

Abstract
Opioid μ- and δ-receptors are present on the central terminals of primary afferents, where they are thought to inhibit neurotransmitter release. This mechanism may mediate analgesia produced by spinal opiates; however, when they used neurokinin 1 receptor (NK1R) internalization as an indicator of substance P release, Trafton et al. (1999) noted that this evoked internalization was altered only modestly by morphine delivered intrathecally at spinal cord segment S1-S2. We reexamined this issue by studying the effect of opiates on NK1R internalization in spinal cord slices and in vivo. In slices, NK1R internalization evoked by dorsal root stimulation at C-fiber intensity was abolished by the μ agonist [d-Ala2, N-Me-Phe4, Gly-ol5]-enkephalin (DAMGO) (1 μm) and decreased by the δ agonist [d-Phe2,5]-enkephalin (DPDPE) (1 μm). In vivo, hindpaw compression induced NK1R internalization in ipsilateral laminas I-II. This evoked internalization was significantly reduced by morphine (60 nmol), DAMGO (1 nmol), and DPDPE (100 nmol), but not by the κ agonist trans-(1S,2S)-3,4-dichloro-N-mathyl-N-[2-(1-pyrrolidinyl)cyclohexyl]-benzeneacetamide hydrochloride (200 nmol), delivered at spinal cord segment L2 using intrathecal catheters. These doses of the μ and δ agonists were equi-analgesic as measured by a thermal escape test. Lower doses neither produced analgesia nor inhibited NK1R internalization. In contrast, morphine delivered by percutaneous injections at S1-S2 had only a modest effect on thermal escape, even at higher doses. Morphine decreased NK1R internalization after systemic delivery, but at a dose greater than that necessary to produce equivalent analgesia. All effects were reversed by naloxone. These results indicate that lumbar opiates inhibit noxious stimuli-induced neurotransmitter release from primary afferents at doses that are confirmed behaviorally as analgesic.
Keywords: C-fibers, dorsal horn, internalization, neurokinin, morphine, spinal cord
Introduction
Spinal administration of μ and δ opiates produces a potent analgesia in animal models and in humans at doses that have little effect on other sensory functions (Yaksh, 1981). Consistent with this analgesic effect, opioids inhibit dorsal horn neuron responses evoked by stimulating C- but not A-fibers (Le Bars et al., 1976; Jurna and Heinz, 1979; Grudt and Williams, 1994). Immunohistochemistry and receptor autoradiography show that opioid receptors are located on small dorsal root ganglion cells and in laminas I-II of the dorsal horn (Fields et al., 1980; Cheng et al., 1996; Abbadie et al., 2001), suggesting that these receptors are present presynaptically on C-fiber terminals. This localization is confirmed by the reduction in receptor numbers after rhizotomy or neonatal capsaicin (Gamse et al., 1979; Besse et al., 1990); however, opioid receptors are also located in dorsal horn neurons, because rhizotomy does not completely eliminate opiate binding, and opioid receptor mRNA is found in the dorsal horn (Todd and Spike, 1993; Mansour et al., 1995; Kemp et al., 1996). Postsynaptically, opioid receptors decrease neuronal excitability by opening potassium channels (Yoshimura and North, 1983; Grudt and Williams, 1994). Presynaptically, opioids inhibit neurotransmitter release by inactivating voltage-gated calcium channels (VGCCs) (Schroeder et al., 1991; Soldo and Moises, 1998). Indeed, local opioids reduce increases in the extracellular concentrations of substance P (SP) or calcitonin gene-related peptide evoked by local depolarization in vitro (Jessell and Iversen, 1977; Chang et al., 1989; Pohl et al., 1989) or by C-fiber stimulation in vivo (Yaksh et al., 1980; Go and Yaksh, 1987; Aimone and Yaksh, 1989).
Activation of neurokinin 1 receptors (NK1Rs) in dorsal horn by neurokinins (e.g., SP and neurokinin A), released from small primary afferents produces NK1R internalization in lamina I neurons (Mantyh et al., 1995; Marvizon et al., 1997). Accordingly, NK1R internalization indicates SP release evoked by noxious stimuli (Abbadie et al., 1997; Allen et al., 1997; Liu et al., 1997; Honore et al., 1999; Trafton et al., 1999, 2001) or by primary afferent stimulation (Allen et al., 1999; Marvizon et al., 1999b, 2003a,b). Trafton et al. (1999), using this elegant methodology, reported that percutaneous intrathecal delivery of morphine, at doses said to be analgesic, produced only a modest (≤20%) block of NK1R internalization evoked by noxious stimuli. Systemic morphine also failed to reduce NK1R internalization, unless it was administered with an NK1R antagonist. These authors concluded that “opioid analgesia predominantly involves postsynaptic inhibitory mechanisms and/or presynaptic control of non-SP-containing primary afferent nociceptors.” Their conclusion prompted us to reexamine this issue similarly using NK1R internalization to measure neurokinin release in both ex vivo and in vivo spinal models. Here, we find that NK1R internalization evoked by C-fiber stimulation in spinal cord slices or by noxious stimulation in vivo was indeed blocked in a naloxone-reversible manner by agonists of μ- and δ-opioid receptors delivered spinally, and these effects occurred readily in vivo at intrathecal doses defined as analgesic.
Materials and Methods
Animals
For ex vivo studies, 3- to 4-week-old male Sprague Dawley rats (Harlan Sprague Dawley, Indianapolis, IN) were used in accordance with a protocol approved by the Animal Research Committee of the University of California-Los Angeles. For in vivo experiments, male Holtzman Sprague Dawley rats (300-350 g; Harlan Sprague Dawley) were used in accordance with a protocol approved by the Institutional Animal Care and Use Committee of the University of California-San Diego. The rats were housed in individual standard cages at room temperature on a 12 h light/dark cycle (lights on at 7 A.M.). Testing was performed during the light cycle at 12 noon. Animals had ad libitum access to food and water.
Ex vivo studies
Spinal cord slices. Spinal cords were obtained from 3- to 4-week-old Sprague Dawley rats anesthetized with isoflurane (Halocarbon Laboratories, River Edge, NJ). Coronal slices (400 μm) with one dorsal root were cut with a Vibratome (Technical Products International, St. Louis, MO) from a lumbar spinal cord segment (L2-L4), as described previously (Randic et al., 1993; Marvizon et al., 1997; Sandkuhler et al., 1997; Lao et al., 2003; Song and Marvizon, 2003). Fiber continuity between the dorsal root and the dorsal horn was preserved by aiming the blade of the Vibratome at the point of entrance of the root with the help of a stereo microscope. Only slices with >80% of the dorsal funiculus continuous with the root were used.
Dorsal root stimulation. The dorsal root attached to the slice was stimulated electrically using a custom-made chamber, as described previously (Song and Marvizon, 2003). Slices were superfused (3-6 ml/min) with artificial CSF containing the following (in mm): 124 NaCl, 1.9 KCl, 26 NaHCO3, 1.2 KH2PO4, 1.3 MgSO4, 2.4 CaCl2, and 10 glucose at 35°C, bubbled with 95% O2/5% CO2. Drugs were present in the superfusate for 5 min before, during, and 10 min after root stimulation. The root was placed on a bipolar stimulation electrode (0.5 mm diameter platinum wire; 1 mm pole separation) in a compartment separated from the superfusion chamber by a grease bridge. The root and electrodes were covered with mineral oil, and any excess CSF around them was suctioned away. Electrical stimulation was provided by a Master-8 stimulator and an SIU5A stimulus-isolating unit (AMP Instruments, Jerusalem, Israel) and consisted of 1000 square pulses of 20 V and 0.4 ms (C-fiber intensity) delivered at 100 Hz. The side of the slice ipsilateral to the stimulated root was identified by punching a round hole in its ventral horn. Treatments were randomized between slices, and no more than two slices from the same animal received the same treatment.
