

Abstract
Filamentous tau inclusions in neurons and glia are neuropathological hallmarks of sporadic and familial tauopathies. Because tau gene mutations are pathogenic for the autosomal dominant tauopathy “frontotemporal dementia and parkinsonism linked to chromosome 17,” tau abnormalities are implicated directly in the onset and/or progression of disease. Although filamentous tau aggregates are acknowledged to play roles in degenerative mechanisms resulting in neuron loss, the contributions of glial tau pathology to neurodegeneration remain essentially unexplored. To begin to elucidate the role of glial pathology in tauopathies, we generated a transgenic (Tg) mouse model of astrocytic tau pathology by expressing the human tau protein driven by the glial fibrillary acidic protein (GFAP) promoter. Whereas endogenous tau was not detected in astrocytes of control mice, in GFAP/tau Tg mice there was robust astrocytic tau expression that was associated with a redistribution of the GFAP network. Subsequently, there was an age-dependent accumulation of tau pathology in astrocytes that was Gallyas and variably thioflavine S positive as observed in many tauopathies. The tau pathology in these Tg mice was abnormally phosphorylated, ubiquitinated, and filamentous, and the emergence of this pathology coincided with accumulation of insoluble tau protein. Furthermore, in regions with robust astrocytic tau pathology, there was mild blood- brain barrier disruption, induction of low-molecular-weight heat shock proteins, and focal neuron degeneration. Thus, these Tg mice recapitulate key features of astrocytic pathology observed in human tauopathies and demonstrate functional consequences of this pathology including neuron degeneration in the absence of neuronal tau inclusions.
Keywords: tau protein, astrocytes, neurodegeneration, tauopathy, transgenic mice, blood- brain barrier
Introduction
Filamentous inclusions composed of the microtubule-associated protein (MAP) tau are defining pathological hallmarks of neurodegenerative diseases referred to as tauopathies, including Pick's disease (PiD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and Alzheimer's disease (AD) (Lee et al., 2001). The pathological description of tauopathies other than AD that manifest tau pathology in the absence of Aβ deposition provided indirect evidence for a causative role of tau in neurodegeneration (Feany and Dickson, 1996). However, the discovery of mutations in the tau gene in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) provided unequivocal confirmation of the central role of tau abnormalities in the pathogenesis of tauopathies (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998).
Tau is a low-molecular-weight MAP that is abundant in the CNS, where it is expressed predominantly in axons (Cleveland et al., 1977). Tau regulates the assembly and stability of microtubules (Weingarten et al., 1975; Cleveland et al., 1977), and this microtubule-binding function of tau is regulated negatively by phosphorylation (Drechsel et al., 1992; Biernat et al., 1993; Bramblett et al., 1993). However, tau protein in the filamentous inclusions is abnormally phosphorylated and highly insoluble (Lee et al., 2001).
In many tauopathies, the tau pathology occurs not only in neurons but also in astrocytes and oligodendrocytes (Nishimura et al., 1992; Feany and Dickson, 1995; Komori, 1999). This contrasts with normal CNS expression, wherein tau is present at low levels in glia (Shin et al., 1991; LoPresti et al., 1995). The distribution of tau pathology in astrocytes does not correlate with the pattern of neuron loss, suggesting that this pathology reflects an independent degenerative change rather than a reactive process (Togo and Dickson, 2002). Therefore, the expression or catabolism of tau protein is altered in glial cells by unknown mechanisms leading to the accumulation of aggregated tau. Whereas the role of tau pathology in neurons is mostly accepted, the role of the glial tau pathology in neurodegeneration is completely unknown. Thus, there are numerous transgenic (Tg) models of tau pathology in neurons, some of which recapitulate many of the features of tauopathies, including the age-dependent accumulation of filamentous tau pathology associated with behavioral abnormalities and variable neuron loss (Götz et al., 2004). In contrast, there are few models systems that address the specific contribution of glial tau pathology to the pathogenesis of tauopathies (Götz et al., 2001; Higuchi et al., 2002a; Lin et al., 2003). However, in all of these systems, there is tau expression and pathology in multiple cell lineages, thereby limiting insights into the pathobiology of tau pathology, specifically in astrocytes.
To study the specific contribution of astrocytic tau pathology to the pathogenesis of tauopathies, we developed a Tg mouse model of tau pathology restricted to astrocytes. These Tg mice express the T34 human tau isoform (Goedert and Jakes, 1990) under the control of the astrocyte-specific glial fibrillary acidic protein (GFAP) promoter, resulting in an age-dependent accumulation of tau pathology in astrocytes that recapitulates the astrocytic pathology observed in human tauopathies. Furthermore, functional consequences of the astrocytic tau pathology are described.
Materials and Methods
Generation of GFAP/tau Tg mice. A wild-type tau cDNA construct containing the T34 human tau isoform (Goedert and Jakes, 1990) was subcloned into the eukaryotic expression vector pGfa2-expression vector at the BamHI restriction site that uses the astrocyte-specific GFAP promoter (Brenner et al., 1994). T34, one of the most abundant tau isoforms, contains both exon 2, the first of two alternatively spliced N-terminal exons, and exon 10, encoding for the second of four microtubule-binding repeats that is also alternatively spliced (Goedert and Jakes, 1990). Linearized constructs were microinjected into fertilized C57BL/6 × C3H (B6C3/F1) mouse embryos and then implanted into pseudopregnant females. Genomic DNA was isolated from mouse tails of pups with the Puregene DNA Isolation kit (Gentra Systems, Minneapolis, MN), and potential founders were identified by Southern blotting with 32P-labeled T34 cDNA probes. Candidate founder lines were backcrossed to non-Tg mice to establish germline transmission. Human tau heterozygous mice were bred subsequently to homozygosity, and non-Tg littermates were inbred and maintained as control mice.
Histochemical and immunohistochemical analysis. Tg and non-Tg mice were anesthetized lethally by intraperitoneal injection of ketamine hydrochloride (1 mg/10 g) and xylazine (0.1 mg/10 g) and perfused intracardially with 15 ml of PBS followed by 15 ml of 10% neutral buffered formalin or 70% ethanol in isotonic saline in accordance with protocols approved by the University of Pennsylvania. The brains and spinal cords were removed and fixed for an additional 24 h, processed by sequential dehydration in ethanol, paraffin-embedded, and cut into 6-μm-thick sections. Sections were stained with Gallyas silver, thioflavine S, and hematoxylin/eosin stains using standard protocols. For immunohistochemical analysis, sections were rehydrated and endogenous peroxide blocked by incubation with methanol/hydrogen peroxide. Sections were blocked with 2% fetal bovine serum in 50 mm Tris, pH 7.4, 150 mm NaCl, and incubated with primary antibody (Table 1) overnight at 4°C. Subsequently, sections were incubated with horseradish peroxidase-conjugated anti-Ig antibodies (Vector Laboratories, Burlingame, CA) followed by visualization with the avidin-biotin peroxidase method with 3,3′-diaminobenzidine as chromagen (Vectastain ABC kit; Vector Laboratories).
Table 1.
Antibodies
|
Antibody |
Source |
Specificity |
Reference/source |
|---|---|---|---|
| Ntau | Rabbit | Tau; 1-12 | Dabir et al., 2004 |
| OT12 | Mouse | Tau; human specific, exon 2 | V.M.-Y. Lee, unpublished observation |
| T14 | Mouse | Tau; human specific, 141-178 | Kosik et al., 1988 |
| AT8 | Mouse | Tau; pSer202/pThr205 | Goedert et al., 1993 |
| AT10 | Mouse | Tau; pThr212/pSer214 | Mercken et al., 1992 |
| PHF6 | Mouse | Tau; pThr231 | Hoffmann et al., 1997 |
| PHF41 | Mouse | Tau; pThr231 | Hoffmann et al., 1997 |
| PHF1 | Mouse | Ptau; pSer396/pSer404 | Greenberg and Davies, 1990 |
| T46 | Mouse | Pl tau; 404-441 | Kosik et al., 1988 |
| 17026 | Rabbit | Recombinant tau | Ishihara et al., 1999 |
| T49 | Mouse | Pl murine tau | Mawal-Dewan et al., 1994 |
| GFAP | Rabbit | GFAP | DakoCytomation (Carpinteria, CA) |
| RM024 | Mouse | NFH; multiphosphorylation repeats | Lee et al., 1988 |
| RM0189 | Mouse | NFM; multiphosphorylation repeats | Lee et al., 1988 |
| RM0217 | Mouse | NFH; multiphosphorylation repeats | Lee et al., 1988 |
| 1510 | Mouse | Ubiquitin | Chemicon (Temecula, CA) |
| Hsp27 | Goat | Hsp27 | StressGen Biotechnologies (Victoria, British Columbia, Canada) |
| αB-crystallin | Rabbit | αB-crystallin | StressGen Biotechnologies |
| Mouse lg | Rabbit | Mouse lg | Jackson ImmunoResearch |
| Albumin | Sheep | Albumin | Biogenesis (Sandown, NH) |
| NeuN |
Mouse |
NeuN |
Chemicon |
Pl, Phosphorylation independent; NFH, neurofilament heavy molecular weight; NFM, neurofilament middle molecular weight.
