

Abstract
Microglial activation and inflammation are associated with progressive neuronal apoptosis in neurodegenerative human brain disorders. We sought to investigate molecular signaling mechanisms that govern activation of microglia in apoptotic neuronal degeneration. We report here that the active form of matrix metalloproteinase-3 (MMP-3) was released into the serum-deprived media (SDM) of PC12 cells and other media of apoptotic neuronal cells within 2-6 h of treatment of the cells, and SDM and catalytic domain of recombinant MMP-3 (cMMP-3) activated microglia in primary microglia cultures as well as BV2 cells, a mouse microglia cell line. Both SDM and cMMP-3 induced generation of tumor necrosis factor α (TNF-α), interleukin-6 (IL-6), IL-1β, and interleukin-1 receptor antagonist but not IL-12 and inducible nitric oxide synthase, which are readily induced by lipopolysaccharide, in microglia, suggesting that there is a characteristic pattern of microglial cytokine induction by apoptotic neurons. Neither glial cell line-derived neurotrophic factor nor anti-inflammatory cytokines, such as IL-10 and transforming growth factor-β1, were induced. SDM and cMMP-3 extensively released TNF-α from microglia and activated the nuclear factor-κB pathway, and these microglial responses were totally abolished by preincubation with an MMP-3 inhibitor, NNGH [N-isobutyl-N-(4-methoxyphenylsulfonyl)-glycylhydroxamic acid]. MMP-3-mediated microglial activation mostly depended on ERK (extracellular signal-regulated kinase) phosphorylation but not much on either JNK (c-Jun N-terminal protein kinase) or p38 activation. Conditioned medium of SDM- or cMMP-3-activated BV2 cells caused apoptosis of PC12 cells. These results strongly suggest that the distinctive signal of neuronal apoptosis is the release of active form of MMP-3 that activates microglia and subsequently exacerbates neuronal degeneration. Therefore, the release of MMP-3 from apoptotic neurons may play a major role in degenerative human brain disorders, such as Parkinson's disease.
Keywords: microglia, apoptosis, neurodegeneration, cytokines, matrix metalloproteinase-3, MMP-3
Introduction
Apoptosis contributes to pathogenesis of neurodegenerative disease such as Parkinson's disease (PD) and Huntington's disease (Goldberg et al., 1996; Mochizuki et al., 1996; Gervais et al., 1999; Nicotera et al., 1999; Wu et al., 2000; Andersen, 2001). Apoptotic cells trigger the recruitment of phagocytic cells to be removed quickly and efficiently and modulate immune responses by either direct contact with phagocytes and subsequent engulfment or releasing chemoattractants. In the brain, apoptotic neuronal cells are phagocytosed and degraded by microglia, the resident macrophage of the brain (Stolzing and Grune, 2004).
However, a recent study demonstrates that, in addition to phagocytosing apoptotic cells, microglia promote the death of Purkinje cells in the developing mouse cerebellum. Apoptosis of Purkinje cells is strongly reduced by selective elimination of microglia (Marin-Teva et al., 2004). This study suggests that microglia may exacerbate neuronal degeneration under neuronal apoptotic conditions. The implications of activated microglia in the pathogenesis of many neurodegenerative diseases that involve apoptosis, including PD, acquired immunodeficiency syndrome (AIDS) dementia complex, multiple sclerosis, and prion-related diseases (McGeer et al., 1988; Rogers et al., 1988; Dickson et al., 1993; Raine, 1994; Brown, 2001), suggest that the aberrant interaction between apoptotic cells and microglia may aggravate neurodegenerative conditions.
Compared with the extensive studies on the molecular mechanisms underlying neuronal apoptosis, signals from apoptotic neurons that might initiate microglial response remain to be elucidated. We present here a novel signaling mechanism of apoptotic neuronal cells in activating microglia. Our data show that matrix metalloproteinase-3 (MMP-3) is a microglia-activating signaling molecule that is released from apoptotic neurons in early apoptosis. MMP-3 is a member of matrix metalloproteinases family that is mainly responsible for the remodeling of extracellular macromolecules. Recent evidence has linked MMPs to various pathologic conditions in the CNS, including ischemia, multiple sclerosis, Alzheimer's disease, and malignant glioma (Cuzner et al., 1996; Gottschall and Deb, 1996; Maeda and Sobel, 1996; Yamamoto et al., 1996). This implies that, in addition to its known function of extracellular macromolecule degradation, MMP-3 could serve as a signaling molecule that mediates the interaction between apoptotic neurons and microglia in early apoptosis. Neurotrophic factors, such as glial cell line-derived neurotrophic factor (GDNF), or anti-inflammatory cytokines, such as interleukin-10 (IL-10) or transforming growth factor-β1 (TGF-β1), were not detected in MMP-3-activated microglia in early apoptosis, suggesting that microglial activation by MMP-3 from apoptotic neuronal cells is characteristic microglial responses to neuronal apoptosis for inflammatory reactions as well as exacerbation of neuronal cell apoptosis. Our data provide a novel mechanism in neuronal apoptosis that might modulate immune reactions by releasing potential soluble mediators such as MMP-3.
Materials and Methods
Materials. Cell-culture ingredients were obtained from Mediatech (Herndon, VA). Both catalytic domain and proform of MMP-3 were purchased from Calbiochem (La Jolla, CA). NNGH [N-isobutyl-N-(4-methoxyphenylsulfonyl)-glycylhydroxamic acid] and MMP-3 assay kit were from Biomol (Plymouth, PA). Lipopolysaccharide (LPS) was from Sigma (St. Louis, MO). MEK1/2 [MAP (mitogen-activated protein) kinase kinase] inhibitor U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto) butadiene] and p38 inhibitor SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)imidazole] were purchased from Biomol. JNK (c-Jun N-terminal protein kinase) inhibitor SP600125 (anthra[1,9-cd]pyrazol-6(2H)-one) was from Calbiochem. Tumor necrosis factor-α (TNF-α) ELISA kit was obtained from eBioscience (San Diego, CA). Precast gels for Western blot and zymography were from Invitrogen (San Diego, CA). Polyclonal antibody against MMP-3 (SC-7383) and phosphorylated ERK (extracellular signal-regulated kinase) (SC-6839) from Santa Cruz Biotechnology (Santa Cruz, CA) were used. TNF-α antibody was from Santa Cruz Biotechnology. ECL+ Western blot detection system was from Amersham Biosciences (Piscataway, NJ). Multiprobe RNase protection assay (RPA) kit was purchased from BD PharMingen (San Diego, CA). Rneasy kit for total RNA preparation was from Qiagen (Valencia, CA). Cytotox 96 kit for lactate dehydrogenase (LDH) assay was purchased from Promega (Madison, WI). Apoptosis detection kit was from Sigma. Nuclear factor-κB (NF-κB) oligonucleotide for gel shift assay was obtained from Santa Cruz Biotechnology. YM-10 molecular weight filter for the concentration of serum-deprived medium (SDM) from PC12 cells was purchased from Millipore (Billerica, MA). Apparatus and materials for two-dimensional electrophoresis were purchased from Bio-Rad (Hercules, CA).
Cell culture, SDM preparation, and treatment of microglia. PC12 cells were grown in RPMI (Roswell Park Memorial Institute medium) containing 10% horse serum, 5% fetal bovine serum, and 1% penicillin/streptomycin at 37°C in a humidified incubator under 5% CO2. For the preparation of SDM, 80% confluent monolayer cells were washed with serum-free RPMI four times to remove serum components. The fourth washed medium consisting of diluted serum was used for control. Supernatant was collected at various time points from 2 to 48 h, followed by centrifugation for 1 h at 40,000 × g to remove insoluble debris and then concentrated using the 10 kDa molecular weight cutoff filter YM-10. Supernatant collected from one 10 cm dish was concentrated and reconstituted with 100 μl of PBS. Differentiated PC12 cells with neural cell phenotype were generated by growing 1 × 105 PC12 cells in 60 mm polyethyleneimmine-coated dishes for 7 d in RPMI containing 50 ng/ml nerve growth factor (NGF). BV2 murine microglia cell line were grown in DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C in a humidified incubator under 5% CO2. Cells were treated with SDM, LPS, or MMP-3.
Primary microglia-enriched cultures. Microglia were prepared from whole brains of 1-d-old mice pups as described previously (Liu et al., 2001). Briefly, after removing meninges and blood vessels, the brain tissue was gently triturated and seeded (5 × 107) in 150 cm2 flasks. One week after seeding, the media were replaced. Two weeks after seeding when the cells had reached a confluent monolayer of glial cells, microglia were separated from astrocytes by shaking the flasks for 5 h at 150 rpm and were replated at 1 × 105 in a 96-well plate precoated with poly-d-lysine. The enriched microglia were >95% pure, as determined by immunostaining with antibodies against F4/80 (pan macrophage marker; eBioscience) and glial fibrillary acidic protein. Enriched primary microglia were treated with either LPS (10 ng/ml) or catalytic domain of MMP-3 (125, 250, or 400 ng/ml).
Preparation of recombinant catalytic domain of MMP-3- or SDM-conditioned media from BV2 microglia and treatment. BV2 microglial cells were cultured in 100 mm dishes at 80% confluency. Either 400 ng/ml recombinant catalytic domain of MMP-3 (cMMP-3) or 100 μg/ml concentrated SDM was added in 5 ml of culture media for 3 h. After washing three times with fresh media, BV2 cells were incubated for 16 h. Supernatant was collected and centrifuged at 40,000 × g for 1 h. The neuronal toxicity of conditioned media (CM) was evaluated by using differentiated PC12 cells as target neuron. Medium was replaced with CM containing NGF. Neuronal death was detected by LDH assay. For the TNF-α inhibition study, CM was incubated with anti-TNF-α antibody 1 h before CM was added to differentiated PC12 cells.