In vivo studies
Intrathecal catheter implantation. Drugs were delivered through a single chronically implanted catheter (Yaksh and Rudy, 1976) to assess their effects on behavior and NK1R internalization. In brief, the rats were anesthetized with 4% isoflurane in a room air/oxygen mixture (1:1), and the backs of the head and neck were shaved. Anesthesia was maintained with 2% isoflurane delivered by mask. The animal was placed in a stereotaxic head holder with the head flexed forward. A midline incision was made on the back of the neck, and the cisternal membrane was exposed by dissection. The membrane was opened with a stab blade, and a 7.5 cm polyethylene catheter (stretched PE-10; outer diameter, 0.015; Becton Dickinson, Sparks, MD) was then inserted through the cisternal opening and passed into the intrathecal space to the Th11-L2 spinal segment. The other end of the catheter was tunneled subcutaneously to exit through the top of the head, flushed with 10 μl of saline, and then plugged. The rats were given 5 ml of lactated Ringer's solution subcutaneously and allowed to recover under a heat lamp; those showing motor weakness or signs of paresis on recovery from anesthesia were killed immediately. The rats were allowed to recovered for 5-7 d before the experiment.
Percutaneous intrathecal injections. In separate experiments, we determined the effects on behavior of drugs delivered percutaneously at the S1-S2 spinal interspace using the method described by Trafton et al. (1999). First, we undertook initial dissections to define the approach to the site in male Holtzman rats weighting 265-350 g. Second, to determine initially the characteristic distribution of the percutaneous delivery, a series of rats were anesthetized with isoflurane by inhalation (4%) and maintained at 2% in an air/O2 mixture. Each of these rats received an injection of methylene blue (5%) delivered in a volume of 20 μl through a 30 gauge needle connected to a 50 μl Hamilton syringe. Third, to validate the reliability of our injection technique, five rats were prepared as described to receive methylene blue in 20 μl and were killed at 10 min; the lumbar sacral cord was carefully dissected, and the distribution of dye was assessed. We paid particular attention to the needle bevel at the time of injection to ensure that it was facing rostrally. Finally, an additional group of 25 rats received percutaneous intrathecal injections of vehicle (saline) or different doses of morphine sulfate. These animals were removed from the gas flow at the moment of injection and allowed to recover. Spontaneous righting reflexes began to appear within several minutes, and normal thermal escape latencies in the saline-treated animals were observed within 20 min. Escape latencies were then followed for 1 additional hour.
Hindpaw compression injury. Rats were anesthetized with sodium pentobarbital (100 mg/kg, i.p.), one of the hindpaws was positioned perpendicularly across the jaws of a 6 inch mosquito forceps with nonserrated jaws, and the jaws were closed to the first click of the hemostat ratchet. Compression was applied for 60 s. Five minutes after compression, the rats were fixed by aortal perfusion, and the spinal cords were harvested for NK1R immunocytochemistry (see below, NK1R immunohistochemistry and assessment of internalization).
Assessment of thermal nociception. The latency of the hindpaw withdrawal evoked by thermal stimulation was determined using a modified Hargreaves Box (Dirig et al., 1997). In the absence of a response within 20 s, the stimulus was terminated (cutoff time). Thermal escape latency data were expressed as percentage maximum possible effect (% MPE), which was calculated as follows: % MPE = (post-drug latency - baseline) × 100/(cutoff - baseline).
NK1R immunohistochemistry and assessment of internalization Immunohistochemistry. For in vivo studies, anesthetized rats were perfused intracardially with saline 5 min after hindpaw stimulation, followed by 4% paraformaldehyde in 0.1 m PBS, pH 7.4. The lumbar spinal cord was dissected out, fixed overnight, and cryoprotected in 30% sucrose. Immunostaining was performed on 30 μm lumbar spinal cord sections (segments L1-L6) sectioned in the coronal plane using a cryostat. The sections were washed with PBS and incubated with primary antibody for 24 h at room temperature. Primary antibody was a rabbit anti-NK1R polyclonal antibody (Advanced Targeting Systems, San Diego, CA) diluted 1:3000 in PBS, 10% normal goat serum, and 0.2% Triton X-100. After washing with PBS, the sections were incubated with secondary antibody (Alexa-488 goat anti-rabbit IgG) diluted 1:1000 in PBS, 5% normal goat serum, and 0.2% Triton X-100 for 2 h. The sections were washed and mounted in Prolong (Molecular Probes, Eugene, OR). For ex vivo studies, similar procedures were followed as described previously (Marvizon et al., 1997, 1999b; Lao et al., 2003; Song and Marvizon, 2003), with the following exceptions. Slices were fixed, cryoprotected, frozen on dry ice, and sectioned with a cryostat at 25 μm. Submerged sections were washed four times and incubated overnight at room temperature with the NK1R primary antibody (a gift from Dr. Nigel Bunnett, University of California-San Francisco, San Francisco, CA) (Grady et al., 1996) and diluted 1:2000 in PBS containing 0.3% Triton X-100, 0.001% thimerosal, and 5% normal goat serum. Although two NK1R antibodies were used in these studies, both NK1R antibodies were raised against the same epitope (immunizing peptide), and they produced identical staining of the spinal cord.
Quantification of NK1R internalization. The amount of NK1R internalization was quantified using a standard method (Mantyh et al., 1995; Abbadie et al., 1997; Marvizon et al., 1997, 1999b; Honore et al., 1999; Trafton et al., 1999, 2001; Riley et al., 2001; Lao et al., 2003; Song and Marvizon, 2003). This consisted of visual counting of NK1R immunoreactive neurons in lamina I, with and without NK1R internalization, to calculate the percentage of NK1R-positive neurons with internalization. Neuronal somas and contiguous proximal dendrites with ≥10 endosomes were considered to have internalized receptors. In each section, all lamina I NK1R neurons in the dorsal horns ipsilateral and contralateral to stimulation were counted. The person counting the neurons was blinded to the treatment given to the slice or the animal. In the study ex vivo, at least three sections per slice were counted at 100× using a Zeiss Axiovert 135 (Carl Zeiss, Thornwood, NY) fluorescence microscope. In the study in vivo, three to five sections per segment from the lumbar spinal segments (L1-L2, L3-L4, and L5-L6) were counted at 40× using an Olympus BX-51 fluorescence microscope (Olympus Optical, Tokyo, Japan). At least three animals were used for each experiment.
Confocal microscopy and images. In the figures, images obtained at 20× consist of two optical sections (2.53 μm thick; full width half-maximum) separated by 2.48 μm. Images at 100× consist of two or three optical sections (full width half-maximum, 0.62 μm) typically separated by 0.57 μm. Images were processed using Adobe Photoshop 5.5, using the “curves” feature of the program to slightly adjust the contrast. Images were initially acquired at a digital size of 1024 × 1024 pixels and later were cropped to the relevant part of the field.
Statistical analysis
Data were analyzed using Prism 3.0 for Macintosh or 4.01 for the personal computer (GraphPad Software, San Diego, CA). All data are presented as mean ± SEM. The statistical significance was calculated using the t test or one-way or two-way ANOVA and Bonferroni's post-test. Differences are considered to be significant when the critical value reaches a level of p < 0.05. For two-way ANOVA, the two variables were “drugs” (or drug combinations) and “stimulation” (ipsilateral or contralateral to stimulation), and the Bonferroni's post-test was applied to the variable drugs to compare effects on the ipsilateral side.
