

Abstract
G-protein modulation of neuronal nicotinic acetylcholine receptor (nAChR) channels in rat intrinsic cardiac ganglia was examined using dialyzed whole-cell and excised membrane patch-recording configurations. Cell dialysis with GTPγS increased the agonist affinity of nAChRs, resulting in a potentiation of nicotine-evoked whole-cell currents at low concentrations. ACh- and nicotine-evoked current amplitudes were increased approximately twofold in the presence of GTPγS. In inside-out membrane patches, the open probability (NPo) of nAChR-mediated unitary currents was reversibly increased fourfold after bath application of 0.2 mm GTPγS relative to control but was unchanged in the presence of GDPβS. The modulation of nAChR-mediated whole-cell currents was agonist specific; currents evoked by the cholinergic agonists ACh, nicotine, and 1,1-dimethyl-4-phenylpiperazinium iodide, but not cytisine or choline, were potentiated in the presence of GTPγS. The direct interaction between G-protein subunits and nAChRs was examined by bath application of either Goα or Gβγ subunits to inside-out membrane patches and in glutathione S-transferase pull-down and coimmunoprecipitation experiments. Bath application of 50 nm Gβγ increased the open probability of ACh-activated single-channel currents fivefold, whereas Goα (50 nm) produced no significant increase in NPo. Neuronal nAChR subunits α3-α5 and β2 exhibited a positive interaction with Goα and Gβγ, whereas β4 and α7 failed to interact with either of the G-protein subunits. These results provide evidence for a direct interaction between nAChR and G-protein subunits, underlying the increased open probability of ACh-activated single-channel currents and potentiation of nAChR-mediated whole-cell currents in parasympathetic neurons of rat intrinsic cardiac ganglia.
Keywords: cholinergic, nicotinic receptor, G-protein subunits, parasympathetic, intracardiac ganglia, patch clamp
Introduction
Neuronal nicotinic acetylcholine receptors (nAChRs) are pentameric ion channels composed of α and β subunits and play an important role in signal transmission in the mammalian CNS and peripheral nervous system. To date, eight α (α2-α7, α9, and α10) and three β (β2-β4) subunits have been identified in the mammalian nervous system. Neuronal nAChRs on intrinsic cardiac ganglion (ICG) neurons mediate ganglionic transmission and are composed of α3, α5, α7, β2, and β4 subunits (Poth et al., 1997; Bibevski et al., 2000). In contrast to the metabotropic muscarinic AChR (mAChR), the nAChR does not require the activation of guanine nucleotide-binding proteins (G-proteins) to open its associated ion channel. However, nAChRs have been reported to be regulated by protein kinases (Swope et al., 1995); for example, inhibition of Src family tyrosine kinases has been shown recently to reduce the amplitude of nicotine-induced currents in chromaffin cells and recombinant α3β4α5 nAChRs expressed in human embryonic kidney 293 (HEK293) cells (Wang et al., 2004).
G-proteins consist of Gαβγ heterotrimers (Clapham and Neer, 1997), and the conformational change in the G-protein-coupled receptor (GPCR) induced by ligand binding leads to the exchange of GDP for GTP on the Gα subunit and the dissociation or rearrangement of Gα-GTP from its Gβγ dimer (Bunemann et al., 2003). Free Gβγ dimers have been shown to modulate the activity of an array of effector proteins, including voltage-gated Ca2+ and K+ channels, by direct interaction with intracellular domains of these channels (Reuveny et al., 1994; Krapivinsky et al., 1995; Herlitze et al., 1996; De Waard et al., 1997). A direct interaction of the Gβγ dimer has been shown recently with native and recombinant glycine receptor channels (Yevenes et al., 2003). The response of glycine, but not GABAA, receptor channels could be enhanced by the activation of GPCRs or activation with the Gβγ dimer itself. In the present study, we show direct modulation of neuronal nAChR channels by the nonhydrolyzable GTP analog GTPγS and the Gβγ subunit, consistent with the idea of direct G-protein activation of neuronal nAChR subtypes.
Materials and Methods
Preparation of dissociated intrinsic cardiac ganglia neuron cultures. Parasympathetic neurons from neonatal rat ICGs were isolated and placed in tissue culture. The procedures for the isolation of ICG neurons have been described previously (Xu and Adams, 1992) and were in accordance with guidelines of the University of Queensland Animal Experimentation Ethics Committee. Briefly, Wistar rats (2-12 d of age) were killed by stunning and decapitation, and the heart was excised and placed in HBSS. Clusters of ICGs surrounding the pulmonary veins were dissected from the atria and transferred to HBSS containing collagenase (300 U/ml; Worthington Biochemical, Lakewood, NJ). After 1 h of digestion at 37°C, the ganglia were rinsed twice in HBSS and triturated in culture medium (DMEM with 10% v/v fetal calf serum and 1% v/v penicillin-streptomyocin; Invitrogen, Mount Waverley, Victoria, Australia) using a fine-bore Pasteur pipette The dissociated neurons were plated on sterile 12 mm glass coverslips and incubated at 37°C for 12-24 h in a 95% O2-5% CO2 atmosphere.