Assessment of cell death by LDH assay and detection of apoptosis by annexin V staining. At various time points after CM treatment, culture medium was collected for LDH assay with Cytotox 96 kit (Promega). Collected medium (50 μl) was transferred to 96-well plates, and 50 μl of substrate solution was added. After 30 min incubation in dark room, LDH level was determined at 540 nm in plate reader (Molecular Devices, Sunnyvale, CA). Apoptotic cells were detected by annexin V staining kit (Sigma). After washing with PBS, cells were incubated in double-label staining solution containing 1 μg/ml annexin V-cyanine 3 (Cy3) and 500 μm 6 carboxyfluorescein diacetate (CFDA) for 10 min in dark conditions. After washing with binding buffer, apoptotic cells were determined by both annexin V-Cy3 (red) and CFDA (green) staining under fluorescence microscopy.
Rnase protection assay. For RNA collection, BV2 cells were cultured in 10 cm dishes and then treated with various stimuli at 80% confluency (total of ∼107 cells per dish). After 6 h, the cells were harvested by scrapping, and total RNA was extracted by Rneasy kit. Cytokine mRNA levels were analyzed by RNase protection assay using the RiboQuant multiprobe set (Pharmingen, San Diego, CA) following the instructions of the supplier. In brief, 10 μg of RNA obtained from cells were hybridized overnight to the 32P-labeled RNA probe, which had been synthesized previously from the supplied template set (containing 10 different templates). Single-stranded RNA was phenolized, precipitated, and analyzed on 6% denaturing polyacrylamide gel. The quantity of protected RNAs was determined using a phosphorimager (Fuji, Stamford, CT). The cytokine transcripts were identified by the length of the respective fragments. For quantitation, cytokine values were expressed as a percentage of the mean values of the housekeeping genes L32 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Measurement of TNF-α levels by ELISA. The concentration of TNF-α in microglial culture media was measured using Ready-Set-Go mouse TNF-α ELISA kit (eBioscience). Briefly, a 96-well plate was coated with coating buffer containing capture antibody for overnight. After washing three times with PBS-T (PBS containing 0.1% Tween 20), wells were blocked with assay diluent. After the same washing step, standards and culture media were added to the appropriate wells and incubated for 2 h. After washing five times with PBS-T, each well was incubated with detection antibody for 1 h. After washing, avidin-HRP was added for 30 min. After thorough washing, each well was incubated with substrate solution for 15 min. Reaction was stopped by 1 m H3PO4. Plate was read at 450 nm. The concentration of TNF-α was calculated according to the standard curve.
Western blot analysis. To obtain total cell lysates, 0.5 ml (or 0.2 ml) of RIPA buffer (1× PBS, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS, with freshly added 100 ng/ml PMSF, 30 μl/ml aprotinin, and 10 μl/ml of 100 mm sodium orthovanadate) was added to cells in a 10 cm (or 6 cm) cell-culture plate. Cells were scraped, incubated for 30-60 min on ice, and centrifuged at 15,000 × g for 20 min at 4°C. Protein concentrations were determined, and 40-60 μg of whole-cell lysate was loaded for SDS-PAGE. Electrophoresis was performed, and proteins were transferred to polyvinylidene difluoride membrane using an electroblotting apparatus. Membranes were blocked for 1 h in TBS containing 0.1% Tween 20 and 5% dry milk and then incubated overnight with primary antibodies [anti-phospho-specific ERK, 1:2000; anti-MMP-3, 1:1000], followed by incubation for 1 h with horseradish peroxidase-conjugated secondary antibodies. Membranes were washed three times (30 min each) with TBS containing 0.1% Tween 20. Bands were visualized using ECL+ Western blotting detection system.
Zymographic analysis. The casein lytic activities in the SDM were determined by zymography using 12% zymogram casein gels (Invitrogen). Concentrated SDM and control medium were mixed with 2× Tris-glycine SDS sample buffer and incubated for 10 min at room temperature (not heated). Samples were applied on the casein gel and separated according to their molecular weight until bromophenol blue dye reached the bottom of the gel. After running, the gels were incubated in the renaturing buffer for 30 min with gentle agitation at room temperature. After removing the renaturing buffer, the gels were equilibrated in the developing buffer for 30 min at room temperature. After replacing with fresh developing buffer, the gels were incubated overnight at 37°C with gentle agitation. On next day, the gels were stained with 0.5% Coomassie blue R-250 for 30 min. Area of proteinase activity was visualized as clear bands by destaining.
Measurement of NF-κB activation by electrophoresis mobility shift assay. Double-stranded consensus oligonucleotide was end labeled with [γ-32P]ATP. Binding reactions containing equal amounts of protein (3 μg) and 10 fmol (∼10,000 cpm, Cherenkov counting) of oligonucleotide were performed for 30 min in binding buffer (10 mm Tris, pH 7.5, 75 mm NaCl, 2 μg of poly dI-dC, 5% glycerol, 1 mm EDTA, and 1 mm DTT) at room temperature. Total reaction volumes were held at 20 μl. Reaction products were separated in 6% polyacrylamide gel and analyzed by PhosphorImager.
Two-dimensional electrophoresis and silver staining. Both SDM and serum diluent were concentrated using YM-10. For the isoelectric focusing, concentrated samples were reconstituted with rehydration buffer: 8 m urea, 2% CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate), 0.2% ampholine pH3-10, 65 mm DTT, and 0.001% bromophenol blue. After 12 h in-gel rehydration with samples, immobilized pH gradient (IPG) strip pH 3-10 was focused at 250 V for 30 min, 250-4000 V for 2 h, and then finally for 70,000 volt-hours by using Protean IEF cells (Bio-Rad). For SDS-PAGE, focused IPG strip was incubated with equilibration buffer I (6 m urea, 2% SDS, 0.375 m Tris-HCl, pH 8.8, 20% glycerol, and 130 mm DTT) for 10 min and subsequent equilibration in buffer II (6 m urea, 2% SDS, 0.375 m Tris-HCl, pH 8.8, 20% glycerol, and 135 mm iodoacetamide) for 15 min. Then proteins were separated according to their molecular weight using 4-15% gradient gel. After electrophoresis, proteins were detected using SilverQuest silver staining kit (Invitrogen) according to the user's manual. Briefly, gel was rinsed with ultra-pure water and then fixed in 100 ml of fixative for 20 min. After washing with 30% ethanol, gel was incubated in sensitizing solution for 10 min. After washing with 30% ethanol and ultra-pure water, gel was incubated in 100 ml of staining solution for 15 min. After washing with ultra-pure water, gel was incubated in developing solution for 4-8 min until spots started to appear. Reaction was stopped by 10 ml of stopper. After washing with ultra-pure water, gel was scanned and analyzed.
In-gel tryptic digestion and matrix-assisted laser desorption ionization time-of-flight mass spectrometry and data analysis. Silver-stained gel electrophoresis slices trimmed to ∼1 mm2 were destained in a 0.5 ml conical test tube containing 100 μl of destain solution (equal volumes of 30 mm potassium ferrocyanide and 100 mm sodium thiosulfate) with vortexing for 15 min. The destaining solution was removed by vortexing the gel slice pieces for 15 min in 400 μl of deionized water three times. The gel slice pieces were dehydrated in 400 μl of acetonitrile with vortexing for 15 min. The gel slice pieces were vortexed in 100 μl of 10 mm dithiothreitol in 25 mm ammonium bicarbonate 30 min at 50°C for reduction of disulfide bonds, and 100 μl of 100 mm iodoacetamide in 25 mm ammonium bicarbonate for 30 min at room temperature in the dark for alkylation of cysteines. The gel slice pieces were vortexed for 15 min in 400 μl of 50% acetonitrile in 25 mm ammonium bicarbonate at pH 8 three times. The gel slice pieces were again dehydrated in 400 μl of acetonitrile with vortexing for 15 min and dried in a vacuum centrifuge for 3 min. The gel slice pieces were rehydrated with 50 μl of cold 15 mg/ml sequencing grade modified trypsin (V5111; Promega) in 25 mm ammonium bicarbonate at pH 8 and incubated at 37°C overnight for proteolytic digestion. The digestion supernatant was transferred to another 0.5 ml conical test tube, and the gel slice pieces were extracted with 50 μl of 50% acetonitrile in 1% trifluoroacetic acid by sonication for 5 min and vigorous vortexing for 30 min. The extract solution was added to the digestion supernatant, and the volume was completely dried by vacuum centrifugation. The dried residue was reconstituted with 100 μl of 0.1% trifluoroacetic acid by vortexing for 5 min. c18 ZipTips were wetted with 50 μl of 50% acetonitrile in deionized water three times and equilibrated with 50 μl of 0.1% trifluoroacetic acid three times. The reconstituted sample peptides were adsorbed to the reverse-phase sorbent by slowly aspirating and dispensing a 50 μl volume to another conical test tube 10 times and repeating the aspirating and dispensing with the remaining 50 μl of reconstituted sample. The ZipTip was washed with three 50 μl volumes of 0.1% trifluoroacetic acid, and the peptides were eluted by aspirating and dispensing in a 0.2 ml PCR test tube with 10 μl of 50% acetonitrile in 0.1% trifluoroacetic acid three times. In a 0.2 ml PCR test tube, 1 ml of the eluted peptides was mixed by vortexing for 15 min with 1, 5, or 10 μl of 10 mg/ml 3,5-dimethoxy-4-hydroxy-cinnamic acid in 50% acetonitrile/0.1% trifluoroacetic acid. To the matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) target plate, 0.75 μl of the eluted peptides in the organic acid was aliquoted and allowed to dry at room temperature in the dark. MALDI-TOF mass spectrometry was performed using a Voyager-DE PRO mass spectrometer (Applied Biosystems, Foster City, CA) in either the linear or ion reflector modes. Applied Biosystems Voyager 5.1 software was used for data acquisition, and Data Explorer 4.0 software was used for spectrum processing. Database searching for mass fingerprint matching was performed using MS-FIT (http://prospector.ucsf.edu/ucsfhtml4.0/msfit.htm) and referencing the NCBInr.10.25.2002 protein database.
