

Abstract
Receptors with tyrosine kinase activity (RTKs) control tissue growth and development in metazoans. How they generate cell-specific responses remains essentially unknown; one model proposes that distinct RTKs activate different second-messenger pathways, whereas a second proposes that all RTKs deliver a generic “go” signal to these pathways that is uniquely interpreted by downstream, cell-specific response competence factors. We examine pathway activation and pathway-specific responses downstream of PDGFα receptors, whose expression in the developing CNS identifies oligodendrocyte progenitor cells (OPCs) and whose activation controls OPC proliferation, migration, survival, and maturation. PDGFRα-null mice die in utero, and OPCs that emerge before their demise have migration and proliferation defects and rapidly differentiate into postmitotic oligodendrocytes in vitro. OPCs from hemizygous mice also undergo precocious differentiation, indicating a role for PDGFRα gene dosage in timing OPC maturation. The rescue of PDGFRα-null OPCs with PDGFRα transgenes revealed specific roles for the phosphatidylinositol 3-kinase (PI3K) and phospholipase Cγ (PLCγ) pathways and a distinct ligand concentration dependence. Activation of the PI3K pathway is required for PDGFRα-induced migration, whereas activation of both PI3K and PLCγ are required for PDGFRα-induced proliferation. For proliferation, PI3K activation is required at low ligand concentration, whereas PLCγ is required at high signal strength. Dose-response studies further demonstrate that PDGFRα activates PI3K at low ligand concentrations, whereas PLCγ is activated at high signal strength. Thus, PDGFRα signaling acts like a rheostat rather than generic ON switch, with signal strength dictating pathway activation during OPC maturation.
Keywords: development, glia, growth factor, myelin, oligodendrocyte, PDGF α-receptor, RTK, signal transduction
Introduction
Receptor tyrosine kinases (RTKs) control intercellular communication in metazoans, coupling distinct extracellular ligands to common intracellular signaling pathways to regulate key cellular processes in both development and homeostasis (van der Geer et al., 1994). A clear picture of the biochemical pathways that can be activated by RTKs has emerged during the past decade (Heldin and Westermark, 1999). In contrast, how individual RTKs generate ligand-specific biological responses remains primarily unknown. Two contrasting models have emerged to explain RTK specificity (Simon, 2000). One proposes that all RTKs generate a generic “ON” signal, interpreted by individual cell types based on signaling history and competence to respond, supported by studies indicating that the signaling domains of distinct Drosophila RTKs can be functionally substituted (Dossenbach et al., 2001). A second proposes that distinct RTKs activate different signaling pathways and generate specific responses based on strength of signal and which pathways are engaged, as suggested by differential activation of the mitogen-activated protein kinase (MAPK) pathway by nerve growth factor (NGF) and epidermal growth factor (EGF) receptors in PC12 cells (Marshall, 1995) and phosphoinositol 3′-kinase (PI3K) by PDGF but not fibroblast growth factor (FGF) receptors in vertebrate glia (Ebner et al., 2000).
We examined PDGF signaling in oligodendrocytes, myelinating glial cells of the vertebrate CNS, and tested the hypothesis that unique pathways are coupled to separate biological outcomes. Oligodendrocytes are generated from progenitors [oligodendrocyte progenitor cells (OPCs)] in the embryonic neuroepithelium, migrate into brain parenchyma, exit the cell cycle, and then assemble myelin sheaths to insulate neuronal axons. OPCs and their neuroepithelial precursors isolated from brain and spinal cord further present a unique opportunity to examine signaling specificity in primary, non-immortal cells in vitro. Studies have defined discrete stages of maturation in this lineage from neural stem cells in the ventricular zone to tripotential glial restricted progenitors (GRPs) (Liu and Rao, 2004) and OPCs (Pfeiffer et al., 1993). PDGF promotes OPC proliferation, migration, survival, and maturation via PDGFRα, the only PDGF receptor isoform in these cells detected by ligand binding (Pringle et al., 1989) and molecular expression (McKinnon et al., 1990). Transgenic studies further indicate that PDGF levels can be limiting for OPC pool expansion (van Heyningen et al., 2001), demonstrating the importance of PDGFRα signaling in these cells.
We used a transgene rescue approach to examine signaling pathways in OPC proliferation, migration, and maturation. OPCs from PDGFRα-null mice failed to migrate or proliferate and underwent an accelerated maturation. Transfection with wild-type PDGFRα expression vectors rescued the migration defect, whereas transgenes incapable of activating PI3K did not. Because the PI3K pathway was otherwise intact, this demonstrates a specific requirement for PDGFRα-PI3K coupling in migration. Mutant transgenes unable to activate either PI3K or the phospholipase C-γ (PLCγ) pathway gave only partial rescue of the proliferation defect. Finally, dose-response studies revealed that PI3K is both activated and required specifically at low ligand concentrations, whereas PLCγ is activated and required at high ligand concentrations. Together, these results indicate that PDGFRα signaling in OPCs results from specific pathways engaged at distinct signal strengths and is more like a graded rheostat than a generic “ON-OFF” response.
Materials and Methods
Animals. PDGFRα alleles examined included the chromosomal deficiency Patch (Ph; The Jackson Laboratory, Bar Harbor, ME) and a PDGFRα-null targeted disruption (Soriano, 1997). Both alleles are viable as hemizygotes and cause embryonic lethality in homozygotes. Ph has additional phenotypes (such as coat color) attributable to effects on the downstream White locus encoding c-kit. Patch (C57BL/6J background) were outbred to DBA mice (The Jackson Laboratory) and hemizygous (Ph/+) animals identified by their dominant white spotting coat color. The Soriano αR2-4 allele was identified by PCR analysis of DNA extracted from tails as described previously (Soriano, 1997).
Immunochemistry. Monoclonal antibodies A2B5, O4, and O1 were obtained as supernatant fluids from hybridoma cell lines, polyclonal anti-Olig2 antisera was obtained from Dr. Hideaki Yokoo (Gunma University School of Medicine, Maebashi, Gunma, Japan), and anti-NG2 antisera was obtained from Dr. Joel Levine (State University of New York at Stony Brook, Stony Brook, NY). Other antibodies included anti-myelin basic protein (MBP) (Chemicon, Temecula, CA), anti-PDGFRα and active caspase 3 (R & D Systems, Minneapolis MN), and anti-Akt (phosphoserine 473) and PLCγ1 (phosphotyrosine 783) (Santa Cruz Biotechnology, Santa Cruz, CA). Alexa-conjugated fluorescent secondary antibodies were from Molecular Probes (Eugene, OR). For histology, animals were perfused with 4% paraformaldehyde in phosphate buffer, the tissue was cryoprotected with 20% sucrose, and then 20 μm frozen sections were mounted on glass slides for staining.
Cell culture. Myelinating cocultures were established from embryonic mouse spinal cords as described previously (Vartanian et al., 1999) with dissected cords cleaned of peripheral nerve roots, minced, and then cultured in DMEM (Invitrogen, Gaithersburg, MD) containing 50 U/ml penicillin, 50 μg/ml streptomycin, and 10% fetal bovine serum (Invitrogen). Dissociated tissue from individual brain and spinal cords was divided equally among six wells of a 24-well Costar (Cambridge, MA) plate, and the media were replaced twice weekly. These studies included mixed cultures without further purification of GRPs or OPCs from the other cell types present. The Soriano allele was used for all embryonic studies described, with the genotype of pups determined by PCR analysis as outlined above. For mixed glial cultures established from Sprague Dawley rat pups, the OPCs were further purified by A2B5 immune selection (McKinnon et al., 1990), plated on dishes precoated with 100 μg/ml poly-l-ornithine (Sigma, St. Louis MO), and cultured in DMEM with 100 μg/ml transferrin, 30 nm sodium selenite, 50 ng/ml bovine insulin, 10 μm forskolin, 60 μg/ml N-acetyl cystein, 100 μg/ml BSA, 10 ng/ml PDGF, and 5 ng/ml FGF2 (Invitrogen). For staining, cells on coverslips were fixed in 4% paraformaldehyde and then permeabilized in 0.1% Triton X-100 in PBS.