Double-labeling immunofluorescence studies were performed by coincubating sections with antibodies as indicated (Table 1). After extensive washes, sections were labeled using Alexa Fluor 488- and 594-conjugated secondary antibodies (Molecular Probes, Eugene, OR), washed, and coverslipped with Vectashield 4′,6′-diamidino-2-phenylindole mounting medium (Vector Laboratories). The sections were viewed with an Olympus (Melville, NY) PX51 microscope equipped with bright-field and fluorescence light sources. Both bright-field and fluorescent images were obtained from the same field using a ProGres C14 Jenoptik camera (Laser Optik Systeme, Mainz, Germany).
Biochemical analysis of tau expression and solubility. The cerebral hemispheres, brainstem including cerebellum, and spinal cord were dissected from mice after they were anesthetized lethally in accordance with protocols approved by the University of Pennsylvania. The tissue samples were homogenized in 2 ml/g wet tissue in high-salt Tris-buffered saline (HS-TBS; composed of the following: 50 mm Tris, pH 7.6, 750 mm NaCl, 1 mm EGTA, 0.5 mm MgS04, 20 mm NaF, 100 μm EDTA) supplemented with protease inhibitors (1 mm PMSF and 100 μg/ml each of pepstatin A, leupeptin, soybean trypsin inhibitor, N-tosyl-l-phenylalanyl chloromethyl ketone, and N-tosyl-lysine chloromethyl ketone) and centrifuged at 100,000 × g for 30 min at 4°C. The supernatant was boiled for 5 min and centrifuged at 15,000 × g for 20 min at 4°C. The resulting supernatant contains the soluble tau fraction. To remove myelin and associated lipids, the insoluble pellets were reextracted with 1 m sucrose in HS-TBS and centrifuged at 100,000 × g for 30 min at 4°C. The resulting pellets were homogenized in radioimmunoprecipitation assay buffer (composed of the following: 50 mm Tris, pH 8.0, 150 mm NaCl, 5 mm EDTA, 0.5% sodium deoxycholate, 1% NP-40, 0.1% SDS supplemented with protease inhibitors) at 2 ml/g starting tissue and centrifuged as before. The supernatant were saved as the detergent-soluble samples, which contained only low levels of tau protein. The detergent-insoluble pellets were extracted with 70% formic acid (FA) at 1 ml/g starting wet tissue and disrupted with sonication. FA was evaporated in an Automatic Environmental Speed-Vac system (Savant Instruments, Holbrook, NY). The dried pellets were resuspended in SDS sample buffer at a concentration of 1 ml/g starting wet tissue. Where indicated, tau was dephosphorylated by treatment with Escherichia coli alkaline phosphatase (Sigma, St. Louis, MO) at 67°C for 1 h. For Western blot analysis, nitrocellulose replicas were prepared from 7.5% SDS polyacrylamide slab gels containing either the soluble, detergent-soluble or -insoluble (FA) tau extracts and probed with a panel of anti-tau antibodies (Table 1). Primary antibodies were detected with horseradish peroxidase-conjugated anti-mouse and antirabbit IgG, respectively (Jackson ImmunoResearch, West Grove, PA). Immunoreactive proteins were revealed using ECL chemiluminescence (NEN, Boston, MA). Quantitative Western blot analysis was performed using either T46 or 17026 followed by 1 μCi/ml I125-labeled goat anti-mouse or goat anti-rabbit Ig, respectively (NEN) as secondary antibodies. The radiolabeled blots were exposed to PhosphorImager plates, and the protein bands were visualized and quantified with ImageQuant software using endogenous tau protein expression as an internal standard (Molecular Dynamics, Sunnyvale, CA).
Ultrastructural analysis. Transmission electron microscopy (EM) was performed for the Tg and control mice at 20-24 months of age (n = 3). These mice were anesthetized deeply and perfused intracardially with 10 ml of 0.1 m cacodylate buffer, pH 7.4, containing 0.05% glutaraldehyde and 0.5% paraformaldehyde, followed by 50 ml of 0.1 m cacodylate buffer containing 2% glutaraldehyde and 2% paraformaldehyde. The brain and spinal cord were removed, fixed in 2% glutaraldehyde/2% paraformaldehyde overnight, and postfixed in 2% osmium tetroxide for 60 min at 4°C. After dehydration with graded alcohol and propylene oxide, the tissues were embedded in Epon-812 and polymerized at 60°C for 72 h. Sections (65 nm thin) were cut and mounted on 200 mesh copper grids. The sections were stained with 1% uranyl acetate in 50% ethanol and bismuth subnitrate and examined with a JEM1010 electron microscope (Jeol, Peabody, MA) at 80 kV.
Preembedding immuno-EM was performed on 20- to 24-month-old Tg and control mice (n = 2 per group). The mice were anesthetized deeply and perfused intracardially with 10 ml of 0.05% glutaraldehyde, 0.5% paraformaldehyde in 0.1 m cacodylate buffer, pH 7.4, followed by 50 ml of 0.2% glutaraldehyde, 2% paraformaldehyde in 0.1 m cacodylate buffer. The brains and spinal cords were removed and postfixed with 4% paraformaldehyde, 0.2% glutaraldehyde, 0.2% picric acid in 0.1 m cacodylate buffer overnight. Representative regions from the brainstem and spinal cord were dissected and cut into 50-μm-thick vibratome sections. Sections were quenched in 0.1% sodium borohydride in TBS for 10 min followed by 20% ethanol for 10 min. Nonspecific staining was blocked with 5% donor horse serum in PBS with 0.1% cold-water fish skin gelatin and 1% chicken egg albumin for 60 min, and sections were incubated subsequently with primary antibody in 0.1% bovine serum albumin/PBS overnight at 4°C. Sections were washed and incubated with biotinylated secondary antibodies for 2 h at room temperature followed by visualization with diaminobenzidine and silver-gold intensification using silver methenamine developer containing 3% methenamine, 5% silver nitrate, and 1% sodium tetraborate at 60°C for 10 min as described previously (Ishihara et al., 2001). The reaction was stopped with 2% sodium acetate and then stabilized in 3% sodium thiosulfate for 5 min. Gold toning was performed by incubating the sections in 0.1% gold chloride for 5 min, followed by stabilizing with 3% sodium thiosulfate for 5 min and overnight fixation with 2% glutaraldehyde in PBS buffer. Sections with positive staining were postfixed in 0.5% osmium tetroxide for 20 min at 4°C, dehydrated with graded series of ethanols, and embedded in Epon-Araldite resin at 60°C for 48 h. Ultrathin sections of these blocks were cut and mounted on 100 mesh EM grids and examined using a JEM1010 electron microscope at 80 kV.
Results
Generation of Tg mice that express human tau specifically in astrocytes
Two founder lines (lines 4 and 7) of GFAP/tau Tg mice were obtained that expressed the human tau gene driven by the GFAP promoter stably with germline transmission, and both lines were bred to homozygosity (Fig. 1). Regional tau protein expression from cortex, brainstem, and spinal cord was determined by immunoblot analysis of the soluble protein extract from 2-month-old mice. In both line 4 and line 7, tau expression levels was region dependent, with the highest levels consistently observed in the spinal cord (Fig. 1A). The expression of human tau protein did not affect the expression of endogenous mouse tau (Fig. 1B), and the relative overexpression of human tau protein versus murine tau is depicted in Figure 1, C and D. Quantitative Western blot analysis indicated an increase of ∼25, 50, and 100% of total tau protein expression in cerebral cortex, brainstem, and spinal cord, respectively, relative to endogenous mouse tau. The relative overexpression of human tau compared with endogenous mouse tau levels was consistently highest in the spinal cord of both lines as a result of the lower levels of endogenous tau in the spinal cord and the enigmatic increased human tau expression in the spinal cord driven by the GFAP promoter. Dephosphorylation of the extracted tau protein with alkaline phosphatase demonstrated a unique tau protein isoform in the GFAP/tau Tg versus non-Tg mice that comigrated with recombinant T34 human tau (Fig. 1D, arrow). Because both GFAP/tau Tg lines showed comparable tau expression, the majority of the studies to follow, except where indicated, were performed using line 4. However, both line 4 and line 7 showed comparable results.
Figure 1.

Expression of human tau in GFAP/tau Tg mice. A-C, Western blot analysis of HS-TBS-soluble proteins (10 μg per lane) extracted from cortex, brainstem (BS), and spinal cord (SC) samples of 2-month-old GFAP/tau Tg and control mice [non-Tg (NTg)]. Immunoblots were detected with antibodies that detect human tau only (T14; A), murine tau only (T49; B), or both human and murine tau (T46; C). D, Western blot analysis of native (-) and dephosphorylated (+) HS-TBS-soluble proteins extracted from brainstem and probed with T46 that recognizes both mouse and human tau. Arrows in C and D indicate migration of T34 human tau that is specifically expressed in GFAP/tau Tg mice. Molecular weight standards are indicated to the left of each panel. WT, Wild type; DeP, dephosphorylated.