Statistical analysis. Statistical significance was assessed by ANOVA, followed by Bonferroni's t test using the StatView program (Abacus Concepts, Berkeley, CA). A value of p < 0.05 was considered statistically significant.
Results
Induction of cytokines in BV2 cells by supernatant of the apoptotic neuronal cell-culture media
To test the hypothesis that apoptotic neurons release soluble molecules that evoke microglial responses, the supernatants of apoptotic neuronal cell cultures were added to BV2 murine microglia cell cultures. Three neuronal cells, PC12, SK-N-BE(2)C, and SH-SY5Y cells, were used. PC12 cells underwent apoptosis in the SDM, and apoptosis of other two neuronal cells was also observed when they were treated with staurosporine (10 μm). Apoptosis of each cell line was confirmed by the presence of nuclear clumping after 4′,6′-diamidino-2-phenylindole staining (data not shown). To determine various cytokines, including IL-12, TNF-α, IL-6, IL-1β, IL-1Ra, TGF-β1, and inducible nitric oxide (NO) synthase (iNOS) mRNA in microglia at the same time, multi-prove RPA was used. Supernatants from all three cell lines undergoing apoptosis produced a similar pattern of cytokine mRNA induction (Fig. 1A, B). However, similar experiments with multi-prove RPA showed no induction of GDNF or anti-inflammatory cytokines, such as IL-10 or TGF-β (data not shown). To identify cytokine-inducing molecules in these apoptotic cell-culture media, the serum-deprived medium of PC12 cell culture was used because they had much less proteins than other two cell-culture media. SDM was collected at various time points from 2 to 48 h after serum deprivation, each SDM sample was concentrated, and 50 μg/ml proteins were used for each experiment. Because induction of cytokines was near maximum by using the SDM that was collected at 6 h after serum deprivation (6 h SDM), and also to avoid contamination by cellular components that might leak from secondary necrotic cells after longer serum deprivation, 6 h SDM was used for the rest of experiments. The 6 h SDM (50 μg/ml) induced cytokine mRNAs in microglia, and the induction pattern was comparable with LPS (100 ng/ml), except that IL-12 and iNOS were not induced by the 6 h SDM (Fig. 1A, B). The supernatant of serum-containing PC12 cell-culture medium was ineffective (Fig. 1A). TNF-α release from microglia measured using ELISA showed that only SDM induced TNF-α release but not by normal PC12 media (Fig. 1C). Whereas 6 h SDM increased the levels of TNF-α in the media from 19.0 ± 4.4 pg/ml (untreated control) to 928.8 ± 189.4 pg/ml for 24 h, microglial TNF-α release by the treatment of the supernatant of PC12 cells in normal serum-containing media for 6 h was negligible.
Figure 1.
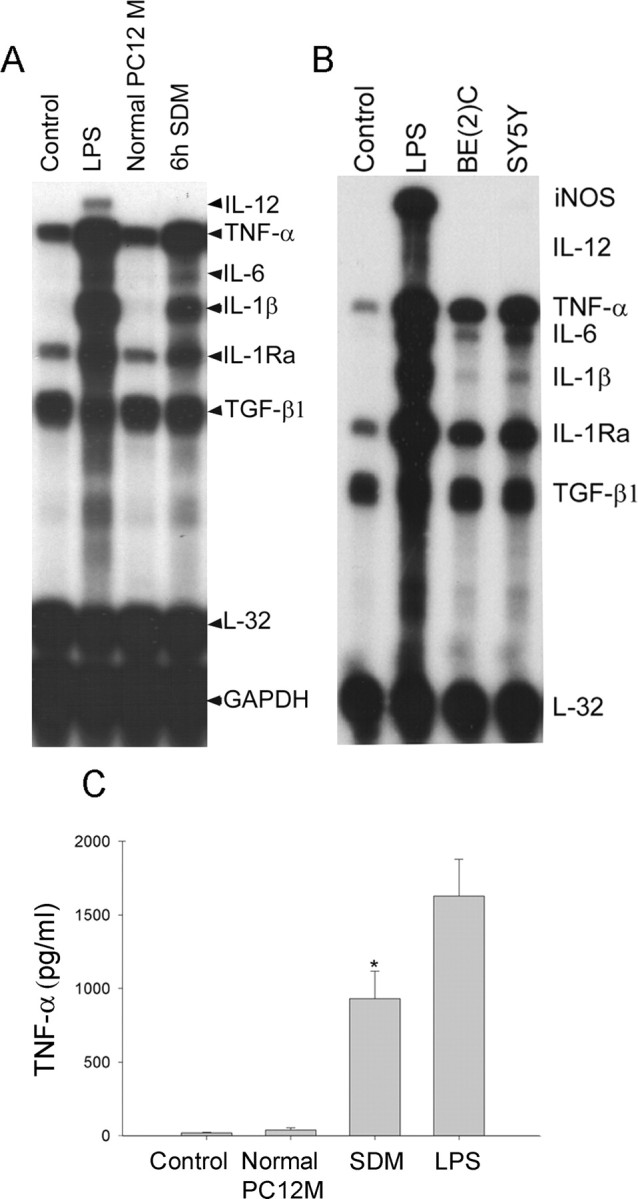
The supernatants from apoptotic neuronal-mediated induction of cytokine genes in BV2 cells and TNF-α release. A, SDM from apoptotic PC12 culture strongly induced microglial cytokine genes. BV2 cells were treated for 6 h with either concentrated 6 h SDM (50 μg of proteins) or equivalents from normal PC12 culture. Various cytokine mRNA levels, including IL-12, TNF-α, IL-6, IL-1β, IL-1Ra, TGF-β1, and iNOS, were determined using RPA. L32 and GAPDH were used as internal controls. B, Supernatants from staurosporine-induced apoptotic SK-N-BE(2)C and SH-SY5Y also resulted in a similar pattern of cytokine induction in microglia. Apoptosis of both SK-N-BE(2)C and SH-SY5Y cells was induced by 4 h staurosporine treatment, and then, to remove staurosporine completely, cells were washed three times with fresh media. After the last wash, cells were incubated in fresh media for 6 h, and then supernatant was collected for microglial treatment. After 6 h treatment with supernatants, cytokine mRNA levels of BV2 cells were analyzed. Pictures shown are representative autoradiographs of the four independent RPAs. C, SDM caused TNF-α release from microglia. TNF-α levels were measured in the microglial culture media after 24 h treatments with each condition using ELISA. Values represent the mean ± SD (n = 4). Control, Untreated; Normal PC12 M, supernatant from PC12 cells in serum-containing normal medium for 6 h; SDM, supernatant from PC12 cells in serum-deprived medium for 6 h; LPS, 100 ng/ml LPS; SY5Y, supernatant from apoptotic SH-SY5Y cells that were treated with staurosporine (10 μm) for 4 h and then incubated with fresh medium for 6 h; BE(2)C, supernatant from apoptotic SK-N-BE(2)C cells that were treated with staurosporine (10 μm) for 4 h and then incubated with fresh medium for 6 h.
Release of matrix metalloproteinase-3 from apoptotic PC12 cells into the serum-deprived media
SDM was subject to be analyzed by two-dimensional gel electrophoresis, followed by mass spectrometry. Because SDM contained high serum components even after PC12 monolayer was thoroughly washed for four times with serum-free media, the last wash was used as control. Proteins from both 6 h SDM and control medium were concentrated by using 10 kDa cutoff filter and separated by two-dimensional electrophoresis. Among several protein spots exclusively expressed in 6 h SDM, highly reproducible three spots (arrowhead) were analyzed by MALDI-TOF mass spectrometry (Fig. 2A). Obtained mass information was analyzed by peptide fingerprint matching by referencing the NCBInr. protein database using MS-FIT in ProteinProspector software. The possible molecules from three spots included MMP-3, cytoskeletal proteins (desmin and plectin), and cell adhesion molecules [intracellular adhesion molecules (ICAMs)]. MMP-3 was the one with highest probability. To confirm the release of MMP-3 from serum-deprived apoptotic PC12 cells, MMP-3 levels in SDM and whole lysates were determined by Western blot assay. The results showed that active MMP-3 (aMMP-3) of 48 kDa, but not proform of MMP-3 (pMMP-3) (55 kDa), was detected in the SDM as early as 2 h, and amounts of aMMP-3 in the media steadily increased to 24 h (Fig. 2B, top). Similar analysis of whole lysates of PC12 cells showed that both pMMP-3 (55 kDa) and aMMP-3 (48 kDa) were present in PC12 cells and that both pMMP-3 and aMMP-3 were steadily decreased in time-dependent manner by serum deprivation (Fig. 2B, bottom). aMMP-3 in the SDM was enzymatically active as demonstrated by zymographic analysis in which aMMP-3 appeared as a clear band by degrading casein in the gel as early as 2 h after serum deprivation (Fig. 2C).
Figure 2.