Drugs
Morphine (morphine sulfate) was from Merck (West Point, PA), naloxone hydrochloride was from DuPont Pharmaceuticals (Garden City, NJ), and [d-Ala2, N-Me-Phe4, Gly-ol5]-enkephalin (DAMGO), [d-Phe2,5]-enkephalin (DPDPE), trans-(1S,2S)-3,4-dichloro-N-mathyl-N-[2-(1-pyrrolidinyl)cyclohexyl]-benzeneacetamide hydrochloride (U50488H), and substance P were from Sigma (St. Louis, MO). All drugs for intrathecal injection were freshly prepared in physiologic saline, and the required dose was delivered in an injection volume of 10 μl.
Results
Ex vivo NK1R internalization
Dorsal root stimulation
Electrical stimulation of the root at an intensity (20 V) sufficient to recruit both A- and C-fibers (Song and Marvizon, 2003) increased the number of lamina I NK1R neurons with internalization ipsilateral to the stimulated root (43 ± 6%) (Figs. 1A, 2) as compared with the contralateral dorsal horn (16 ± 5%; p < 0.01) (Figs. 1B, 2). No NK1R internalization was found in deeper laminas. Importantly, our stimulation method isolated the spinal cord from the electrical pulses delivered to the root, and therefore all of the evoked SP release had to be mediated by primary afferent firing. Figure 1A shows confocal images of six NK1R neurons with internalization and their location in the dorsal horn ipsilateral to the stimulated root, whereas Figure 1B shows three NK1R neurons without internalization in the contralateral dorsal horn.
Figure 1.
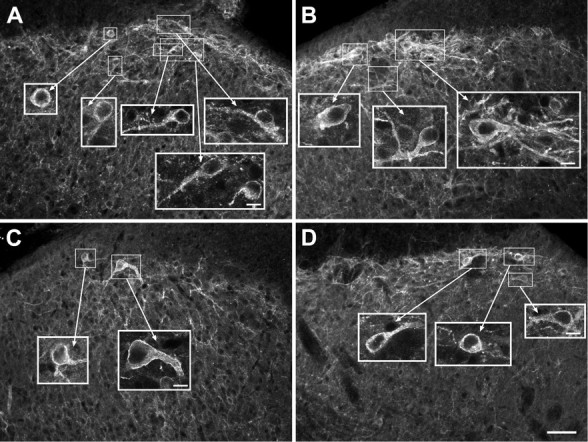
Effect of DAMGO on NK1R internalization evoked by dorsal root stimulation in spinal cord slices. The dorsal root attached to spinal cord slices was stimulated at 100 Hz. The large panels are confocal images taken at 20×, consisting of two optical sections separated by 2.48 μm. Scale bar, 50 μm. The insets are confocal images taken at 100×, consisting of two to three optical sections separated by 0.57 μm. Scale bars, 10 μm. A, Dorsal horn ipsilateral to the stimulated root in a slice superfused with artificial CSF (control); all neurons show NK1R internalization. B, Contralateral dorsal horn in a control slice; neurons show no NK1R internalization. C, Ipsilateral dorsal horn in a slice superfused with 1 μm DAMGO; neurons show no NK1R internalization. D, Ipsilateral dorsal horn in a slice superfused with 1 μm DAMGO plus 10 μm naloxone; neurons show NK1R internalization.
Figure 2.

DAMGO and DPDPE decreased NK1R internalization in spinal cord slices. Slices were superfused with the drugs indicated, and the dorsal root was stimulated at 100 Hz (1000 pulses of 20 V; 0.4 ms). DAMGO and DPDPE were 1 μm, and naloxone was 10 μm. Data are the mean ± SEM of four to five slices. Two-way ANOVA yielded p < 0.001 for the variables drugs and “side.” Contralateral to the stimulated root, NK1R internalization was low. Ipsilateral to the stimulated root, Bonferroni's post-test revealed significant decreases by DAMGO (***p < 0.001) and DPDPE (*p < 0.05) and significant differences between DAMGO and DAMGO plus naloxone (†††p < 0.001).
Effects of opiates on NK1R internalization evoked by root stimulation
NK1R internalization evoked by dorsal root stimulation was abolished by the selective μ-opioid agonist DAMGO (1 μm) (9 ± 4%; p < 0.001) (Fig. 1C) and significantly decreased by the selective δ-opioid agonist DPDPE (1 μm) (25 ± 7%; p < 0.05). The opioid receptor antagonist naloxone (10 μm) reversed to levels not significantly different from control, i.e., the inhibition produced by DAMGO (37 ± 3%) or DPDPE (32 ± 6%). The concentration of DAMGO (1 μm) was selected because it is just enough to saturate μ-opioid receptors inside the spinal cord, as observed using μ-opioid receptor internalization in rat spinal cord slices (Marvizon et al., 1999a; Trafton et al., 2000). In contrast, 1 μm DPDPE did not produce μ-opioid internalization, indicating that an effect is produced through δ- and not μ-opioid receptors. Figure 1C shows examples of NK1R neurons in the dorsal horn ipsilateral to the stimulated root in a slice superfused with DAMGO; both neurons have no NK1R internalization. Figure 1D shows NK1R neurons ipsilateral to stimulation in a slice superfused with DAMGO and naloxone; it can be seen that naloxone restored NK1R internalization. These results show that activation of μ- and δ-opioid receptors inhibits SP release from the central terminals of primary afferents, which are primarily C-fibers (McCarthy and Lawson, 1989; Allen et al., 1999).
In vivo NK1R internalization
Effects of paw compression
In naive rats without noxious stimulation, most of the cells displayed NK1R immunoreactivity on the cell membrane. Under such nonstimulated conditions, a small fraction (typically <10-15%) was judged as showing some evidence of internalization. Five minutes after compression of the hindpaw, NK1R internalization was observed in lamina I of the ipsilateral, but not the contralateral, lumbar (L1-L6) spinal cord (Fig. 3A, B). NK1R internalization was not observed in laminas III-V. Figure 3A shows NK1R-immunoreactive neurons of the ipsilateral dorsal horn induced by hindpaw compression; much of the NK1R immunoreactivity is present in endosomes in the cytoplasm of the neurons. NK1R internalization was quantified and presented as a percentage of NK1R-positive neurons showing internalization in lamina I (Fig. 3C). NK1R neurons with internalization were more numerous in the lower lumbar segments (e.g., L5-L6: 65 ± 6%) and diminished rostrally (e.g., L1-L2: 40 ± 12%).
Figure 3.

NK1R internalization induced by noxious stimulation. Confocal images (A, B) show lamina I NK1R-immunoreactive neurons in animals that received noxious stimulation in the hindpaw at approximately the L5 spinal level. A, Low-magnification image (10×) of the dorsal horn ipsilateral to the stimulated paw in a rat that received intrathecal saline. Scale bar, 60 μm. Inset, High-magnification image (40×) of the ipsilateral lamina I showing neurons with NK1R internalization. Scale bar, 12 μm. B, Similar images of the contralateral lamina I; there was no NK1R internalization. C, Quantification of the NK1R internalization induced by hindpaw compression for 60 s. NK1R internalization was measured in spinal segments L1-L2, L3-L4, and L5-L6. One-way ANOVA and Bonferroni's post-test indicated significant differences between the contralateral and ipsilateral sides (**p < 0.01).