Electrophysiological recordings from ICG neurons. Membrane currents were recorded from isolated ICG neurons using standard whole-cell patch clamp and inside-out excised membrane patch-recording techniques. Electrodes for both recording techniques (model GF150F-7.5; Harvard Apparatus, Edenbridge, UK) were pulled and fire-polished and had resistances of 1-2 MΩ when filled with intracellular solution. For single-channel experiments, pipettes were coated with dental wax to within 100 μm of the tip. Membrane currents evoked by agonist application were amplified and low-pass filtered (10 kHz) using an Axopatch 200A patch-clamp amplifier (Axon Instruments, Union City, CA), and voltage steps were generated using a Pentium personal computer, pClamp 9.0 software, and a Digidata 1322A interface (Axon Instruments).
Solutions and drug application. Agonist-activated currents in ICG neurons were recorded using patch pipettes filled with an internal solution containing the following (in mm): 140 KCl, 2 CaCl2, 11 EGTA, 2 MgATP, and 10 HEPES-KOH, pH 7.2. The bath solution contained the following (in mm): 140 NaCl, 3 KCl, 1.2 MgCl2, 2.5 CaCl2, 7.7 glucose, and 10 HEPES-NaOH, pH 7.35. Agonists were applied to cells by brief pressure ejection (15 psi; Picospritzer II; General Valve, Fairfield, NJ) from an extracellular patch pipette positioned 50-100 μm from the cell soma to evoke maximal responses to agonists. For whole-cell recordings, the pipette solution contained 0.2 mm GTP sodium salt, GDPβS trilithium salt, or GTPγS tetralithium salt. For inside-out excised membrane patch recordings, both extracellular and pipette solutions were the bath solution. Acetylcholine (5 μm) was added to the pipette solution, and 0.2 mm GTP, GDPβS, or GTPγS was bath applied to the inside-out membrane patches. Recombinant rat G-protein subunits Goα, myristolated, and βγ dimer (Calbiochem, La Jolla, CA) were also bath applied. All experiments were performed at room temperature (22°C). Data were analyzed using Clampfit 9.0 software (Axon Instruments) and are expressed as means ± SE, with n indicating the number of neurons studied. The significance of differences was evaluated using the Student's t test. Concentration-response curves were analyzed using SigmaPlot 8.0 (SPSS, Chicago, IL) and fitted by unweighted nonlinear regression to the Hill equation.
Plasmid constructs. The large intracellular loop region of rat nAChR subunits was identified via analysis of the amino acid sequence on TMHMM server version 2.0 (for the prediction of transmembrane helices in proteins; http://www.cbs.dtu.dk/services/TMHMM-2.0/). Amino acids α3 (321-471), α4 (331-603), α5 (321-411), α7 (300-472), β2 (321-458), β3 (325-433), and β4 (320-463) were amplified by PCR from rat nAChR clones (provided by Dr. J. Patrick, Baylor University, Waco, TX) using Elongase (Invitrogen) and cloned in frame into the PGEX2TK vector (Amersham Biosciences, Castle Hill, New South Wales, Australia). All constructs were verified by sequencing at the Australian Genome Research Facility (St. Lucia, Queensland, Australia).
Glutathione S-transferase pull-down assays. Glutathione S-transferase (GST) fusion proteins were generated in Escherichia coli BL21 by induction with 0.5-1.5 mm isopropylthiogalactoside in superoptimal catabolite media (2% tryptone, 0.5% yeast extract, 10 mm NaCl, 2.5 mm KCl, 10 mm MgCl2, 10 mm MgSO4, and 20 mm glucose) at 18°C. After 6-18 h, the cells were collected by centrifugation at 2000 rpm and lysed by sonication in 7 ml of NETN lysis buffer containing 100 mm NaCl, 1 mm EDTA, 20 mm Tris, pH 8.0, and 0.5% NP-40, with protease inhibitor cocktail set II (Calbiochem). The fusion proteins were purified using glutathione-Sepharose 4B beads (Amersham Biosciences). The expression levels of the GST fusion proteins were monitored on 12% SDS-polyacrylamide gels by Coomassie blue staining, and equivalent amounts of proteins were used in the pull-down assays. Normalized amounts of GST fusion proteins were incubated with 300 ng of each G-protein subunit. For the Gβγ complex (Calbiochem), incubations were in 1 ml of binding buffer A (100 mm NaCl, 1 mm EDTA, 20 mm Tris, pH 8.0, 1% Lubrol, and protease inhibitor cocktail set II) at 4°C overnight. For the Goα subunit (Calbiochem), incubations took place in a more stringent binding buffer B (200 mm NaCl, 1 mm EDTA, 20 mm Tris, pH 8.0, 1% Lubrol, 1% milk, and protease inhibitor cocktail set II) at 4°C for 4 h. The beads were washed three times in either binding buffer A or B, and bound proteins were separated on 12% SDS-polyacrylamide gels and transferred to a 0.45 μm nitrocellulose membrane (Bio-Rad, Regents Park, New South Wales, Australia). The membrane was blocked overnight in PBS, pH 7.4, and 3% milk at 4°C. The membranes were then probed with rabbit anti-Gβγ complex (Calbiochem) or rabbit anti-Goα subunit (Calbiochem) (at 1:1000 in PBS/3% milk/0.05% Tween 20) for 2 h at room temperature. Subsequently, membranes were incubated in goat anti-rabbit IgG peroxidase conjugate (Calbiochem) (1:4000 dilution in PBS/0.3% milk/0.1% Tween 20) for 1 h at room temperature. Bound G-proteins were visualized using an Immun-Star HRP chemiluminescence kit (Bio-Rad). Rabbit anti-GABAA α1 antibody (provided by Dr. W. Sieghart, Brain Research Institute, Medical University of Vienna, Vienna, Austria) was used in control experiments.