Active MMP-3 release from apoptotic PC12 cells into the serum-deprived media. A, Two-dimensional gel electrophoresis identified several protein spots exclusively expressed in SDM over control medium. Concentrated 6 h SDM and fourth wash were separated using two-dimensional gel electrophoresis. After silver staining, gels were scanned, and protein patterns were compared with each other. Three spots (arrowheads) that were exclusively shown in 6 h SDM were analyzed by in-gel tryptic digestion, followed by MALDI-TOF mass spectrometry and peptide fingerprint-matching data analysis. Concentrated 6 h SDM, 6 h SDM concentrated with 10 kDa cutoff filter; Concentrated 4th wash, concentrated last washed medium that might contain diluted serum proteins. B, Time course release of active MMP-3 was confirmed in both SDM (top) and whole lysates of serum-deprived PC12 cells (bottom). 4th wash, Concentrated fourth washed medium; 2 h, 6 h, and 24 h SDM, concentrated SDM at 2, 6, and 24 h of serum deprivation, respectively; 2 h, 6 h, and 24 h SD, whole lysates from corresponding cultures of serum deprivation for 2, 6, and 24 h, respectively; normal PC12, whole lysates from PC12 cells grown in serum-containing normal medium. C, SDM contained enzymatically active form of MMP-3. Catalytic activity of concentrated 2 h and 6 h SDM was analyzed using casein zymography. Clear bands of MMP-3 (48 kDa) after Coomassie blue staining of the gel represent proteolytically active MMP-3. 4th wash, Concentrated fourth washed medium; 2 h and 6 h SDM, concentrated SDM at 2 and 6 h of serum deprivation, respectively. Pictures are representative of three independent experiments.
Activation of microglia by cMMP-3
To confirm that active MMP-3 is the soluble mediator causing microglial cytokine induction, both cMMP-3 and pMMP-3 were added to microglial culture. The profiles of cMMP-3-elicited cytokine mRNA expression in BV2 cells were the same as those of SDM-elicited expression (data not shown). pMMP-3 had no effect. TNF-α was used as a marker for quantitative determination of induced cytokines (Fig. 3A, top). The densitometric measurements of RPA showed a significant increase of TNF-α in microglia induced by cMMP-3 (Fig. 3A, bottom) but not by pMMP-3. Pretreatment with NNGH (60 μm), an inhibitor of MMP-3, for 30 min decreased cMMP-3-elicited increase of TNF-α. The level of TNF-α in BV2 cells was measured by ELISA after treatment with MMP-3 for 24 h (Fig. 3B). Release of TNF-α into the media by cMMP-3 (400 ng/ml) was increased from 19 ± 4.4 pg/ml (untreated control) to 1244 ± 328.7 pg/ml, whereas NNGH (60 μm) diminished the TNF-α release and pMMP-3 had no effects (Fig. 3B). These results implied that MMP-3 might induce cytokines in microglia by its proteolytic activity rather than simple molecular binding.
Figure 3.

Induction of TNF-α in BV2 cells by cMMP-3. MMP-3 increased TNF-α mRNA levels (A) and released TNF-α protein (B) in BV2 cells through its enzymatic activity. BV2 cells were treated with cMMP-3 (400 ng/ml), pMMP-3 (400 ng/ml), or NNGH (60 μm, 30 min pretreatment) plus cMMP-3 for 6 h, and then cytokine mRNAs were analyzed by RPA. A, Top, Representative autoradiograph of TNF-α mRNA expression in microglia from four independent RPA experiments. Bottom, Densitometric measurement of TNF-α levels was normalized by GAPDH levels. Values represent the mean ± SD (n = 4) (A). Under the same conditions, TNF-α release from BV2 cells was determined by ELISA. Values represent the mean ± SD (n = 4) (B).
To assess whether MMP-3 was an active component responsible for microglial activation among SDM constituents, BV2 cells were pretreated with NNGH (60 μm) for 30 min before addition of 6 h SDM (50 μg/ml), and TNF-α release was measured by ELISA (Fig. 4A). SDM increased TNF-α release from 150.8 ± 51.6 pg/ml at 6 h to 928.9 ± 189.3 pg/ml at 24 h and 1319.4 ± 288.1 pg/ml at 48 h. Whereas NNGH pretreatment decreased SDM-mediated TNF-α release at 6 h (20.5 ± 5.9 pg/ml), 24 h (100.3 ± 38.1 pg/ml), and 48 h (322.6 ± 164.8 pg/ml), NNGH without SDM had no effect on BV2 cells. NNGH reduced SDM-induced TNF-α release by ∼85% for 48 h (Fig. 4A). Similar experiments using cMMP-3 (400 ng/ml) resulted in similar results (Fig. 4B). The levels of TNF-α in the microglial culture media were 166.2 ± 77.9, 1244.5 ± 328.6, and 1802.1 ± 387.1 pg/ml at 6, 24, and 48 h after cMMP-3 treatments, respectively, demonstrating that cMMP-3 was capable of inducing TNF-α in activated microglia. NNGH pretreatment abolished cMMP-3-mediated TNF-α release. pMMP-3 failed to increase TNF-α level. The results indicated that the enzymatically active form of MMP-3 in the SDM induced TNF-α release from microglia.
Figure 4.
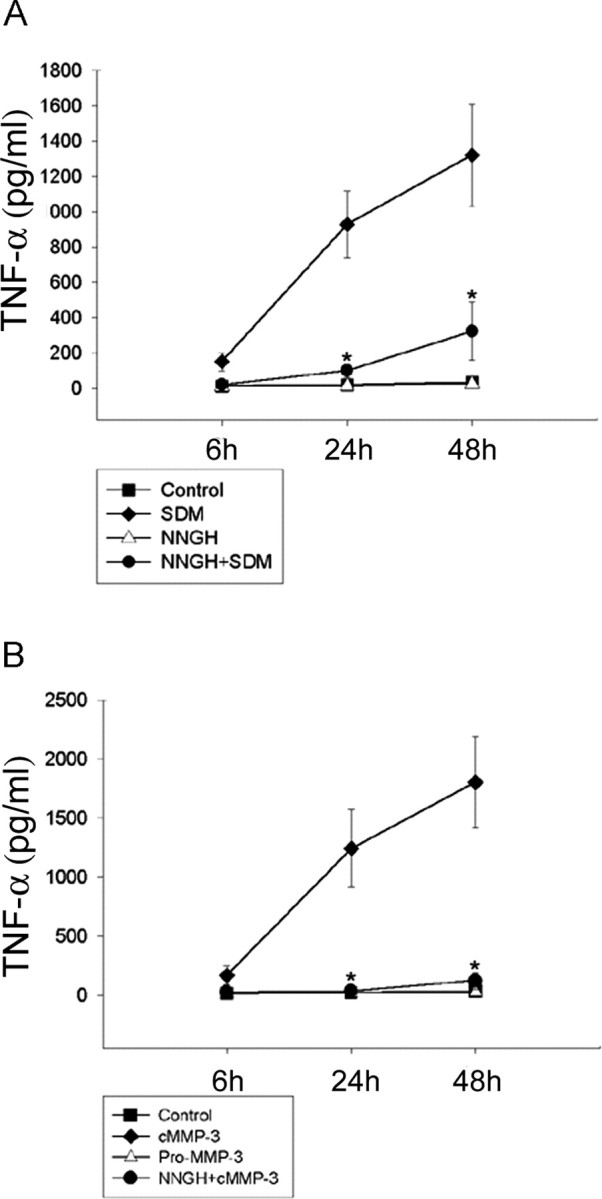
Inhibition of SDM-induced TNF-α release into the BV2 cell-culture media by the MMP-3 inhibitor NNGH. A, SDM-mediated TNF-α release from microglia was abolished by NNGH pretreatment. TNF-α levels were measured in BV2 cell-culture media after 24 h incubation with either SDM or SDM with NNGH. Control, Untreated; SDM, concentrated SDM (50 μg/ml); NNGH+SDM, 30 min pretreatment of NNGH (60 μm) and then SDM (50 μg/ml). B, Enzymatic activity of MMP-3 was essential to induce TNF-α in microglia. TNF-α levels were measured in BV2 cell-culture media after 24 h incubation with pMMP-3, cMMP-3, or cMMP-3 with NNGH. Control, Untreated; pMMP-3, pMMP-3 at 400 ng/ml; cMMP-3, cMMP-3 at 400 ng/ml; NNGH+cMMP-3, 30 min pretreatment of NNGH (60 μm) and then cMMP-3 (400 ng/ml). Values represent the mean ± SD (n = 4).
Other known proteases that activate microglia are thrombin through protease-activated receptor 1 (PAR1) (Suo et al., 2002) and tissue plasminogen activator (tPA) through nonproteolytic mechanism by binding annexin II (Siao and Tsirka, 2002). PAR1 antibody inhibited thrombin activation of microglia but was ineffective on cMMP-3-mediated microglial activation (data not shown).