Molecular biology. Plasmids included wild-type and mutant versions of the PDGFRα expression vector pMo.PDGFRα.iresNeo (Osterhout et al., 1997), encoding either wild-type or point mutant forms of the intracellular signaling domain of PDGFRα (C. Edwards and R. D. McKinnon, unpublished observation), and a chimeric Fms/PDGFRα vector pSVfms/ PDGFRα (Yu et al., 1994, 1995) from M. Heidarin (Celgene Cellular Therapeutics, Cedar Knolls, NJ). DNA was purified by chromatography (Qiagen, Valencia, CA), and constructs were verified by sequence analysis (DNA core facility of Robert Wood Johnson Medical School) of fragments generated by PCR amplification (Ebner et al., 2000) using upstream primers (5′-3′) agggtcattgaatcaatcagcccggatgg and actacgtggacatgaagcagg and the downstream primer aaagtggaactactggaacccg. Immunoblot analyses were performed as described previously (McKinnon et al., 1990).
DNA-mediated gene transfer. DNA transfections were performed using LipofectAMINE reagent (Invitrogen), with 3 μg/ml LipofectAMINE plus 32 μg transferrin (Cheng, 1996) preincubated with the indicated amounts of plasmid DNA, and then aliquots were added to wells for 4 h at 37°C. Cells were then fed media plus supplements and, for proliferation studies, incubated an additional 36 h before assays. The efficiency of transient transfection determined with a green fluorescent protein reporter was at least 30%. OPC lines with stable transgene expression were selected in media containing 400 μg/ml G418 and amplified as described for the CG4 cell line (Louis et al., 1992; Ebner et al., 2000).
Proliferation assay. Thymidine incorporation was determined by incubating cells in 96-well plates (5 × 103 OPCs per well) for 24 h without growth factors and then in media containing the indicated concentrations of either PDGF-AA chain homodimer or the fms ligand hCSF1 (R & D Systems) for an additional 24 h, with 0.1 μCi [3H]thymidine present for the final 4 h (specific activity 48 Ci/mmol; Amersham Biosciences, Arlington Heights, IL). DNA was harvested onto Whatman (Clifton, NJ) GF/C filters, and 3H incorporation was determined by liquid scintillation counting. All assays were conducted at multiple input DNA concentrations (0.1, 0.5, and 1.0 μg/well) for each construct and growth factor condition described and were repeated at least three independent times per construct.
Migration. Spinal cords minced to a uniform size (∼0.5 mm) were plated at low density on 12 mm coverslips and processed for histochemical analysis after 15 d. Explant edges were identified in phase contrast, and the dispersion of OPCs from the edge was determined by counting O4+ and MBP+ cells in radial “bins” (0-50, 50-100, 100-200, 200-500, and >500 μm) from the edge. The radial gradient of OPCs from their source explants was unambiguous, and explants separated by <2 mm were excluded from analysis.
Biochemistry. PI3K activation was monitored by phosphorylation status of the PI3K target protein kinase B/Akt (Ebner et al., 2000). Cells on coverslips were cultured for either 5 h (primary OPCs) or 18 h (OPC line) in the absence of mitogens and then triggered with PDGF for 15 min immediately before harvesting for analysis by anti-Akt (phosphoserine 473) histochemistry. Activation of PLCγ1 was monitored at 5 and 15 min after ligand with anti-PLCγ1 (phosphotyrosine 783), and the resultant intracellular Ca2+ mobilization was monitored in fura-2-loaded cells by real-time image analysis using an automated imaging system (Attofluor, Rockville, MD). Cells were loaded with 8 μm fura-2 AM (37°C 30 min; Calbiochem, La Jolla, CA), and then multiple cells (n > 40 per sample) were visualized during sequential addition of agonists (hCSF1 and then PDGF-AA, 100 μg/ml). Changes in emission intensity (580 nm) were monitored for >30 min with sequential excitation at 340 and 380 nm. Values represent the relative levels of intracellular free calcium.
Results
Oligodendrocyte progenitors in PDGFRα homozygous-null mice
We examined two protein-null alleles of pdgfra, a targeted disruption αR2-4 (Soriano, 1997) and the spontaneous Patch (Ph) deletion that affects pdgfra and the 3′-proximal gene c-kit (Gruneberg and Truslove, 1960; Smith et al., 1991; Stephenson et al., 1991). Both alleles are recessive lethal, with 100% penetrance of severe developmental phenotypes, including a cleft face (Fig. 1a). Patch on an inbred C57BL background is lethal between embryonic day 9.5 (E9.5) and E14 (Gruneberg and Truslove, 1960; Schatteman et al., 1992), and Ph/Ph mutants are observed at only low frequency up to E14 (Table 1). The aR2-4 allele mutants survive up to but not beyond day 16 (Soriano, 1997) and, unlike Ph, can be rescued with a wild-type PDGFRα transgene (Sun et al., 2000), suggesting a less severe chromosomal disruption. We extended the survival of homozygous-null embryos through extensive outbreeding of the Ph and αR2-4 alleles (Table 1). Ph/Ph mutants were now identified as late as E18, and, to date, three αR2-4/αR2-4 mutants survived gestation through to stillbirth. Thus, the penetrance of both null alleles is background sensitive, suggesting strain-specific modifiers.
Figure 1.
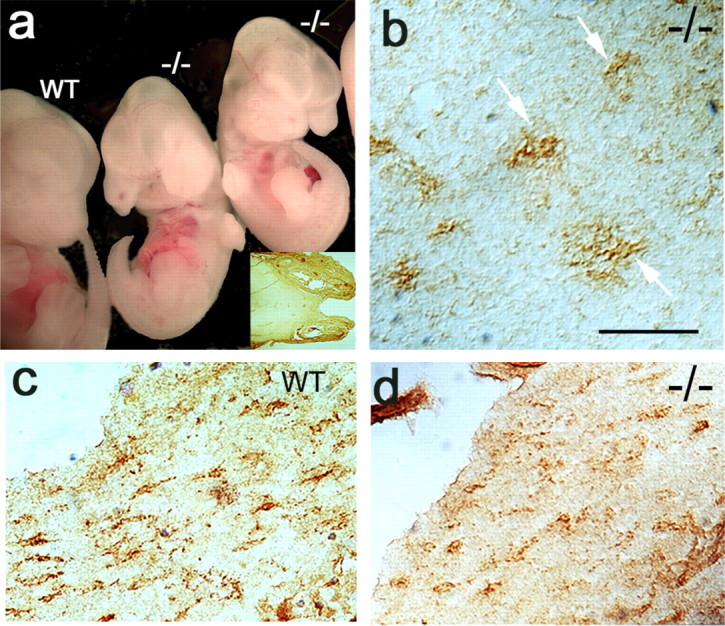
Emergence of NG2-immunoreactive cells in pdgfra-null mouse brain. a, Wild-type (wt) and homozygous αR2-4-null (-/-) littermates at embryonic day 13.5; the inset is a horizontal section of mutant cranium showing the prominent cleft palate. b, Horizontal section of day 16.5 pdgfra-null mutant showing highly branched NG2+ cells in brain cortex (arrows). c, d, NG2 cells with morphology of migrating OPCs in day 19 wild-type and mutant littermates. Scale bar, 100 μm.
Table 1.