Human tau expression in astrocytes is associated with altered GFAP expression
To analyze the cell-type-specific tau expression in the GFAP/tau Tg mice, an immunohistochemical analysis was performed on 6-month-old Tg and control mice using the human-specific monoclonal antibody (MAb) OT12 (Fig. 2). There was robust tau expression throughout the brain and spinal cord in the GFAP/tau Tg mice, predominantly in areas of gray matter (Fig. 2E-H). High magnification revealed tau-positive cells with morphology of protoplasmic astrocytes (Fig. 2E, inset). Furthermore, consistent with the biochemistry data above (Fig. 1), tau staining was more robust in spinal cord relative to cerebral cortex, subcortical nuclei, and brainstem regions. In contrast, in non-Tg mice, there was no immunoreactivity with human tau-specific antibodies (Fig. 2A-D), whereas antibodies that detect human or murine tau revealed diffuse weak neuropil immunoreactivity consistent with axonal staining of endogenous mouse tau (data not shown). To confirm the astrocyte-specific tau expression, two-color immunofluorescent staining was performed with antibodies specific for tau and GFAP (Fig. 2). In non-Tg mice, there was no detectable tau immmunostaining using a human-specific anti-tau antibody and GFAP staining preferentially in white matter (Fig. 2I-K). In contrast, human tau protein colocalized in GFAP-positive astrocytes, predominantly within gray matter in the GFAP/tau Tg mice (Fig. 2L-Q). These astrocytes resemble reactive astrocytes observed in many pathological conditions of the CNS. Thus, the expression of tau protein in astrocytes leads to the altered expression of GFAP, a change that was observed at all ages analyzed from 1 to 24 months of age (data not shown), although this did not correspond to increased total GFAP by Western blot analysis (data not shown). This change in GFAP staining may reflect a redistribution of the intermediate filament network similar to that described with the overexpression of tau protein in primary rat astrocyte cultures (Yoshiyama et al., 2003).
Figure 2.
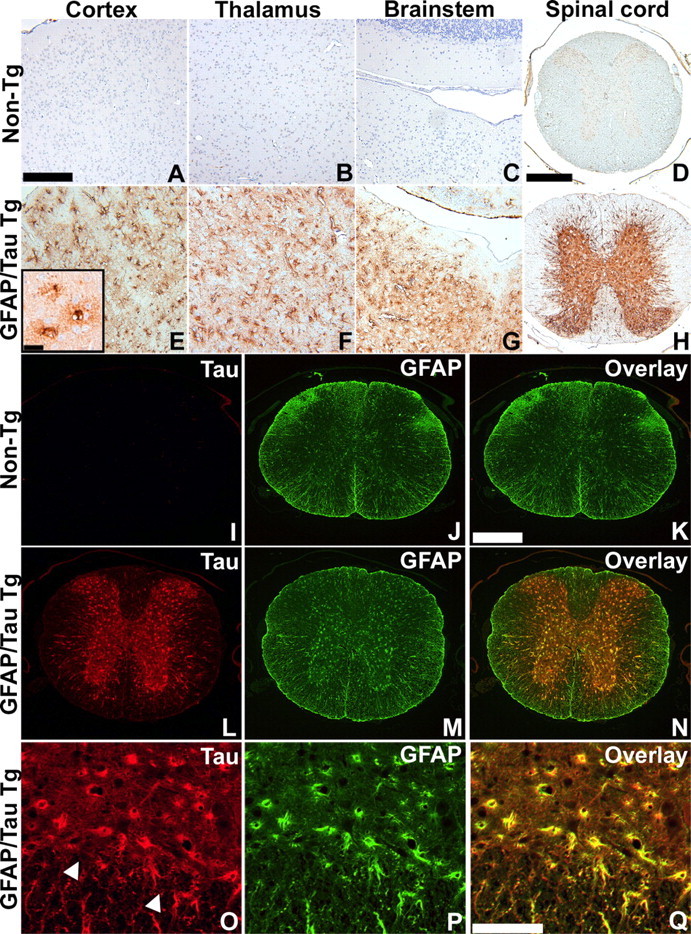
Astrocyte-specific regional tau expression in GFAP/tau Tg mice. A-H, Immunohistochemistry was performed with the human tau-specific MAb OT12 on 6-month-old non-Tg (A-D) and GFAP/tau Tg line 4 (E-H) mice from the CNS regions as indicated. Consistent with the biochemical analysis (Fig. 1), the highest levels of expression were observed consistently in the gray matter of the spinal cord. The inset in E shows morphology of cortical astrocytes at high magnification. Scale bars: (in A) A-C, E-G, 200 μm; (in D) D, H, 400 μm; E, inset, 25 μm. I-Q, Spinal cord sections from 6-month-old non-Tg (I-K) and GFAP/tau Tg, line 4 (L-Q) mice were double-labeled with OT12 (green; I, L, O) and GFAP (red; J, M, P). Merged images are depicted in K, N, and Q. Prominent GFAP staining is observed in astrocytes within the gray matter of Tg mice that is not detected in non-Tg animals. Arrowheads in O indicate the junction of the gray and white matter in the anterior horn of the spinal cord. Scale bars: (in K) I-N, 400 μm; (in Q) O-Q, 100 μm.
Tau pathologies in GFAP/tau Tg mice resembles astrocytic pathology in human tauopathies
Human tauopathies such as PSP and CBD are characterized by the accumulation of abnormally phosphorylated and biochemically insoluble tau protein in astrocytes, oligodendrocytes, and neurons (Feany and Dickson, 1996; Lee et al., 2001). To further characterize the GFAP/tau Tg mice, immunostaining was performed on mice from 6 to 24 months of age with antibodies that detect recombinant tau as well as phosphorylation-dependent tau epitopes characteristic of the human diseases (Figs. 3, 4). Whereas there was little change in staining pattern of young and old mice with antibodies generated to recombinant tau (Fig. 3A-C), there was an age-dependent increase in expression of phosphorylation-dependent tau epitopes in GFAP/tau Tg mice (Figs. 3D-F, 4). These phosphoepitopes epitopes were first detected in brainstem and spinal cord of ∼30% of Tg mice by 12 months of age, and by 18 months, the phosphoepitopes were observed in >90% of animals in brainstem, spinal cord, and subcortical nuclei, especially thalamus. By 24 months of age, there was extensive tau pathology in all animals throughout these same regions, as well as cortical pathology in a subset of animals (Fig. 5). Of note, the astrocytic tau pathology was often asymmetric in distribution, similar to that described in many of the tauopathies and in particular CBD (see Fig. 8). And at high magnification, the astrocytic tau pathology resembled the tufted astrocytes, astrocytic plaques, and thread pathology observed in the human tauopathies (Fig. 3G-L). Furthermore, the detection of pathological tau phosphoepitopes coincided with positive Gallyas silver impregnation stains (Figs. 4F, 5E-H) (Ikeda et al., 1994; Takahashi et al., 1996). A subset of these tau pathologies were both ubiquitin positive (Fig. 4E) and stained by thioflavine S, a histochemical dye that binds amyloid fibrils (Fig. 4G-I). Thus, the GFAP/tau Tg mice develop an age-dependent accumulation of tau pathology in astrocytes that histochemically and immunohistochemically resembled that observed in tauopathies.
Figure 3.
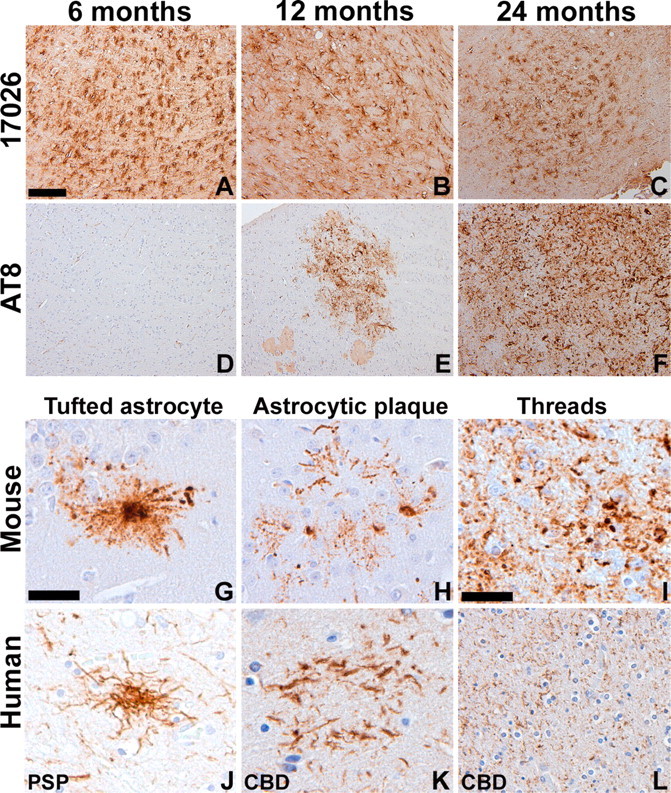
Accumulation of tau pathology in aged GFAP/tau Tg mice that resembles astrocytic pathology in human tauopathies. A-F, Immunohistochemical analysis was performed on sections of medulla from 6-month-old (A, D), 12-month-old (B, E), and 24-month-old (C, F) GFAP/tau Tg mice, line 4, as indicated with antibodies to recombinant tau (17026; A-C) or tau phosphorylated at Ser202 and Thr205 (AT8; D-F). There is an age-dependent accumulation of phosphorylated tau epitopes detected in GFAP/tau Tg mice. G-L, High-power photomicrographs of astrocytic pathology of Tg mice (G-I) and human tauopathies (J-L). The astrocytic pathology in the Tg mice resembles the tufted astrocytes (G, J), astrocytic plaques (H, K), and thread pathology (I, L) observed in tauopathies such as PSP and CBD. Scale bars: (in A) A-F, 200 μm; (in G) G, H, J, K, 40 μm; (in I) I, L, 80 μm.