Both SDM- and cMMP-3-mediated induction of cytokines in microglia depend on ERK activation
Because MAPK (mitogen-activated protein kinases) have been reported to be involved in the activation of both macrophage and microglia under different stimuli, such as LPS, interferon-β, chemokines, β-amyloid peptide, and CD40L (Weinstein et al., 1992; Tan et al., 2000a,b; Cambien et al., 2001; Kim et al., 2002), we examined time course activation of three MAPKs, including ERK, p38, and JNK, in SDM-treated BV2 cells. Both LPS (100 ng/ml) and 6 h SDM (50 μg/ml) significantly induced phosphorylation of ERK42/44 within 5 min, and activation was sustained up to 2 h. Phosphorylation of p38 was detected only in LPS-treated BV2 cells within 5 min and reached maximal level at 2 h. Activation of JNK was detected in both LPS- and SDM-treated BV2 microglia. Whereas LPS induced phosphorylation of JNK within 5 min and it was sustained up to 2 h, SDM relatively weakly activated JNK at 10 min and phosphorylation disappeared within 1 h (Fig. 5A). To test whether the inhibition of ERK activation resulted in abolition of SDM-mediated microglial activation, the specific MEK inhibitor U0126 was used. Pretreatment of U0126 (20 μm) almost completely reversed SDM-activated microglial genes to control levels. However, it failed to completely block the LPS-induced mRNA levels, although it decreased TNF-α, IL-6, and IL-1β by ∼40%. Interestingly, iNOS gene was increased in U0126-pretreated BV2 cells (Fig. 5B). SDM-induced TNF-α release was also completely blocked by U0126 (data not shown). Next, we examined ERK activation in cMMP-3-treated BV2 cells. Strong activation of ERK was found in cMMP-3 (400 ng/ml)-treated BV2 cells, and NNGH (60 μm) and U0126 (20 μm) pretreatment lowered the levels of phosphorylated ERK42/44. Compared with cMMP-3, pMMP-3 treatment did not induce ERK phosphorylation (Fig. 5C). Pretreatment of U0126 (20 μm) for 30 min completely inhibited cMMP-3-mediated TNF-α mRNA induction (Fig. 5D). U0126 pretreatment also completely blocked cMMP-3-mediated TNF-α release in 24 h treated BV2 cells. Inhibitors of JNK (SP600125) and p38MAPK (SB203580), however, had not completely blocked TNF-α release (Fig. 5E). The data suggest that active MMP-3 contained in SDM evokes cytokine gene induction in microglia through activation of ERK pathway.
Figure 5.

ERK-dependent activation of microglia treated with SDM and cMMP-3. A, ERK pathway was most strongly activated in SDM-mediated microglial activation. BV2 cells were treated with either LPS (100 ng/ml) or 6 h SDM (50 μg/ml) for 5 min to 2 h. Phosphorylation of three MAPKs (ERK, p38, and JNK) was detected by Western blot analysis. B, U0126, a specific MEK1/2 inhibitor, abolished SDM-mediated microglial cytokine inductions. After 30 min pretreatment of U0126 (20 μm), BV2 cells were incubated with either LPS (100 ng/ml) or SDM (50 μg/ml) for 6 h. Cytokine mRNAs were determined using RPA. C, cMMP-3 also activated ERK pathway in microglia. BV2 cells were pretreated with either U0126 (20 μm) or NNGH (60 μm) for 30 min and then incubated with cMMP-3 (400 ng/ml) for 30 min. Phospho-ERK was detected by Westernblot analysis. β-Actin was shown as internal control. D, E, U0126 completely blocked cMMP-3-mediated microglial TNF-α induction and release. However, SP600125 and SB203580 did not completely inhibit TNF-α release. After 30 min pretreatment of U0126 (20 μm), BV2 cells were incubated with cMMP-3 (400 ng/ml) for 6 h. TNF-α mRNA was determined using RPA. The top panel shows autoradiograph of TNF-α RPA. Densitometric measurement of TNF-α level normalized by GAPDH level is shown in the bottom panel (D). TNF-α release was determined after 24 h treatment with either cMMP-3 or U0126 plus cMMP-3 by ELISA (E). Control, Untreated; pMMP-3, pMMP-3 at 400 ng/ml; cMMP-3, cMMP-3 at 400 ng/ml; U0126+cMMP-3, 30 min pretreatment of U0126 (20 μm) and then cMMP-3 (400 ng/ml); NNGH+cMMP-3, 30 min pretreatment of NNGH (60 μm) and then cMMP-3 (400 ng/ml); SP+cMMP-3, 30 min pretreatment of SP600125 (20 μm) and then cMMP-3 (400 ng/ml); SB+cMMP-3, 30 min pretreatment of SB203580 (20 μm) and then cMMP-3 (400 ng/ml). Pictures shown are representative of four independent experiments.
To verify our findings in primary culture system, we used both neuronal culture and microglial culture. We confirmed that active MMP-3 was released from degenerating dopaminergic neurons using primary mesencephalic neuronglia cultures in the presence of 1-methyl-4-phenylpyridinium (MPP+). In the primary mesencephalic culture media undergoing degeneration by MPP+, MMP activity was increased, and an active MMP-3 was detected by immunoprecipitation (data not shown). Primary microglia were used to confirm cMMP-3-mediated microglial activation. cMMP-3 induced TNF-α activation in a dose-dependent manner, and pretreatment of either NNGH or U0126 completely abolished cMMP-3-mediated TNF-α release (Fig. 6).
Figure 6.

Activation of primary microglia by cMMP-3. Primary mouse microglia were also activated by cMMP-3 in an ERK-dependent manner. Primary mouse microglia were treated with either various concentration of cMMP-3 alone (125, 250, and 400 ng/ml) or combined pretreatment with inhibitors. U0126 (20 μm) or NNGH (60 μm) were 30 min pretreated before cMMP-3 (400 ng/ml). TNF-α release was measured at 3 and 8 h after treatment. Control, Untreated; LPS, LPS at 10 ng/ml; 125, 250, 400, cMMP-3 at 125, 250, and 400 ng/ml, respectively; NNGH+cMMP-3, 30 min pretreatment of NNGH (60 μm) and then cMMP-3 (400 ng/ml); U0126+cMMP-3, 30 min pretreatment of U0126 (20 μm) and then cMMP-3 (400 ng/ml). Values represent the mean ± SD (n = 4).
NF-κB activation in microglia by cMMP-3 is through the ERK pathway
We also investigated NF-κB activation because NF-κB regulation of various cytokine genes, such as iNOS, TNF-α, and IL-6, has been reported previously (Carter et al., 1996; Kleinert et al., 1996). NF-κB activation in microglia by cMMP-3 was determined by the electrophoresis mobility shift assay. The 6 h treatment of cMMP-3 (400 ng/ml) prominently induced NF-κB nuclear translocation, and NNGH (60 μm) pretreatment reduced nuclear NF-κB level by ∼65%. Pretreatment of U0126 (20 μm) for 30 min also reduced the nuclear translocation of NF-κB. This result suggests that NF-κB activation is downstream to ERK activation in cMMP-3-treated microglia. In the presence of excess amounts of unlabeled oligonucleotides, the shifted bands disappeared (Fig. 7).
Figure 7.
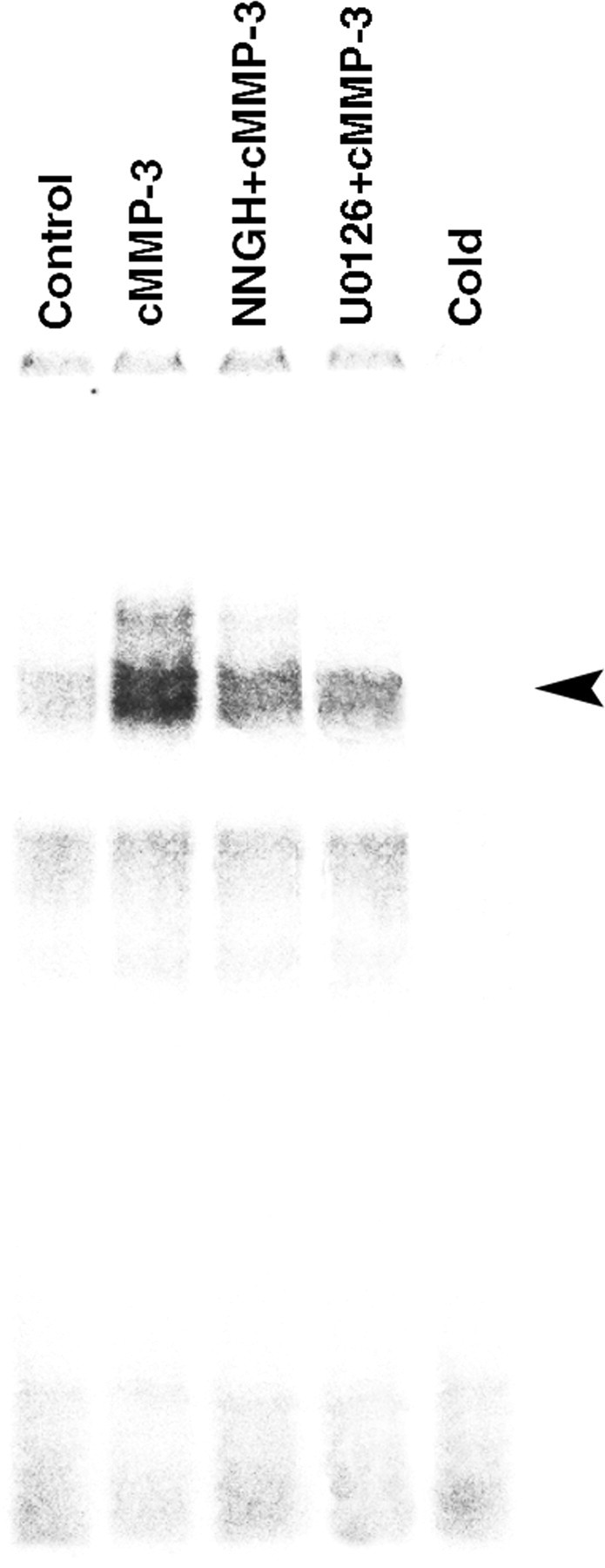
ERK-dependent activation of NF-κ B by cMMP-3. cMMP-3 induced translocation of NF-κ B through ERK pathway in BV2 cells. BV2 cells were incubated with either cMMP-3 alone (400 ng/ml) or combined pretreatment with inhibitors for 6 h. Nuclear translocation of NF-κ B was examined by gel mobility shift assay. Control, Untreated; cMMP-3, cMMP-3 at 400 ng/ml; U0126+cMMP-3, 30 min pretreatment of U0126 (20 μm) and then cMMP-3 (400 ng/ml); NNGH+cMMP-3, 30 min pretreatment of NNGH (60 μm) and then cMMP-3 (400 ng/ml); Cold, excessive amounts of unlabeled NF-κ B oligonucleotide with cMMP-3-treated nuclear extract.