Viability of PDGFRα homozygous mutant mice: effect of genetic background
|
PDGFRα allele |
E13.5 |
E14.5 |
E16.5 |
E18.5 |
P0 |
|---|---|---|---|---|---|
| Patch (C57BL/6J) | 3/43 (0.07) | 1/10 (0.10) | (0.00) | (0.00) | (0.00) |
| Patch (C57 × DBA) | — | — | 8/41 (0.20) | 6/22 (0.27) | (0.00) |
| αR2-4 (C57 × DBA) |
29/190 (0.16) |
12/99 (0.12) |
3/75 (0.04) |
1/11 (0.09) |
3a
|
Numbers of homozygous PDGFRα-null mutants per total embryos observed at E13.5 through postnatal day 0 (birth) (P0) are shown. The Patch deletion was maintained on a C57BL/6J background or outbred for at least five generations to DBA, and Ph/Ph mutants were identified by their small size and cleft face. The αR2-4 allele was outbred to DBA, and genotypes were confirmed by genomic DNA analysis. Numbers in parentheses represent the frequency of homozygous-null embryos; 0.00, null embryos have not been detected at these gestation ages; —, gestation ages not examined in this study.
Three confirmed PDGFRα-null pups survived gestation to term from the same (αR2-4/+) cross.
Extending the survival of PDGFRα-null embryos allowed us to examine late neurogenesis and directly test an unresolved question in glial lineage (Liu and Rao, 2004): whether oligodendrocytes can be specified from neuroepithelial precursors in the absence of PDGFRα. We first examined homozygous-null embryos obtained from pdgfra+/- matings. The NG2 proteoglycan is expressed by cells with the morphology and distribution of OPCs in embryonic and adult CNS, coincident with PDGFRα (van Heyningen et al., 2001), and, although not all NG2-cells acquire OPC markers (Mallon et al., 2002), they generate OPCs in vitro (Nishiyama et al., 1996). In homozygous-null animals, NG2-positive cells were evident in forebrain at E16.5 through E19 within gray (Fig. 1b) and white (Fig. 1d) matter. Their density in the hippocampal fimbria was lower than wild-type littermates (Fig. 1c). In explants established from E13.5 forebrain and maintained 7 d in vitro (Table 2), OPC numbers were ∼40% lower in homozygous-null than wild-type cultures (pdgfra+/+, 80; pdgfra-/-, 48; n = 5 independent samples). O4+ cell numbers were similarly lower in spinal cord explants from PDGFRα-null embryos (data not shown). The reduced numbers in mutant cultures may in part reflect a lower plating density attributable to the smaller size of null embryos (Fig. 1a). However, these results demonstrate that although the absence of PDGFRα reduced the census, it did not prevent the genesis of OPCs in the neural axis. Of interest, the absence of PDGFRα also did not appear to affect the migration of OPCs in vivo.
Table 2.
Emergence, expansion, and maturation of OPCs from PDGFRα mutant mice
|
|
OPC (04+) |
Oligos (04+, MBP+) |
||||||
|---|---|---|---|---|---|---|---|---|
| Genotype |
Day 7 |
Day 14 |
Day 21 |
Day 14 |
Day 21 |
|||
| +/+ | 80 ± 17 | 1333 ± 330 | 1497 ± 242 | 80 ± 29 | 271 ± 119 | |||
| +/− | 76 ± 10 | 229 ± 53* | 201 ± 64* | 384 ± 36* | 1110 ± 336 | |||
| −/− |
48 ± 13 |
28 ± 5*
|
25 ± 3*
|
35 ± 11 |
16 ± 4 |
|||
Numbers of 04-positive OPCs and MBP+ oligodendrocytes (oligos) in forebrain explants established from E13.5 αR2-4 mice are shown. Explants plated on cover slips (6 wells per pup) were maintained from 1 to 3 weeks in culture, and values represent total cell counts (mean ± SEM) per well from at least three experiments for each genotype and time point. Animals were genotyped as described in Materials and Methods: +/+, wild type; +/−, pdgfra hemizygous; −/−, pdgfra homozygous null. *p < 0.05, differences between wild type were statistically significant (Student's t test).
Accelerated oligodendrocyte maturation in PDGFRα haploid insufficiency
We also examined the maturation of OPCs in PDGFRα-deficient embryos. Because the null mutants die before oligodendrocytes emerge, we first considered hemizygous mice with one copy of pdgfra in which haploid insufficiency may uncover PDGFRα effects dependent on gene dosage. These studies used both the αR2-4 and Ph alleles. Hemizygosity for pdgfra results in 50% lower PDGFRα protein levels in whole embryos (Soriano, 1997), and, by quantitative Northern blot analysis, PDGFRα levels are 50% lower in cell lines established from hemizygous versus wild-type mice (McKinnon, unpublished observation).
We examined both the OPC marker NG2 and the mature oligodendrocyte marker MBP. NG2+ cells in newborns were observed in white matter of wild-type but not hemizygous littermates, which in principal could represent delayed OPC genesis or an acceleration in their maturation, because NG2 is progressively lost as OPCs mature (Nishiyama et al., 1996). Consistent with the latter, MBP staining was higher in day 8 hemizygotes compared with wild-type littermates (Fig. 2a). Blot analysis with normalized levels of whole-brain extracts also revealed fourfold higher levels of MBP in postnatal day 8 hemizygotes (Fig. 2b). Thus, the temporal pattern of MBP expression suggested an accelerated maturation of OPCs in pdgfra haploid insufficiency. By postnatal day 14, levels of MBP were equivalent in wild-type and hemizygous pups (Fig. 2b), indicating that this precocious emergence did not affect the ultimate levels of myelin proteins.
Figure 2.

Emergence of oligodendrocyte marker MBP in PDGFRα hemizygous mice. a, Myelin basic protein immunoreactivity in cerebellar folia of day 8 wild-type (+/+) and PDGFRα hemizygous (pdgfraPh/+) littermates; rostral is top. b, Immunoblot analysis of MBP at postnatal days 5-14 in load-normalized extracts from wild-type and hemizygous littermates; arrows identify the four major MBP isoforms that were fourfold higher in PDGFRα hemizygous mice at day 8 but equivalent for both genotypes at day 14.
PDGF receptor levels also affected the maturation of OPCs when isolated from the brain (Table 2). In cultures established from wild type embryos, the O4+ pool rapidly expanded (15-fold increase). Only low numbers of MPB+ oligos emerged after 14 d, equivalent to the time of their first emergence (postnatal day 8) in vivo. In contrast, in PDGFRα+/- cultures, OPC pool expansion was dramatically reduced (Table 2). This again may represent precocious differentiation, because fivefold more MBP+ oligodendrocytes were present than in age-matched wild-type sibling cultures (Table 2). Similar results were also observed in spinal cord cultures. Together with precocious OPC maturation in vivo, these results reveal a critical role for pdgfra gene dose, and its correlated PDGFRα receptor levels, in OPC pool dynamics and the onset of oligodendrocyte maturation.
Oligodendrocyte maturation in the absence of PDGFRα
We next considered the maturation of OPCs from PDGFRα-null embryos in vitro. In both brain and spinal cord cultures, homozygous-nulls had a distinct phenotype from wild-type consistent with precocious OPC maturation. Wild-type cells (Fig. 3a) expressed early progenitor antigens (PDGFRα+ and NG2+), whereas homozygous-null cultures lacked PDGFRα reactivity but did contain cells with characteristic early progenitor cell morphology, including elaborate processes (Fig. 3c, arrows). These cultures also generated cells expressing O4 antigens (Fig. 3d) found on late-maturation OPCs and young oligodendrocytes. This population did not increase in brain (Table 2) or spinal cord cultures, indicating that PDGFRα is essential for the in vitro expansion of OPC pools from these distinct levels of the neural axis.
Figure 3.

Emergence of OPCs and oligodendrocytes in pdgfra-null mice. Explant cultures established from spinal cord of αR2-4 mice genotyped as wild-type (a), hemizygous (b), and homozygous-null (c-e), immunostained with anti-PDGFRα antibody (a-c), monoclonal antibody O4 (d), and anti-MBP (e) are shown. Wild-type but not pdgfra-/- cultures contained anti-PDGFRα reactive cells, and cultures from pdgfra-null embryos contained O4+ OPCs (d) and MBP+ oligodendrocytes (e). MBP+ cell soma in pdgfra-null cultures (arrow in e) but not in wild-type cultures extended numerous processes that terminate at T-intersections on axons and myelin-like sheaths. Scale bars: (in c) a-c, 50 μm; (in e) d, e, 100 μm.