Figure 4.
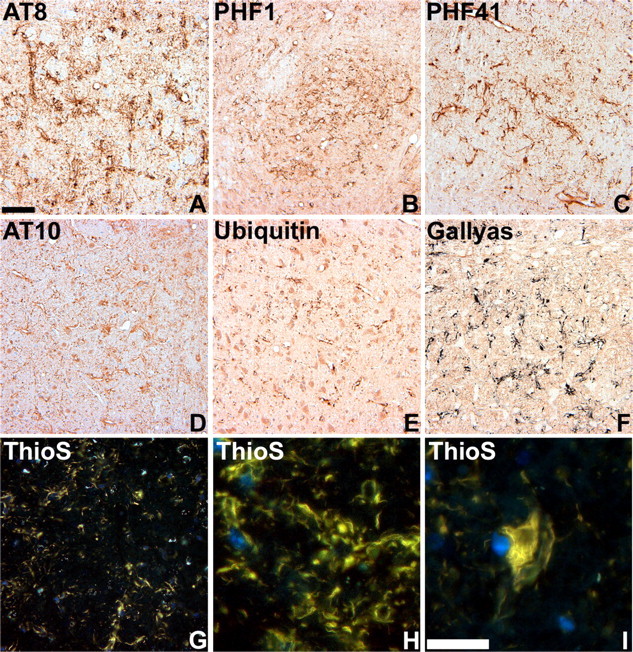
Characterization of tau pathology in GFAP/tau Tg mice. A-E, Immunohistochemical analysis was performed on sections of pons (A, B) and thalamus (C-E) from 22-month-old GFAP/tau Tg mice, line 4 with a panel of antibodies to distinct phosphorylated tau epitopes (A-D) and ubiquitin, as indicated. Ubiquitin (E) is detected only in a subset of tau pathologies. F-I, Gallyas silver impregnation (F) and thioflavine S (ThioS) (G-I) staining of astrocytic tau pathology in brainstem of 22-month-old GFAP/tau Tg mice. There is robust Gallyas-positive tau pathology in both astrocytic processes and cell soma. The thioflavine S stain detects only a subset of the pathology observed with antibodies to tau phosphoepitopes or Gallyas stains. Scale bars: (in A) A-G, 80 μm; (in I) H, I, 25 μm.
Figure 5.
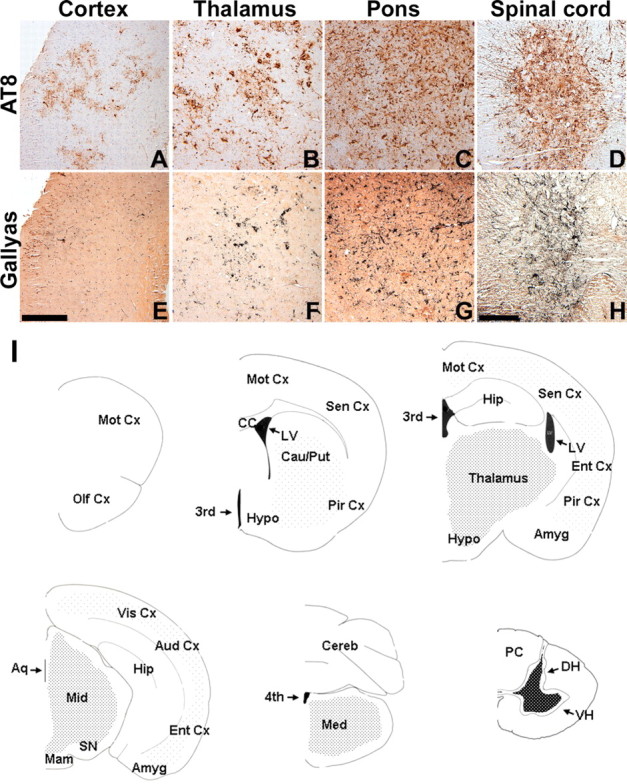
Distribution of tau pathology in GFAP/tau Tg mice. A-H, Immunohistochemical analysis with AT8 (A-D) and Gallyas silver impregnation stain (E-H) of CNS regions indicated from 24-month-old GFAP/tau Tg mouse. Robust pathology was observed consistently in the spinal cord, brainstem, and thalamus, whereas cortical pathology was highly variable. Scale bars: (in E) A-C, E-G, 100 μm; (in H) D, H, 200 μm. I, Diagrammatic summary of astrocytic tau pathology shown as coronal sections of the mouse neuroaxis. The density of dots corresponds to frequency and quantity of observed pathology in aged Tg mice. Amyg, Amygdala; Aq, aqueduct; Aud Cx, auditory cortex; Cau/Put, caudate-putamen; CC, corpus callosum; Cereb, cerebellum; DH, dorsal horn; Ent Cx, entorhinal cortex; Hip, hippocampus; Hypo, hypothalamus; LV, lateral ventricle; Mam, mamillary body; Med, medulla; Mid, midbrain; Mot Cx, motor cortex; Olf Cx, olfactory cortex; PC, posterior column; Pir Cx, piriform cortex; Sen Cx, sensory cortex; SN, substantia nigra; Vis Cx, visual cortex; VH, ventral horn; third, third ventricle; fourth, fourth ventricle.
Figure 8.
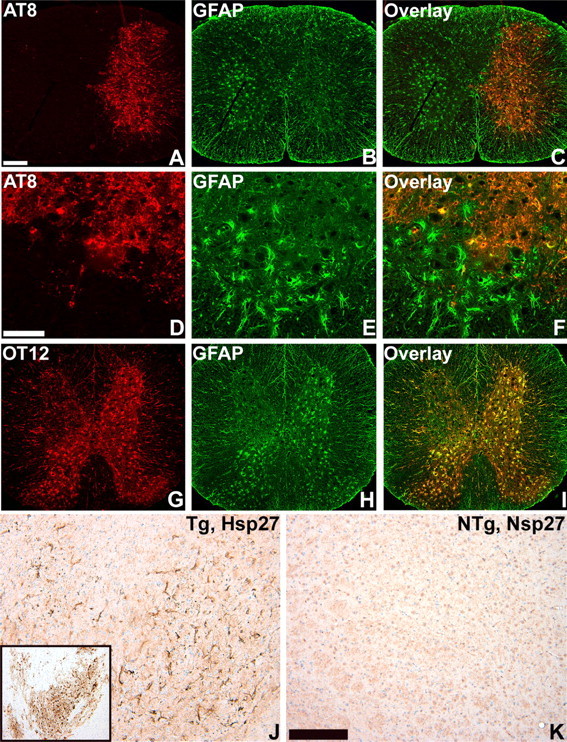
Phenotypic changes associated with tau pathology in astrocytes. A-I, Two-color immunofluorescence staining of spinal cord from 24-month-old GFAP/tau Tg mice. OT12, which is specific for exon 2 of human tau, colocalizes with GFAP (G-I). In contrast, in regions with AT8-positive tau pathology in astrocytes, there is a relative reduction of stainable GFAP (A-F). J, K, Immunohistochemistry for hsp27, from the thalamus of 24-month-old GFAP/tau Tg and non-Tg (NTg) mice. Tau pathology (J, inset) is associated with increased staining of hsp27. Scale bars: (in A) A-C, G-I, 400 μm; (in D) D-F, 100 μm; (in K) J, K, 200 mm.
Because aggregated pathological tau in human tauopathies is biochemically insoluble (Lee et al., 2001), we assessed the solubility of the tau in our GFAP/tau Tg mice by sequential extraction of CNS tissue with buffers of increasing protein solubilization strength followed by Western blot analysis of soluble (Fig. 6A) and insoluble FA-extractable tau proteins (Fig. 6B-D). Specifically, cortex, brainstem, and spinal cord extracts were analyzed from pairs of GFAP/tau Tg and non-Tg mice at 6, 12, 18, and 24 months of age. Soluble extracts from the brain and spinal cord showed only mild and variable changes in tau expression with aging (Fig. 6A and data not shown). However, there was a progressive accumulation of insoluble and heavily aggregated tau protein in the brainstem and spinal cord of GFAP/tau Tg mice (Fig. 6). There was little insoluble tau in cortex, consistent with the low and variable cortical tau pathology detected by immunohistochemistry. In non-Tg mice, endogenous tau protein was not detected in the insoluble fraction of brain and spinal cord (Fig. 6C and data not shown). Furthermore, insoluble tau was abnormally phosphorylated similar to human tauopathies (Fig. 6D) (Lee et al., 2001). Finally, the insoluble tau protein was detected with human tau-specific antibodies (T14) but not antibodies that detect only rodent tau (T49), indicating that the astrocytic tau pathology is composed exclusively of human tau (Fig. 6D), which presumably reflects low levels of endogenous tau expression in murine astrocytes (Couchie et al., 1985, 1988).
Figure 6.