Effects of intrathecal opiates on injury-induced NK1R internalization
To determine whether intrathecal opioids alter SP release from the primary afferent, we examined the effect of intrathecal morphine on NK1R internalization induced by paw compression. Morphine (60 nmol, i.t.) administrated 10 min before compression decreased NK1R internalization in the ipsilateral lamina I (Fig. 4A). Quantification of the internalization after 20 and 60 nmol of intrathecal morphine revealed a dose-dependent suppression (Fig. 4C, D) of compression-induced NK1R internalization. Importantly, there was no effect on the magnitude of internalization measured in the contralateral paw across treatments as compared with vehicle-treated animals. To determine whether this inhibitory effect of intrathecal morphine acts via an opioid receptor, naloxone (1 mg/kg, i.p.) was administrated 15 min before intrathecal morphine. Naloxone completely reversed this inhibitory effect on the NK1R internalization (Fig. 4B, D).
Figure 4.

Intrathecal (IT) morphine inhibits NK1R internalization induced by noxious stimulation. A, Rats received 60 nmol of IT morphine and hindpaw compression 10 min later; confocal images taken at approximately the L5 spinal level show NK1R neurons in ipsilateral lamina I with no internalization. B, Rats received naloxone (1 mg/kg, i.p.), 60 nmol of IT morphine 15 min later, and hindpaw compression 10 min after morphine; NK1R neurons in the ipsilateral lamina I show internalization. C, Amount of NK1R internalization ipsilateral to hindpaw compression delivered 10 min after intrathecal saline (control) or morphine (20 or 60 nmol); *p < 0.05 compared with control (ANOVA and Bonferroni's post-test). D, Amounts of NK1R internalization (average of L1-L2, L3-L4, and L5-L6) produced by hindpaw compression. Naloxone (1 mg/kg, i.p.) reversed the inhibition produced by IT morphine; *p < 0.05 compared with morphine (ANOVA and Bonferroni's post-test). Data are the mean ± SEM of three to five rats per group. IP, Intraperitoneal.
The μ-agonist DAMGO (1 nmol, i.t.) produced a significant suppression of compression-evoked NK1R internalization (Fig. 5), as did morphine. The δ-opiate agonist DPDPE (100 nmol, i.t.) also inhibited NK1R internalization. The effects of both DAMGO and DPDPE were reversed by naloxone (1 mg/kg, i.p.). In contrast, the κ-opioid receptor agonist U50488H (200 nmol, i.t.) did not significantly alter NK1R internalization (Fig. 5). These findings provide additional evidence that activation of δ- and μ- but not κ-opioid receptors inhibits SP release from primary afferents.
Figure 5.

DPDPE and DAMGO, but not U50488H, inhibit NK1R internalization induced by noxious stimulation. A, Rats received intrathecal injections of vehicle (10 μl of saline; control) or DPDPE (50 or 100 nmol) 10 min before hindpaw compression. Naloxone (1 mg/kg, i.p.; 15 min before DPDPE) reversed the inhibition of NK1R internalization in lamina I neurons produced by 100 nmol of DPDPE. One-way ANOVA and Bonferroni's post-test indicated significant differences between control and 100 nmol of DPDPE (**p < 0.01) and between 100 nmol of DPDPE and DPDPE plus naloxone (*p < 0.05). B, Rats received intrathecal injections of vehicle (control), DAMGO (1 nmol), or U50488H (200 nmol) 10 min before hindpaw compression. Naloxone (1 mg/kg, i.p.; 15 min before DAMGO) reversed the inhibition of NK1R internalization in lamina I neurons produced by DAMGO. One-way ANOVA and Bonferroni's post-test indicated significant differences between control and DAMGO (**p < 0.01) and between DAMGO and DAMGO plus naloxone (**p < 0.01). Data are the mean ± SEM of three to eight rats per group. IP, Intraperitoneal.
Effect of intrathecal morphine on NK1R internalization induced by exogenous SP
To rule out the possibility that morphine directly blocks the NK1R internalization mechanism, we examined the effect of morphine on NK1R internalization induced by exogenous SP (intrathecal injection). Figure 6A presents confocal images of NK1R neurons in lamina I of the lumbar spinal cord after SP (30 nmol, i.t.); Figure 6B images are after SP plus morphine (30 and 60 nmol, i.t., respectively). Intrathecal SP produced widespread NK1R internalization. Quantification of the internalization revealed that it was greater than that observed after intrathecal saline (intrathecal SP, 59 ± 12%, vs intrathecal saline, 10 ± 4%; p < 0.01). Intrathecal morphine at 60 nmol, a dose that completely blocked paw compression-induced NK1R internalization (Fig. 4), did not affect exogenous SP-induced NK1R internalization (intrathecal SP plus morphine, 67 ± 5%, vs intrathecal SP, 59 ± 12%; p > 0.05) (Fig. 6B).
Figure 6.

Morphine does not block NK1R internalization induced by SP. Rats received intrathecal injections of saline (10 μl), SP (30 nmol), or morphine (60 nmol) followed by SP (30 nmol); 30 min later they were killed and fixed. A, B, Confocal images of NK1R neurons in lamina I after SP alone (A) or SP after morphine (B). Scale bar, 10 μm. Quantification of NK1R internalization in lamina I showed the following: after intrathecal saline, 10 ± 4% (n = 5); after intrathecal SP, 59 ± 12% (n = 5); and after intrathecal SP plus intrathecal morphine, 67 ± 5% (n = 4). One-way ANOVA and Bonferroni's post-test indicated significant differences between intrathecal saline and intrathecal SP with and without morphine (p < 0.01).
Effect of systemic morphine on injury-induced NK1R internalization
Morphine was administrated (10-30 mg/kg, s.c.) 15 min before the noxious stimulation. Morphine, at a dose of 30 mg/kg, attenuated NK1R internalization (p < 0.05) evoked by compression injury (Fig. 7A), and this effect was reversed by naloxone (1 mg/kg, i.p.) (Fig. 7B); however, systemic morphine given at a lower dose (10 mg/kg, s.c.) did not significantly reduce NK1R internalization in the dorsal horn (Fig. 7C).
Figure 7.
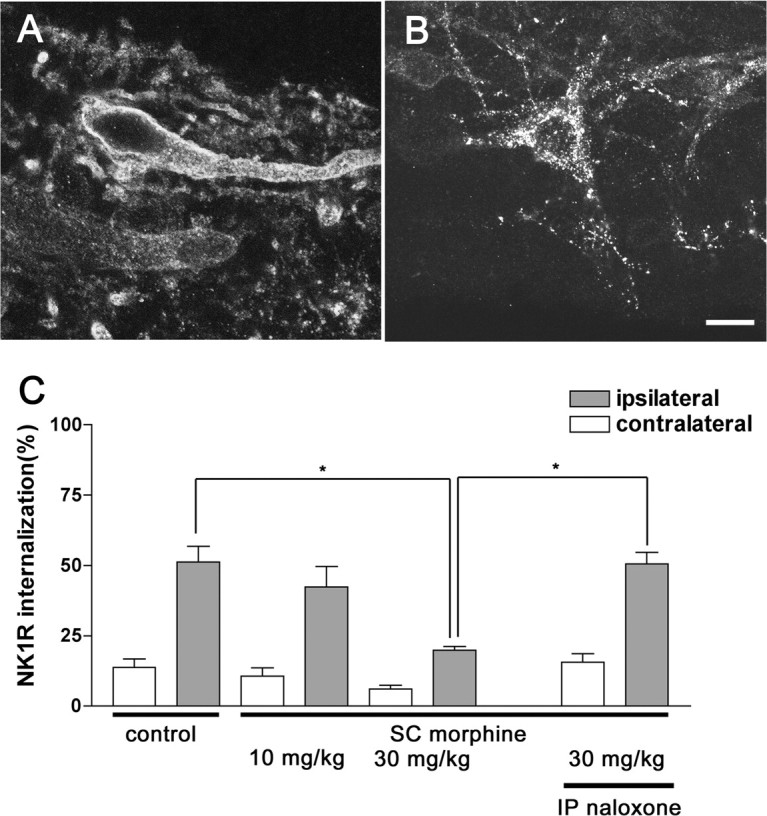
Effect of systemic morphine on NK1R internalization induced by noxious stimulation. Systemic morphine (10 or 30 mg/kg) was given by subcutaneous injection, and paw compression was applied 15 min after the morphine injection. Naloxone (1 mg/kg, i.p.) was given 15 min before morphine. The control group received an injection of saline (1 ml/kg, s.c.) 15 min before paw compression. A, B, Confocal images of NK1R-immunoreactive neurons in lamina I after 30 mg/kg morphine (A) or after 1 mg/kg naloxone and 30 mg/kg morphine (B). Scale bar, 10 μm. C, Amount of NK1R internalization induced by noxious stimulation in the ipsilateral lamina I after saline, morphine, or morphine and naloxone. Data are the mean ± SEM of three to five rats per group. One-way ANOVA and Bonferroni's post-test indicated significant differences between ipsilateral control and 30 mg/kg morphine and between 30 mg/kg morphine alone and with naloxone (*p < 0.05). IP, Intraperitoneal; SC, subcutaneous.