The absence of PDGFRα also accelerated the maturation of OPCs. In 14 d cultures from wild-type embryos, <10% of O4+ cells coexpressed MBP (Table 2). In sibling hemizygous and homozygous-null cultures, in contrast, all O4-cells were MBP+ (Table 2). Sun et al. (2000) also reported oligos in cultures from αR2-4/αR2-4 mice. In spinal cord cultures from wild-type embryos, oligodendrocytes were abundant, and, as described previously, ventral but not dorsal spinal cord explants were oligo competent (Noll and Miller, 1993). Rather surprisingly, oligodendrocytes in spinal cord cultures from PDGFRα-null but not wild-type or hemizygous cultures were also competent to elaborate myelin-like axonal sheaths (Fig. 3e). Thus, the absence of PDGFRα signaling appears essential for the final stages of oligodendrocyte maturation in vitro.
PDGFRα-coupled signaling pathways in OPC maturation
The above studies define an obligate cell-autonomous role for PDGFRα in timing OPC maturation. We next addressed the role of specific downstream signaling pathways. Our approach was by transfection of PDGFRα-null OPCs with expression vectors encoding wild-type (PDGFRαwt) or signaling mutant (tyrosine-to-phenylalanine substitution) receptors. These mutations simply prevent the transgene-encoded receptors from activating specific downstream signaling molecules while leaving the intracellular pathways otherwise intact. To test their efficacy, we first examined the ability to activate specific pathways in OPC lines stably transfected with constructs encoding chimeric versions of these mutant receptors (described in detail below). Mutations uncoupling these receptors from PI3K blocked receptor-dependent PI3K-Akt activation but not the ability to activate other pathways, including PLCγ (Ebner et al., 2000). Constructs tested included both single (Y731F and Y742F) and double (Y731F/Y742F) point mutations, indicating that both sites are required to couple PDGFRα to the PI3K cascade. In contrast, mutations uncoupling PDGFRα from PLCγ (Y1018F) generated receptors intact for PI3K-Akt signaling but defective for intracellular calcium release (Fig. 4a). Thus, these mutations generated receptors whose activation defects are pathway specific.
Figure 4.

Rescue of PDGFRα-null OPCs with wild-type and signaling uncoupled receptor transgenes. a, Point mutations uncouple PDGFRα from specific second-messenger pathways. Intracellular free calcium in control OPCs (top), OPCs expressing wild-type receptor transgenes (middle), and OPCs expressing mutant receptor transgenes defective for PLCγ activation (bottom). The analysis used chimeric fms:pdgfra transgenes, and tracings represent the emission ratio (560/520 nm) from individual fura-2-loaded cells (>40 cells imaged per field) sequentially stimulated with the Fms-specific ligand hCSF and then with PDGF-AA to activate endogenous PDGFRα. Vertical arrows in the top panel denote the time of stimulation, and an absence of hCSF response in the bottom panel identifies PDGFRα tyrosine 1018 as essential for PLCγ activation. b, c, Impaired rescue of OPCs and oligodendrocytes (OL) with point-mutant receptor transgenes. Mean ± SEM number of O4+ OPCs (b) and MBP+ oligodendrocytes (c) per well in PDGFRα-null spinal cord explant cultures (1) and parallel cultures after transfection with expression vectors encoding wild-type (wt) PDGFRα (2), PDGFRαY742F (3), or PDGFRαY1018F (4). Values in parentheses represent the number of independent experiments, and asterisks indicate values that are statistically significant from nontransfected control cultures (*p < 0.05; **p < 0.01).
In studies with αR2-4/αR2-4 cultures, transgenes encoding PDGFRαwt generated sevenfold more O4+ OPCs than mock-transfected cultures (157 ± 31 vs 22 ± 3; p < 0.02; Student's t test) and rescued their pool expansion defect (Fig. 4b). Rescue with either PI3K or PLCγ activation-defective transgenes, in contrast, gave only threefold higher OPCs (Fig. 4b), both of which were significantly different from control and wild-type rescued cultures (p < 0.01). This attenuated rescue with either PI3K or PLCγ activation-defective transgenes was pathway specific, because transgenes carrying a distinct mutation (PDGFRαY762F) rescued OPC pool sizes to the same level as wild-type constructs (data not shown). Thus, both PI3K and PLCγ activation are necessary, and neither appears sufficient, for PDGF-mediated OPC pool expansion.
The rescue of MBP+ oligodendrocytes with wild-type and PLCγ activation-defective transgenes paralleled the OPC pool expansion (Fig. 4c), consistent with OPC pool dynamics regulating the generation of oligodendrocytes. Both wild-type and PLCγ-defective transgenes generated twofold higher numbers of MBP-oligos than control cultures, and both were significantly different from the nontransfected controls (p < 0.02). In contrast, PI3K activation-defective transgenes gave no net increase in oligodendrocytes (Fig. 4c, lane 3). Because the OPC expansion was rescued by these PI3K-uncoupled receptors (Fig. 4b), this suggests an additional role for PI3K activation in oligodendrocytes. One interpretation is prevention of cell death, because PI3K activity is essential for oligodendrocyte survival (Vemuri and McMorris, 1996), although we have not quantified cell death in these cultures.
PDGFRα-coupled PI3K is obligate for OPC migration in vitro
In addition to proliferation defects, OPCs from PDGFRα-null embryos also had a migration defect consistent with a role for PDGF in chemotaxis (Armstrong et al., 1990). Whereas OPCs in wild-type cultures migrated away from the spinal cord explants (Fig. 5a), MBP+ cells in PDGFRα-null cultures were located within or immediately adjacent to the spinal cord tissue (Fig. 5b, arrows), with a mean radial dispersion from the center (238 ± 17 μm) that was less than the average diameter of the tissue (350 μm). Transfection of these cultures with PDGFRα transgenes rescued OPC migration, with many O4+ MBP+ cells located distal from the explants. Rescue of migration was obtained with wild-type PDGFRα, with the PLCγ activation-defective transgene PDGFRαY1018F and with the PDGFRαY762F construct (Fig. 5c,d,f). The mean distance from center for cells in PDGFRαY1018F cultures (350 ± 12 μm) was significantly larger than the nontransfected controls (p < 0.01). In contrast, the radial dispersion of cells in the PI3K activation-defective cultures (Fig. 5e) was significantly lower than the PDGFRαY1018F cultures (268 ± 18 μm; p < 0.01) and not significantly different from nontransfected controls (p = 0.3). This apparent failure to rescue the migration defect was unlikely to be an indirect effect on proliferation or survival, because PI3K-defective transgenes rescued OPC proliferation (Fig. 4b) and because MBP+ oligos survived in these cultures (Fig. 5e). These results indicate that PI3K coupling to PDGFRα is necessary for OPC migration in vitro. Although this does not determine whether PI3K is sufficient, the results further indicate that PLCγ coupling is not required (Fig. 5f).
Figure 5.

PDGFRα activation of PI3K is necessary for OPC migration. The radial dispersion of OPCs in cultures of wild-type (a) or pdgfra-/- (b-f) spinal cord explants is shown. In c-f, pdgfra-/- cultures were transfected with expression vectors encoding wild-type PDGFRα (d) or signaling uncoupled PDGFRαY762F (c), PDGFRαY742F (e), or PDGFRαY1018F (f). The percentage total of O4+ and MBP+ cells was determined in concentric circles radial from the edge of individual explants (inner circle in a), and the graph shows their cumulative numbers in wild-type (circles), homozygous-null (triangles), and transgene rescued null cultures (squares). OPC/oligodendrocytes (OLs) in pdgfra-null cultures are located within or proximal to spinal cord explants, and the PDGFRαY742F transgene was unable to rescue this migration defect. Scale bars, 100 μm.