Age-dependent accumulation of insoluble, aggregated, and phosphorylated tau protein in GFAP/tau Tg mice. Cortex (Cx), brainstem (BS), and spinal cord (SC) from pairs of non-Tg (NTg) and GFAP/tau Tg mice at 6, 12, 18, and 24 months of age were sequentially extracted as described in Materials and Methods. A, Immunoblot analysis of 10 μg of soluble protein extracted from the brainstem of GFAP/tau Tg mice at the age indicated and detected with the human-specific anti-tau MAb T14. There is only mild and variable change in tau protein expression with increasing age. B, Immunoblot analysis of insoluble tau corresponding to 25 mg of starting wet tissue weight extracted from GFAP/tau Tg mice at the age indicated and detected with the human tau-specific MAb T14. C, Immunoblot analysis of insoluble tau corresponding to 25 mg of starting wet tissue weight extracted from the spinal cord of non-Tg and GFAP/tau Tg mice at the age indicated and detected with polyclonal antibody 17026. Insoluble and heavily aggregated tau is detected in brainstem and spinal cord by 18 months of age. Insoluble tau is not detected in non-Tg mice. Arrowheads indicate nonspecific bands that are not recognized by other tau antibodies. D, Immunoblot analysis of insoluble tau extracted from cortex, brainstem, and spinal cord of 24-month-old GFAP/tau Tg mice and detected with a panel of tau-specific MAb as indicated. In contrast to B and C above, samples corresponding to only 5 mg starting wet tissue weight were loaded on the gels to reduce the smearing associated with heavily aggregated, insoluble tau protein, which facilitated the visualization of the predominant tau protein band at ∼66 kDa. Insoluble tau is phosphorylated at multiple sites (PH1, AT8, and PHF6) and is composed of human tau only (compare human tau-specific antibody T14 to murine tau-specific antibody T49). The position of molecular weight markers are depicted on the left. WT, Wild type.
Formation of tau filaments in astrocytes of aged GFAP/tau Tg mice
Ultrastructurally, tau inclusions in astrocytes of human tauopathies exhibit various morphologies ranging from poorly formed fibrils to straight tubules with a diameter of 15-20 nm (Komori, 1999). To determine whether the astrocytic tau pathology in the GFAP/tau Tg mice was filamentous, immuno-EM was performed with antibodies specific for tau phosphoepitopes (Fig. 7). In the thalamus, brainstem, and spinal cord of 20- to 24-month-old Tg mice, we detected accumulations of tau-positive fibrils ranging from 15 to 25 nm in diameter irregularly arranged in the soma and processes of cells with the morphology of astrocytes (Fig. 7A-D). These filaments were composed of either straight fibrils or filaments with a subtle twist; however, AD-like paired helical filaments were not identified. Furthermore, the fibrils did not form large bundles as is typical of the neuronal and oligodendroglial pathology in tauopathies (Komori, 1999). In contrast, the coarsely bundled astrocyte-specific intermediate filaments were distinguished easily from the tau filaments and labeled avidly with antibodies to GFAP but were nonreactive with a panel of anti-tau antibodies (Fig. 7E,F).
Figure 7.
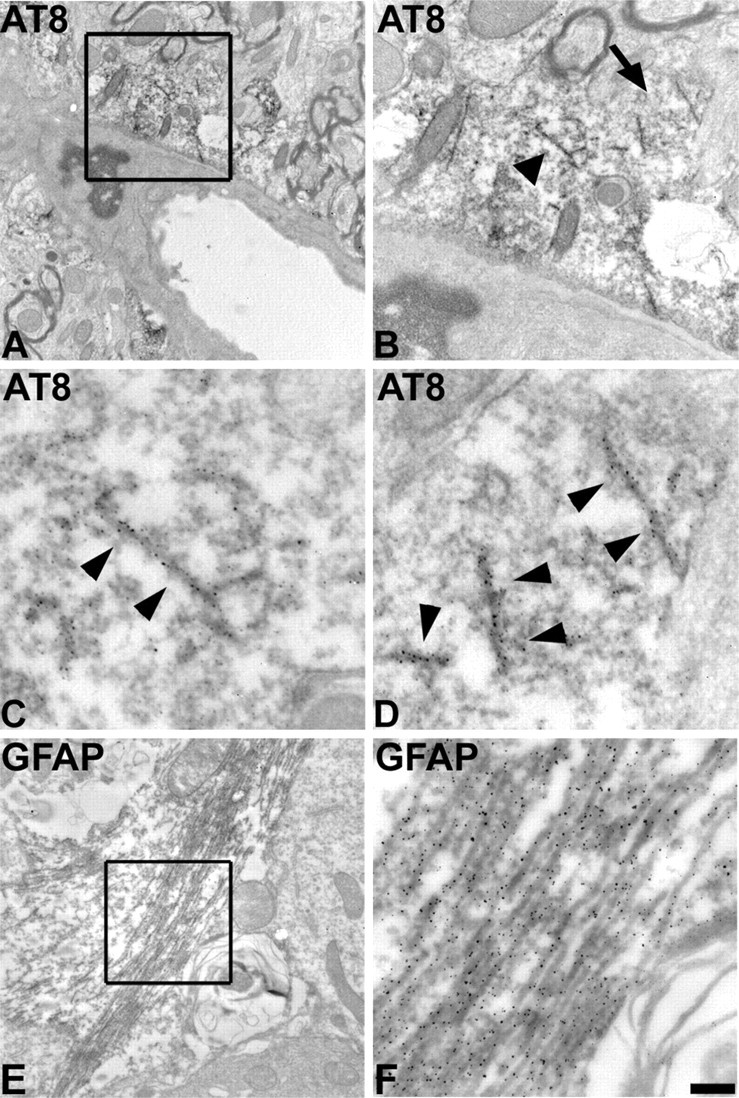
Immuno-EM demonstrates filamentous tau accumulation in astrocytes of aged GFAP/tau Tg mice. A, Immuno-EM of filamentous inclusion in spinal cord gray matter of a 22-month-old GFAP/tau Tg mouse labeled with AT8. B, High-power photomicrograph of the boxed area in A showing tau-positive filaments. C, D, High-power photomicrographs of the tau-positive filaments depicted by the arrowhead (C) and the arrow (D) in B. Arrowheads in C andD indicate filaments that are immunolabeled with the AT8. E, Immuno-EM of GFAP-positive filaments in astrocytes of spinal cord from same mouse depicted in A-D. F, High-power photomicrograph of the boxed area in E showing coarsely bundled GFAP-positive intermediate filaments. Scale bar: (in F) A, 1.25 mm; B, E, 500 nm; C, D, F, 100 nm.
Tau pathology induces phenotypic changes in astrocytes
In neurodegenerative diseases, tau pathology is associated with reactive changes in affected brain regions including gliosis and expression of specific heat shock proteins (HSPs) such as hsp70 in neurons (Dou et al., 2003) and the low-molecular-weight HSPs hsp27 and αB-crystallin in astrocytes and oligodendrocytes (Dabir et al., 2004). To address specifically the role of the astrocytic tau pathology in these phenotypic changes, we analyzed the GFAP/tau Tg mice before and after the development of astrocytic tau inclusions. As described above, the initial expression of tau in astrocytes before the onset of pathological inclusions was associated with an increase in the somatic expression of GFAP (Figs. 2, 8). In contrast, the development of pathological tau inclusions resulted in a redistribution of stainable GFAP in affected astrocytes (Fig. 8). This was associated with a remarkable masking of tau epitopes such as exon 2 (Fig. 8) and the N terminus (data not shown) that presumably was caused by the altered folding and/or fibrillization of the tau protein (Fig. 8). Furthermore, tauopathies with prominent glial pathology are associated with the induction of the low-molecular-weight HSPs such as hsp27 and αB-crystallin (Dabir et al., 2004). We detected a prominent increase in staining of hsp27 (Fig. 8), and, to a lesser extent, αB-crystallin (data not shown), in regions with astrocytic tau pathology that colocalized with phosphorylated tau epitopes (data not shown). In contrast, there were no changes in the staining pattern of hsp70 that is associated with the formation of insoluble tau protein in neurons (Dou et al., 2003) but not glia (data not shown) (Dabir et al., 2004). Thus, tau protein expression in astrocytes leads to two distinct stages in the astrocytic response. Initially, there is an alteration of the intermediate filament network with an increase in immunohistochemically detectable GFAP. This is followed by the accumulation of phospho-tau epitopes, which coincides with the detection of insoluble tau aggregates, a second alteration in the GFAP network and the induction of low-molecular-weight heat shock proteins.
Astrocytic tau pathology is associated with mild disruption of the blood- brain barrier
Astrocytes now are known to play roles in response to CNS injury, maintenance of the blood- brain barrier (BBB), immune modulation, and modulation of synaptic activity (Ransom et al., 2003). We examined the GFAP/tau Tg mice for impairments in BBB function because we observed frequent pathology in astrocytic end feet that abut endothelial cells to form the BBB (Fig. 9). There was evidence of mild BBB disruption by staining brain and spinal cord sections from GFAP/tau Tg animals for serum proteins including mouse immunoglobulin and albumin before and after the development of pathology. For example, in non-Tg mice of all ages and GFAP/tau Tg mice before the development of tau pathology, there was no extravascular immunoglobulin or albumin detected. In contrast, in brain regions with robust tau pathology, we detected low levels of murine immunoglobulin (Fig. 9) and albumin (data not shown) in the brain parenchyma, typically associated with cell soma. Although an ultrastructural analysis of aged GFAP/tau Tg mice did not reveal overt alterations of the astrocyte foot processes and endothelial cell tight junctions (data not shown), these findings suggest the pathology causes a subtle alteration in astrocyte structure and/or function that leads to a mild disruption of the BBB. Similar alterations in the BBB are seen occasionally in the tauopathies, which over the course of the disease, might contribute to the process of neurodegeneration (Claudio, 1996; Ujiie et al., 2003).