Coimmunoprecipitation and Western blot analysis. Rat ICGs were homogenized in immunoprecipitation (IP)-lysis buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Triton X-100, and protease inhibitor mixture (Roche Diagnostics, Mannheim, Germany). After gentle rotation for 4 h at 4°C, homogenates were centrifuged at 27,000 × g for 20 min at 4°C, and the supernatant was collected. Lysate (∼2 mg) was then incubated with ∼10 μg of anti-Goα or anti-Gβγ antibody (Calbiochem) overnight at 4°C, after which 50 μl of protein A-agarose beads (Roche Diagnostics) was added, and the samples were rocked at 4°C for 6 h. The protein A-agarose was then washed four times in IP-lysis buffer. SDS sample buffer (50 μl) was added to the pellet and heated to 95°C for 5 min. Proteins were separated on a 7% SDS-polyacrylamide gel and then transferred to nitrocellulose membrane (Bio-Rad) by electroblotting. Blots were incubated with either monoclonal antibody 35 (mAb35) or mAb270 (500 ng/ml; Sigma, St. Louis, MO) overnight at 4°C in 5% nonfat milk, 100 mm Tris, pH 7.5, 306 mm NaCl, and 0.1% Tween 20. Negative controls were performed using rabbit nonimmune IgG (Alpha Diagnostic International, San Antonio, TX). The blot was washed four times with Tris-NaCl-Tween buffer, incubated for 1 h with horseradish peroxidase-conjugated anti-rat IgG (Pierce Biotechnology, Rockford, IL) at 10 ng/ml, and washed again. Immunoreactive proteins were detected by enhanced chemiluminescence using the SuperSignal kit (Pierce Biotechnology).
Results
G-protein activation of nAChRs in neurons of the intrinsic cardiac plexus
Previous results from our laboratory have shown that activation of members of the pituitary adenylate cyclase-activating polypeptide (PACAP)/vasoactive intestinal peptide (VIP) receptor family potentiated ACh- and nicotine-evoked currents in parasympathetic neurons dissociated from neonatal rat intracardiac and submandibular ganglia (Cuevas and Adams, 1996; Liu et al., 2000). This modulation was mediated by a pertussis toxin-sensitive Go-protein pathway, was unaffected by cell dialysis (Cuevas and Adams, 1996), and could be mimicked by intracellular perfusion with the nonhydrolyzable GTP analog GTPγS (Liu et al., 2000). To further examine the mechanism of G-protein modulation of neuronal nAChRs, intracardiac neurons were dialyzed with 0.2 mm GTP (control), GTPγS, or the nonhydrolyzable GDP analog GDPβS. GPCR-induced potentiation of nAChRs could be mimicked by cell dialysis with 0.2 mm GTPγS (Fig. 1). Representative whole-cell membrane currents evoked by 30 μm nicotine in the presence of different GTP/GDP analogs are shown in Figure 1A. Current-voltage relationships obtained for the peak nicotine-evoked current density from neurons dialyzed with different GTP/GDP analogs are shown in Figure 1B. GTPγS potentiated nicotine-evoked current amplitude at all membrane potentials, increasing peak current density more than threefold from -23.8 ± 2.4 to -76.1 ± 7.2 pA/pF (n = 6) in the presence of GTPγSat -50 mV. GDPβS in the pipette had no significant effect on peak current amplitude; the peak current density at -50 mV was -26.2 ± 6.3 pA/pF (n = 6). G-protein potentiation of nicotine-evoked current amplitude was dependent on nicotine concentration. A normalized plot of the peak nicotine-evoked current density as a function of nicotine concentration is shown in Figure 1C. The data were fit using the following equation: y = 1/1 + ([A]/EC50)h, where y is the normalized response, [A] is the agonist concentration, EC50 is the concentration that gives a half-maximal response, and h is the slope factor (Hill coefficient). The fit of the data indicates a shift toward higher affinity of the nAChR with an EC50 of 10.0 ± 1.2 μm obtained for neurons dialyzed with GTPγS compared with 48.6 ± 1.7 μm obtained for control. However, dialysis with GDPβS did not significantly change the EC50 for nicotine (43.5 ± 4.7 μm).
Figure 1.