Analgesic effects of intrathecal drugs
Thermal escape latencies were measured after intrathecal (via implanted lumbar catheter) or subcutaneous administration of opiates. As observed previously with this injection technique, intrathecal doses of 60 nmol of morphine (Fig. 8A), 1 nmol of DAMGO, or 100 nmol of DPDPE (Fig. 9) were found to uniformly produce a significant but just submaximal effect on thermal escape (e.g., % MPE = 70-80). At lower intrathecal doses of morphine (20 nmol) or DPDPE (50 nmol) or at the maximum dose of U50488H (200 nmol), there was a minor to modest increase in thermal escape latency (Figs. 8, 9) that did not reach statistical significance. Subcutaneous administration of systemic morphine at 10 mg/kg had no statistically significant effect on NK1R internalization but produced a significant analgesia equivalent to the high doses of intrathecal μ and δ agonists (i.e., % MPE = 70-80) (Fig. 8B). As noted above, only after a supra-maximal analgesic dose (30 mg/kg, s.c.) (Fig. 8) was a pronounced block of compression-induced NK1R internalization observed (Fig. 7).
Figure 8.

Analgesic effects of morphine. Peak paw withdrawal responses to thermal stimulation were expressed as percentage maximum (% Max) possible effect. A, Spinal morphine. Rats received intrathecal injections of vehicle (Veh; saline) or different doses of morphine sulfate through chronically implanted intrathecal catheters (L2/3 IT catheter) or by an acute percutaneous needle placed at the S1/S2 interspace (S1/2 Percutan). B, Systemic morphine. Rats received subcutaneous injections of morphine (Morphine s.c.) or vehicle. Naloxone (+ Nal; 1 mg/kg, i.p.) was injected 15 min before the opiate injection. Data are the mean ± SEM of five to eight rats per group. One-way ANOVA and Bonferroni's post-test indicated significant differences with saline (*p < 0.01) or naloxone (#p < 0.01).
Figure 9.

Analgesic effects of opiates. Peak paw withdrawal responses to thermal stimulation were expressed as percentage maximum (% Max) possible effect. Rats with intrathecal catheters received intrathecal injections of saline (10 μl), DAMGO (1 nmol), DPDPE (50 or 100 nmol), or U50488H (200 nmol). Naloxone (Nal; 1 mg/kg, i.p.) was injected 15 min before the opiate injection. Data are the mean ± SEM of three to eight rats per group. One-way ANOVA and Bonferroni's post-test indicated significant differences with saline (*p < 0.01) or naloxone (#p < 0.01). IT, Intrathecal.
Percutaneous intrathecal injections
To address the differences between the prominent effect of intrathecal morphine on NK1R internalization that we observed and the marginal effect reported by Trafton et al. (1999), we considered differences in the methods used for intrathecal injections. Our studies were undertaken in rats implanted with chronic L2 intrathecal catheters, and drug volumes and doses were demonstrated to produce a potent analgesia in response to hindpaw stimulation. Previous work has shown that intrathecal injections of 10 μl using a chronic catheter with a tip at approximately L2 are effective in distributing the injected fluid two to three segments rostral and caudal to the catheter tip (Yaksh and Rudy, 1976). In contrast, Trafton et al. (1999) used a S1-S2 percutaneous injection in a barbiturate-anesthetized rat, a procedure that precludes the behavioral assessment of analgesia. To assess whether this method of injection is effective in delivering drugs to the lumbar spinal cord, where primary afferents activated by the paw injury project, we obtained dose-response curves for analgesia produced by morphine injected percutaneously in isoflurane-anesthetized rats. Saline injected percutaneously at S1-S2 had no effect on thermal escape latency as compared with baseline 20-60 min after injection (Fig. 8A). Morphine injected percutaneously at 90 nmol [30 μg, the dose used in the study by Trafton et al. (1999)] also failed to alter the thermal escape latency. An increase in the morphine dose to 300 nmol (100 μgin 20 μl) resulted in a small but statistically significant increase in the thermal escape latency. This thermal escape latency was similar to that obtained with a morphine dose of 20 nmol (6 μg) when a chronic catheter was used for the intrathecal injections; therefore, morphine was more effective against a hindpaw stimulus when injected using a lumbar catheter than when injected at S1-S2, suggesting that the latter method does not produce an adequate lumbar distribution of injectate. To confirm this, we systematically examined the rostrocaudal distribution of dye injected percutaneously in necropsies undertaken 10 min after the injection. With a percutaneous injection at S1-S2, we observed a reliable staining of the sacral and lumbar roots at the sacral level; however, the most rostral appearance of staining did not reach the lumbar enlargement of the spinal cord in five of five rats. In contrast, injection of dye using an L2 catheter revealed a clear lumbar distribution of the dye; therefore, it is likely that morphine injected by the S1-S2 route in this volume does not reach the lumbar spinal cord at concentrations sufficient to inhibit lumbar spinal nociceptive processing evoked by hindpaw stimulation.
Discussion
Our study, using ex vivo and in vivo models, demonstrates that spinal μ- and δ-opioid agonists prevent afferent-evoked NK1R internalization, indicating that the local release of SP from the central terminals of primary afferents was inhibited. In the in vivo model, the delivery system, dose-response, and pharmacological profiles of the effect on NK1R internalization matched the analgesic effects of opiates, suggesting that inhibition of afferent neurotransmitter release contributes to intrathecal opiate analgesia.
Spinal effects of opiates
Presynaptic localization of opioid receptors on high-threshold primary afferent terminals (Cheng et al., 1996; Abbadie et al., 2001), along with the negative coupling of these receptors to the opening of VGCCs (Schroeder et al., 1991; Soldo and Moises, 1998), provides the mechanism for the observed inhibition by opiates of SP release in spinal slices (Jessell and Iversen, 1977; Chang et al., 1989; Pohl et al., 1989) or in vivo superfusates (Yaksh et al., 1980; Go and Yaksh, 1987; Aimone and Yaksh, 1989). Our results show that spinal μ- and δ-opioid agonists block NK1R internalization evoked by C-fiber stimulation or paw compression. Importantly, in vivo, morphine, DAMGO, or DPDPE at intrathecal doses that produced similar degrees of antinociception also inhibited NK1R internalization to a similar extent. Lower intrathecal doses of these opioids produced only moderate analgesia and a smaller inhibition in the evoked internalization. In both ex vivo and in vivo models, opioid effects were reversed by naloxone. Additional evidence of specificity was provided by failure of a κ-opioid agonist to decrease internalization or induce thermal analgesia (Schmauss and Yaksh, 1984). Because morphine had no effect on internalization evoked by exogenous intrathecal SP, the reduction of NK1R internalization was not caused by an effect on the internalization process.