Figure 9.
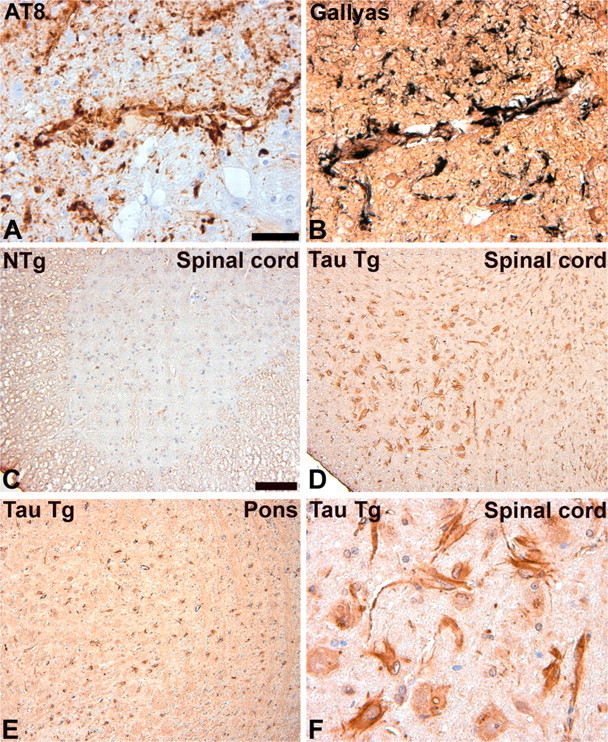
Astrocytic tau pathology is associated with mild BBB disruption. Histochemical analysis of 24-month-old GFAP/tau Tg (A, B, D-F) and non-Tg (NTg; C). A, B, There is prominent tau pathology in astrocytic foot processing surrounding blood vessels detected by both immunostaining with AT8 (A) and Gallyas silver stains (B). C-F, The perivascular astrocytic tau pathology is associated with amild disruption of the BBB. Immunohistochemistry performed on the indicated brain regions with prominent AT8-positive tau pathology showed focal somatic and perivascular staining with a polyclonal rabbit anti-mouse IgG antibody (D-F). No staining was detectable with age-matched control non-Tg mice (C) or in brain regions of Tg mice without tau pathology (data not shown). Scale bars: (in A) A, B, F, 40 μm; (in C) C-E, 100 μm.
Neuron degeneration associated with astrocytic tau pathology
To assess the impact of the astrocytic tau pathology on neurons and their processes, we performed immunohistochemistry on young and aged GFAP/tau Tg and non-Tg mice with panel of antibodies to phosphorylated neurofilament (NF) epitopes. Phosphorylated NF is normally expressed in axons, and detection of these epitopes in neuronal perikarya is indicative of neuronal injury (Julien and Mushynski, 1998). In aged non-Tg mice, all antibodies to phosphorylated NFs stained axons, whereas the cell bodies were nonreactive (Fig. 10). In contrast, in regions with abundant astrocytic tau pathology, there were frequent neurons that were stained with multiple antibodies to these phosphorylated NF epitopes, thereby indicating neuronal injury, features similar to that observed in ballooned neurons characteristic of a variety of neurodegenerative diseases including CBD (Dickson et al., 1986). To further characterize the changes in neurons, we performed transmission EM on Tg and non-Tg mice. In aged GFAP/tau Tg mice (>20 months of age), we observed frequent axonal degeneration with disruption of the associated myelin (Fig. 10). Occasional axons showed well circumscribed inclusions composed of amorphous granular material that on immuno-EM were negative for antibodies to tau (Fig. 10H). In addition, there were numerous degenerative foci with myelin breakdown, macrophage infiltration, and electron-dense bodies. These changes were not observed in Tg mice <12 months of age or in agematched non-Tg animals. To determine whether the neuronal degeneration was associated with neuron loss, neuronal-specific nuclear protein (NeuN)-positive neurons were quantified in the spinal cord of aged (20-24 months of age) Tg and non-Tg mice. However, there was no statistically significant difference in the number of NeuN-positive neurons between the two groups (data not shown).
Figure 10.
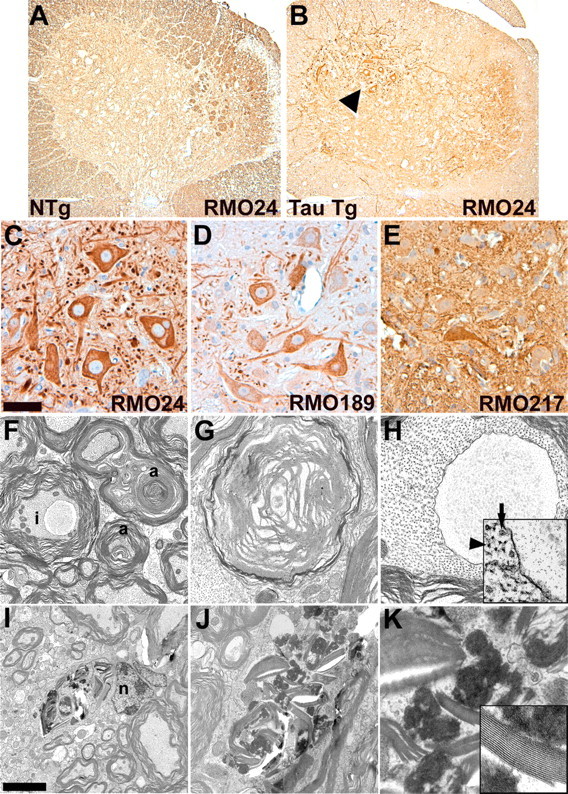
Neuronal degeneration associated with tau pathology in GFAP/tau Tg mice. A-E, Immunohistochemical analysis of spinal cord from 24-month-old GFAP/tau Tg (B-E) and non-Tg (NTg; A) mice with a panel of antibodies to phosphorylated neurofilament epitopes. In regions with robust astrocytic tau pathology, there is abnormal expression of phosphorylated neurofilament epitopes (C-E). The arrowhead in B indicates region examined at higher magnification in C-E. Scale bars: (in C) A, B, 160 μm; C-E, 40 μm. F-K, Transmission EM demonstrating axonal degeneration in 24-month-old GFAP/tau Tg mouse. F, There is prominent axonal degeneration (a) with loss of compact myelin and occasional axonal inclusions (i). G, H, High-power photomicrographs of axonal degeneration with myelin breakdown (G) and axonal inclusion (H). Inset shows edge of axonal inclusion with axonal inclusion on right and comparatively normal axon on left with both neurofilaments (arrowhead) and microtubules (arrow). These inclusions did not stain with antibodies to the tau protein. I, Axonal degeneration with myelin breakdown in association with putative astrocyte nucleus (n). J, K, High-power photomicrographs of degenerative changes shown in I. Inset in K shows fragment of degenerating myelin sheath. Scale bars (in I) I, 1.5 μm; F, 1 μm; J, 600 nm; G, H, 250 nm; K, H, inset, 150 nm; K, inset, 75 nm.
Discussion
The defining feature of tauopathies is filamentous tau inclusions in neurons and glia. It is mostly accepted that neuronal tau inclusions play a pathogenic role in neurodegeneration, especially in view of data from animal models of neuronal tau pathology in species ranging from C. elegans to mouse (Götz et al., 2004). However, the contribution of the glial tau pathology, particularly that in astrocytes, to brain degeneration remains a mystery. In fact, it is still debated whether the glial tau pathology is a reactive or a degenerative process (Ikeda et al., 1995; Komori, 1999). However, it was demonstrated recently that the astrogliosis observed in PSP was associated with neurofibrillary tangle formation and not astrocytic tau pathology (Togo and Dickson, 2002), and preliminary evidence implicates the astrocytic pathology in tauopathies with glutamate excitotoxicity and oxidative stress (Komori et al., 1998; Komori, 1999). To address specifically the role of the astrocytic tau pathology in pathogenesis of neurodegeneration in tauopathies, we sought to model this astrocytic pathology in mice, and we report data from this model implicating astrocytic tau pathology in brain generation.