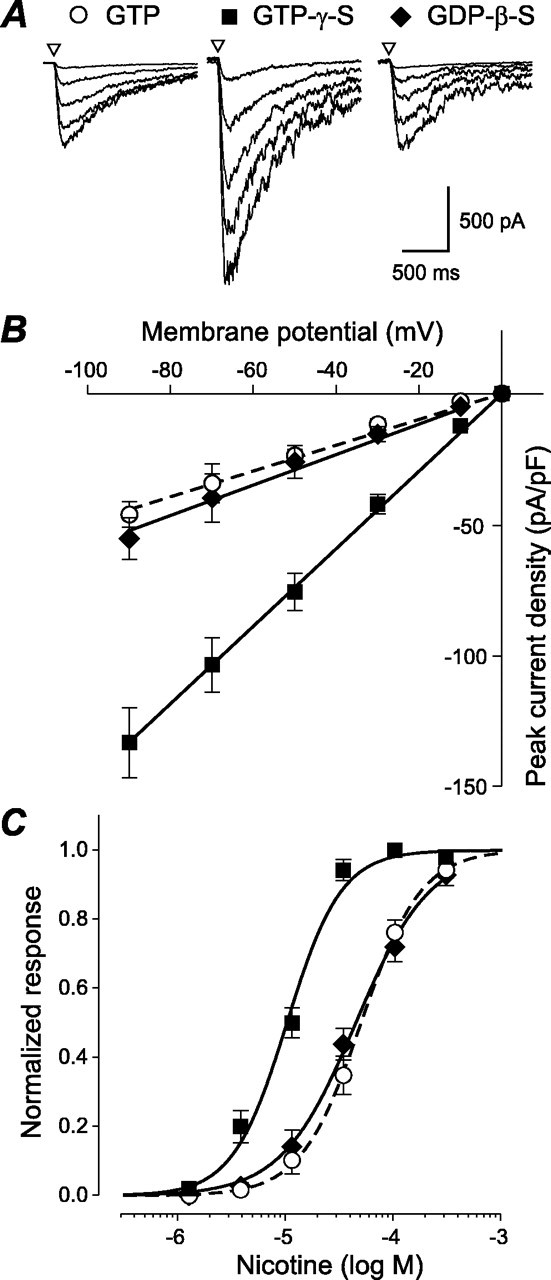
GTPγS-induced activation of G-protein modulation of nAChR channels. A, Families of nicotine-evoked currents elicited by brief pulses (50 ms) of 30 μm nicotine from isolated neurons, voltage clamped at membrane potentials from -90 to -10 mV in 20 mV increments. Currents were recorded using the conventional whole-cell recording technique using 0.2 mm GTP, GTPγS, or GDPβS in the pipette solution. B, Whole-cell current-voltage relationships obtained for peak current density (picoampere per picofarad) evoked by 30 μm nicotine, 10 min after perfusion of the cell using 0.2 mm GTP, GTPγS, or GDPβS. C, Normalized peak nicotine-evoked currents at -70 mV obtained 10 min after internal perfusion with 0.2 mm GTP, GTPγS, or GDPβS, plotted as a function of nicotine concentration. Data points represent mean ± SEM for six to eight cells.
G-protein activation affects the open probability of nAChRs
Single-channel recording was performed using excised inside-out membrane patches to examine the mechanism(s) underlying the potentiation of nicotine-evoked whole-cell currents by GTPγS in rat intracardiac neurons. The product of the number of functional channels and single-channel open probability (NPo) for the inside-out patches was determined using the following equation: NPo = T′/T, where T′ is the total open time for a patch over time T. Consecutive recordings of ∼30 s in duration were made in the presence of a control solution containing 0.2 mm GTP, GTPγS, or GDPβS.
Single-channel currents were only evoked when ACh (5 μm) was present in the pipette. ACh elicited unitary currents occurring as clusters of bursts of openings separated by long closed intervals, whereas bursts within a cluster were separated by closings of intermediate length (Fig. 2A). ACh-evoked channel activity recorded from the same patch increased in the presence of bath-applied 0.2 mm GTPγS. This potentiation of single-channel activity could be reversed after washout (data not shown). The unitary nAChR current traces from a patch in the presence of 0.2 mm GDPβS are similar to those obtained under control conditions (Fig. 2A). The NPo obtained in the presence of GTPγS was 0.86 ± 0.14, which was four times larger than ACh (5 μm) alone (0.24 ± 0.10), whereas bath application of 0.2 mm GTP or GDPβS did not significantly alter the open probability (0.27 ± 0.1 and 0.15 ± 0.04) compared with control conditions (Fig. 2B). The mean unitary current amplitudes were 1.52 ± 0.02 pA (control) and 1.49 ± 0.03 pA (GTPγS), respectively, and were not statistically different (p < 0.01) (Fig. 2C). However, the normalized distribution of channel closings recorded from eight membrane patches in the presence and absence of bath-applied GTPγS reflects the significant differences shown for NPo (Fig. 2 D).
Figure 2.