Comparisons with previous work
In contrast to the near-maximal, dose-dependent block of internalization noted in our study, Trafton et al. (1999), reported that spinal morphine at doses higher than those used here (30 vs 20 μg) had only a modest effect (≤20%) on NK1R internalization. Several variables were considered. First, our study used chronic intrathecal catheters, whereas Trafton et al. (1999) used percutaneous injection. Morphine (60 nmol) delivered at L2 by an intrathecal catheter produces robust analgesia in a wide variety of tests (Yaksh, 1999). Here, μ and δ agonists given intrathecally at L2 produced near-maximum analgesia at ∼10 min. The same doses and pretreatment times were used to assess NK1R internalization. In the study by Trafton et al. (1999), the percutaneous intrathecal injection was given at the S1-S2 vertebral level to avoid injury to the spinal cord; however, percutaneous injections required anesthetizing the rat. The use of a barbiturate precluded acute recovery to determine the degree of antinociception achieved with the S1-S2 intrathecal drug. Our study examined the effects of S1-S2 injections of morphine in isoflurane-anesthetized rats and found no analgesic effect at the dose used. Modest analgesia was noted at a dose even higher than that used in the S1-S2 intrathecal injection study. Importantly, dye distribution confirmed that the S1-S2 intrathecal injections were indeed in the sacral spinal sac but reliably showed an absence of rostral distribution; therefore, the absence of an effect of morphine on either pain behavior or lumbar NK1R internalization may reflect an absence of adequate rostral redistribution to lumbar segments. In this regard, Trafton et al. (1999) did observe a small but statistically significant suppression of NK1R internalization after S1-S2 delivery of 2 nmol of DAMGO. In rats with lumbar catheters, half of that dose produces a very robust analgesia. The higher dose likely forced the distribution of DAMGO to the lumbar level, accounting for its modest effects. Second, Trafton et al. (1999) used a strong compression that produced >80% NK1R internalization. In contrast, paw compression in our study produced less internalization: 60-70%. Thus, the minimal inhibition of NK1R internalization produced by intrathecal morphine in the study by Trafton et al. (1999) may have resulted from using a stimulus that released neurokinins at a concentration greater than that needed to saturate available NK1Rs. We have shown previously that an increase in stimulus intensity shifts opiate dose-response curves to the right (Saeki and Yaksh, 1993; Dirig and Yaksh, 1995). Accordingly, had we delivered the drug at a distant site or used a stronger stimulus, higher doses of morphine would likely have been required to decrease the evoked NK1R internalization.
Systemic opiate action
Consistent with previous work (Trafton et al., 1999), we found that systemic morphine (10 mg/kg) at a dose shown to be equi-analgesic to lumbar intrathecal morphine (60 nmol) did not decrease NK1R internalization; however, we did observe internalization blockade at 30 mg/kg. These observations suggest that μ-opioid receptors in different locations in the CNS may play complementary roles in regulating pain processing. Thus, systemic morphine exerts potent antinociceptive effects by both spinal and supraspinal mechanisms (Yaksh, 1997). Analgesic effects of systemic opiates are reversed by supraspinal (Vigouret et al., 1973) and spinal (Yaksh and Rudy, 1977) opiate antagonists. These observations led to the hypothesis that the concurrent activation of spinal and supraspinal sites altered pain behavior by a synergic interaction (Yaksh and Rudy, 1978). This was confirmed in isobolographic interaction studies examining the analgesic effects of intracerebroventricular and intrathecal morphine (Yeung and Rudy, 1980). Accordingly, intrathecal opiates produce analgesia through effects on primary afferent terminals. Systemic opiates can also inhibit release from primary afferent terminals but only at doses greater than those necessary to produce analgesia.
Presynaptic and postsynaptic contributions to spinal opiate analgesia
Our main observation is that intrathecal opiates diminish SP release at doses that also produce potent analgesia. These actions argue that opioid inhibition of afferent transmitter release plays an important role in the modulation of spinal nociceptive processing. Because superficial dorsal horn neurons bearing NK1R do not coexpress μ-opioid receptors (Spike et al., 2002; Song and Marvizon, 2003), μ agonists cannot produce analgesia by directly inhibiting these neurons. The importance of these NK1R-bearing neurons is highlighted by the fact that many of these neurons are projection neurons (Spike et al., 2002; Todd et al. (1999), 2002; Song and Marvizon, 2003) and that their destruction by intrathecal SP-saporin leads to a block of hyperalgesia (Mantyh et al., 1997), which is mediated in part by a spinobulbospinal facilitatory loop (Suzuki et al., 2002). Interestingly, Trafton et al. (1999) reported that although systemic morphine at 10 mg/kg alone did not inhibit evoked NK1R internalization, it did when coinjected with a minimal dose of NK1R antagonist. Independent of the fact that NK1R antagonists compete with endogenous SP to prevent internalization, these observations raise the possibility that inhibition of NK1R could have reduced the activation of spinofugal pathways, which can facilitate dorsal horn nociceptive processing (Suzuki et al., 2002).
Although analgesic doses of spinal opiates indeed suppress SP release, this effect may be quite complex. Although a direct effect on afferent terminals appears likely, we cannot rule out indirect mechanisms. Excitatory interneurons express μ-opioid receptors (Kemp et al., 1996) and may facilitate SP release through axo-axonic synapses with primary afferents (Trafton and Basbaum, 2000). Glutamate released at these synapses might activate presynaptic NMDA receptors that stimulate SP release (Grady et al., 1996; Liu et al., 1997; Marvizon et al., 1997, 1999b; Malcangio et al., 1998). In this case, inhibition of the excitatory interneurons by μ-opioid receptors would result in a reduction in SP release. This presynaptic effect itself may also be complicated. Activation of presynaptic NMDA receptors initiates primary afferent depolarization and inhibits glutamate release (Bardoni et al., 2004). Alternatively, presynaptic NMDA receptors may facilitate SP release by providing another pathway for Ca2+ entry to mobilize transmitter release. Under such conditions, NMDA receptor-enhanced SP release would not be susceptible to inhibition by presynaptic opioid receptors. Thus, GABAB receptors, which inactivate Ca2+ channels, inhibited SP release when evoked by dorsal root stimulation but not by NMDA application (Marvizon et al., 1999b). NMDA receptors thus might provide a potential mechanism whereby hyperalgesic states involving increased glutamate release could result in an SP release refractory to opioid inhibition.
In conclusion, local μ and δ agonists at analgesic doses suppress afferent-evoked NK1R internalization. Our study does not argue for the specific importance of SP as a “pain transmitter.” Rather, inhibition of the release of multiple primary afferent transmitters may account for an important part of analgesia produced by spinal opiates. Finally, we believe that these observations provide an insight into the clinical actions of opiates. Given that intrathecal but not systemic morphine at analgesic doses blocks spinal terminal release, and because we believe that such release is what drives downstream events, including release of spinal lipid mediators (Svensson and Yaksh, 2002) and activation of non-neuronal cells (Svensson et al., 2003), there are likely important differences in the efficacy of opiates given by these two routes. Such downstream processes are believed to be modified in preemptive modes of analgesic therapy (Moiniche et al., 2002). Our work suggests differences between the preemptive effects initiated by equi-analgesic opiate doses given by the two routes.
Footnotes
This work was supported by National Institutes of Health Grants DA02110 (X.-Y.H., T.L.Y.) and DA12609 (J.C.G.M.). We thank Shelle Malkmus for outstanding technical support.