Conditioned medium from cMMP-3-stimulated microglia causes neuronal death
Because a number of studies have demonstrated that activated microglia induce neural cell degeneration, we investigated whether microglia activated by either SDM or cMMP-3 could induce neural cell death. Differentiated PC12 cells were exposed to cell-free CM collected from either untreated resting BV2 cells or BV2 cells treated with cMMP-3, SDM, or LPS. CM from cMMP-3-stimulated BV2 microglia induced significant neural cell death with increasing LDH release to ∼160% of untreated PC12 cells (Fig. 8). In contrast, CM from resting BV2 microglia did not trigger significant neural cell death. SDM and LPS also caused differentiated PC12 cell death through microglial activation, as indicated by the 140 and 150% increases in LDH release compared with untreated control, respectively. Consistent with the results that demonstrated the involvement of ERK pathway in cMMP-3- and SDM-mediated BV2 microglial activation, CM collected from U0126-pretreated BV2 cells failed to induce neurodegeneration. To further investigate whether CM from cMMP-3 caused apoptosis, CM-treated PC12 cells were stained with annexin V, an early apoptosis marker, over time. Annexin V-positive cells appeared as early as 6 h after treatment with cMMP-3 CM. TNF-α at 1000 pg/ml, a concentration equivalent to that in 12 h cMMP-3 CM, also caused apoptosis of PC12 cells. However, blocking of TNF-α in cMMP-3 CM by preincubation of a specific antibody (2 μg/ml) did not completely inhibit PC12 apoptosis. This result implies that substances other than TNF-α could be involved in CM-mediated PC12 degeneration.
Figure 8.
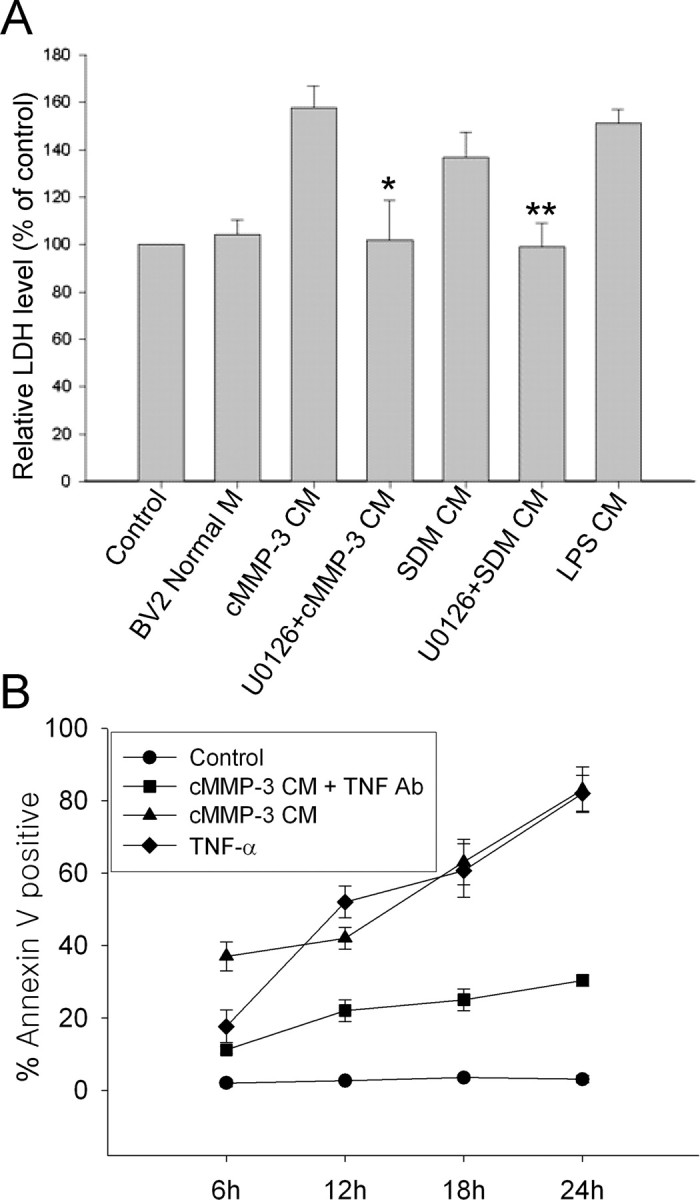
Neurodegeneration induced by SDM- or cMMP-3-activated BV2 cells. A, LDH release was measured from the supernatant of differentiated PC12 cells incubated for 12 h in the various conditioned media from BV2 cells. Control, Untreated; BV2 Normal M, medium from untreated normal BV2 cells; cMMP-3 CM, conditioned medium from BV2 incubated with cMMP-3 (400 ng/ml) for 3 h; U0126+cMMP-3 CM, CM from 30 min U0126 (20 μm) pretreatment, followed by cMMP-3 for 3 h; SDM CM, CM from concentrated SDM (100 μg/ml) for 3 h; U0126 + SDM CM, CM from U0126 (20 μm) pretreatment, followed by SDM (100 μg/ml) for 3 h; LPS CM, CM from LPS (100 ng/ml) for 3 h. The data were obtained from triplicate of two independent experiments. *p < 0.01 versus cMMP-3 CM; **p < 0.05 versus SDM CM. B, Apoptosis of differentiated PC12 cells was determined by annexin V staining at 6, 12, 18, and 24 h after treatment of cells with cMMP-3 CM, TNF-α, or cMMP-3 CM preincubated with TNF-α antibody. Control, Untreated; cMMP-3 CM, conditioned medium from BV2 incubated with cMMP-3 (400 ng/ml) for 3 h; TNF-α, recombinant TNF-α at 1000 pg/ml; cMMP-3 CM + TNF Ab, cMMP-3 CM preincubated with TNF-α antibody (2 μg/ml) for 1 h. Values represent the mean ± SD (n = 4).
Discussion
A number of studies have demonstrated that apoptotic cells modulate immune responses by direct contact with phagocytes and subsequent engulfment. Apoptotic cells trigger the recruitment of phagocytic cells to be removed quickly and efficiently. Apoptotic cells release chemoattractants, such as fractalkine, a chemokine that is predominantly expressed in the brain (Chapman et al., 2000), or the phospholipid lysophosphatidylcholine (Lauber et al., 2003), which promotes chemotaxis of primary microglia and monocytes. However, in addition to phagocytosing apoptotic cells, phagocytes actively promote cell death by engulfment. In the developmental process, apoptosis of Purkinje cells in the cerebellum can be prominently reduced by selective elimination of microglia (Marin-Teva et al., 2004). In the developing nematode, caspase [CED-3 (Caenorhabditis elegans cell death protein)]-dependent cellular degeneration can be reversed by inhibition of engulfment by neighboring cells (Hoeppner et al., 2001; Reddien et al., 2001). Our recent studies also showed that microglia were rapidly activated by apoptotic dopamine neurons in the substantia nigra after axon transection and that phagocytosis of apoptotic neurons by activated microglia occurred at early stages of apoptosis (Cho et al., 2003; Sugama et al., 2003). The question addressed by the present study was whether early apoptotic neurons release soluble signaling molecules that induce microglial activation and induce production of inflammatory cytokines in activated microglia. The answers for this question may provide valuable information regarding mechanisms that underlie microglial activation and phagocytosis in neuronal apoptotic processes.
By identifying the active form of MMP-3 as a signaling molecule from apoptotic neuronal cells that activates microglia and induces inflammatory cytokines in microglia, we established an important sequence of inflammatory reactions that occur in neuronal apoptosis. Additional evidence for MMP-3 release from apoptotic neuronal cells was that MMP-3 was identified in degenerating primary mouse mesencephalic cultures treated with MPP+, a selective dopaminergic neuronal toxin, and inhibition of TNF-α release from SDM-activated microglia by the specific MMP-3 inhibitor NNGH. Increased TNF-α expression has been implicated in the pathogenesis of several degenerative human brain diseases, including multiple sclerosis (Raine, 1994), Alzheimer's disease (Fillit et al., 1991; Meda et al., 1995), Parkinson's disease (Boka et al., 1994; Mogi et al., 1994; Sriram et al., 2002), and AIDS dementia (Akassoglou et al., 1997). Studies have shown that TNF-α directly induces neuronal death (Talley et al., 1995; D'Souza et al., 1996; Batchelor et al., 1999; Haviv and Stein, 1999). These suggest that cMMP-3-activated microglia might cause neuronal degeneration via releasing proinflammatory cytokines. Cytokine induction in microglia by active forms of MMP-3 was dependent of ERK and NF-κB intracellular signal transduction pathway. Moreover, activated microglia in the presence of SDM or cMMP-3 caused differentiated PC12 cell death. Apoptosis is known as silent cell death, and activation of microglia or macrophages in apoptosis was thought to be a null event in cell death. Although recent evidence strongly suggested that various immunological events take place in apoptosis (Savill and Fadok, 2000), mechanisms that trigger immune responses in apoptosis have not been clearly identified. Our results are consistent with the idea that apoptotic neuronal cells release microglia-activating molecules, and apoptosis is not an immunologically null event.