A PDGFRα-coupled rheostat controls OPC proliferation
To this point, our results support a critical role for PDGFRα in the control of OPC pool expansion (proliferation) rather than early genesis (fate specification) or late maturation (myelination) and to the coupling of specific pathways to unique responses. We next examined in more detail the PDGFRα-activated pathways required for OPC proliferation. Because the numbers of OPCs from PDGFRα-null cultures were limiting, these studies used wild-type cells transfected with chimeric (Fms:PDGFRα) receptor constructs with the ligand binding domain of c-fms linked to the signaling domain of PDGFRα (Yu et al., 1994; Ebner et al., 2000). The Fms:PDGFRα chimeras thus circumvent endogenous PDGFRα and give a PDGF-like signaling response to human CSF1. Transgene expression was confirmed by anti-Fms immune staining, and CSF1-dependent chimera stimulation by PI3K activation (Ebner et al., 2000), intracellular calcium mobilization (Fig. 4a, middle panel), and thymidine incorporation (Fig. 6). The analysis was also performed with both primary OPCs isolated from neonatal rat brain (Fig. 6a,c) and with an OPC line conditioned for growth in culture (Fig. 6b,d). For both cell populations, PDGFRα-activated proliferation required PI3K coupling at low ligand concentrations and required PLCγ-coupling at high concentrations.
Figure 6.

Combinatorial signaling pathways are required for PDGFRα mitogenic response. Thymidine incorporation in primary brain OPCs (a, c) and in an OPC line (b, d) after transfection with expression vectors encoding either PI3K-uncoupled (left) or PLCγ-uncoupled (right) PDGFRα transgenes is shown. To circumvent endogenous PDGFRα, the analysis used chimeric fms:pdgfra transgenes, and cells in individual wells were exposed to the indicated levels of the Fms ligand hCSF. Values (mean ± SEM) are representative of at least three independent trials, and asterisks indicate values for activation-defective chimeras that are significantly different from their zero ligand controls (p < 0.05). a, Intact fms transgenes (filled circles), fms:pdgfra chimeras (open circles), and chimeras with mutations of PDGFRα tyrosine residues 731 and 742 (triangles); b, fms:pdgfra chimeras (filled circles) and chimeras with mutations at PDGFRα tyrosines 731 (open circles), residues 731 and 742 (filled triangles), and with a deletion spanning both of these tyrosines (open triangles); c, d, chimeras encoding a mutation at PDGFRα tyrosine 1018 (triangles). For each construct examined, the response was similar in both primary OPCs and the OPC line, with a lower proliferative index in primary cultures reflecting limiting cell numbers and transfection efficiency. TdR, Thymidine deoxyribose.
Both intact fms and the chimeric fms:PDGFRα receptors gave a response to CSF comparable with PDGF activation via endogenous PDGFRα, with half-maximal induction of proliferation at 5-10 ng/ml CSF1 (Fig. 6). No CSF response was observed in nontransfected controls. PI3K activation-defective receptors (Fig. 6a,b) gave significantly lower thymidine incorporation than wild-type transgenes at each ligand concentration tested (p < 0.05). At low ligand levels (5 and 10 ng/ml), these receptors gave only marginal response, and differences with zero ligand controls were not significant (p = 0.56; p = 0.74). At higher ligand levels (20 ng/ml), these receptors gave a thymidine incorporation response that was significantly greater than zero ligand controls (p < 0.01), representing up to 54% maximal response of the wild-type chimeras. Comparable results were obtained with Fms: PDGFRα chimeras encoding either single (Y731F) or double (Y731F/Y742F) mutations and encoding a deletion of residues 710-789 spanning the kinase insert domain (Fig. 6b). In contrast, PLCγ activation-defective receptors gave an apparent wild-type response at subthreshold levels but no response at the higher ligand concentrations (Fig. 6c,d). At low ligand levels (5 and 10 ng/ml), the response was significantly greater than zero ligand control (p < 0.05) and up to 61% maximal response of wild-type chimeras. At high ligand levels (20 ng/ml), the response was not different from zero ligand control (p = 0.11). Thus, PI3K activation appears to contribute more to OPC proliferation at low than at high levels, whereas PLCγ-activation was essential at high but not at low ligand levels.
The cumulative response of OPCs to PI3K and PLCγ activation-defective transgenes predicts that other pathways also participate in OPC proliferation. Combined thymidine incorporation (counts per minute minus background) of both transgenes at the lowest (5 ng/ml) and highest (40 ng/ml) levels tested represented 85 and 82% of net thymidine incorporation of the wild-type transgenes, whereas the cumulative response at 10 and 20 ng/ml represented 51 and 63% of the wild-type chimeras. Of a number of other fms:PDGFRα constructs tested, one deletion (C′ residues 977-1089) and one substitution (Y993F) abrogated mitogenic signaling (data not shown). Although the signaling pathways linked to Y993 have not been identified (Eriksson et al., 1995), these results suggest a role for pathways associated with this PDGFRα domain in OPC proliferation.
Our analysis suggests a “rheostat” model for PDGFRα signaling (Fig. 7c) and predicts that the activation of these pathways through endogenous PDGFRα would be sensitive to ligand levels. We directly tested this by examining the activation (phosphorylation) status of immediate response transducers in both the PI3K (Akt serine 473) and PLCγ1 (tyrosine 783) pathways (Fig. 7a). For both, we detected a prominent cytoplasmic ring of immunofluorescence in responsive cells after acute activation (Fig. 7a), with the numbers of OPCs responding concentration dependent (Fig. 7b). For Akt, we detected serine 473 phosphorylation in 50% of cells at 0.4-1.0 ng/ml ligand, with no significant increase above background levels at higher ligand concentrations (Fig. 7b). This cannot reflect a rapid activation then decay of phosphorylated Akt at higher ligand levels, because decay takes several hours (Downward, 2004). For PLCγ1, maximal phosphotyrosine response was observed at the higher levels examined (Fig. 7b). These results demonstrate that the PI3K and PLCγ pathways are activated at distinct ligand concentrations, and, together with the proliferation studies (Fig. 6), they suggest that both pathways are independently required to deliver a full mitogenic response. Thus, in OPCs, mitogenic signaling through PDGFRα acts more like a pathway specific rheostat than a generic ON-OFF signal.
Figure 7.
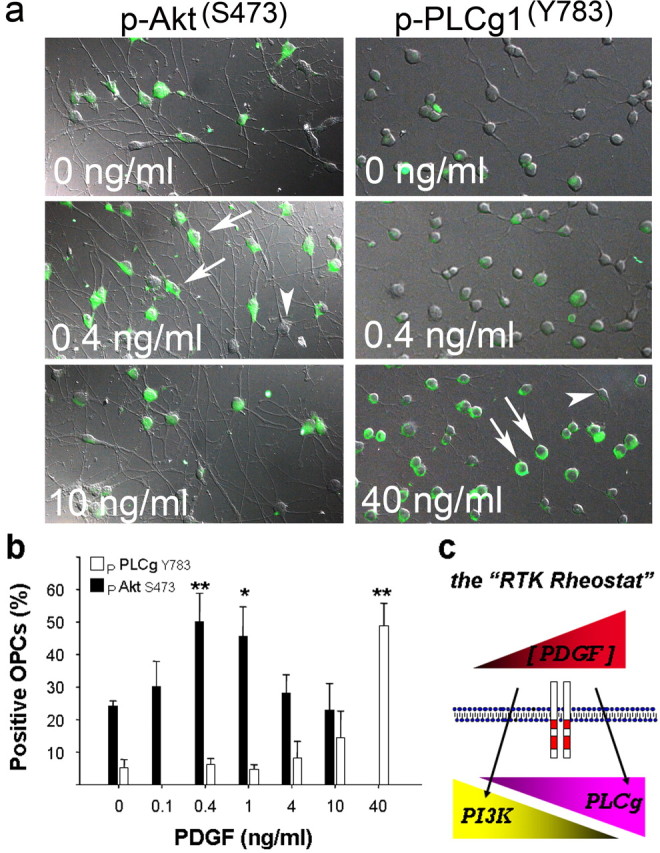
Activation of PI3K and PLCγ at distinct ligand levels. a, Phosphorylation of Akt serine 473 (left) and PLCγ1 tyrosine 783 (right) in OPCs exposed to PDGF-AA. Activation was scored as positive for individual cells in which immunoreactivity was concentrated at the plasma membrane (arrows) and negative if diffuse or nondetectable (arrowheads). b, Dose-dependent phosphorylation of Akt and PLCγ; the maximal response observed was at 0.4 ng/ml PDGF for Akt (filled bars) and 40 ng/ml for PLCγ1 (open bars). Values are mean ± SEM from four independent trials with 200-400 cells per condition, and asterisks indicates a significant difference from mock-treated control cultures (*p < 0.05; **p < 0.01). c, The RTK rheostat; the top symbol depicts relative extracellular ligand levels (low to high, in arbitrary units), and the bottom symbols depict the activation of PI3K and PLCγ at distinct ligand levels. The model predicts that, at low levels, PDGFRα uses PI3K (which promotes proliferation and chemotactic migration), whereas at high levels, PDGFRα engages PLCγ (which promotes proliferation but is not required for migration). The model offers a simple mechanism for how OPCs generate directional migration and sustained proliferation in a gradient of PDGF (see Discussion).