Correspondence should be addressed to Dr. Tony L. Yaksh, Department of Anesthesiology 0818, University of California-San Diego, 9500 Gilman Drive, La Jolla, CA 92093. E-mail: tyaksh@ucsd.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253651-10$15.00/0
References
- Abbadie C, Trafton J, Liu H, Mantyh PW, Basbaum AI (1997) Inflammation increases the distribution of dorsal horn neurons that internalize the neurokinin-1 receptor in response to noxious and non-noxious stimulation. J Neurosci 17: 8049-8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbadie C, Pasternak GW, Aicher SA (2001) Presynaptic localization of the carboxy-terminus epitopes of the μ opioid receptor splice variants MOR-1C and MOR-1D in the superficial laminae of the rat spinal cord. Neuroscience 106: 833-842. [DOI] [PubMed] [Google Scholar]
- Aimone LD, Yaksh TL (1989) Opioid modulation of capsaicin-evoked release of substance P from rat spinal cord in vivo. Peptides 10: 1127-1131. [DOI] [PubMed] [Google Scholar]
- Allen BJ, Rogers SD, Ghilardi JR, Menning PM, Kuskowski MA, Basbaum AI, Simone DA, Mantyh PW (1997) Noxious cutaneous thermal stimuli induce a graded release of endogenous substance P in the spinal cord: imaging peptide action in vivo J Neurosci 17: 5921-5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BJ, Li J, Menning PM, Rogers SD, Ghilardi J, Mantyh PW, Simone DA (1999) Primary afferent fibers that contribute to increased substance P receptor internalization in the spinal cord after injury. J Neurophysiol 81: 1379-1390. [DOI] [PubMed] [Google Scholar]
- Bardoni R, Torsney C, Tong CK, Prandini M, MacDermott AB (2004) Presynaptic NMDA receptors modulate glutamate release from primary sensory neurons in rat spinal cord dorsal horn. J Neurosci 24: 2774-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse D, Lombard MC, Zajac JM, Roques BP, Besson JM (1990) Pre- and postsynaptic distribution of μ, δ and κ opioid receptors in the superficial layers of the cervical dorsal horn of the rat spinal cord. Brain Res 521: 15-22. [DOI] [PubMed] [Google Scholar]
- Chang HM, Berde CB, Holz GG, Steward GF, Kream RM (1989) Sufentanil, morphine, met-enkephalin, and κ-agonist (U-50,488H) inhibit substance P release from primary sensory neurons: a model for presynaptic spinal opioid actions. Anesthesiology 70: 672-677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng PY, Moriwaki A, Wang JB, Uhl GR, Pickel VM (1996) Ultrastructural localization of μ-opioid receptors in the superficial layers of the rat cervical spinal cord: extrasynaptic localization and proximity to Leu5-enkephalin. Brain Res 731: 141-154. [DOI] [PubMed] [Google Scholar]
- Dirig DM, Yaksh TL (1995) Differential right shifts in the dose-response curve for intrathecal morphine and sufentanil as a function of stimulus intensity. Pain 62: 321-328. [DOI] [PubMed] [Google Scholar]
- Dirig DM, Salami A, Rathbun ML, Ozaki GT, Yaksh TL (1997) Characterization of variables defining hindpaw withdrawal latency evoked by radiant thermal stimuli. J Neurosci Methods 76: 183-191. [DOI] [PubMed] [Google Scholar]
- Fields HL, Emson PC, Leigh BK, Gilbert RF, Iversen LL (1980) Multiple opiate receptor sites on primary afferent fibres. Nature 284: 351-353. [DOI] [PubMed] [Google Scholar]
- Gamse R, Holzer P, Lembeck F (1979) Indirect evidence for presynaptic location of opiate receptors on chemosensitive primary sensory neurones. Naunyn Schmiedebergs Arch Pharmacol 308: 281-285. [DOI] [PubMed] [Google Scholar]
- Go VL, Yaksh TL (1987) Release of substance P from the cat spinal cord. J Physiol (Lond) 391: 141-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady EF, Baluk P, Bohm S, Gamp PD, Wong H, Payan DG, Ansel J, Portbury AL, Furness JB, McDonald DM, Bunnett NW (1996) Characterization of antisera specific to NK1, NK2, and NK3 neurokinin receptors and their utilization to localize receptors in the rat gastrointestinal tract. J Neurosci 16: 6975-6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudt TJ, Williams JT (1994) μ-Opioid agonists inhibit spinal trigeminal substantia gelatinosa neurons in guinea pig and rat. J Neurosci 14: 1646-1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore P, Menning PM, Rogers SD, Nichols ML, Basbaum AI, Besson JM, Mantyh PW (1999) Spinal substance P receptor expression and internalization in acute, short-term, and long-term inflammatory pain states. J Neurosci 19: 7670-7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessell TM, Iversen LL (1977) Opiate analgesics inhibit substance P release from rat trigeminal nucleus. Nature 268: 549-551. [DOI] [PubMed] [Google Scholar]
- Jurna I, Heinz G (1979) Differential effects of morphine and opioid analgesics on A and C fibre-evoked activity in ascending axons of the rat spinal cord. Brain Res 171: 573-576. [DOI] [PubMed] [Google Scholar]
- Kemp T, Spike RC, Watt C, Todd AJ (1996) The μ-opioid receptor (MOR1) is mainly restricted to neurons that do not contain GABA or glycine in the superficial dorsal horn of the rat spinal cord. Neuroscience 75: 1231-1238. [DOI] [PubMed] [Google Scholar]
- Lao LJ, Song B, Marvizon JC (2003) Neurokinin release produced by capsaicin acting on the central terminals and axons of primary afferents: relationship with N-methyl-d-aspartate and GABAB receptors. Neuroscience 121: 667-680. [DOI] [PubMed] [Google Scholar]
- Le Bars D, Guilbaud G, Jurna I, Besson JM (1976) Differential effects of morphine on responses of dorsal horn lamina V type cells elicited by A and C fibre stimulation in the spinal cat. Brain Res 115: 518-524. [DOI] [PubMed] [Google Scholar]
- Liu H, Mantyh PW, Basbaum AI (1997) NMDA-receptor regulation of substance P release from primary afferent nociceptors. Nature 386: 721-724. [DOI] [PubMed] [Google Scholar]
- Malcangio M, Fernandes K, Tomlinson DR (1998) NMDA receptor activation modulates evoked release of substance P from rat spinal cord. Br J Pharmacol 125: 1625-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Akil H, Watson SJ (1995) Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci 18: 22-29. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, DeMaster E, Malhotra A, Ghilardi JR, Rogers SD, Mantyh CR, Liu H, Basbaum AI, Vigna SR, Maggio JE, Simone DA (1995) Receptor endocytosis and dendrite reshaping in spinal neurons after somatosensory stimulation. Science 268: 1629-1632. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, Rogers SD, Honore P, Allen BJ, Ghilardi JR, Li J, Daughters RS, Lappi DA, Wiley RG, Simone DA (1997) Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science 278: 275-279. [DOI] [PubMed] [Google Scholar]
- Marvizon JC, Martinez V, Grady EF, Bunnett NW, Mayer EA (1997) Neurokinin 1 receptor internalization in spinal cord slices induced by dorsal root stimulation is mediated by NMDA receptors. J Neurosci 17: 8129-8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvizon JC, Grady EF, Waszak-McGee J, Mayer EA (1999a) Internalization of μ-opioid receptors in rat spinal cord slices. NeuroReport 10: 2329-2334. [DOI] [PubMed] [Google Scholar]
- Marvizon JC, Grady EF, Stefani E, Bunnett NW, Mayer EA (1999b) Substance P release in the dorsal horn assessed by receptor internalization: NMDA receptors counteract a tonic inhibition by GABAB receptors. Eur J Neurosci 11: 417-426. [DOI] [PubMed] [Google Scholar]