Apoptosis of adult neurons has been observed in various neurodegenerative disorders (Goldberg et al., 1996; Mochizuki et al., 1997; Gervais et al., 1999). Recent studies clearly demonstrate that apoptotic neurons and other cells could be phagocytosed by activated microglia in early neuronal apoptosis (Savill and Fadok, 2000; Cho et al., 2003; Sugama et al., 2003). The engulfment of apoptotic cells elicit important consequences for immune responses, such as induction of anti-inflammatory mediators (Voll et al., 1997; Fadok et al., 1998; Huynh et al., 2002). Considering the proximity of microglial location to apoptotic neurons in the brain and rapid activation of microglia, secretion of signaling molecules that are not necessarily chemoattractants is a reasonable assessment. This type of releasing mediator might have a different function from chemotactic factors. We report here MMP-3 as a potential candidate. Active MMP-3 release from apoptotic cells might play important roles in cellular apoptosis and subsequent engulfment by phagocytes. First, it may disrupt physical connections between apoptotic cells and the surrounding extracellular matrix, which might facilitate cellular shrinkage and promote their engulfment by phagocytes. Second, as we shown here, aMMP-3 may induce microglial activation near the site at which apoptosis occurs. Activated microglia may release various cytokines and express receptors responsible for the recognition and engulfment of apoptotic cells. The early activation of microglia might be terminated by engulfment of apoptotic cells. The deregulation of this sequential event may cause pathologic conditions.
The different patterns of signal transduction pathway activation and cytokine induction in microglia by the SDM and LPS are also noteworthy. In the present study, both SDM and cMMP-3 rapidly activated the ERK pathway, followed by proinflammatory cytokine induction, but no significant activation of JNK and p38 MAPK were observed. By blocking MEK1/2, an immediate upstream activator of ERK, both SDM- and cMMP-3-induced cytokine genes and TNF-α release were almost completely inhibited. JNK inhibitor and p38 inhibitor, however, did not completely block MMP-3-mediated TNF-α release. IL-12 and iNOS mRNA expressed by LPS were not induced by the SDM. Because IL-12 is a strong inflammatory cytokine and NO is a neurotoxin, the lack of expression of these two genes by the SDM may represent the differential immunological consequences by neuronal degeneration processes between apoptosis and necrosis or direct injury of neurons. LPS is a potent immunostimulant activating a number of signal transduction pathways, including MAPKs, PKA, PKC, and NF-κB, which coordinate the induction of many genes encoding inflammatory mediators (Guha and Mackman, 2001). In our hands, however, LPS-mediated iNOS induction in BV2 cells was actually increased by inhibition of ERK activation (Fig. 5). In addition, cMMP-3 activated NF-κB pathway in microglia. The inhibition of NF-κB nuclear translocation by a MEK1/2 inhibitor, U0126, suggests that phosphorylation of ERK is upstream of NF-κB activation. Although NF-κB was activated by both LPS and MMP-3 and NF-κB is an essential component in iNOS induction, other transcriptional elements such as LPS-responding elements were also shown to be important (Xie et al., 1994; Xie, 1997). In addition, iNOS induction is mainly regulated by p38 pathway but not by ERK in macrophage and C6 glioma cells (Da Silva et al., 1997; Xu and Malave, 2000). MMP-3 did not significantly activate the p38 MAPK pathway, as shown in Results. These results suggest that, in neuronal apoptosis, the ERK pathway is the predominant signal transduction pathway in activated microglia and that inhibition of the ERK pathway may be an important inhibitory treatment for microglia-induced immune responses.
MMPs are proteolytic enzymes that degrade extracellular macromolecules. Physiologically, MMPs are important in tissue remodeling, cell migration, wound healing, and angiogenesis. MMP-3 has broad substrates, including collagen, laminin, fibronectin, and other latent-type MMPs, such as MMP-7, MMP-8, MMP-9, and MMP-13 (Chandler et al., 1997). MMP-3 has been studied in several CNS diseases, including ischemia, Alzheimer's disease, and multiple sclerosis (Rosenberg et al., 2001). These data suggest that MMP-3 may play an important role in the process of neurodegeneration by disrupting the blood-brain barrier, followed by aggravating inflammatory reactions (Maeda and Sobel, 1996; Yong et al., 1998, 2001). In the present study, we showed that the active form of MMP-3, which includes recombinant catalytic domain but not proform, induced microglial activation. This suggests that, in addition to its known function in extracellular macromolecule degradation, microglia may be activated by enzymatic activity of MMP-3 on the membrane of BV2 cells. Enzymatic cleavages of cellular membrane surface proteins that activate cellular activity are known, and MMP-3 may cleave chemokines or cytokines on the surface of the microglial membrane, or cell-cell interaction proteins that include ICAMS, members of the Ig superfamily that can bind β2 integrin molecules (Hubbard and Rothlein, 2000). Both ICAM-1 and β2 integrin molecules are expressed in BV2 cells (data not shown). MMP-3 may cleave specific receptor(s), such as PAR-1, like thrombin-mediated microglial activation (Suo et al., 2002). However, PAR-1 antibody had no effect on MMP-3-elicited microglia activation. tPA can also cause microglial activation via a nonproteolytic mechanism (Tsirka et al., 1997; Siao and Tsirka, 2002). Molecular mechanisms that govern microglial activation by enzymatic activity of MMP-3 remain to be elucidated.
In addition, by demonstrating differentiated PC12 cell degeneration induced by cMMP-3-activated microglia, this study supports a number of previous studies showing that activated microglia are significant components of the pathology in the brain of chronic neurodegenerative diseases. We demonstrated in this study molecular mechanisms that govern microglial activation in neuronal cell apoptosis. Because MMP-3 did not induce neurotrophic factor, GDNF, or anti-inflammatory cytokines, TGF-β, and IL-10, in activated microglia in early neuronal apoptosis, MMP-3 signaling may trigger inflammatory reactions as well as exacerbation of neuronal apoptosis that lead to rapid phagocytosis of apoptotic neurons. The present study provides important clues to our understanding of the communication between apoptotic neurons and microglia. Early interventions into this interaction may be used as important therapeutic strategies to prevent the propagation of microglia-mediated neurodegeneration.
Footnotes
This research was supported by the Parkinson's Foundation (Y.S.K., M.F.B.), the Burke Foundation and the Goldsmith Foundation (T.H.J.), the Brain Research Center of the 21st Century Frontier Program in Neuroscience of the Ministry of Science and Technology, Korea Grants M103KV010006 and 03K2201 00630 (O.H., T.H.J.), and the Department of Defense (M.F.B.). We thank Drs. Harriet Baker, Soo Yul Kim, Lori DeGiorgio, and Sung Hee Cho for helpful discussions and support and Jessica Lynch for her careful editing of this manuscript.