Discussion
Our results demonstrate that PDGFRα signaling generates a graded response rather than a generic “go” and define three novel aspects of cell-autonomous PDGFRα signaling in OPCs from the rodent brain and spinal cord. First, the emergence of OPCs in mice homozygous for a pdgfra-null allele demonstrates that PDGFRα is dispensable for their genesis. Second, the early emergence of oligodendrocytes in mice heterozygous for a null allele, and their accelerated maturation in vitro, demonstrates that PDGF receptors can be limiting for cell cycle progression and thus part of the machinery regulating the timing of oligodendrocyte maturation. Third, the requirement of multiple PDGFRα-coupled pathways at distinct signal strengths demonstrates that combinatorial signaling pathways contribute to PDGFRα-promoted OPC proliferation. Together, these observations support a role for PDGFRα signal strength, coupled to distinct downstream signaling pathways, in regulating OPC proliferation and their subsequent maturation.
OPC origins and PDGFRα
The genesis of OPCs in that absence of PDGFRα was surprising given the early expression of PDGFRα in SVZ precursors, the multiple roles of PDGF in OPC maturation, and the critical role of PDGF in regulating OPC pool dynamics in vivo (van Heyningen et al., 2001). Both ligand binding and receptor expression studies reveal that PDGFRα is the only PDGF receptor isoform expressed on OPCs (Hart et al., 1989a; McKinnon et al., 1990). In the early embryonic spinal cord, PDGFRα expression is restricted to a narrow ventral lateral focus proximal to the motor neuron domain pMN (Pringle and Richardson, 1993; Richardson et al., 1997), and pMN has been proposed to be the singular origin of OPCs and dedicated to motoneurons and oligodendrocytes (Lu et al., 2002). Their codependence on the bHLH (basic helix-loop-helix) protein Olig2 further suggests coupled mechanisms for specification (Zhou and Anderson, 2002) and a common motor neuron-oligodendrocyte ancestry (Lu et al., 2002; Rowitch et al., 2002). If these pMN-OP progenitors are the sole source of spinal cord OPCs, then the current study certainly indicates that PDGFRα is dispensable for their genesis. However, our results also lend caution that PDGFRα expression is not necessarily a valid marker for identifying all OPCs in vivo.
An alternative view of glial genesis challenges a singular origin of oligos in pMN-OP and is supported by several studies (Liu and Rao, 2004; Miller, 2005). First, glial progenitors that can generate oligodendrocytes are present at E13 in dorsal spinal cord (Lee et al., 2000; Gregori et al., 2002), indicating that non-pMN foci also have at least the potential to generate oligodendrocytes. Second, PDGFRα expression may not be obligate for all oligodendrocyte progenitors, because a population of OPCs emerge from the olfactory anlage independent of PDGFRα (Spassky et al., 2001). This is also consistent with studies identifying cells expressing early OPC markers in non-pMN regions of the subventricular zone, including more dorsal (Spassky et al., 1998) and ventral (Fu et al., 2002; Rowitch et al., 2002) regions. Finally, evidence for two distinct pools of OPCs as defined by plp/NG2 expression (Mallon et al., 2002) is consistent with the potential contribution of multiple populations to the generation of oligodendrocytes. The present study and that of Klinghoffer et al. (2002) demonstrate OPC and oligodendrocyte formation in the absence of PDGFRα and formally raise the possibility that PDGFRα expression neither identifies all OPCs nor is obligate for OPC development. However, studies to date have not ruled out at least transient expression of PDGFRα in all OPC pools, and the contribution of “non-PDGFRα”-expressing cells to oligodendrocyte formation remains unresolved.
PDGFRα levels and the timing of oligodendrocyte maturation
Our results also indicate that PDGFRα may be essential for OPC pool expansion. Although OPCs emerged in PDGFRα-null mice, their census was lower than wild type, their ability to expand in vitro was dramatically impaired, and we did not see the amplification of a significant pool of MBP+ oligodendrocytes in either brain or spinal cord cultures (Table 2). These results extend the findings that PDGF ligand levels are limiting for OPC pool expansion in vivo (van Heyningen et al., 2001). Our population studies were by necessity performed in vitro, an environment that in principal could lack necessary mitogens for the amplification of a PDGFRα-negative OPC pool. Thus, we cannot directly address the relative contribution of potentially distinct (PDGFRα-positive, PDGFRα-negative) pools to oligodendrocyte formation in vivo. The accelerated maturation of oligodendrocytes observed in PDGFRα hemizygous mice also can only be interpreted by considering PDGFRα-positive OPC pool dynamics, because a potential PDGFRα-negative pool would presumably not be affected by this mutation. However, the current data are most consistent with the interpretation that cells that express PDGFRα during at least some phase of their maturation constitute a significant fraction of the oligodendrocyte generating pool in vivo.
The mitogenic response of OPCs to PDGF in vitro is limited, and the number of mitotic cycles controls the timing of OPC maturation (Raff et al., 1988). Experimental manipulation of ligand levels demonstrated a pivotal role for PDGF levels in myelination, because transgenic mice lacking PDGF A-chain isoform have fewer OPCs (Fruttiger et al., 1999), whereas excess PDGF-A gives overproduction of OPCs that are pruned by cell death (Calver et al., 1998). Richardson and colleagues have proposed a mitogen depletion model to account for this (van Heyningen et al., 2001). PDGF receptor levels also decrease as OPCs mature in vitro (Hart et al., 1989a,b), and the current study further demonstrates that PDGFRα gene dosage, and by extension PDGFRα levels, affect OPC maturation in vitro and in vivo. These results implicate PDGFRα levels in OPC pool dynamics, and PDGFRα levels may be an intrinsic component of the biological clock regulating oligodendrocyte maturation. PDGF signaling is not likely the singular mechanism regulating final oligodendrocyte numbers, because experimental manipulations of ligand levels do not affect the levels of myelin at maturity (Calver et al., 1998).
The elaboration of myelin sheaths by PDGFRα-null oligodendrocytes in vitro (Fig. 3e) was another unexpected outcome of this study. Previous attempts to establish efficient, myelin-competent oligo-neuron cocultures have had relatively limited success (Vartanian et al., 1999). Oligodendrocytes normally downregulate PDGFRα at differentiation, and our results now indicate that downregulation may be critical for myelination. The lack of ensheathment in control cultures further suggests that wild-type oligodendrocytes may retain some level of PDGFRα signaling in vitro that prevents their final maturation. This may reflect the presence of factors in neuronal cocultures that would not emanate from myelinated axons in vivo.
