presynaptic and postsynaptic formation via distinct mechanisms. Determining the soluble factors released by astrocytes that modulate neurotrophin signaling in the developing hippocampus, and thereby postsynaptic neurotransmitter receptor clusters, will be a focus of future work.
Differential modulation of postsynaptic GABAAR clusters by opposing actions of BDNF and NT3 in the presence of astrocytes
Currently, our understanding of the specific roles that astrocytes play in modulating the development of postsynaptic specializations is limited. In purified retinal ganglion cell cultures, increased AMPA-mediated EPSC amplitude was observed in the presence of ACM and reflects an increased number of AMPA receptor clusters at postsynaptic sites (Ullian et al., 2001). Consistent with this work, Liu et al. (1996, 1997) reported that astrocytes promote the maintenance of GABAA, glycine, and NMDA receptors expressed in dissociated hippocampal neurons after initial isolation from embryonic rats. Here, we provide evidence that astrocytes enhance the formation of GABAAR clusters during the first week in vitro and continue to promote the synaptic localization of these clusters as cultures mature.
Astrocytes regulate the stability of inhibitory synapses in hippocampal networks by dynamically regulating the assembly, delivery, and/or removal of postsynaptic GABAAR clusters. Our data support a role for astrocytes in promoting the stability of GABAAR clusters and the maintenance of inhibitory synapses via BDNF signaling. Scavenging BDNF in astrocyte cocultures or ACM-treated cultures after 1 week in vitro, when increased levels of postsynaptic GABAAR clusters are already apparent, results in a reduction of postsynaptic GABAAR clusters. Thus, in the absence of BDNF, GABAAR clusters are disassembled and/or removed from synaptic sites, consistent with previous observations in purified retinal ganglion cell cultures, in which the removal of ACM after 5 div led to the loss of glutamatergic synapses (Ullian et al., 2001).
We also provide evidence that astrocytes can actively promote the disassembly of inhibitory synapses by inducing the removal GABAAR clusters from synaptic sites. When neurons are cultured with astrocytes, the addition of NT3 reduces the number of postsynaptic GABAAR clusters and attenuates the BDNF-induced increase in postsynaptic clusters. Moreover, scavenging endogenous NT3 at 8-10 div resulted in an increase in GABAAR cluster localization to synapses. These results are consistent with previous findings that NT3 potentiates neuronal activity by downregulating GABAergic synaptic transmission in dissociated cortical neurons (Kim et al., 1994). In addition, Paul et al. (2001) have shown that NT3 attenuates the BDNF-induced increase in EPSCs and IPSCs and c-fos expression in dissociated cultures of hippocampal neurons. NT3 is produced and released by embryonic hippocampal neurons as well as astrocytes (Rudge et al., 1992; Blondel et al., 2000; Wu et al., 2004), but the relevant source of NT3 for modulating GABAARs in neuron-astrocyte cocultures remains to be determined. Manipulation of NT3 signaling in pure neuronal cultures has no effect on the synaptic localization of GABAAR clusters, suggesting that NT3 might be acting on astrocytes, provoking their release of other synaptic modulators, or that astrocytes modulate TrkC expression and responsivity to NT3 in neurons. Regardless, these results show that neurotrophins play an important role in mediating the effects by which astrocytes modulate postsynaptic GABAAR clusters and thus the formation of functional inhibitory synapses.
Glial modulation of neurotrophin signaling
Our work supports the idea that factors intrinsic to and exchanged between neurons, such as BDNF, are necessary for initial synapse formation and maintenance. In addition to providing extrinsic cues that directly modulate synapse formation and maturation, astrocytes may also alter the expression or activity of neuronal factors, such as neurotrophins and Trks, and thereby indirectly influence synaptic development. Consistent with this hypothesis, vasoactive intestinal polypeptide-stimulated astrocytes induce postsynaptic hippocampal neurons to release NT3, which acts presynaptically to promote the formation of glutamatergic synapses (Blondel et al., 2000). Because neuronal-derived BDNF is required for glial effects on postsynaptic GABAAR clusters, astrocytes may similarly promote BDNF or TrkB expression by neurons, which in turn induces the formation of inhibitory synapses. The role of astrocytes in directly stimulating neurotrophin expression has not yet been evaluated. Astrocytes may also influence the availability of neurotrophins and their receptors by altering spontaneous neuronal activity, itself a potent stimulator of neurotrophin and Trk expression (Lindholm et al., 1994a,b; Tongiorgi et al., 1997). How astrocytes release neurotrophins and/or modulate the expression and activity of BDNF/TrkB and NT3/TrkC in neurons remains to be explored. Finally, determining the mechanisms by which glia modulate Trk signaling will be essential for understanding how glia contribute to the establishment of mature neural networks.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS/AR40763, National Science Foundation Grant 0130822 (R.J.B.-G.), and NIH National Research Service Award NS43821 (S.B.E.). We thank M. Lightner, M. Maronski, M. O. Scott, and H. Y. Zhou for technical assistance and Drs. M. Dichter, P. G. Haydon, S. Moss, and members of the Balice-Gordon laboratory for helpful discussions.