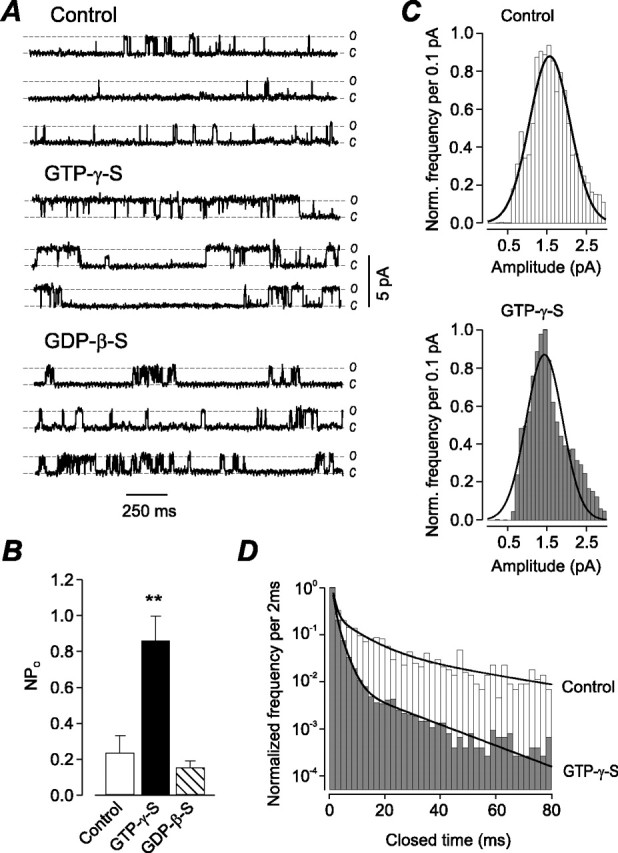
G-protein activation increases the single-channel open probability of nAChRs. A, Direct activation of G-proteins on nAChR single-channel activity recorded from an excised membrane patch in the inside-out configuration (6 s trace). Single-channel currents were evoked in the presence, but not the absence, of ACh (5 μm) in the pipette for control and effects of GTPγS and GDPβS application. B, Bar graph of the effect of G-protein activation on NPo of nAChR channels (n = 8). Holding potential, -60 mV. C, D, Unitary current amplitudes (C) and closed-time distributions (D) from eight inside-out patches exposed to control buffer and 0.2 mm GTPγS. Frequency of events was normalized (Norm. frequency) to the total number of events for each condition. Data points represent mean ± SEM. **p < 0.01.
Agonist-specific modulation of nAChR currents
Pharmacological profiles of nAChR agonists vary among different subunit compositions and provide a useful tool for distinguishing between receptor populations. To determine whether whole-cell currents evoked by particular nicotinic agonists can be differentially modulated by G-proteins, we used 0.2 mm GTPγS or GTP (control) in the pipette solution and applied focal pulses (50 ms) of half-maximal concentrations of agonists every 30 s (except for choline, which was applied every 60 s to avoid receptor desensitization). The peak amplitude of each agonist-evoked current was measured and normalized to the initial current obtained after cell dialysis. Currents evoked by the endogenous neurotransmitter ACh (100 μm) (Fig. 3Ai), nicotine (30 μm) (Fig. 3Aii), and the synthetic compound 1,1-dimethyl-4-phenylpiperazinium iodide (DMPP) (100 μm) could be potentiated by whole-cell dialysis with 0.2 mm GTPγS. Currents evoked by cytisine (30 μm) (Fig. 3Aiii) and choline (10 mm) could not be modulated by intracellular dialysis with either 0.2 mm GTP or GTPγS. The time course of modulation followed a sigmoidal regression analogous to receptor activation, with the slope coefficient indicating the rate of activation. ACh-evoked peak current amplitude was increased by 199 ± 11% (n = 10) (Fig. 3Bi) and reached a maximum after 5 min with a slope factor of -4.3 ± 0.5. Similarly, a maximum increase of 221 ± 14% (n = 10) was observed for nicotine-evoked currents with a slope factor of -6.15 ± 0.8. The potentiation of DMPP-evoked currents was 145 ± 8% (n = 8), with a slope factor of -5.2 ± 2.2. The relative current amplitudes evoked by application of cytisine and nicotine on the same neuron after break-in (0 s) and after 5 min are shown in Figure 3Bii.
Figure 3.

Effect of G-protein modulation on native nAChR channels in rat intracardiac neurons. A, Time course of control (open symbols; 0.2 mm GTP in the intracellular solution) and GTPγS (filled symbols; 0.2 mm) on nAChR-mediated currents obtained in response to ACh (Ai), nicotine (Aii), and cytisine (Aiii). The amplitude of the agonist-induced current was assayed using focal pulses (50 ms) of agonist (concentration as indicated) applied every 30 s. Insets, Representative whole-cell currents elicited by focal application (50 ms) of the relevant agonist in the presence of 0.2 mm GTPγS, 30 s and 10 min after breaking in to the cell. Bi, Bar graph of the maximum effect of all agonists tested on relative whole-cell current amplitude (n = 8-10). Bii, Relative whole-cell current amplitude evoked by 30 μm nicotine and cytisine on the same neuron (n = 5). Data points represent mean ± SEM. **p < 0.01.
Go-protein subunits differentially interact with nAChR subunits
Given the agonist-dependent potentiation of nAChR-mediated currents, the molecular specificity of G-protein modulation of particular nAChR subunits was examined. We had shown previously that VIP- and PACAP-mediated potentiation of ACh-evoked currents in rat parasympathetic neurons is mediated only by Goα and Gβγ subunits (Liu et al., 2000). Bath application of either Goα or Gβγ to the excised inside-out membrane patch increased the single-channel open probability. Representative single-channel recordings obtained after bath application of either Goα or Gβγ to the excised membrane patch are shown in Figure 4A. Gβγ (50 nm) significantly increased NPo fivefold from 0.23 ± 0.12 to 1.15 ± 0.12 (n = 4) by reducing the distribution of channel closings (Fig. 4B). Application of Goα increased NPo, but it was not statistically significant (0.41 ± 0.13; n = 4) from control.