PDGFRα signaling: combinatorial pathways
Our observation that a full mitogenic response of OPCs via PDGFRα requires multiple signaling pathways differs from studies in hepatoma, hematopoetic, and fibroblast cell lines in which PI3K and PLCγ are dispensable (Yu et al., 1994). This may in part reflect differences in signaling requirements of primary explant cultures versus immortal cell lines. OPCs require PI3K but not PLCγ activation at low ligand concentrations, although both are required at high concentrations (Fig. 6). The requirement for PI3K but not PLCγ activation for migration (Fig. 5) further links specific signaling pathways to unique biological outcomes and suggests that RTK signaling specificity may be controlled in part at the level of second-messenger activation. This contrast with studies in fibroblasts, in which FGF and PDGF activate the same immediate response genes (Fambrough et al., 1999). Our results are more consistent with RTKs having intrinsic differences in the signaling pathways they activate and generate specific responses based on strength of signal and which pathways are engaged (Simon, 2000). This model is supported by studies in a number of systems. Quantitative differences in RTK signaling include roles for receptor density and strength of signal in invertebrates (Rebay, 2002) and different levels of MAPK activation by NGF and EGF during differentiation and proliferation of the pheochromocytoma PC12 cell line (Marshall, 1995). Qualitative differences include activation of distinct pathways in different cell types by the EGF receptor in Caenorhabditis elegans (Clandinin et al., 1998) and activation of PI3K by PDGF but not FGF in OPCs (Ebner et al., 2000). Substitution studies in mice also demonstrate that the signaling domains of distinct PDGF receptors are qualitatively different. The signaling domain of PDGFRα cannot replace that of PDGFRβ in development (Klinghoffer et al., 2001), and the signaling domains from FGFR1 or Drosophila Tor cannot replace that of PDGFRα, although it can be replaced by PDGFRβ (Klinghoffer et al., 2002; Hamilton et al., 2003). These results thus contrast with studies in Drosophila, in which the signaling domains of distinct RTKs can be functionally substituted during development (Dossenbach et al., 2001) and suggest that RTK signaling systems have gained both specificity and complexity with tissue specialization during metazoan evolution.
Our findings may also explain why oligodendrocyte survival requires 200-fold lower levels of PDGF than that required to promote proliferation (Barres et al., 1993). PDGF engages PDGFRα to activate PI3K at low ligand levels (Fig. 7), and PI3K is required for oligodendrocyte survival (Vemuri and McMorris, 1996). Thus, survival in very low levels of PDGF may represent specific activation of the PI3K-Akt pathway. This is also consistent with our finding that we could not rescue PDGFRα-null oligodendrocytes with PI3K activation-defective receptor transgenes (Fig. 4c).
Finally, the RTK rheostat model suggests a novel interpretation for PDGF-mediated chemoattraction. At low ligand levels, PDGF activates PI3K, which stimulates OPC migration, whereas PDGF at high levels such as within axonal domains secreting PDGF would not. Thus, chemoattraction of OPCs to PDGF may in part reflect motility at low but not high ligand levels. Together, these observations suggest that RTKs operate as a rheostat (Fig. 7c) activating specific biological responses through combinatorial signaling pathways proportional to ligand and receptor density-dependent strength of signal.
Footnotes
This work was supported by United States Public Health Service Grant MH54652 from the National Institute of Mental Health. We thank Phil Soriano for αR2-4 (PDGFRα-null) mice, Joel Levine, Hideaki Yokoo, and Dave Coleman for antibodies, and Sylvie Ebner, Hongwei Rao, and Maryse Dunbar for excellent technical assistance.
Correspondence should be addressed to R. D. McKinnon, University of Medicine and Dentistry of New Jersey, Robert Wood Johnson Medical School, 675 Hoes Lane, S-225, Piscataway, NJ 08854. E-mail: mckinnon@umdnj.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253499-10$15.00/0
References
- Armstrong RC, Harvath L, Dubois-Dalcq M (1990) Type 1 astrocytes and oligodendrocyte-type 2 astrocyte glial progenitors migrate toward distinct molecules. J Neurosci Res 27: 400-407. [DOI] [PubMed] [Google Scholar]
- Barres BA, Schmid R, Sendnter M, Raff MC (1993) Multiple extracellular signals are required for long-term oligodendrocyte survival. Development 118: 283-295. [DOI] [PubMed] [Google Scholar]
- Calver AR, Hall AC, Yu WP, Walsh FS, Heath JK, Betsholtz C, Richardson WD (1998) Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron 20: 869-882. [DOI] [PubMed] [Google Scholar]
- Cheng PW (1996) Receptor ligand-facilitated gene transfer: enhancement of liposome-mediated gene transfer and expression by transferrin. Hum Gene Ther 7: 275-282. [DOI] [PubMed] [Google Scholar]
- Clandinin TR, DeModena JA, Sternberg PW (1998) Inositol trisphosphate mediates a RAS-independent response to LET-23 receptor tyrosine kinase activation in C. elegans Cell 92: 523-533. [DOI] [PubMed] [Google Scholar]
- Dossenbach C, Rock S, Affolter M (2001) Specificity of FGF signaling in cell migration in Drosophila Development 128: 4563-4572. [DOI] [PubMed] [Google Scholar]
- Downward J (2004) PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol 15: 177-182. [DOI] [PubMed] [Google Scholar]
- Ebner S, Dunbar M, McKinnon RD (2000) Distinct roles for PI3K in proliferation and survival of oligodendrocyte progenitor cells. J Neurosci Res 62: 336-345. [DOI] [PubMed] [Google Scholar]
- Eriksson A, Nanberg E, Ronnstrand L, Engstrom U, Hellman U, Rupp E, Carpenter G, Heldin CH, Claesson-Welsh L (1995) Demonstration of functionally different interactions between phospholipase C-gamma and the two types of platelet-derived growth factor receptors. J Biol Chem 270: 7773-7781. [DOI] [PubMed] [Google Scholar]
- Fambrough D, McClure K, Kazlauskas A, Lander ES (1999) Diverse signaling pathways activated by growth factor receptors induce broadly overlapping, rather than independent, sets of genes. Cell 97: 727-741. [DOI] [PubMed] [Google Scholar]
- Fruttiger M, Karlsson L, Hall AC, Abramsson A, Calver AR, Bostrom H, Willetts K, Bertold CH, Heath JK, Betsholtz C, Richardson WD (1999) Defective oligodendrocyte development and severe hypomyelination in PDGF-A knockout mice. Development 126: 457-467. [DOI] [PubMed] [Google Scholar]
- Fu H, Qi Y, Tan M, Cai J, Takebayashi H, Nakafuku M, Richardson W, Qiu M (2002) Dual origin of spinal oligodendrocyte progenitors and evidence for the cooperative role of Olig2 and Nkx2.2 in the control of oligodendrocyte differentiation. Development 129: 681-693. [DOI] [PubMed] [Google Scholar]
- Gregori N, Proschel C, Noble M, Mayer-Proschel M (2002) The tripotential glial-restricted precursor (GRP) cell and glial development in the spinal cord: generation of bipotential oligodendrocyte-type-2 astrocyte progenitor cells and dorsal-ventral differences in GRP cell function. J Neurosci 22: 248-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruneberg H, Truslove GM (1960) Two closely linked genes in the mouse chromosome. Genet Res 1: 69-90. [Google Scholar]
- Hamilton TG, Klinghoffer RA, Corrin PD, Soriano P (2003) Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Mol Cell Biol 23: 4013-4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart IK, Richardson WD, Bolsover SR, Raff MC (1989a) PDGF and intracellular signaling in the timing of oligodendrocyte differentiation. J Cell Biol 109: 3411-3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart IK, Richardson WD, Heldin C, Westermark B, Raff MC (1989b) PDGF receptors on cells of the oligodendrocyte-type-2 astrocyte (O-2A) cell lineage. Development 105: 595-603. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Westermark B (1999) Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 79: 1283-1316. [DOI] [PubMed] [Google Scholar]