Correspondence should be addressed to Dr. Rita Balice-Gordon, Department of Neuroscience, University of Pennsylvania School of Medicine, 215 Stemmler Hall, Philadelphia, PA 19104-6074. E-mail: rbaliceg@mail.med.upenn.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253638-13$15.00/0
References
- Alderson RF, Curtis R, Alterman AL, Lindsay RM, DiStefano PS (2000) Truncated TrkB mediates the endocytosis and release of BDNF and neurotrophin-4/5 by rat astrocytes and Schwann cells in vitro. Brain Res 871: 210-222. [DOI] [PubMed] [Google Scholar]
- Binder DK, Routbort MJ, Ryan TE, Yancopoulos GD, McNamara JO (1999) Selective inhibition of kindling development by intraventricular administration of TrkB receptor body. J Neurosci 19: 1424-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel O, Collin C, McCarran WJ, Zhu S, Zamostiano R, Gozes I, Brenneman DE, McKay RD (2000) A glia-derived signal regulating neuronal differentiation. J Neurosci 20: 8012-8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunig I, Penschuck S, Berninger B, Benson J, Fritschy JM (2001) BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABA(A) receptor surface expression. Eur J Neurosci 13: 1320-1328. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CCA, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA (2005) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120: 421-433. [DOI] [PubMed] [Google Scholar]
- Condorelli DF, Dell'Albani P, Mudo G, Timmusk T, Belluardo N (1994) Expression of neurotrophins and their receptors in primary astroglial cultures: induction by cyclic AMP-elevating agents. J Neurochem 63: 509-516. [DOI] [PubMed] [Google Scholar]
- Du J, Feng L, Yang F, Lu B (2000) Activity- and Ca2+-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. J Cell Biol 150: 1423-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S, Anderson CM, Keung EC, Chen Y, Swanson RA (2003) P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J Neurosci 23: 1320-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmariah SB, Crumling MA, Parsons TD, Balice-Gordon RJ (2004) Postsynaptic TrkB-mediated signaling modulates excitatory and inhibitory neurotransmitter receptor clustering at hippocampal synapses. J Neurosci 24: 2380-2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernfors P, Wetmore C, Olson L, Persson H (1990) Identification of cells in rat brain and peripheral tissues expressing mRNA for members of the nerve growth factor family. Neuron 5: 511-526. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Jaenisch R (1994) Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature 368: 147-150. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD (2004) Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J Neurosci 24: 722-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslin K, Schreyer DJ, Skene JH, Banker G (1988) Development of neuronal polarity: GAP-43 distinguishes axonal from dendritic growth cones. Nature 336: 672-674. [DOI] [PubMed] [Google Scholar]
- Hama H, Hara C, Yamaguchi K, Miyawaki A (2004) PKC signaling mediates global enhancement of excitatory synaptogenesis in neurons triggered by local contact with astrocytes. Neuron 41: 405-415. [DOI] [PubMed] [Google Scholar]
- Ivanova T, Beyer C (2001) Pre- and postnatal expression of brain-derived neurotrophic factor mRNA/protein and tyrosine protein kinase receptor B mRNA in the mouse hippocampus. Neurosci Lett 307: 21-24. [DOI] [PubMed] [Google Scholar]
- Jovanovic JN, Thomas P, Kittler JT, Smart TG, Moss SJ (2004) Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABAA receptor phosphorylation, activity, and cell-surface stability. J Neurosci 24: 522-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M (1998) Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci 1: 683-692. [DOI] [PubMed] [Google Scholar]
- Kim HG, Wang T, Olafsson P, Lu B (1994) Neurotrophin 3 potentiates neuronal activity and inhibits gamma-aminobutyratergic synaptic transmission in cortical neurons. Proc Natl Acad Sci USA 91: 12341-12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M (1993) Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell 75: 113-122. [PubMed] [Google Scholar]
- Kohara K, Kitamura A, Morishima M, Tsumoto T (2001) Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science 291: 2419-2423. [DOI] [PubMed] [Google Scholar]
- Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N (1998) Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci 18: 10231-10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm D, Castren E, Berzaghi M, Blochl A, Thoenen H (1994a) Activity-dependent and hormonal regulation of neurotrophin mRNA levels in the brain—implications for neuronal plasticity. J Neurobiol 25: 1362-1372. [DOI] [PubMed] [Google Scholar]