Figure 4.
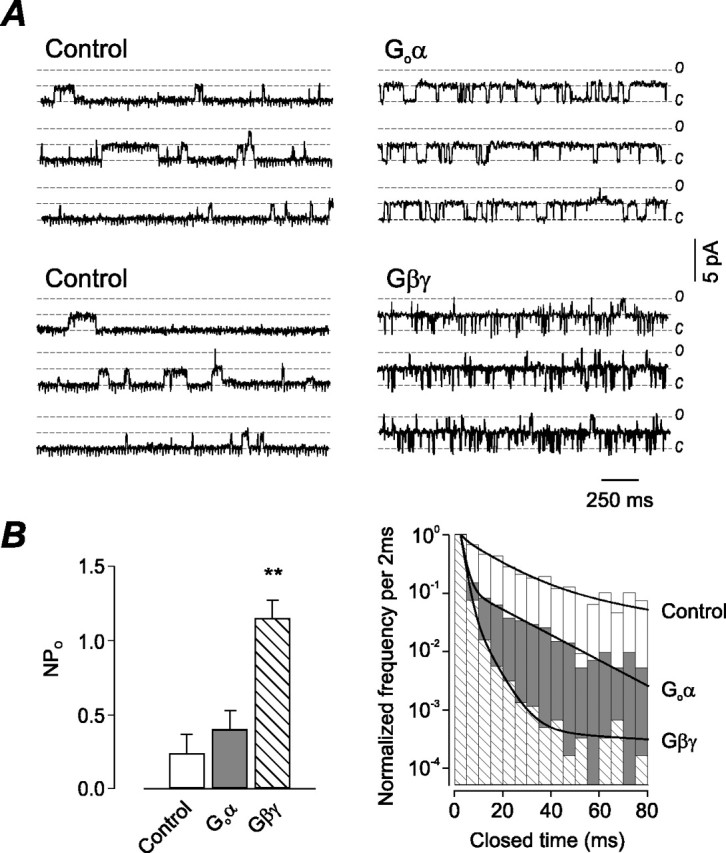
Go-protein subunits modulate neuronal nAChR channel activity. A, Modulation of nAChR single-channel activity recorded from an inside-out membrane patch during receptor activation with 5 μm ACh in the absence (control) and presence of 50 nm Goα or Gβγ subunits. Holding potential was -60 mV. B, Effect of both G-protein subunits on NPo and closed-time distributions obtained from four excised membrane patches (mean ± SEM; **p < 0.01).
To examine the direct interaction of G-protein subunits with nAChR subunits, in vitro GST pull-down experiments were performed. The intracellular loop region of the rat nAChR subunits of α3 (321-471), α4 (331-603), α5 (321-411), α7 (300-472), β2 (321-458), β3 (325-433), and β4 (320-463) were tested for their ability to bind either to the Goα subunit or to the Gβγ dimer. We found a positive interaction with Goα for the nAChR subunits α3-α5, β2, and β3. All of the nAChR subunits tested, except for β3, also exhibited a positive interaction with the Gβγ dimer. The β4 and α7 subunits failed to interact with any of the G-protein subunits tested, and the control GST alone failed to bind the G-protein subunits (Fig. 5A).
Figure 5.

Go-protein subunits interact directly with neuronal nAChR subunits. A, GST pull-down experiments in which glutathione-Sepharose-bound GST, GST-α3 (321-471), GST-α4 (331-603), GST-α5 (321-411), GST-β2 (321-458), GST-β3 (325-433), GST-β4 (320-463), and GST-α7 (300-472) were incubated with either 300 ng of rat recombinant Goα subunit or 300 ng of bovine brain Gβγ dimer. Bound proteins were eluted and analyzed by SDS-PAGE and Western blotting, using antibodies specific to the G-protein subunits, as described in Materials and Methods. One-tenth of the total G-protein used in each binding reaction is shown for comparison. Similar results were obtained in triplicate experiments. B, Experiment similar to that shown in A, but GST-α4 (331-603) and GST-α7 (300-472) were incubated with rat ICG lysate and tested for their ability to bind to the Gβγ dimer or the GABAA receptor subunit α1. C, Lysates from rat intracardiac ganglia were immunoprecipitated (IP) with (+) or without (-) the anti-Goα/Gβγ antibody. The precipitated immunocomplexes were probed for the presence of nAChR-α1, nAChR-α3, nAChR-α5 (mAb35), or nAChR-β2 (mAb270).
Given that nAChR GST-α4 was consistently shown to give the strongest signals in in vitro pull-down assays with Goα and Gβγ, we tested the ability of this fusion protein to bind other proteins. Therefore, we incubated GST-α4 (331-603) and GST-α7 (300-472) with rat ICG lysate (∼2 mg) and tested for their ability to bind to the Gβγ dimer or the GABAA receptor subunit α1. We found a positive interaction of α4, but not α7, with endogenous Gβγ in the lysate, but both nAChR subunits failed to bind to the α1 subunit of the GABAA receptor (Fig. 5B). Coimmunoprecipitation of ICG lysate with G-protein subunits and selective nAChR antibodies showed that both Goα and Gβγ bind α3/α5- and β2-containing native nAChRs in rat ICG neurons (Fig. 5C). Reverse experiments using the same antibodies were negative and thus excluded cross-reactivity between the antibodies. Furthermore, failure of coimmunoprecipitation using a rabbit preimmune control IgG also excludes unspecific binding of the Ig-heavy chain (data not shown).