There are few reports of model systems enabling investigation of the role of tau pathology in glia. In 2001, Götz et al. described tau inclusions in oligodendrocytes as well as neurons in Tg mice expressing the G272V FTDP-17 mutation in an inducible system using the prion protein promoter. The oligodendrocytic inclusions were reported as thioflavine S positive, immunoreactive with antibodies to phosphorylated tau protein, and ultrastructurally, composed of tubulofilamentous aggregates that were 17-20 nm in diameter with a periodicity of 75 nm. However, in contrast to authentic tauopathies, the inclusions were not Gallyas silver positive, tau showed only a limited shift to insoluble species, and there were no functional consequences of these tau pathologies. More recently, Higuchi and colleagues described tau pathology in oligodendrocytes, and to a lesser extent, in astrocytes, in aged Tg mice using a tau minigene construct combined with the murine Tα1 tubulin promoter leading to expression of tau isoforms with three microtubule-binding repeats (Higuchi et al., 2002a). Aged mice accumulated thioflavine S- and Gallyas-positive filamentous aggregates in oligodendrocytes and, to a lesser extent, astrocytes, in association with the accumulation of insoluble tau protein. Furthermore, the pathology was associated with glial cell degeneration and motor deficits, providing the first direct evidence linking glial pathology to neurodegeneration. However, the relative contribution of the tau pathology in oligodendrocytes versus astrocytes to the observed phenotype was difficult to determine, especially because neuronal tau abnormalities also occurred in this model. Finally, tau inclusions were reported to occur in oligodendrocytes and astrocytes of Tg mice expressing four microtubule-binding repeat tau isoforms with the P301L mutation driven by the prion protein promoter in association with amyotrophy and progressive motor dysfunction (Lin et al., 2003). But, as in the other models systems described above, it was difficult to dissect out the independent contributions of glial tau pathologies to the observed phenotype.
Recent in vitro studies have contributed to understanding the biological consequences of tau abnormalities in astrocytes by using adenoviral-mediated gene transfer to overexpress the longest human tau isoform in primary rat astrocytes that led to decreases in stable, detyrosinated microtubules and the accumulation of destabilized (acetylated and tyrosinated) tubulin (Yoshiyama et al., 2003). Because these changes also were associated with a disruption of the intermediated filaments, kinesin-dependent organelle trafficking, and Golgi fragmentation culminating in nonapoptotic cell death, it is plausible that tau accumulation in astrocytes disrupts microtubule stability. Similarly, in the GFAP/tau Tg mice, the expression of tau in astrocytes leads to a redistribution of the intermediate filament GFAP. However, no filamentous tau inclusions occurred in the in vitro model system, and the effects of tau overexpression on the function of astrocytes were not assessed.
Thus, by developing tau pathologies exclusively in astrocytes, the GFAP/tau Tg mice described herein provide unique insights into the role of astrocytic tau abnormalities in mechanisms of neurodegenerative disease. First, we showed that there is somatic expression of the human tau protein in young Tg mice, which initially is not detected with antibodies to phosphorylated tau epitopes, and this early stage is characterized by stable tau expression associated with a redistribution of GFAP resembling reactive astrocytes at this early age (Pekny and Pekna, 2004). Although tau expression in astrocytes is normally low, the specific mechanism leading to the altered tau expression in the disease state is unknown, and tau pathology in astrocytes is not typical of all tauopathies such as AD with extensive neuronal tau pathology (Lee et al., 2001). Thus, the alteration in tau expression most likely reflects a disease-specific alteration in tau protein metabolism.
The GFAP/tau Tg mice develop an age-dependent accumulation of tau pathology restricted to astrocytes that morphologically, histochemically, and biochemically bears verisimilitude to the astrocytic tau pathology characteristic of a subset of tauopathies such as PSP, CBD, PiD, and the autosomal dominant disorder FTDP-17. This second stage of tau pathology is characterized by the emergence of phospho-tau epitopes that coincides with their detection by Gallyas silver stains and the accumulation of insoluble, FA-extractable tau protein. A subset of the tau pathology in the GFAP/tau Tg mice also is detected with antibodies to ubiquitin and, similar to human tauopathies, there is induction of low-molecular-weight HSPs in regions with robust pathology (Dabir et al., 2004). Ultrastructurally, the astrocytic inclusions in the GFAP/tau Tg mice are composed of tau filaments with a diameter of 15-25 nm. Although some of the tau inclusions in these mice resemble the astrocytic plaques and tufted astrocytes typical of CBD and PSP, respectively (Feany and Dickson, 1996; Komori, 1999), the majority of the pathology was similar to the thread pathology characteristic of CBD (Dickson et al., 2001). In addition, similar to CBD, the pathology often showed an asymmetric distribution that is not described in other tau Tg model systems (Götz et al., 2004), suggesting the possibility that astrocytic pathology contributes to the asymmetric pattern of pathology observed in some of the human tauopathies. The astrocytic tau pathology is most prominent in subcortical nuclei, brainstem, deep cerebellar nuclei, and spinal cord, particularly within the gray matter similar to that observed in the tauopathy PSP. However, this topographic distribution of tau pathology most likely reflects the regional variation in tau expression driven by the GFAP promoter in the pGfa2 vector (Brenner et al., 1994), and although robust spinal cord pathology is not typical of tauopathies, tau inclusions do occur in the spinal cord of patients with PSP, CBD, and amyotropic lateral sclerosis/parkinsonism dementia complex of Guam (Mori et al., 1994; Oyanagi et al., 1997; Schmidt et al., 2000; Vitaliani et al., 2002).
Because it is now known that astrocytes contribute to a wide array of CNS functions including the BBB, detoxification, immune modulation, neurogenesis, synaptogenesis, and modulation of synapse function (Ransom et al., 2003), the GFAP/tau Tg mice described here offer opportunities to investigate the consequences of tau pathologies on astrocyte function. We showed that there was evidence of a mild disruption of the BBB in the GFAP/tau Tg mice that coincided with the detection of tau pathology including detection of phospho-tau epitopes and Gallyas-positive staining. Although the significance of the BBB disruption in the Tg mice is unclear, there is evidence in AD that Aβ pathology perturbs the BBB (Claudio, 1996; Ujiie et al., 2003), but there is little data on BBB abnormalities in other tauopathies.
More importantly, the accumulation of astrocytic tau pathology was associated with focal neuronal injury in the GFAP/tau Tg mice as indicated by aberrant expression of phosphorylated NF proteins in neuronal perikarya (Julien and Mushynski, 1998). In addition, there was ultrastructural evidence of axonal degeneration, which is likely to play a pivotal role in the pathogenesis of tauopathies (Higuchi et al., 2002b). Unfortunately, we were not able to document any statistically significant neuron loss in conjunction with these morphological alterations in neurons. The absence of neuron loss may reflect the patchy and asymmetric distribution of astrocytic pathology. Conversely, the tau pathology in astrocytes may have limited impact on neuron survival. However, experiments to assess behavioral abnormalities are currently underway, and preliminary evidence indicates impaired motor function in GFAP/tau Tg mice in comparison to non-Tg age-matched controls (data not shown). Longitudinal studies on a large cohort of animals are currently underway to further characterize these motor deficits and determine the relationship between the behavioral phenotype and tau expression in astrocytes versus the development of bona fide astrocytic tau pathology. Although the specific mechanism underlying the neurodegenerative changes is unknown, it might reflect the toxic effects of the tau aggregates as proposed in Alexander's disease, which is caused by mutations in the GFAP gene leading to filamentous inclusions composed of GFAP in astrocytes (Head et al., 1993; Brenner et al., 2001; Johnson and Brenner, 2003; Mignot et al., 2004). Notably, these astrocytic GFAP aggregates have been proposed to lead to neurodegeneration by a variety of mechanisms including myelin destruction, excitotoxicity, oxidative stress, and disruption of the BBB (Mignot et al., 2004).
In summary, the GFAP/tau Tg mice provide the first model of tau pathology exclusively in astrocytes. As such, these Tg mice will be useful in delineating the role of astrocytic pathology in the pathogenesis of neurodegeneration in a variety of tauopathies including PSP, CBD, PiD, and FTDP-17.
Footnotes
V.M.-Y.L. is The John H. Ware Third Chair of Alzheimer's disease research. J.Q.T. is the William Maul Measey-Truman G. Schnabel Jr Professor of Geriatric Medicine and Gerontology. Technical assistance was provided by personnel in the Biomedical Imaging Core Facility of The University of Pennsylvania. M.S.F. is supported by a mentored Clinical Scientist Development Award (K08 AG20073) from the National Institute on Aging. This work was supported by Grant P01 AG17586 from the National Institute on Aging.