- Marvizon JC, Wang X, Lao LJ, Song B (2003a) Effect of peptidases on the ability of exogenous and endogenous neurokinins to produce neurokinin 1 receptor internalization in the rat spinal cord. Br J Pharmacol 140: 1389-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvizon JC, Wang X, Matsuka Y, Neubert JK, Spigelman I (2003b) Relationship between capsaicin-evoked substance P release and neurokinin 1 receptor internalization in the rat spinal cord. Neuroscience 118: 535-545. [DOI] [PubMed] [Google Scholar]
- McCarthy PW, Lawson SN (1989) Cell type and conduction velocity of rat primary sensory neurons with substance P-like immunoreactivity. Neuroscience 28: 745-753. [DOI] [PubMed] [Google Scholar]
- Moiniche S, Kehlet H, Dahl JB (2002) A qualitative and quantitative systematic review of preemptive analgesia for postoperative pain relief: the role of timing of analgesia. Anesthesiology 96: 725-741. [DOI] [PubMed] [Google Scholar]
- Pohl M, Lombard MC, Bourgoin S, Carayon A, Benoliel JJ, Mauborgne A, Besson JM, Hamon M, Cesselin F (1989) Opioid control of the in vitro release of calcitonin gene-related peptide from primary afferent fibres projecting in the rat cervical cord. Neuropeptides 14: 151-159. [DOI] [PubMed] [Google Scholar]
- Randic M, Jiang MC, Cerne R (1993) Long-term potentiation and long-term depression of primary afferent neurotransmission in the rat spinal cord. J Neurosci 13: 5228-5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley RC, Trafton JA, Chi SI, Basbaum AI (2001) Presynaptic regulation of spinal cord tachykinin signaling via GABA(B) but not GABAA receptor activation. Neuroscience 103: 725-737. [DOI] [PubMed] [Google Scholar]
- Saeki S, Yaksh TL (1993) Suppression of nociceptive responses by spinal μ opioid agonists: effects of stimulus intensity and agonist efficacy. Anesth Analg 77: 265-274. [DOI] [PubMed] [Google Scholar]
- Sandkuhler J, Chen JG, Cheng G, Randic M (1997) Low-frequency stimulation of afferent A δ-fibers induces long-term depression at primary afferent synapses with substantia gelatinosa neurons in the rat. J Neurosci 17: 6483-6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmauss C, Yaksh TL (1984) In vivo studies on spinal opiate receptor systems mediating antinociception. II. Pharmacological profiles suggesting a differential association of μ, δ and κ receptors with visceral chemical and cutaneous thermal stimuli in the rat. J Pharmacol Exp Ther 228: 1-12. [PubMed] [Google Scholar]
- Schroeder JE, Fischbach PS, Zheng D, McCleskey EW (1991) Activation of μ opioid receptors inhibits transient high- and low-threshold Ca2+ currents, but spares a sustained current. Neuron 6: 13-20. [DOI] [PubMed] [Google Scholar]
- Soldo BL, Moises HC (1998) μ-Opioid receptor activation inhibits N- and P-type Ca2+ channel currents in magnocellular neurones of the rat supraoptic nucleus. J Physiol (Lond) 513: 787-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Marvizon JC (2003) Dorsal horn neurons firing at high frequency, but not primary afferents, release opioid peptides that produce micro-opioid receptor internalization in the rat spinal cord. J Neurosci 23: 9171-9184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spike RC, Puskar Z, Sakamoto H, Stewart W, Watt C, Todd AJ (2002) MOR-1-immunoreactive neurons in the dorsal horn of the rat spinal cord: evidence for nonsynaptic innervation by substance P-containing primary afferents and for selective activation by noxious thermal stimuli. Eur J Neurosci 15: 1306-1316. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Morcuende S, Webber M, Hunt SP, Dickenson AH (2002) Superficial NK1-expressing neurons control spinal excitability through activation of descending pathways. Nat Neurosci 5: 1319-1326. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Yaksh TL (2002) The spinal phospholipase-cyclooxygenase-prostanoid cascade in nociceptive processing. Annu Rev Pharmacol Toxicol 42: 553-583. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Fresh-water JD, Catalano R, Feng Y, Protter AA, Scott B, Yaksh TL (2003) Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem 86: 1534-1544. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Spike RC (1993) The localization of classical transmitters and neuropeptides within neurons in laminae I-II of the mammalian spinal dorsal horn. Prog Neurobiol 41: 609-645. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Puskar Z, Spike RC, Hughes C, Watt C, Forrest L (2002) Projection neurons in lamina I of rat spinal cord with the neurokinin 1 receptor are selectively innervated by substance P-containing afferents and respond to noxious stimulation. J Neurosci 22: 4103-4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trafton JA, Basbaum AI (2000) The contribution of spinal cord neurokinin-1 receptor signaling to pain. Pain 1: 57-65. [DOI] [PubMed] [Google Scholar]
- Trafton JA, Abbadie C, Marchand S, Mantyh PW, Basbaum AI (1999) Spinal opioid analgesia: how critical is the regulation of substance P signaling? J Neurosci 19: 9642-9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trafton JA, Abbadie C, Marek K, Basbaum AI (2000) Postsynaptic signaling via the μ-opioid receptor: responses of dorsal horn neurons to exogenous opioids and noxious stimulation. J Neurosci 20: 8578-8584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trafton JA, Abbadie C, Basbaum AI (2001) Differential contribution of substance P and neurokinin A to spinal cord neurokinin-1 receptor signaling in the rat. J Neurosci 21: 3656-3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigouret J, Teschemacher H, Albus K, Herz A (1973) Differentiation between spinal and supraspinal sites of action of morphine when inhibiting the hindleg flexor reflex in rabbits. Neuropharmacology 12: 111-121. [DOI] [PubMed] [Google Scholar]
- Yaksh TL (1981) Spinal opiate analgesia: characteristics and principles of action. Pain 11: 293-346. [DOI] [PubMed] [Google Scholar]
- Yaksh TL (1997) Pharmacology and mechanisms of opioid analgesic activity. Acta Anaesthesiol Scand 41: 94-111. [DOI] [PubMed] [Google Scholar]
- Yaksh TL (1999) Spinal systems and pain processing: development of novel analgesic drugs with mechanistically defined models. Trends Pharmacol Sci 20: 329-337. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA (1976) Chronic catheterization of the spinal sub-arachnoid space. Physiol Behav 17: 1031-1036. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA (1977) Studies on the direct spinal action of narcotics in the production of analgesia in the rat. J Pharmacol Exp Ther 202: 411-428. [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA (1978) Narcotic analgesics: CNS sites and mechanisms of action as revealed by intracerebral injection techniques. Pain 4: 299-359. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Jessell TM, Gamse R, Mudge AW, Leeman SE (1980) Intrathecal morphine inhibits substance P release from mammalian spinal cord in vivo. Nature 286: 155-157. [DOI] [PubMed] [Google Scholar]
- Yeung JC, Rudy TA (1980) Multiplicative interaction between narcotic agonisms expressed at spinal and supraspinal sites of antinociceptive action as revealed by concurrent intrathecal and intracerebroventricular injections of morphine. J Pharmacol Exp Ther 215: 633-642. [PubMed] [Google Scholar]
- Yoshimura M, North RA (1983) Substantia gelatinosa neurones hyperpolarized in vitro by enkephalin. Nature 305: 529-530. [DOI] [PubMed] [Google Scholar]