- Lindholm D, da Penha Berzaghi M, Cooper J, Thoenen H, Castren E (1994b) Brain-derived neurotrophic factor and neurotrophin-4 increase neurotrophin-3 expression in the rat hippocampus. Int J Dev Neurosci 12: 745-751. [DOI] [PubMed] [Google Scholar]
- Liu QY, Schaffner AE, Li YX, Dunlap V, Barker JL (1996) Upregulation of GABAA current by astrocytes in cultured embryonic rat hippocampal neurons. J Neurosci 16: 2912-2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QY, Schaffner AE, Chang YH, Vaszil K, Barker JL (1997) Astrocytes regulate amino acid receptor current densities in embryonic rat hippocampal neurons. J Neurobiol 33: 848-864. [PubMed] [Google Scholar]
- Mammen AL, Huganir RL, O'Brien RJ (1997) Redistribution and stabilization of cell surface glutamate receptors during synapse formation. J Neurosci 17: 7351-7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty S, Wehrle R, Sotelo C (2000) Neuronal activity and brain-derived neurotrophic factor regulate the density of inhibitory synapses in organotypic slice cultures of postnatal hippocampus. J Neurosci 20: 8087-8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A, Pfrieger FW (2001) CNS synaptogenesis promoted by glia-derived cholesterol. Science 294: 1354-1357. [DOI] [PubMed] [Google Scholar]
- Miklic S, Juric DM, Caman-Krzan M (2004) Differences in the regulation of BDNF and NGF synthesis in cultured neonatal rat astrocytes. Int J Dev Neurosci 22: 119-130. [DOI] [PubMed] [Google Scholar]
- Nagler K, Mauch DH, Pfrieger FW (2001) Glia-derived signals induce synapse formation in neurones of the rat central nervous system. J Physiol (Lond) 533: 665-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul J, Gottmann K, Lessmann V (2001) NT-3 regulates BDNF-induced modulation of synaptic transmission in cultured hippocampal neurons. NeuroReport 12: 2635-2639. [DOI] [PubMed] [Google Scholar]
- Palizvan MR, Sohya K, Koharn K, Maruyama A. Yasuda H, Kimura F, Tsumoto T (2004) Brain-derived neurotrophic factor increases inhibitory synapses, revealed in solitary neurons cultured from rat visual cortex. Neurosccience 12: 955-966. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW, Barres BA (1996) New views on synapse-glia interactions. Curr Opin Neurobiol 6: 615-621. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW, Barres BA (1997) Synaptic efficacy enhanced by glial cells in vitro. Science 277: 1684-1687. [DOI] [PubMed] [Google Scholar]
- Rose CR, Blum R, Pichler B, Lepier A, Kafitz KW, Konnerth A (2003) Truncated TrkB-T1 mediates neurotrophin-evoked calcium signalling in glia cells. Nature 426: 74-78. [DOI] [PubMed] [Google Scholar]
- Rudge JS, Alderson RF, Pasnikowski E, McClain J, Ip NY, Lindsay RM (1992) Expression of ciliary neurotrophic factor and the neurotrophins—nerve growth factor, brain-derived neurotrophic factor and neurotrophin 3—in cultured rat hippocampal astrocytes. Eur J Neurosci 4: 459-471. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, DeWan A, Lauer HM, Turrigiano GG (1997) Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. J Neurosci 17: 4527-4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seil FJ, Drake-Baumann R (2000) TrkB receptor ligands promote activity-dependent inhibitory synaptogenesis. J Neurosci 20: 5367-5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tongiorgi E, Righi M, Cattaneo A (1997) Activity-dependent dendritic targeting of BDNF and TrkB mRNAs in hippocampal neurons. J Neurosci 17: 9492-9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA (2001) Control of synapse number by glia. Science 291: 657-661. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Harris BT, Wu A, Chan JR, Barres BA (2004) Schwann cells and astrocytes induce synapse formation by spinal motor neurons in culture. Mol Cell Neurosci 25: 241-251. [DOI] [PubMed] [Google Scholar]
- Vicario-Abejon C, Collin C, McKay RD, Segal M (1998) Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. J Neurosci 18: 7256-7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, W. J. Friedman, Dreyfus CF (2004) Differential regulation of neurotrophin expression in basal forebrain astrocytes by neuronal signals. J Neurosci Res 76: 76-85. [DOI] [PubMed] [Google Scholar]
- Yamada MK, Nakanishi K, Ohba S, Nakamura T, Ikegaya Y, Nishiyama N, Matsuki N (2002) Brain-derived neurotrophic factor promotes the maturation of GABAergic mechanisms in cultured hippocampal neurons. J Neurosci 22: 7580-7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, Wu CP, Poo MM, Duan S (2003) ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron 40: 971-982. [DOI] [PubMed] [Google Scholar]