Correspondence should be addressed to Dr. Tong H. Joh, Molecular Neurobiology, Burke Medical Research Institute, Weill Medical College of Cornell University, 785 Mamaroneck Avenue, White Plains, NY 10021. E-mail: thjoh@mail.med.cornell.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253701-11$15.00/0
References
- Akassoglou K, Probert L, Kontogeorgos G, Kollias G (1997) Astrocyte-specific but not neuron-specific transmembrane TNF triggers inflammation and degeneration in the central nervous system of transgenic mice. J Immunol 158: 438-445. [PubMed] [Google Scholar]
- Andersen JK (2001) Does neuronal loss in Parkinson's disease involve programmed cell death? BioEssays 23: 640-646. [DOI] [PubMed] [Google Scholar]
- Batchelor PE, Liberatore GT, Wong JY, Porritt MJ, Frerichs F, Donnan GA, Howells DW (1999) Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J Neurosci 19: 1708-1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, Hirsch EC (1994) Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson's disease. Neurosci Lett 172: 151-154. [DOI] [PubMed] [Google Scholar]
- Brown DR (2001) Microglia and prion disease. Microsc Res Tech 54: 71-80. [DOI] [PubMed] [Google Scholar]
- Cambien B, Pomeranz M, Schmid-Antomarchi H, Millet MA, Breittmayer V, Rossi B, Schmid-Alliana A (2001) Signal transduction pathways involved in soluble fractalkine-induced monocytic cell adhesion. Blood 97: 2031-2037. [DOI] [PubMed] [Google Scholar]
- Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhauser N, Bohm-Matthaei R, Baeuerle PA, Barde YA (1996) Selective activation of NF-kappa B by nerve growth factor through the neurotrophin receptor p75. Science 272: 542-545. [DOI] [PubMed] [Google Scholar]
- Chandler S, Miller KM, Clements JM, Lury J, Corkill D, Anthony DC, Adams SE, Gearing AJ (1997) Matrix metalloproteinases, tumor necrosis factor and multiple sclerosis: an overview. J Neuroimmunol 72: 155-161. [DOI] [PubMed] [Google Scholar]
- Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ (2000) Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci 20: RC87(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho BP, Sugama S, Shin DH, DeGiorgio LA, Kim SS, Kim YS, Lim SY, Park KC, Volpe BT, Cho S, Joh TH (2003) Microglial phagocytosis of dopamine neurons at early phases of apoptosis. Cell Mol Neurobiol 23: 551-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzner ML, Gveric D, Strand C, Loughlin AJ, Paemen L, Opdenakker G, Newcombe J (1996) The expression of tissue-type plasminogen activator, matrix metalloproteases and endogenous inhibitors in the central nervous system in multiple sclerosis: comparison of stages in lesion evolution. J Neuropathol Exp Neurol 55: 1194-1204. [DOI] [PubMed] [Google Scholar]
- Da Silva J, Pierrat B, Mary JL, Lesslauer W (1997) Blockade of p38 mitogen-activated protein kinase pathway inhibits inducible nitric-oxide synthase expression in mouse astrocytes. J Biol Chem 272: 28373-28380. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C (1993) Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer's disease. Glia 7: 75-83. [DOI] [PubMed] [Google Scholar]
- D'Souza SD, Bonetti B, Balasingam V, Cashman NR, Barker PA, Troutt AB, Raine CS, Antel JP (1996) Multiple sclerosis: Fas signaling in oligodendrocyte cell death. J Exp Med 184: 2361-2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM (1998) Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 101: 890-898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, Lawlor B, Wolf-Klein G (1991) Elevated circulating tumor necrosis factor levels in Alzheimer's disease. Neurosci Lett 129: 318-320. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW (1999) Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 97: 395-406. [DOI] [PubMed] [Google Scholar]
- Goldberg YP, Nicholson DW, Rasper DM, Kalchman MA, Koide HB, Graham RK, Bromm M, Kazemi-Esfarjani P, Thornberry NA, Vaillancourt JP, Hayden MR (1996) Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Genet 13: 442-449. [DOI] [PubMed] [Google Scholar]
- Gottschall PE, Deb S (1996) Regulation of matrix metalloproteinase expressions in astrocytes, microglia and neurons. Neuroimmunomodulation 3: 69-75. [DOI] [PubMed] [Google Scholar]
- Guha M, Mackman N (2001) LPS induction of gene expression in human monocytes. Cell Signal 13: 85-94. [DOI] [PubMed] [Google Scholar]
- Haviv R, Stein R (1999) Nerve growth factor inhibits apoptosis induced by tumor necrosis factor in PC12 cells. J Neurosci Res 55: 269-277. [DOI] [PubMed] [Google Scholar]
- Hoeppner DJ, Hengartner MO, Schnabel R (2001) Engulfment genes cooperate with ced-3 to promote cell death in Caenorhabditis elegans Nature 412: 202-206. [DOI] [PubMed] [Google Scholar]
- Hubbard AK, Rothlein R (2000) Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free Radic Biol Med 28: 1379-1386. [DOI] [PubMed] [Google Scholar]
- Huynh ML, Fadok VA, Henson PM (2002) Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest 109: 41-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MO, Si Q, Zhou JN, Pestell RG, Brosnan CF, Locker J, Lee SC (2002) Interferon-beta activates multiple signaling cascades in primary human microglia. J Neurochem 81: 1361-1371. [DOI] [PubMed] [Google Scholar]
- Kleinert H, Euchenhofer C, Ihrig-Biedert I, Forstermann U (1996) In murine 3T3 fibroblasts, different second messenger pathways resulting in the induction of NO synthase II (iNOS) converge in the activation of transcription factor NF-kappaB. J Biol Chem 271: 6039-6044. [DOI] [PubMed] [Google Scholar]
- Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S (2003) Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 113: 717-730. [DOI] [PubMed] [Google Scholar]
- Liu B, Wang K, Gao HM, Mandavilli B, Wang JY, Hong JS (2001) Molecular consequences of activated microglia in the brain: overactivation induces apoptosis. J Neurochem 77: 182-189. [DOI] [PubMed] [Google Scholar]
- Maeda A, Sobel RA (1996) Matrix metalloproteinases in the normal human central nervous system, microglial nodules, and multiple sclerosis lesions. J Neuropathol Exp Neurol 55: 300-309. [DOI] [PubMed] [Google Scholar]
- Marin-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M (2004) Microglia promote the death of developing Purkinje cells. Neuron 41: 535-547. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology 38: 1285-1291. [DOI] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos Jr L, Baron P, Villalba M, Ferrari D, Rossi F (1995) Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature 374: 647-650. [DOI] [PubMed] [Google Scholar]
- Mochizuki H, Goto K, Mori H, Mizuno Y (1996) Histochemical detection of apoptosis in Parkinson's disease. J Neurol Sci 137: 120-123. [DOI] [PubMed] [Google Scholar]
- Mochizuki H, Mori H, Mizuno Y (1997) Apoptosis in neurodegenerative disorders. J Neural Transm Suppl 50: 125-140. [DOI] [PubMed] [Google Scholar]
- Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T (1994) Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett 165: 208-210. [DOI] [PubMed] [Google Scholar]
- Nicotera P, Leist M, Manzo L (1999) Neuronal cell death: a demise with different shapes. Trends Pharmacol Sci 20: 46-51. [DOI] [PubMed] [Google Scholar]
- Raine CS (1994) Multiple sclerosis: immune system molecule expression in the central nervous system. J Neuropathol Exp Neurol 53: 328-337. [DOI] [PubMed] [Google Scholar]
- Reddien PW, Cameron S, Horvitz HR (2001) Phagocytosis promotes programmed cell death in C. elegans Nature 412: 198-202. [DOI] [PubMed] [Google Scholar]
- Rogers J, Luber-Narod J, Styren SD, Civin WH (1988) Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer's disease. Neurobiol Aging 9: 339-349. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Cunningham LA, Wallace J, Alexander S, Estrada EY, Grossetete M, Razhagi A, Miller K, Gearing A (2001) Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: activation of MMP-9 linked to stromelysin-1 and microglia in cell cultures. Brain Res 893: 104-112. [DOI] [PubMed] [Google Scholar]
- Savill J, Fadok V (2000) Corpse clearance defines the meaning of cell death. Nature 407: 784-788. [DOI] [PubMed] [Google Scholar]
- Siao CJ, Tsirka SE (2002) Tissue plasminogen activator mediates microglial activation via its finger domain through annexin II. J Neurosci 22: 3352-3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O'Callaghan JP (2002) Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson's disease. Faseb J 16: 1474-1476. [DOI] [PubMed] [Google Scholar]
- Stolzing A, Grune T (2004) Neuronal apoptotic bodies: phagocytosis and degradation by primary microglial cells. Faseb J 18: 743-745. [DOI] [PubMed] [Google Scholar]
- Sugama S, Cho BP, Degiorgio LA, Shimizu Y, Kim SS, Kim YS, Shin DH, Volpe BT, Reis DJ, Cho S, Joh TH (2003) Temporal and sequential analysis of microglia in the substantia nigra following medial forebrain bundle axotomy in rat. Neuroscience 116: 925-933. [DOI] [PubMed] [Google Scholar]
- Suo Z, Wu M, Ameenuddin S, Anderson HE, Zoloty JE, Citron BA, Andrade-Gordon P, Festoff BW (2002) Participation of protease-activated receptor-1 in thrombin-induced microglial activation. J Neurochem 80: 655-666. [DOI] [PubMed] [Google Scholar]
- Talley AK, Dewhurst S, Perry SW, Dollard SC, Gummuluru S, Fine SM, New D, Epstein LG, Gendelman HE, Gelbard HA (1995) Tumor necrosis factor alpha-induced apoptosis in human neuronal cells: protection by the antioxidant N-acetylcysteine and the genes bcl-2 and crmA. Mol Cell Biol 15: 2359-2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Town T, Mullan M (2000a) CD45 inhibits CD40L-induced microglial activation via negative regulation of the Src/p44/42 MAPK pathway. J Biol Chem 275: 37224-37231. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Mori T, Wu Y, Saxe M, Crawford F, Mullan M (2000b) CD45 opposes β-amyloid peptide-induced microglial activation via inhibition of p44/42 mitogen-activated protein kinase. J Neurosci 20: 7587-7594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirka SE, Rogove AD, Bugge TH, Degen JL, Strickland S (1997) An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci 17: 543-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I (1997) Immunosuppressive effects of apoptotic cells. Nature 390: 350-351. [DOI] [PubMed] [Google Scholar]
- Weinstein SL, Sanghera JS, Lemke K, DeFranco AL, Pelech SL (1992) Bacterial lipopolysaccharide induces tyrosine phosphorylation and activation of mitogen-activated protein kinases in macrophages. J Biol Chem 267: 14955-14962. [PubMed] [Google Scholar]
- Wu Q, Combs C, Cannady SB, Geldmacher DS, Herrup K (2000) Betaamyloid activated microglia induce cell cycling and cell death in cultured cortical neurons. Neurobiol Aging 21: 797-806. [DOI] [PubMed] [Google Scholar]
- Xie Q (1997) A novel lipopolysaccharide-response element contributes to induction of nitric oxide synthase. J Biol Chem 272: 14867-14872. [DOI] [PubMed] [Google Scholar]
- Xie QW, Kashiwabara Y, Nathan C (1994) Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem 269: 4705-4708. [PubMed] [Google Scholar]
- Xu X, Malave A (2000) P38 MAPK, but not p42/p44 MAPK mediated inducible nitric oxide synthase expression in C6 glioma cells. Life Sci 67: 3221-3230. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Mohanam S, Sawaya R, Fuller GN, Seiki M, Sato H, Gokaslan ZL, Liotta LA, Nicolson GL, Rao JS (1996) Differential expression of membrane-type matrix metalloproteinase and its correlation with gelatinase A activation in human malignant brain tumors in vivo and in vitro. Cancer Res 56: 384-392. [PubMed] [Google Scholar]
- Yong VW, Krekoski CA, Forsyth PA, Bell R, Edwards DR (1998) Matrix metalloproteinases and diseases of the CNS. Trends Neurosci 21: 75-80. [DOI] [PubMed] [Google Scholar]
- Yong VW, Power C, Forsyth P, Edwards DR (2001) Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci 2: 502-511. [DOI] [PMC free article] [PubMed] [Google Scholar]