Correspondence should be addressed to Dr. Mark S. Forman, Department of Pathology and Laboratory Medicine, University of Pennsylvania School of Medicine, 422 Curie Boulevard, 605B Stellar-Chance Building, Philadelphia, PA 19104. E-mail: formanm@mail.med.upenn.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253539-12$15.00/0
References
- Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E (1993) Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron 11: 153-163. [DOI] [PubMed] [Google Scholar]
- Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM-Y (1993) Abnormal tau phosphorylation at Ser396 in Alzheimer's disease recapitulates development and contributes to reduced microtubule binding. Neuron 10: 1089-1099. [DOI] [PubMed] [Google Scholar]
- Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A (1994) GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci 14: 1030-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A (2001) Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet 27: 117-120. [DOI] [PubMed] [Google Scholar]
- Claudio L (1996) Ultrastructural features of the blood-brain barrier in biopsy tissue from Alzheimer's disease patients. Acta Neuropathol (Berl) 91: 6-14. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Hwo SY, Kirschner MW (1977) Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol 116: 207-225. [DOI] [PubMed] [Google Scholar]
- Couchie D, Fages C, Bridoux AM, Rolland B, Tardy M, Nunez J (1985) Microtubule-associated proteins and in vitro astrocyte differentiation. J Cell Biol 101: 2095-2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couchie D, Charriere-Bertrand C, Nunez J (1988) Expression of the mRNA for tau proteins during brain development and in cultured neurons and astroglial cells. J Neurochem 50: 1894-1899. [DOI] [PubMed] [Google Scholar]
- Dabir DV, Trojanowski JQ, Richter-Landsberg C, Lee VM-Y, Forman MS (2004) Expression of the small heat-shock protein alphaB-crystallin in tauopathies with glial pathology. Am J Pathol 164: 155-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Yen SH, Suzuki KI, Davies P, Garcia JH, Hirano A (1986) Ballooned neurons in select neurodegenerative diseases contain phosphorylated neurofilament epitopes. Acta Neuropathol (Berl) 71: 216-223. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian DS, Ikeda K, Jellinger K, Lantos PL, Lippa CF, Mirra SS, Tabaton M, Vonsattel JP, Wakabayashi K, Litvan I (2001) Provisional neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 61: 935-946. [DOI] [PubMed] [Google Scholar]
- Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H (2003) Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci USA 100: 721-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drechsel DN, Hyman AA, Cobb MH, Kirschner MW (1992) Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell 3: 1141-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feany MB, Dickson DW (1995) Widespread cytoskeletal pathology characterizes corticobasal degeneration. Am J Pathol 146: 1388-1396. [PMC free article] [PubMed] [Google Scholar]
- Feany MB, Dickson DW (1996) Neurodegenerative disorders with extensive tau pathology: a comparative study and review. Ann Neurol 40: 139-148. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R (1990) Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J 9: 4225-4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Crowther RA, Six J, Lubke U, Vandermeeren M, Cras P, Trojanowski JQ, Lee VM-Y (1993) The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc Natl Acad Sci USA 90: 5066-5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz J, Tolnay M, Barmettler R, Chen F, Probst A, Nitsch RM (2001) Oligodendroglial tau filament formation in transgenic mice expressing G272V tau. Eur J Neurosci 13: 2131-2140. [DOI] [PubMed] [Google Scholar]
- Götz J, Streffer JR, David D, Schild A, Hoerndli F, Pennanen L, Kurosinski P, Chen F (2004) Transgenic animal models of Alzheimer's disease and related disorders: histopathology, behavior and therapy. Mol Psychiatry 9: 664-683. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P (1990) A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA 87: 5827-5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head MW, Corbin E, Goldman JE (1993) Overexpression and abnormal modification of the stress proteins alpha B-crystallin and HSP27 in Alexander disease. Am J Pathol 143: 1743-1753. [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Ishihara T, Zhang B, Hong M, Andreadis A, Trojanowski J, Lee VM-Y (2002a) Transgenic mouse model of tauopathies with glial pathology and nervous system degeneration. Neuron 35: 433-446. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Lee VM-Y, Trojanowski JQ (2002b) Tau and axonopathy in neurodegenerative disorders. Neuromol Med 2: 131-150. [DOI] [PubMed] [Google Scholar]
- Hoffmann R, Lee VM-Y, Leight S, Varga I, Otvos Jr L (1997) Unique Alzheimer's disease paired helical filament specific epitopes involve double phosphorylation at specific sites. Biochemistry 36: 8114-8124. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, et al. (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393: 702-705. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Akiyama H, Haga C, Kondo H, Arima K, Oda T (1994) Argyrophilic thread-like structure in corticobasal degeneration and supranuclear palsy. Neurosci Lett 174: 157-159. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Akiyama H, Kondo H, Haga C, Tanno E, Tokuda T, Ikeda S (1995) Thorn-shaped astrocytes: possibly secondarily induced taupositive glial fibrillary tangles. Acta Neuropathol (Berl) 90: 620-625. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM-Y (1999) Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron 24: 751-762. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Zhang B, Higuchi M, Yoshiyama Y, Trojanowski JQ, Lee VM-Y (2001) Age-dependent induction of congophilic neurofibrillary tau inclusions in tau transgenic mice. Am J Pathol 158: 555-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AB, Brenner M (2003) Alexander's disease: clinical, pathologic, and genetic features. J Child Neurol 18: 625-632. [DOI] [PubMed] [Google Scholar]
- Julien JP, Mushynski WE (1998) Neurofilaments in health and disease. Prog Nucleic Acid Res Mol Biol 61: 1-23. [DOI] [PubMed] [Google Scholar]
- Komori T (1999) Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick's disease. Brain Pathol 9: 663-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T, Shibata N, Kobayashi M, Sasaki S, Iwata M (1998) Inducible nitric oxide synthase (iNOS)-like immunoreactivity in argyrophilic, tau-positive astrocytes in progressive supranuclear palsy. Acta Neuropathol (Berl) 95: 338-344. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Orecchio LD, Binder L, Trojanowski JQ, Lee VM-Y, Lee G (1988) Epitopes that span the tau molecule are shared with paired helical filaments. Neuron 1: 817-825. [DOI] [PubMed] [Google Scholar]
- Lee VM-Y, Otvos Jr L, Carden MJ, Hollosi M, Dietzschold B, Lazzarini RA (1988) Identification of the major multiphosphorylation site in mammalian neurofilaments. Proc Natl Acad Sci USA 85: 1998-2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM-Y, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24: 1121-1159. [DOI] [PubMed] [Google Scholar]
- Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW (2003) Filamentous tau in oligodendrocytes and astrocytes of transgenic mice expressing the human tau isoform with the P301L mutation. Am J Pathol 162: 213-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI (1995) Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc Natl Acad Sci USA 92: 10369-10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawal-Dewan M, Henley J, Van d V, Trojanowski JQ, Lee VM-Y (1994) The phosphorylation state of tau in the developing rat brain is regulated by phosphoprotein phosphatases. J Biol Chem 269: 30981-30987. [PubMed] [Google Scholar]
- Mercken M, Vandermeeren M, Lubke U, Six J, Boons J, Van d V, Martin JJ, Gheuens J (1992) Monoclonal antibodies with selective specificity for Alzheimer Tau are directed against phosphatase-sensitive epitopes. Acta Neuropathol (Berl) 84: 265-272. [DOI] [PubMed] [Google Scholar]
- Mignot C, Boespflug-Tanguy O, Gelot A, Dautigny A, Pham-Dinh D, Rodriguez D (2004) Alexander disease: putative mechanisms of an astrocytic encephalopathy. Cell Mol Life Sci 61: 369-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H, Nishimura M, Namba Y, Oda M (1994) Corticobasal degeneration: a disease with widespread appearance of abnormal tau and neurofibrillary tangles, and its relation to progressive supranuclear palsy. Acta Neuropathol (Berl) 88: 113-121. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Namba Y, Ikeda K, Oda M (1992) Glial fibrillary tangles with straight tubules in the brains of patients with progressive supranuclear palsy. Neurosci Lett 143: 35-38. [DOI] [PubMed] [Google Scholar]
- Oyanagi K, Makifuchi T, Ohtoh T, Chen KM, Gajdusek DC, Chase TN (1997) Distinct pathological features of the gallyas- and tau-positive glia in the parkinsonism-dementia complex and amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 56: 308-316. [DOI] [PubMed] [Google Scholar]
- Pekny M, Pekna M (2004) Astrocyte intermediate filaments in CNS pathologies and regeneration. J Pathol 204: 428-437. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43: 815-825. [DOI] [PubMed] [Google Scholar]
- Ransom B, Behar T, Nedergaard M (2003) New roles for astrocytes (stars at last). Trends Neurosci 26: 520-522. [DOI] [PubMed] [Google Scholar]
- Schmidt ML, Garruto R, Chen J, Lee VM-Y, Trojanowski JQ (2000) Tau epitopes in spinal cord neurofibrillary lesions in Chamorros of Guam. NeuroReport 11: 3427-3430. [DOI] [PubMed] [Google Scholar]
- Shin RW, Iwaki T, Kitamoto T, Tateishi J (1991) Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer's disease brain tissues. Lab Invest 64: 693-702. [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95: 7737-7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Amano N, Hanihara T, Nagatomo H, Yagishita S, Itoh Y, Yamaoka K, Toda H, Tanabe T (1996) Corticobasal degeneration: widespread argentophilic threads and glia in addition to neurofibrillary tangles. Similarities of cytoskeletal abnormalities in corticobasal degeneration and progressive supranuclear palsy. J Neurol Sci 138: 66-77. [DOI] [PubMed] [Google Scholar]
- Togo T, Dickson DW (2002) Tau accumulation in astrocytes in progressive supranuclear palsy is a degenerative rather than a reactive process. Acta Neuropathol (Berl) 104: 398-402. [DOI] [PubMed] [Google Scholar]
- Ujiie M, Dickstein DL, Carlow DA, Jefferies WA (2003) Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation 10: 463-470. [DOI] [PubMed] [Google Scholar]
- Vitaliani R, Scaravilli T, Egarter-Vigl E, Giometto B, Klein C, Scaravilli F, An SF, Pramstaller PP (2002) The pathology of the spinal cord in progressive supranuclear palsy. J Neuropathol Exp Neurol 61: 268-274. [DOI] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW (1975) A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA 72: 1858-1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Zhang B, Bruce J, Trojanowski JQ, Lee VM-Y (2003) Reduction of detyrosinated microtubules and Golgi fragmentation are linked to tau-induced degeneration in astrocytes. J Neurosci 23: 10662-10671. [DOI] [PMC free article] [PubMed] [Google Scholar]