Discussion
G-protein subunits have been reported recently to directly modulate the glycine receptor, a member of the pentameric ligand-gated ion channel superfamily (Yevenes et al., 2003). The results presented in this study demonstrate another member of this family that is regulated by Goα and Gβγ subunits. Previous experiments demonstrated that nAChR-mediated currents can be potentiated by coactivation of GPCRs by VIP and PACAP (Liu et al., 2000) and that this modulation is mediated by a pertussis toxin-sensitive Go-protein pathway. We now show that this potentiation is attributable to direct binding of G-protein subunits to intracellular regions of specific nAChR subunits. Using rat intracardiac ganglion neurons, activation of G-proteins by intracellular GTPγS shifted the affinity of the nAChR for nicotine to lower concentrations. The increase in agonist potency is attributable to an increase in the ligand affinity for the neuronal nAChR. Inside-out patch recordings indicate an almost fourfold increase in single-channel open probability attributable to a decrease in interburst and intercluster closed times (Cuevas and Adams, 1996). Activation of GPCRs and the release of Go-protein subunits have been shown to be fast enough to increase ACh- and nicotine-evoked currents during coactivation of nAChRs (Liu et al., 2000). Therefore, simultaneous activation of Go-coupled receptors during cholinergic transmission is likely to increase the open probability of presynaptic and postsynaptic nAChRs and increase neuronal excitability. Not all neuronal nAChR subtypes are regulated to the same extent by concurrent G-protein activation. The amplitudes of nAChR-mediated currents elicited by application of half-maximal concentrations of either ACh or nicotine were doubled compared with control, whereas currents evoked by either cytisine or choline were unaffected by G-protein activation. These results suggest nAChR subtype-selective modulation by G-proteins, which was confirmed with GST pull-down experiments using GST-coupled intracellular regions of nAChR subunits and free Go-protein subunits. A robust and direct interaction between specific nAChR subunits and the G-protein subunits was observed. The nAChR and G-protein interaction required neither auxiliary proteins nor additional cytoplasmic factors. These results are consistent with a direct interaction between Go-protein subunits and the intracellular region of specific nAChR subunits to increase the affinity of ACh binding to the receptor.
The high degree of subunit specificity of G-protein modulation of neuronal nAChRs is intriguing, given that neuronal nAChRs of rat intrinsic cardiac ganglia have been shown to be composed mainly of α3, α5, α7, β2, and β4 subunits (Poth et al., 1997; Bibevski et al., 2000) and that a large component of cholinergic (vagal) transmission in intracardiac neurons is mediated by the α3β2 subunit (Bibevski et al., 2000). G-protein activation did not modify currents evoked by the cholinergic agonist, cytisine, which has been shown to be most potent for α3β4 and α3β4α5 subunit combinations (Covernton et al., 1994). Furthermore, no direct interaction between the Go-protein subunits and the intracellular region of the β4 and α7 subunit was observed. In contrast, there is a strong interaction between G-proteins and the α3 subunit, suggesting modulation of α3-containing nAChRs. G-protein interaction with either the β subunit alone or both β and α subunits appears necessary for nAChR modulation. The lack of G-protein binding and potentiation of α7 nAChR-mediated currents by G-protein activation supports the hypothesis that at least one β subunit of the nAChR is required for G-protein activation. The high probability of the occurrence of α3β2-containing nAChRs in mammalian intracardiac neurons (Poth et al., 1997; Bibevski et al., 2000) explains the robust potentiation of ACh- and nicotine-evoked currents by G-protein subunits. The coimmunoprecipitation of native nAChRs with Goα and Gβγ subunits clearly demonstrates direct protein interaction.
In contrast to the present findings, alternative pathways for the modulation of nAChRs by activation of GPCRs in chick ciliary ganglia have been reported previously (Pardi and Margiotta, 1999). Activation of PACAP receptors in chick ciliary ganglion neurons has been shown to modulate nicotine-evoked currents using two different pathways; the first inhibits nAChRs via an unknown G-protein activation that involves phospholipase C, IP3, and mobilization of Ca2+ from internal stores, and a second pathway enhances currents by the activation of stimulatory G-protein and involves the generation of cAMP. A different signaling pathway involving Src family kinases (SFKs) has been described recently, whereby the inhibition of SFKs or the expression of kinase-defective c-Src reduced the peak amplitude of nicotine-induced currents in chromaffin cells or in HEK293 cells expressing the α3β4α5 subunit combination (Wang et al., 2004). In rat parasympathetic neurons, intracellular dialysis failed to antagonize nAChR potentiation (Liu et al., 2000). The rapid onset observed for GPCR-induced potentiation of nAChR-mediated currents and the fact that specific nAChR subunits bind to Go-protein subunits demonstrates the direct activation of nAChRs by G-proteins without involvement of any diffusible factor. The amplitude of glycine-activated Cl- currents in mouse spinal neurons was similarly enhanced after the activation of a GPCR and after application of purified Gβγ (Yevenes et al., 2003). In contrast to glycine receptors, nAChRs are composed of a variety of α and β subunits providing distinct molecular and pharmacological properties. Given the diversity of nAChR subtypes and their importance in both the CNS and peripheral nervous system, the subtype-specific modulation by G-protein subunits may have a significant effect on cholinergic transmission.