- Klinghoffer RA, Mueting-Nelsen PF, Faerman A, Shani M, Soriano P (2001) The two PDGF receptors maintain conserved signaling in vivo despite divergent embryological functions. Mol Cell 7: 343-354. [DOI] [PubMed] [Google Scholar]
- Klinghoffer RA, Hamilton TG, Hoch R, Soriano P (2002) An allelic series at the PDGFalphaR locus indicates unequal contributions of distinct signaling pathways during development. Dev Cell 2: 103-113. [DOI] [PubMed] [Google Scholar]
- Lee JC, Mayer-Proschel M, Rao MS (2000) Gliogenesis in the central nervous system. Glia 30: 105-121. [DOI] [PubMed] [Google Scholar]
- Liu Y, Rao MS (2004) Glial progenitors in the CNS and possible lineage relationships among them. Biol Cell 96: 279-290. [DOI] [PubMed] [Google Scholar]
- Louis JC, Magal E, Muir D, Manthorpe M, Varon S (1992) CG-4, a new bipotential glial cell line from rat brain, is capable of differentiating in vitro into either mature oligodendrocytes or type-2 astrocytes. J Neurosci Res 31: 193-204. [DOI] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH (2002) Common developmental requirement for olig function indicates a motor neuron/oligodendrocyte connection. Cell 109: 75-86. [DOI] [PubMed] [Google Scholar]
- Mallon BS, Shick HE, Kidd GJ, Macklin WB (2002) Proteolipid promoter activity distinguishes two populations of NG2-positive cells throughout neonatal cortical development. J Neurosci 22: 876-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CJ (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80: 179-185. [DOI] [PubMed] [Google Scholar]
- McKinnon RD, Matsui T, Dubois-Dalcq M, Aaronson SA (1990) FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron 5: 603-614. [DOI] [PubMed] [Google Scholar]
- Miller RH (2005) Dorsally derived oligodendroctyes come of age. Neuron 45: 1-3. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Lin X-H, Giese N, Heldin C-H, Stallcup WB (1996) Colocalization of NG2 proteoglycan and PDGF a-receptor on O2A progenitor cells in the developing rat brain. J Neurosci Res 43: 315-330. [DOI] [PubMed] [Google Scholar]
- Noll E, Miller RH (1993) Oligodendrocyte precursors originate at the ventral ventricular zone dorsal to the ventral midline region in the embryonic rat spinal cord. Development 118: 563-573. [DOI] [PubMed] [Google Scholar]
- Osterhout DJ, Ebner S, Xu J, Ornitz DM, Zazanis GA, McKinnon RD (1997) Transplanted oligodendrocyte progenitor cells expressing a dominant negative FGF receptor transgene fail to migrate in vivo J Neurosci 17: 9122-9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer SE, Warrington AE, Bansal R (1993) The oligodendrocyte and its many cellular processes. Trends Cell Biol 3: 191-197. [DOI] [PubMed] [Google Scholar]
- Pringle N, Collarini EJ, Mosley MJ, Heldin C-H, Westermark B, Richardson WD (1989) PDGF-A chain homodimers drive proliferation of bipotential (O-2A) glial progenitor cells in the developing rat optic nerve. EMBO J 8: 1049-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle NP, Richardson WD (1993) A singularity of PDGF alpha-receptor expression in the dorsoventral axis of the neural tube may define the origin of the oligodendrocyte lineage. Development 117: 525-533. [DOI] [PubMed] [Google Scholar]
- Raff MC, Lillien LE, Richardson WD, Burne JF, Noble MD (1988) Platelet-derived growth factor from astrocytes drives the clock that times oligodendrocyte development in culture. Nature 333: 562-565. [DOI] [PubMed] [Google Scholar]
- Rebay I (2002) Keeping the receptor tyrosine kinase signaling pathway in check: lessons from Drosophila Dev Biol 251: 1-17. [DOI] [PubMed] [Google Scholar]
- Richardson WD, Pringle NP, Yu WP, Hall AC (1997) Origins of spinal cord oligodendrocytes: possible developmental and evolutionary relationships with motor neurons. Dev Neurosci 19: 58-68. [DOI] [PubMed] [Google Scholar]
- Rowitch DH, Lu QR, Kessaris N, Richardson WD (2002) An “oligarchy” rules neural development. Trends Neurosci 25: 417-422. [DOI] [PubMed] [Google Scholar]
- Schatteman GC, Morrison-Graham K, van Koppen A, Weston JA, Bowen-Pope DF (1992) Regulation and role of PDGF receptor alpha-subunit during embryogenesis. Development 115: 123-131. [DOI] [PubMed] [Google Scholar]
- Simon MA (2000) Receptor tyrosine kinases: specific outcomes from general signals. Cell 103: 13-15. [DOI] [PubMed] [Google Scholar]
- Smith EA, Seldin MF, Martinez L, Watson ML, Choudhury GG, Lalley PA, Pierce J, Aaronson SA, Barker J, Naylor SL, Sakaguchi AY (1991) Mouse platelet-derived growth factor receptor alpha gene is deleted in W19H and patch mutations on chromosome 5. Proc Natl Acad Sci USA 88: 4811-4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P (1997) The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development 124: 2691-2700. [DOI] [PubMed] [Google Scholar]
- Spassky N, Goujet-Zalc C, Parmantier E, Olivier C, Martinez S, Ivanova A, Ikenaka K, Macklin W, Cerruti I, Zalc B, Thomas JL (1998) Multiple restricted origin of oligodendrocytes. J Neurosci 18: 8331-8343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassky N, Heydon K, Mangatal A, Jankovski A, Olivier C, Queraud-Lesaux F, Goujet-Zalc C, Thomas JL, Zalc B (2001) Sonic hedgehog-dependent emergence of oligodendrocytes in the telencephalon: evidence for a source of oligodendrocytes in the olfactory bulb that is independent of PDGFRalpha signaling. Development 128: 4993-5004. [DOI] [PubMed] [Google Scholar]
- Stephenson DA, Mercola M, Anderson E, Wang C, Stiles CD, Bowen-Pope DF, Chapman VM (1991) Platelet-derived growth factor receptor a-subunit gene (Pdgfra) is deleted in the mouse patch (Ph) mutation. Proc Natl Acad Sci USA 88: 6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Jayatilake D, Afink GB, Ataliotis P, Nister M, Richardson WD, Smith HK (2000) A human YAC transgene rescues craniofacial and neural tube development in PDGFRalpha knockout mice and uncovers a role for PDGFR alpha in prenatal lung growth. Development 127: 4519-4529. [DOI] [PubMed] [Google Scholar]
- van der Geer P, Hunter T, Lindberg RA (1994) Receptor protein-tyrosine kinases and their signal transduction pathways. Annu Rev Cell Biol 10: 251-337. [DOI] [PubMed] [Google Scholar]
- van Heyningen P, Calver AR, Richardson WD (2001) Control of progenitor cell number by mitogen supply and demand. Curr Biol 11: 232-241. [DOI] [PubMed] [Google Scholar]
- Vartanian T, Fischbach G, Miller R (1999) Failure of spinal cord oligodendrocyte development in mice lacking neuregulin. Proc Natl Acad Sci USA 96: 731-735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri GS, McMorris FA (1996) Oligodendrocytes and their precursors require phosphatidylinositol 3-kinase signaling for survival. Development 122: 2529-2537. [DOI] [PubMed] [Google Scholar]
- Yu J-C, Gutkind JS, Mahadevan D, Li W, Meyers KA, Pierce JH, Heidaran MA (1994) Biological function of PDGF-induced PI-3 kinase activity: its role in aPDGF receptor-mediated mitogenic signaling. J Cell Biol 127: 479-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J-C, Li W, Wang L-M, Pierce JH, Heidaran MA (1995) Requirement of a single motif within the carboxy terminal domain of PDGFRa for PDGF focus-forming activity. J Biol Chem 270: 7033-7036. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Anderson DJ (2002) The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell 109: 61-73. [DOI] [PubMed] [Google Scholar]