The present findings also indicate another important effector target for Gα and Gβγ subunits that bind and affect a variety of enzymes and ion channels such as the inward rectifying potassium channels, some voltage-gated N- and P/Q-type calcium channels (for review, see Dascal, 2001), and more recently, the ionotropic glycine receptor (Yevenes et al., 2003). The mechanism has been shown to be fast and reversible and provides a new pathway to describe synaptic processes in neural networks, giving rise to unique receptor cross talk and modulation. Because neuronal nAChRs are found throughout the nervous system, G-protein modulation of specific nAChRs channels may serve as an endogenous regulator of ganglionic transmission in autonomic pathways, as well as central presynaptic nAChRs involved in regulation of drug addiction and neurological disorders such as Alzheimer's disease and epilepsy.
Footnotes
This work was supported by a grant from the Australian Research Council.
Correspondence should be addressed to David J. Adams at the above address. E-mail: dadams@uq.edu.au.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253571-07$15.00/0
References
- Bibevski S, Zhou Y, McIntosh JM, Zigmond RE, Dunlap ME (2000) Functional nicotinic acetylcholine receptors that mediate ganglionic transmission in cardiac parasympathetic neurons. J Neurosci 20: 5076-5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunemann M, Frank M, Lohse MJ (2003) Gi protein activation in intact cells involves subunit rearrangement rather than dissociation. Proc Natl Acad Sci USA 100: 16077-16082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE, Neer EJ (1997) G protein βγ subunits. Annu Rev Pharmacol Toxicol 37: 167-203. [DOI] [PubMed] [Google Scholar]
- Covernton PJ, Kojima H, Sivilotti LG, Gibb AJ, Colquhoun D (1994) Comparison of neuronal nicotinic receptors in rat sympathetic neurones with subunit pairs expressed in Xenopus oocytes. J Physiol (Lond) 481: 27-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas J, Adams DJ (1996) Vasoactive intestinal polypeptide modulation of nicotinic ACh receptor channels in rat intracardiac neurones. J Physiol (Lond) 493: 503-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dascal N (2001) Ion-channel regulation by G proteins. Trends Endocrinol Metab 12: 391-398. [DOI] [PubMed] [Google Scholar]
- De Waard M, Liu H, Walker D, Scott VE, Gurnett CA, Campbell KP (1997) Direct binding of G-protein βγ complex to voltage-dependent calcium channels. Nature 385: 446-450. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA (1996) Modulation of Ca2+ channels by G-protein βγ subunits. Nature 380: 258-262. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Gordon EA, Wickman K, Velimirovic B, Krapivinsky L, Clapham DE (1995) The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature 374: 135-141. [DOI] [PubMed] [Google Scholar]
- Liu DM, Cuevas J, Adams DJ (2000) VIP and PACAP potentiation of nicotinic ACh-evoked currents in rat parasympathetic neurons is mediated by G-protein activation. Eur J Neurosci 12: 2243-2251. [DOI] [PubMed] [Google Scholar]
- Pardi D, Margiotta JF (1999) Pituitary adenylate cyclase-activating polypeptide activates a phospholipase C-dependent signal pathway in chick ciliary ganglion neurons that selectively inhibits α7-containing nicotinic receptors. J Neurosci 19: 6327-6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poth K, Nutter TJ, Cuevas J, Parker MJ, Adams DJ, Luetje CW (1997) Heterogeneity of nicotinic receptor class and subunit mRNA expression among individual parasympathetic neurons from rat intracardiac ganglia. J Neurosci 17: 586-596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuveny E, Slesinger PA, Inglese J, Morales JM, Iniguez-Lluhi JA, Lefkowitz RJ, Bourne HR, Jan YN, Jan LY (1994) Activation of the cloned muscarinic potassium channel by G protein βγ subunits. Nature 370: 143-146. [DOI] [PubMed] [Google Scholar]
- Swope SL, Qu Z, Huganir RL (1995) Phosphorylation of the nicotinic acetylcholine receptor by protein tyrosine kinases. Ann NY Acad Sci 757: 197-214. [DOI] [PubMed] [Google Scholar]
- Wang K, Hackett JT, Cox ME, Van Hoek M, Lindstrom JM, Parsons SJ (2004) Regulation of the neuronal nicotinic acetylcholine receptor by SRC family tyrosine kinases. J Biol Chem 279: 8779-8786. [DOI] [PubMed] [Google Scholar]
- Xu ZJ, Adams DJ (1992) Voltage-dependent sodium and calcium currents in cultured parasympathetic neurones from rat intracardiac ganglia. J Physiol (Lond) 456: 425-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yevenes GE, Peoples RW, Tapia JC, Parodi J, Soto X, Olate J, Aguayo LG (2003) Modulation of glycine-activated ion channel function by G-protein βγ subunits. Nat Neurosci 6: 819-824. [DOI] [PubMed] [Google Scholar]